

Abstract
Controlled mechanical ventilation (MV) is a life-saving measure for patients in respiratory failure. However, MV renders the diaphragm inactive leading to diaphragm weakness due to both atrophy and contractile dysfunction. It is now established that oxidative stress is a requirement for MV-induced diaphragmatic proteolysis, atrophy, and contractile dysfunction to occur. Given that endurance exercise can elevate diaphragmatic antioxidant capacity and the levels of the cellular stress protein heat shock protein 72 (HSP72), we hypothesized that endurance exercise training before MV would protect the diaphragm against MV-induced oxidative stress, atrophy, and contractile dysfunction in female Sprague-Dawley rats. Our results confirm that endurance exercise training before MV increased both HSP72 and the antioxidant capacity in the diaphragm. Importantly, compared with sedentary animals, exercise training before MV protected the diaphragm against MV-induced oxidative damage, protease activation, myofiber atrophy, and contractile dysfunction. Further, exercise protected diaphragm mitochondria against MV-induced oxidative damage and uncoupling of oxidative phosphorylation. These results provide the first evidence that exercise can provide protection against MV-induced diaphragm weakness. These findings are important and establish the need for future experiments to determine the mechanism(s) responsible for exercise-induced diaphragm protection.
Keywords: proteolysis, antioxidants, muscle atrophy, respiratory muscle weakness
mechanical ventilation (MV) is used clinically to achieve adequate pulmonary gas exchange in patients who are incapable of maintaining sufficient alveolar ventilation. Although MV is a life-saving intervention, ∼20–30% of MV patients experience weaning difficulties (7). Failure to wean patients from MV is associated with increased hospital stays along with higher patient morbidity and mortality. Although the failure to wean patients may be due to several factors, MV-induced respiratory muscle weakness is predicted to play an important role (17). In this regard, numerous studies demonstrate that prolonged MV promotes the rapid development of both diaphragm atrophy and contractile dysfunction [collectively referred to as ventilator-induced diaphragm dysfunction (VIDD)] in both humans and animals (10, 18, 35, 38, 41). Therefore, because VIDD is linked to weaning problems, developing a strategy to protect the diaphragm against VIDD is important.
In regard to the signaling pathways responsible for VIDD, growing evidence indicates that MV-induced increased production of reactive oxygen species (ROS) in the diaphragm is a required upstream trigger that initiates signaling events (e.g., protease activation) leading to the rapid development of VIDD (3, 23, 32, 49, 50). Efforts to identify the specific cellular location of MV-induced ROS generation in diaphragm muscle fibers reveal that xanthine oxidase, NADPH oxidase, and the mitochondria are all contributors to ROS production during prolonged MV (16, 24, 32, 36, 48). Therefore, in theory, improving diaphragm antioxidant defenses and/or increasing diaphragm levels of cytoprotective stress proteins could provide protection against MV-induced weakness. Specifically, it was shown clinically that use of an antioxidant supplement containing vitamins E and C can reduce the duration of MV compared with nonsupplemented patients (28, 29). In addition, other strategies have also been used clinically in an attempt to alleviate weaning problems. For example, inspiratory muscle strength training has also been used as a method to combat weaning difficulties (22).
In reference to a protective strategy to shield against VIDD, endurance exercise training has been shown to protect both locomotor and respiratory (e.g., diaphragm) skeletal muscles against a variety of stressors including oxidative stress (30, 31, 44, 46, 47). Indeed, exercise training increases the expression of numerous cytoprotective proteins in muscle including antioxidant enzymes and stress proteins. For example, endurance exercise training increases diaphragmatic antioxidant capacity through the upregulation of antioxidant enzymes [e.g., superoxide dismutase (SOD) and glutathione peroxidase (GPX); Refs. 30, 46, 47]. Also, endurance exercise training elevates the expression of heat shock protein 72 (HSP72) in skeletal muscle (26, 44). This is significant because overexpression of HSP72 can protect locomotor skeletal muscles against disuse muscle atrophy (27, 40). Although exercise training has been shown to provide protection during contraction-induced oxidative damage and against doxorubicin-induced muscle damage (44, 46), it is unknown whether exercise will protect the diaphragm against VIDD. To address this issue, we tested the hypothesis that endurance exercise training will protect the diaphragm against MV-induced oxidative damage, protease activation, myofiber atrophy, and contractile dysfunction. Since exercise training has been show to induce the expression of numerous antioxidant and stress proteins, we used this intervention as a tool to identify cytoprotective proteins that could be used in the future as therapeutic targets to protect against VIDD.
METHODS
Animals
Young adult (∼4–6 mo old) female Sprague-Dawley rats were used in these experiments. Animals were maintained on a 12:12-h light-dark cycle and provided food and water ad libitum throughout the experimental period. The Institutional Animal Care and Use Committee of the University of Florida approved these experiments.
Experimental Design
To test the hypothesis that exercise training can provide a protective effect against MV-induced diaphragm weakness, rats were randomly assigned to one of three experimental groups (n = 8/group): 1) acutely anesthetized sedentary control (CON), 2) sedentary control with 12 h of controlled MV, and 3) endurance exercise trained with 12 h of controlled MV (MV-EX).
Experimental Protocol
Acutely anesthetized controls.
Animals in the control group were acutely anesthetized with an intraperitoneal injection of sodium pentobarbital (60 mg/kg body wt). After reaching a surgical plane of anesthesia, the diaphragms were quickly removed and the costal diaphragm was divided into several segments. A strip of the medial costal diaphragm was immediately used for in vitro contractile measurements, a separate section was stored for histological measurements, and the remaining portions of the costal diaphragm were rapidly frozen in liquid nitrogen and stored at −80°C for subsequent biochemical analyses.
MV.
Numerous modes of MV (e.g., controlled MV and pressure-support MV) are used clinically. In pressure support modes, the patient's inspiratory effort is assisted by the ventilator and in controlled MV the entire breath is delivered by the ventilator. Although diaphragm dysfunction has been shown to occur in all modes of MV, it has been reported that greater weaning problems occur in patients whose respiratory muscles are completely unloaded via controlled MV (9, 39). Therefore, we focused only on the effects of controlled MV in the current experiment.
