

Abstract
Cancer is a complex disease; glioblastoma (GBM) is no exception. Short survival, poor prognosis, and very limited treatment options make it imperative to unravel the disease pathophysiology. The critically important identification of proteins that mediate various cellular events during disease is made possible with advancements in mass spectrometry (MS)-based proteomics. The objective of our study is to identify and characterize proteins that are differentially expressed in GBM to better understand their interactions and functions that lead to the disease condition. Further identification of upstream regulators will provide new potential therapeutic targets. We analyzed GBM tumors by SDS-PAGE fractionation with internal DNA markers followed by liquid chromatography-tandem mass spectrometry (MS). Brain tissue specimens obtained for clinical purposes during epilepsy surgeries were used as controls, and the quantification of MS data was performed by label-free spectral counting. The differentially expressed proteins were further characterized by Ingenuity Pathway Analysis (IPA) to identify protein interactions, functions, and upstream regulators. Our study identified several important proteins that are involved in GBM progression. The IPA revealed glioma activation with z score 2.236 during unbiased core analysis. Upstream regulators STAT3 and SP1 were activated and CTNNα was inhibited. We verified overexpression of several proteins by immunoblot to complement the MS data. This work represents an important step towards the identification of GBM biomarkers, which could open avenues to identify therapeutic targets for better treatment of GBM patients. The workflow developed represents a powerful and efficient method to identify biomarkers in GBM.
Keywords: glioblastoma, mass spectrometry-based proteomics, tumor tissue biomarkers, label-free quantification
glioblastoma (gbm) is the most common and aggressive primary malignant brain tumor affecting ∼10,000 Americans annually. GBMs are characterized as World Health Organization grade IV gliomas due to their extreme invasiveness and poor prognosis. These diffuse and invasive brain tumors result from the transformation of glial cells or their precursors in the central nervous system. Treatment of GBM consists of surgical resection, radiation, and chemotherapy. Although advances have been made in conventional therapies over the past three decades, virtually all GBMs are ultimately resistant to current modalities of treatment and inevitably recur (25). Thus, there might be a common underlying physiological process responsible for tumor recurrences across all GBM patients. The prognosis for patients with GBM is grim, with a median duration of only ∼17 mo after diagnosis, and <5% survival more than 3 years after diagnosis (19, 34). Thus, there is a desperate need to better understand the molecular mechanisms associated with GBM tumorigenesis and to identify therapeutic targets for the treatment of this deadly brain tumor.
Molecular biomarkers are cellular molecules (DNA, mRNA, miRNA, proteins, and metabolites), which can be used as indicators of biological processes and pathogenic states. Therefore, they can provide information regarding diagnosis, prognosis and response to therapeutic intervention. These cellular molecules can be identified in tumor tissue specimens, blood plasma/serum or cerebrospinal fluid. Tumor tissue biomarkers are important as they lend insight into the biological processes involved in tumorigenesis.
Gene expression-based methods provide valuable insights to the genomic alterations in the disease condition; however, they do not necessarily reflect the protein expression profile and cellular signaling pathways activated due to regulation of mRNA translation into protein. Proteomic studies for biomarker discovery provide important insights into the pathophysiology of a disease. Thus recently, mass spectrometry (MS)-based proteomics techniques reflect important emerging frontiers in the field of GBM tumor biomarker discovery. Proteins play a direct role in dictating signaling pathways and cellular activities; thus directly measuring their abundance can provide valuable biological insights. Proteomes, the global expression of proteins, change during progression from a healthy to a diseased state. Proteins found to be differentially expressed compared with control samples are thought to play important and direct roles in the cellular mechanisms related to disease. Thus, many existing drug therapies aim to alter the expression or the activity of a specific protein found to be differentially expressed (6, 11). In addition, differentially expressed proteins that do not play a direct role in disease formation are thought to be the by-products of a disease state, which may be used as potential diagnostic biomarkers of a disease.
Over the last few years, a number of different methods have been developed to quantitatively compare proteomic data sets. The most common approach is by two-dimensional (2D) gel electrophoresis and 2D-differential in-gel electrophoresis (DIGE) for protein quantification followed by MS for protein identification. However, 2D gel-based approaches are limited by poor reproducibility, insufficient resolution of proteins, and low throughput (42). In recent years, several MS-based quantitative techniques have been developed for the direct comparison of protein