All surgical procedures were performed using aseptic techniques. Animals in the MV groups were anesthetized with an intraperitoneal injection of sodium pentobarbital (60 mg/kg body wt), tracheostomized, and mechanically ventilated with a pressure-controlled ventilator (Servo Ventilator 300; Siemens, Munich, Germany) for 12 h with the following settings: upper airway pressure limit: 20 cmH2O; respiratory rate: 80 beats/min; and positive end-expiratorypressure: 1 cmH2O. Animals were typically ventilated at upper airway pressures of 4–8 cmH2O above positive end-expiratory pressure, which was sufficient to maintain blood gas homeostasis.
The carotid artery was cannulated to permit the continuous measurement of blood pressure and the collection of blood during the protocol. Arterial blood samples (100 μl/sample) were removed periodically and analyzed for arterial Po2, Pco2, and pH using an electronic blood-gas analyzer (GEM Premier 3000; Instrumentation Laboratory, Lexington, MA). Ventilator adjustments were made if arterial Pco2 exceeded 40 mmHg. Moreover, arterial Po2 was maintained >60 mmHg throughout the experiment by increasing the FiO2 (22–26% oxygen).
A venous catheter was inserted into the jugular vein for continuous infusion of pentobarbital sodium (∼10 mg·kg−1·h−1). Body temperature was maintained at 37°C by use of a recirculating heating blanket, and heart rate was monitored via a lead II electrocardiograph. Continuous care during the MV protocol included lubricating the eyes, expressing the bladder, removing airway mucus, rotating the animal, and passively moving the limbs. Animals also received an intramuscular injection of glycopyrrolate (0.04 mg/kg) every 2 h during MV to reduce airway secretions. Upon completion of MV, the diaphragm was quickly removed and processed as described above.
Exercise training protocol.
Animals assigned to the exercise-trained group were habituated to treadmill exercise for 5 consecutive days. After 2 days of rest, the endurance trained animals then performed 10 days of treadmill exercise for 60 min/day at 30 m/min, 0% grade (estimated work rate of 70% maximum O2 consumption) with 2 days of rest in the middle (4). This protocol was chosen based on previous work from our laboratory as well as preliminary experiments, indicating that this exercise intensity/duration leads to diaphragmatic adaptations in both antioxidants and heat shock proteins (4). The animals were mechanically ventilated ∼24 h after their final exercise bout.
Mitochondrial Measures
Isolation of mitochondria.
Approximately 500 mg of costal diaphragm muscle were used to isolate diaphragmatic mitochondria using the methods of Makinen and Lee (21) with minor modifications (16).
Mitochondrial respiration.
Mitochondrial oxygen consumption was measured using previously described techniques (16). The maximal respiration (state 3) and basal respiration (state 4) were measured as described previously (8). The respiratory control ratio (RCR) was calculated by dividing state 3 by state 4 respiration.
Mitochondrial ROS emission.
Diaphragmatic mitochondrial ROS emission was determined using Amplex Red (Molecular Probes, Eugene, OR). Details of this assay have been described previously (16). Mitochondrial ROS production was measured using the creatine kinase energy clamp technique to maintain respiration at steady state using previously described methods (25).
Biochemical Measures
Western blot analysis.
Protein abundance was determined in whole diaphragm samples and isolated diaphragm mitochondria via Western blot analysis. Briefly, diaphragm tissue samples were homogenized 1:10 (wt/vol) in 5 mM Tris (pH 7.5) and 5 mM EDTA (pH 8.0) with a protease inhibitor cocktail (Sigma, St. Louis, MO) and centrifuged at 1,500 g for 10 min at 4°C. After the resulting supernatant was collected, diaphragm protein content was assessed by the method of Bradford (Sigma). Proteins from the supernatant fraction of the diaphragm homogenates and isolated diaphragm mitochondria were separated via polyacrylamide gel electrophoresis via 4–20% gradient polyacrylamide gels containing 0.1% SDS for ∼1 h at 200 V. After electrophoresis, the proteins were transferred to nitrocellulose membranes and incubated with primary antibodies directed against proteins of interest. 4-Hydroxynoneal (4-HNE; Abcam, Cambridge, MA) was probed as a biomarker of oxidative stress. In addition, HSP72 (Abcam), levels were obtained. Copper-zinc SOD1 (Santa Cruz Biotechnology, Santa Cruz, CA), manganese SOD2 (Santa Cruz), GPX1 (Abcam), and catalase (Abcam) were measured to assess changes in antioxidant potential in both whole diaphragm homogenate as well as in isolated diaphragm mitochondria. Proteolytic activity was assessed by analyzing cleaved (active) calpain-1 (Cell Signaling Technology, Carlsbad, CA), cleaved caspase-3 (Cell Signaling), and α-II spectrin (Santa Cruz). Finally, α-tubulin (Santa Cruz) was probed, which served as a loading control to normalize equal protein loading and transfer. Following incubation with primary antibodies, membranes were washed extensively with PBS-Tween and then incubated with secondary antibodies (GE Healthcare, Piscataway, NJ). After being washed, a chemiluminescent system was used to detect labeled proteins (GE Healthcare). Membranes were developed using autoradiography film, and images of the film were captured and analyzed using the 440CF Kodak Imaging System (Kodak, New Haven, CT).
RNA isolation and cDNA synthesis.
Total RNA was isolated from muscle tissue with TRIzol reagent (Life Technologies, Carlsbad, CA) according to the manufacturer's instructions. Total RNA and RNA contents (μg/mg muscle) were evaluated by spectrophotometry. Total RNA (5 μg) was then reverse transcribed with the Superscript III first-strand synthesis system for RT-PCR (Life Technologies), using oligo(dT)20 primers and the protocol outlined by the manufacturer.
Real-time PCR.
One microliter of cDNA was added to a 25 μl PCR reaction for real-time PCR using Taqman chemistry and the ABI Prism 7000 Sequence Detection system (ABI, Foster City, CA) as previously described (24). β-Glucuronidase, a lysosomal glycoside hydrolase, was chosen as the reference gene for diaphragm muscle samples based on previous work (5, 6, 45) showing unchanged expression with our experimental manipulations. HSP72, atrogin-1, MuRF-1, and proliferator-activated receptor-coactivator-1α (PGC-1α) mRNA transcripts were assayed using predesigned rat primer and probe sequences commercially available from Applied Biosystems (Assays-on-Demand).
Functional Measures
Measurement of in vitro diaphragmatic contractile properties.
Upon death, a muscle strip, including the tendinous attachments at the central tendon and rib cage, was dissected from the midcostal region. The strip was suspended vertically with one end connected to an isometric force transducer (model FT-03, Grass Instruments, Quincy, MA) within a jacketed tissue bath and diaphragm skeletal, and muscle contractile properties were measured (35).
Histological Measures
Myofiber cross-sectional area.
Sections from frozen diaphragm samples were cut at 10 μm using a cryotome (Shandon, Pittsburgh, PA) and stained for dystrophin, myosin heavy chain I, and myosin heavy chain type IIa proteins for fiber cross-sectional area analysis as described previously (23). Cross-sectional area was determined using Scion software (National Institutes of Health).
Statistical Analysis
Comparisons between groups for each dependent variable were made by a one-way ANOVA and, when appropriate, a Tukey's honestly significant difference test was performed post hoc. Significance was established at P < 0.05. Data are presented as means ± SE.
RESULTS
Exercise Training and Body Weights
All animals in the exercise group successfully completed the 10-day exercise training protocol. No mean differences existed in animal body weights between the experimental groups.
Systemic and Biologic Response to MV
Before the initiation of MV, no significant differences existed in body weight between the groups (CON = 299 ± 4 g, MV = 293 ± 5 g, and MV-EX = 286 ± 7 g). Importantly, 12 h of MV and exercise training did not significantly alter body weight of the MV or MV-EX group (P < 0.05). Our results also indicate that arterial blood pressure (MV = 105 ± 6 and MV-EX = 108 ± 8 mm/Hg), PaO2 (MV = 74 ± 5 and MV-EX = 82 ± 4 mm/Hg), PaCO2 (MV = 39 ± 2 and MV-EX = 39 ± 2 mm/Hg), and pH (MV = 7.42 ± 0.01 and MV-EX = 7.41 ± 0.01) were not significantly different (P > 0.05) between the ventilated groups. Furthermore, at the completion of 12 h of MV, there were no visual abnormalities of the lungs or peritoneal cavity, no visible indication of major lung injury, and no evidence of infection, indicating that our aseptic surgical technique was successful.
Mitochondrial Respiration
Our data indicate that 12 h of MV results in a significant reduction of the RCR in mitochondria isolated from diaphragms of MV animals. Specifically, this RCR decline is due to a significant increase in state 4 respiration in MV animals. Importantly, our results reveal that endurance exercise training before MV is sufficient to attenuate this decrease in the RCR (Table 1).
Table 1.
Mitochondrial respiratory function in CON, MV, and MV-EX animals
| CON | MV | MV-EX | |
|---|---|---|---|
| State 3 respiration, nmol O2 · mg−1 · min−1 | 234.3 ± 13.5 | 212.4 ± 7.5 | 192 ± 2.1* |
| State 4 respiration, nmol O2 · mg−1 · min−1 | 43.4 ± 1.3 | 61.8 ± 5.6† | 38.5 ± 3.1 |
| RCR | 5.4 ± 0.33 | 3.6 ± 0.33† | 5.1 ± 0.32 |
Values are means ± SE. These data were obtained using pyruvate/malate as substrate. CON, control; MV, mechanical ventilation; MV-EX, mechanical ventilation and exercise trained; RCR, respiratory control ratio.
P < 0.05, MV-EX significantly different from CON.
P < 0.05, MV significantly different than CON and MV-EX.
MV-Induced Oxidative Stress in the Diaphragm
Our findings indicate that endurance exercise training prevented the MV-induced increase in diaphragmatic mitochondrial H2O2 release (Fig. 1). Further, to determine if exercise training can protect the diaphragm against MV-induced oxidative damage, we measured 4-HNE as a biomarker of lipid peroxidation. Specifically, 4-HNE is formed during the oxidant-triggered lipid peroxidation cascade and 4-HNE/protein adducts are excellent biomarkers of lipid peroxidation in cells. Our results reveal that 12 h of MV results in a significant increase in 4-HNE-modified proteins. In addition, compared with untrained animals, our data also indicate that endurance exercise training before MV results in a significant reduction of MV-induced 4-HNE/protein adducts in the diaphragm.
Fig. 1.
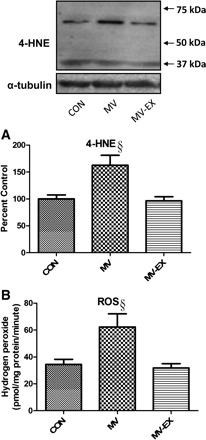
A: levels of 4-hydroxynonenal (4-HNE) were analyzed as an indicator of lipid peroxidation via Western blotting. A representative blot for 4-HNE protein conjugates is shown at top. Values are mean percent change ± SE and normalized to α-tubulin. §P < 0.05, mechanical ventilation (MV) significantly different from control (CON) and MV-exercise (EX). B: rates of hydrogen peroxide (H2O2) release from isolated mitochondria of diaphragm muscle. Values are means ± SE. §P < 0.05, MV significantly different from CON and MV-EX.
Antioxidant Enzyme Levels in Diaphragm Muscle
Antioxidant enzymes serve as a protective barrier against ROS-induced oxidative damage in cells. Therefore, we measured the protein abundance of key antioxidant enzymes in both whole diaphragm homogenate and isolated mitochondria. Compared with diaphragms from both control animals and sedentary animals exposed to MV, the protein levels of SOD1, SOD2, GPX1, and catalase were significantly increased in diaphragm homogenates from exercised animals exposed to MV (Fig. 2). Note that during the process of tissue homogenization, mitochondrial membranes are disrupted the protein contents are released. Therefore, the increase in the protein abundance of SOD2 from diaphragm homogenates of trained animals is likely due to the training-induced increase in mitochondrial levels of SOD2. Indeed, protein expression of both GPX1 and SOD2 was significantly increased in mitochondria isolated from diaphragms of exercised animals compared with both control and sedentary animals exposed to MV (Fig. 3). These results indicate that exercise training increases mitochondrial capacity to remove both superoxide and hydrogen peroxide.
Fig. 2.
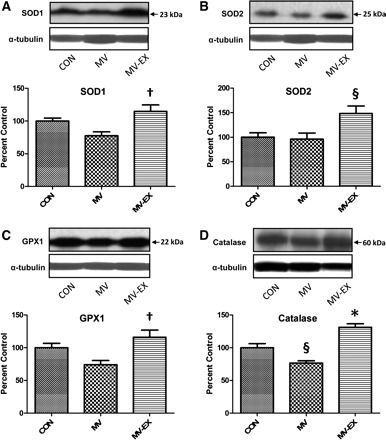
Protein abundance of primary antioxidants in homogenates from diaphragm muscle. Representative Western blots for the antioxidants analyzed are shown at top. Values are mean percent change ± SE and normalized to α-tubulin. A: protein levels of SOD1 (CuZnSOD). †P < 0.05, MV-EX significantly greater than MV. B: protein levels of SOD2 (MnSOD). §P < 0.05, MV-EX significantly greater than MV and CON. C: protein levels of GPX 1. †P < 0.05, MV-EX significantly greater than MV. D: protein levels of catalase. §P < 0.05, MV significantly different from MV-EX and CON. *P < 0.05, MV-EX significantly greater than CON.
Fig. 3.
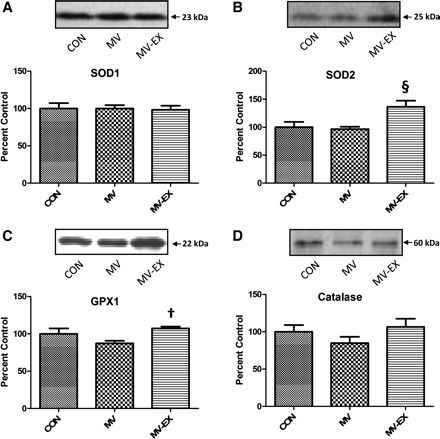
Protein levels of primary antioxidants in isolated mitochondria. Representative Western blots for the antioxidants analyzed are shown at top. Values are mean percent change ± SE. A: protein levels of SOD1 (CuZnSOD). B: protein levels of SOD2 (MnSOD). §P < 0.05, MV-EX significantly greater than MV and CON. C: protein levels of GPX 1. †P < 0.05, MV-EX significantly greater than MV. D: protein levels of catalase.
HSP72 Expression in Diaphragm Muscle
Compared with control and sedentary animals exposed to MV, exercise training significantly increased diaphragm levels of both HSP72 mRNA and protein abundance (Fig. 4).
Fig. 4.
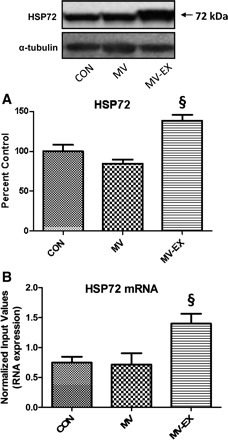
Assessment of heat shock protein 72. A: HSP72 protein levels. Values are mean percent change ± SE and normalized to α-tubulin. A representative Western blot is shown at top. §P < 0.05, MV-EX significantly greater than MV and CON. B: HSP72 mRNA expression. §P < 0.05, MV-EX significantly greater than MV and CON.
Proteolytic Activity
To determine the effect of exercise on several proteolytic systems, we measured the activity of calpain and caspase-3, as well as the change in expression of key E3 ubiquitin-ligases. Compared with diaphragms from control animals, MV resulted in significant increases in both calpain 1 and caspase-3 activity. In contrast, endurance exercise training before MV caused a significant attenuation in the activation of both these important proteases (Fig. 5). In addition, atrogin-1 and MuRF-1 are two muscle-specific E3 ligases that play an important role in protein degradation by the ubiquitin-proteasome system. Our results show that the mRNA levels of both of these proteins were increased in the diaphragm as a result of MV, and that endurance exercise training before MV attenuated the expression of atrogin-1 and MuRF-1 (Fig. 6, A and B).
Fig. 5.
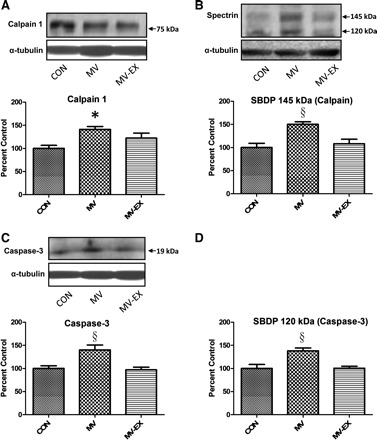
Calpain and caspase-3 activation in diaphragm was determined via Western blotting. Representative Western blots are shown at top. Values are mean percent change ± SE and normalized to α-tubulin. Assessment of calpain activity via assessment of active calpain 1 (A) and the calpain specific spectrin breakdown product (SBDP; B). *P < 0.05, MV significantly greater than CON and §MV significantly greater than MV-EX and CON. Assessment of caspase-3 activity via cleaved caspase-3 (C) and the caspase-3 SBDP (D). §P < 0.05, MV significantly greater than MV-EX and CON.
Fig. 6.
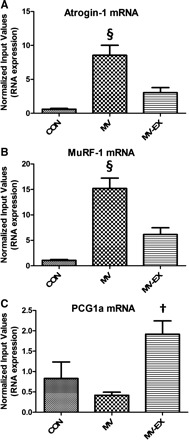
Values are fold difference ± SE. A: atrogin-1 mRNA expression. §P < 0.05, MV significantly different from MV-EX and CON. B: MuRF-1 mRNA expression. §P < 0.05, MV significantly different from MV-EX and CON. C: proliferator-activated receptor-coactivator-1α (PGC-1α) mRNA expression. †P < 0.05, MV-EX significantly greater than MV.
PGC-1α Expression in Diaphragm Muscle
In addition, endurance exercise training can result in an increase in both PGC-1α and mitochondrial content in skeletal muscle. In this regard, PGC-1α is an important regulator of mitochondria biogenesis and our data reveal that endurance training before MV resulted in an increase in the mRNA expression of PGC-1α compared with MV (Fig. 6C).
MV-Induced Diaphragm Contractile Dysfunction
To determine if exercise training can protect the diaphragm against MV-induced contractile dysfunction, we measured both the in vitro maximal isometric twitch force and force-frequency responses of strips of diaphragm muscle. Compared with control, 12 h of MV results in a significant reduction in diaphragm muscle force production at all stimulation frequencies. However, endurance exercise training before MV resulted in significant protection against MV-induced contractile dysfunction at all stimulation frequencies >15 Hz (Fig. 7).
Fig. 7.
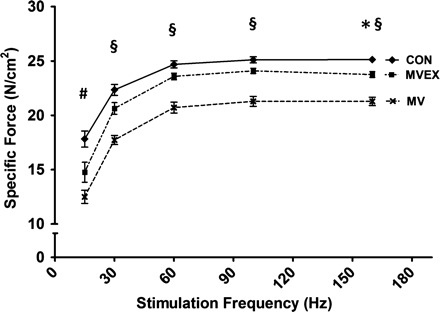
Diaphragmatic force-frequency response (in vitro). Values are means ± SE. §P < 0.05, MV significantly different from MV-EX and CON. *P < 0.05, CON significantly greater than MV-EX. #P < 0.05, CON significantly greater than MV-EX and MV.
MV-Induced Diaphragm Atrophy
Myofiber cross-sectional area was determined for individual fiber types in diaphragm muscle strips from all experimental groups. Prolonged MV resulted in significant atrophy of type I, type IIa, and type IIb/IIx diaphragm myofibers. Endurance exercise training before MV protected against MV-induced atrophy in all diaphragm muscle fiber types (Fig. 8).
Fig. 8.
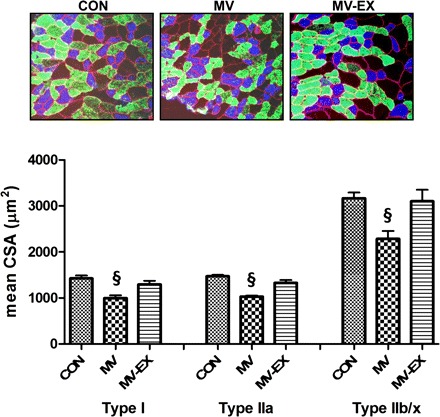
Fiber cross-sectional area (CSA) in diaphragm muscle myofibers expression myosin heavy chain (MHC) I (type I), MHC IIa (type IIa), and MHC IIb/IIx (type IIb/IIx). Representative fluorescent staining of MHC I (DAPI filter/blue), MHC IIa (RITC filter/green), and dystrophin (rhodamine filter/red) proteins in diaphragm samples are shown at top. Values are means ±SE. §P < 0.05, MV significantly different from MV-EX and CON.
DISCUSSION
Overview of Principle Findings
These experiments support the hypothesis that endurance exercise training performed before MV protects the diaphragm from MV-induced mitochondrial oxidative damage/dysfunction, protease activation, fiber atrophy, and contractile dysfunction. This exercise-induced protection against VIDD was associated with an increase in diaphragm levels of both antioxidant enzymes and HSP72, both of which could serve as possible therapeutic targets to minimize VIDD. A detailed discussion of these novel findings follows.
Endurance Exercise Training Protects the Diaphragm Against MV-Induced Mitochondrial Dysfunction and Oxidative Damage
We have established that mitochondria are an important source of ROS production in the diaphragm during MV and that prolonged MV is associated with both mitochondrial damage and respiratory dysfunction (16, 32). Because endurance exercise training alters the mitochondrial phenotype in cardiac muscle to protect against damaging stimuli (13–15), we hypothesized that exercise would protect diaphragm mitochondria from oxidative damage and dysfunction, and our results support this prediction. For example, compared with control animals, mitochondria isolated from diaphragms of the MV group exhibited impaired respiratory function (e.g., decreased RCR) and oxidative damage (Fig. 1 and Table 1). In contrast, endurance exercise training performed before MV protected diaphragm mitochondria against MV-induced oxidative damage and respiratory dysfunction. Also, exercise training abated the MV-induced increase in diaphragm mitochondrial ROS emission (Fig. 1). During periods of skeletal muscle inactivity, a calcium overload can occur in the mitochondria leading to increase ROS emission (12). It is possible that exercise training prevented this inactivity induced calcium accumulation in diaphragmatic mitochondria and thus attenuated ROS emission.
Another potential mechanism to explain the protective effects of exercise training on the diaphragm is the exercise-induced increase in the antioxidant capacity of diaphragm muscle fibers (30, 37, 44, 47). In this regard, SOD, GPX, and catalase are antioxidant enzymes that form a network to scavenge cellular ROS and all three enzymes were increased in diaphragm muscle fibers with exercise training (33). Two isoforms of SOD exist in mammalian cells; SOD1 (CuZnSOD) is located in both the cytosol and mitochondrial intermembrane space whereas SOD2 (MnSOD) is located exclusively in the mitochondrial matrix. Both isoforms defend against ROS by dismutating the superoxide radical to form hydrogen peroxide and oxygen. Additionally, GPX 1 is located in both the mitochondria and cytosol of muscle fibers and GPX1 promotes the reduction of hydrogen peroxide to water and oxygen (20). Catalase is also distributed throughout the cytosol and catalyzes the breakdown of hydrogen peroxide to water and oxygen. Our data reveal that exercise training increased the protein abundance of SOD1, GPX1, and catalase in the cytosol of diaphragm fibers while only SOD2 and GPX1 were increased in the diaphragm mitochondria (Figs. 2 and 3). Therefore, it appears likely that the exercise-induced increases in these antioxidant enzymes contributed to the protection against MV-induced increases in mitochondrial ROS emission and oxidative damage.
Endurance Exercise Training Protects Against MV-Induced Proteolysis and Atrophy
It is well established that prolonged MV results in a rapid activation of proteases and atrophy in diaphragm muscle fibers (23, 42, 49). Importantly, the current experiments demonstrate that endurance exercise training can impede MV-induced protease activation and atrophy in the diaphragm. Previous work from our group (3, 32, 49) has demonstrated that MV-induced oxidative stress is a requirement for both protease activation and diaphragm fiber atrophy. Hence, it appears likely that the exercise-mediated improvements in diaphragm antioxidant capacity contributed to the exercise-associated protection against MV-induced protease activation. In addition to protection against protease activation, exercise can also contribute to the defense against MV-induced diaphragm atrophy by preventing oxidation of diaphragm muscle proteins. Indeed, it is well established that oxidatively modified proteins are more susceptible to proteolytic degradation (2, 11, 43).
Along with the exercise-induced increases in diaphragmatic antioxidant capacity, endurance exercise training also increased the abundance of a key stress protein, HSP72. Increased diaphragm levels of HSP72 can contribute to the exercise-induced protection against MV-induced proteolysis and atrophy in several ways. For example, HSP72 can maintain mitochondrial integrity by protecting mitochondria against apoptotic stimuli and contribute to the repair of damaged proteins (1, 34). Further, HSP72 can prevent proteolysis by binding to oxidized proteins and assisting in their refolding (19, 27). Evidence also indicates that overexpression of HSP72 in limb muscles can prevent disuse atrophy, in part, by inhibiting the activity of two transcriptional activators, FOXO3a and NF-κB, that participate in the atrophic process (40). Specifically, inhibition of FOXO3a activity can prevent the expression of two muscle-specific E3 ligases involved in the ubiquitin-proteasome system (atrogin-1 and MuRF-1). Further, NF-κB has been shown to be required for disuse muscle atrophy and activation of NF-κB is sufficient to increase MuRF-1 expression in muscle (40). Therefore, in theory, the exercise-induced increase in diaphragmatic levels of HSP72 could contribute, at least in part, to the protection against MV-induced atrophy. Nonetheless, it is unclear if the significant but relatively small exercise-induced increases in diaphragmatic levels of HSP72 are adequate to contribute to the observed protection against MV-induced diaphragm atrophy.
Endurance Exercise Training Protects Against MV-Induced Contractile Dysfunction
Mechanical ventilation-induced diaphragm weakness occurs due to both diaphragm atrophy and contractile dysfunction. Indeed, it is well-known that prolonged MV results in a progressive impairment in diaphragm specific force production at all stimulation frequencies (3, 35). Importantly, our results reveal that endurance training before MV protected the diaphragm against MV-induced contractile dysfunction at all stimulation frequencies >15 Hz. The molecular mechanism(s) responsible for MV-induced contractile dysfunction in the diaphragm remain unknown. Nonetheless, it is clear that increased ROS production is a required upstream signal to promote MV-induced contractile dysfunction. Therefore, it seems likely that exercise-induced protection against contractile dysfunction in the diaphragm is due, in part, to protection against MV-induced oxidative damage in the diaphragm. As discussed previously, exercise training increased the levels of antioxidant enzymes in the diaphragm and protected against the MV-induced increases in mitochondrial ROS production. Collectively, these changes could play an important role in the observed protection against MV-induced diaphragm dysfunction.
Conclusions
In summary, the investigation provides the first evidence that exercise training is an effective intervention against MV-induced contractile dysfunction and atrophy in the diaphragm. Specifically, our findings reveal that endurance exercise training results in a decrease in MV-induced mitochondrial ROS emission and respiratory dysfunction along with decreased activation of key proteases in the diaphragm. Our results also disclose that exercise training promotes increased diaphragm levels of both antioxidant enzymes and HSP72. However, while endurance exercise training before MV protected the diaphragm against MV-induced weakness, the circumstances preceding most patients' need for MV do not allow for such an intervention. Nonetheless, our results suggest that the antioxidant pathways and/or HSP72 signaling are potential therapeutic targets to protect against VIDD. Collectively, our experiments establish the platform for future experiments directed toward the discovery of the mechanism(s) responsible for exercise-induced diaphragm protection. Indeed, we predict that exercise can be used as a novel experimental tool to identify molecular targets that can be manipulated pharmacologically resulting in new approaches for the prevention of VIDD. Finally, while exercise training before MV may be an unlikely clinical strategy to combat weaning difficulties, the results of the current study suggest that patient physical fitness levels has the potential to be a predictor of weaning outcome. This is a testable hypothesis worthy of further research.
DISCLOSURES
Conflict of interest statement: no conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: A.J.S. and S.K.P. conception and design of research; A.J.S., K.M., M.B.H., A.N.K., O.-s.K., and W.B.N. performed experiments; A.J.S., K.M., and A.N.K. analyzed data; A.J.S. interpreted results of experiments; A.J.S. prepared figures; A.J.S. drafted manuscript; A.J.S., K.M., M.B.H., A.N.K., O.-s.K., W.B.N., and S.K.P. edited and revised manuscript; A.J.S., K.M., M.B.H., A.N.K., O.-s.K., W.B.N., and S.K.P. approved final version of manuscript.
REFERENCES
- 1. Ascensao A, Magalhaes J, Soares JM, Ferreira R, Neuparth MJ, Marques F, Oliveira PJ, Duarte JA. Moderate endurance training prevents doxorubicin-induced in vivo mitochondriopathy and reduces the development of cardiac apoptosis. Am J Physiol Heart Circ Physiol 289: H722–H731, 2005. [DOI] [PubMed] [Google Scholar]
- 2. Bader N, Grune T. Protein oxidation and proteolysis. Biol Chem 387: 1351–1355, 2006. [DOI] [PubMed] [Google Scholar]
- 3. Betters JL, Criswell DS, Shanely RA, Van Gammeren D, Falk D, Deruisseau KC, Deering M, Yimlamai T, Powers SK. Trolox attenuates mechanical ventilation-induced diaphragmatic dysfunction and proteolysis. Am J Respir Crit Care Med 170: 1179–1184, 2004. [DOI] [PubMed] [Google Scholar]
- 4. Criswell D, Powers S, Dodd S, Lawler J, Edwards W, Renshler K, Grinton S. High intensity training-induced changes in skeletal muscle antioxidant enzyme activity. Med Sci Sports Exerc 25: 1135–1140, 1993. [PubMed] [Google Scholar]
- 5. Deruisseau KC, Kavazis AN, Powers SK. Selective downregulation of ubiquitin conjugation cascade mRNA occurs in the senescent rat soleus muscle. Exp Gerontol 40: 526–531, 2005. [DOI] [PubMed] [Google Scholar]
- 6. DeRuisseau KC, Shanely RA, Akunuri N, Hamilton MT, Van Gammeren D, Zergeroglu AM, McKenzie M, Powers SK. Diaphragm unloading via controlled mechanical ventilation alters the gene expression profile. Am J Respir Crit Care Med 172: 1267–1275, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Esteban A, Frutos F, Tobin MJ, Alia I, Solsona JF, Valverdu I, Fernandez R, de la Cal MA, Benito S, Tomas R, Carriedo D, Macias S, Blanco J. A comparison of four methods of weaning patients from mechanical ventilation Spanish Lung Failure Collaborative Group. N Engl J Med 332: 345–350, 1995. [DOI] [PubMed] [Google Scholar]
- 8. Estrabrook R. Mitochondrial respiratory control and the polarographic measurement of ADP/O ratios. Methods Enzymol 10: 41–47, 1967. [Google Scholar]
- 9. Futier E, Constantin JM, Combaret L, Mosoni L, Roszyk L, Sapin V, Attaix D, Jung B, Jaber S, Bazin JE. Pressure support ventilation attenuates ventilator-induced protein modifications in the diaphragm. Crit Care 12: R116, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gayan-Ramirez G, Testelmans D, Maes K, Racz GZ, Cadot P, Zador E, Wuytack F, Decramer M. Intermittent spontaneous breathing protects the rat diaphragm from mechanical ventilation effects. Crit Care Med 33: 2804–2809, 2005. [DOI] [PubMed] [Google Scholar]
- 11. Grune T, Merker K, Sandig G, Davies KJ. Selective degradation of oxidatively modified protein substrates by the proteasome. Biochem Biophys Res Commun 305: 709–718, 2003. [DOI] [PubMed] [Google Scholar]
- 12. Kandarian SC, Stevenson EJ. Molecular events in skeletal muscle during disuse atrophy. Exerc Sport Sci Rev 30: 111–116, 2002. [DOI] [PubMed] [Google Scholar]
- 13. Kavazis AN, Alvarez S, Talbert E, Lee Y, Powers SK. Exercise training induces a cardioprotective phenotype and alterations in cardiac subsarcolemmal and intermyofibrillar mitochondrial proteins. Am J Physiol Heart Circ Physiol 297: H144–H152, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kavazis AN, McClung JM, Hood DA, Powers SK. Exercise induces a cardiac mitochondrial phenotype that resists apoptotic stimuli. Am J Physiol Heart Circ Physiol 294: H928–H935, 2008. [DOI] [PubMed] [Google Scholar]
- 15. Kavazis AN, Smuder AJ, Min K, Tumer N, Powers SK. Short-term exercise training protects against doxorubicin-induced cardiac mitochondrial damage independent of HSP72. Am J Physiol Heart Circ Physiol 299: H1515–H1524, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kavazis AN, Talbert EE, Smuder AJ, Hudson MB, Nelson WB, Powers SK. Mechanical ventilation induces diaphragmatic mitochondrial dysfunction and increased oxidant production. Free Radic Biol Med 46: 842–850, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Laghi F, Cattapan SE, Jubran A, Parthasarathy S, Warshawsky P, Choi YS, Tobin MJ. Is weaning failure caused by low-frequency fatigue of the diaphragm? Am J Respir Crit Care Med 167: 120–127, 2003. [DOI] [PubMed] [Google Scholar]
- 18. Levine S, Nguyen T, Taylor N, Friscia ME, Budak MT, Rothenberg P, Zhu J, Sachdeva R, Sonnad S, Kaiser LR, Rubinstein NA, Powers SK, Shrager JB. Rapid disuse atrophy of diaphragm fibers in mechanically ventilated humans. N Engl J Med 358: 1327–1335, 2008. [DOI] [PubMed] [Google Scholar]
- 19. Locke M, Tanguay RM, Klabunde RE, Ianuzzo CD. Enhanced postischemic myocardial recovery following exercise induction of HSP 72. Am J Physiol Heart Circ Physiol 269: H320–H325, 1995. [DOI] [PubMed] [Google Scholar]
- 20. Lubos E, Loscalzo J, Handy DE. Glutathione peroxidase-1 in health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal 15: 1957–1997, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Makinen MW, Lee CP. Biochemical studies of skeletal muscle mitochondria. I. Microanalysis of cytochrome content, oxidative and phosphorylative activities of mammalian skeletal muscle mitochondria. Arch Biochem Biophys 126: 75–82, 1968. [DOI] [PubMed] [Google Scholar]
- 22. Martin AD, Smith BK, Davenport PD, Harman E, Gonzalez-Rothi RJ, Baz M, Layon AJ, Banner MJ, Caruso LJ, Deoghare H, Huang TT, Gabrielli A. Inspiratory muscle strength training improves weaning outcome in failure to wean patients: a randomized trial. Crit Care 15: R84, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McClung JM, Kavazis AN, DeRuisseau KC, Falk DJ, Deering MA, Lee Y, Sugiura T, Powers SK. Caspase-3 regulation of diaphragm myonuclear domain during mechanical ventilation-induced atrophy. Am J Respir Crit Care Med 175: 150–159, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McClung JM, Van Gammeren D, Whidden MA, Falk DJ, Kavazis AN, Hudson MB, Gayan-Ramirez G, Decramer M, DeRuisseau KC, Powers SK. Apocynin attenuates diaphragm oxidative stress and protease activation during prolonged mechanical ventilation. Crit Care Med 37: 1373–1379, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Messer JI, Jackman MR, Willis WT. Pyruvate and citric acid cycle carbon requirements in isolated skeletal muscle mitochondria. Am J Physiol Cell Physiol 286: C565–C572, 2004. [DOI] [PubMed] [Google Scholar]
- 26. Naito H, Powers SK, Demirel HA, Aoki J. Exercise training increases heat shock protein in skeletal muscles of old rats. Med Sci Sports Exerc 33: 729–734, 2001. [DOI] [PubMed] [Google Scholar]
- 27. Naito H, Powers SK, Demirel HA, Sugiura T, Dodd SL, Aoki J. Heat stress attenuates skeletal muscle atrophy in hindlimb-unweighted rats. J Appl Physiol 88: 359–363, 2000. [DOI] [PubMed] [Google Scholar]
- 28. Nathens AB, Neff MJ, Jurkovich GJ, Klotz P, Farver K, Ruzinski JT, Radella F, Garcia I, Maier RV. Randomized, prospective trial of antioxidant supplementation in critically ill surgical patients. Ann Surg 236: 814–822, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Petrof BJ, Jaber S, Matecki S. Ventilator-induced diaphragmatic dysfunction. Curr Opin Crit Care 16: 19–25. [DOI] [PubMed] [Google Scholar]
- 30. Powers SK, Criswell D, Lawler J, Ji LL, Martin D, Herb RA, Dudley G. Influence of exercise and fiber type on antioxidant enzyme activity in rat skeletal muscle. Am J Physiol Regul Integr Comp Physiol 266: R375–R380, 1994. [DOI] [PubMed] [Google Scholar]
- 31. Powers SK, Criswell D, Lieu FK, Dodd S, Silverman H. Exercise-induced cellular alterations in the diaphragm. Am J Physiol Regul Integr Comp Physiol 263: R1093–R1098, 1992. [DOI] [PubMed] [Google Scholar]
- 32. Powers SK, Hudson MB, Nelson WB, Talbert EE, Min K, Szeto HH, Kavazis AN, Smuder AJ. Mitochondria-targeted antioxidants protect against mechanical-ventilation-induced diaphragm weakness. Crit Care Med 39: 1749–1759, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev 88: 1243–1276, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Powers SK, Locke, Demirel HA. Exercise, heat shock proteins, and myocardial protection from I-R injury. Med Sci Sports Exerc 33: 386–392, 2001. [DOI] [PubMed] [Google Scholar]
- 35. Powers SK, Shanely RA, Coombes JS, Koesterer TJ, McKenzie M, Van Gammeren D, Cicale M, Dodd SL. Mechanical ventilation results in progressive contractile dysfunction in the diaphragm. J Appl Physiol 92: 1851–1858, 2002. [DOI] [PubMed] [Google Scholar]
- 36. Powers SK, Talbert EE, Adhihetty PJ. Reactive oxygen and nitrogen species as intracellular signals in skeletal muscle. J Physiol 589: 2129–2138, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Quintanilha AT. Effects of physical exercise and/or vitamin E on tissue oxidative metabolism. Biochem Soc Trans 12: 403–404, 1984. [DOI] [PubMed] [Google Scholar]
- 38. Radell PJ, Remahl S, Nichols DG, Eriksson LI. Effects of prolonged mechanical ventilation and inactivity on piglet diaphragm function. Intensive Care Med 28: 358–364, 2002. [DOI] [PubMed] [Google Scholar]
- 39. Sassoon CS, Zhu E, Caiozzo VJ. Assist-control mechanical ventilation attenuates ventilator-induced diaphragmatic dysfunction. Am J Respir Crit Care Med 170: 626–632, 2004. [DOI] [PubMed] [Google Scholar]
- 40. Senf SM, Dodd SL, McClung JM, Judge AR. Hsp70 overexpression inhibits NF-kappaB and Foxo3a transcriptional activities and prevents skeletal muscle atrophy. FASEB J 22: 3836–3845, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shanely RA, Coombes JS, Zergeroglu AM, Webb AI, Powers SK. Short-duration mechanical ventilation enhances diaphragmatic fatigue resistance but impairs force production. Chest 123: 195–201, 2003. [DOI] [PubMed] [Google Scholar]
- 42. Shanely RA, Zergeroglu MA, Lennon SL, Sugiura T, Yimlamai T, Enns D, Belcastro A, Powers SK. Mechanical ventilation-induced diaphragmatic atrophy is associated with oxidative injury and increased proteolytic activity. Am J Respir Crit Care Med 166: 1369–1374, 2002. [DOI] [PubMed] [Google Scholar]
- 43. Smuder AJ, Kavazis AN, Hudson MB, Nelson WB, Powers SK. Oxidation enhances myofibrillar protein degradation via calpain and caspase-3. Free Radic Biol Med 49: 1152–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Smuder AJ, Kavazis AN, Min K, Powers SK. Exercise protects against doxorubicin-induced oxidative stress and proteolysis in skeletal muscle. J Appl Physiol 110: 935–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Staib JL, Tumer N, Powers SK. Increased temperature and protein oxidation lead to HSP72 mRNA and protein accumulation in the in vivo exercised rat heart. Exp Physiol 94: 71–80, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vincent HK, Powers SK, Demirel HA, Coombes JS, Naito H. Exercise training protects against contraction-induced lipid peroxidation in the diaphragm. Eur J Appl Physiol Occup Physiol 79: 268–273, 1999. [DOI] [PubMed] [Google Scholar]
- 47. Vincent HK, Powers SK, Stewart DJ, Demirel HA, Shanely RA, Naito H. Short-term exercise training improves diaphragm antioxidant capacity and endurance. Eur J Appl Physiol 81: 67–74, 2000. [DOI] [PubMed] [Google Scholar]
- 48. Whidden MA, McClung JM, Falk DJ, Hudson MB, Smuder AJ, Nelson WB, Powers SK. Xanthine oxidase contributes to mechanical ventilation-induced diaphragmatic oxidative stress and contractile dysfunction. J Appl Physiol 106: 385–394, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Whidden MA, Smuder AJ, Wu M, Hudson MB, Nelson WB, Powers SK. Oxidative stress is required for mechanical ventilation-induced protease activation in the diaphragm. J Appl Physiol 108: 1376–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zergeroglu MA, McKenzie MJ, Shanely RA, Van Gammeren D, DeRuisseau KC, Powers SK. Mechanical ventilation-induced oxidative stress in the diaphragm. J Appl Physiol 95: 1116–1124, 2003. [DOI] [PubMed] [Google Scholar]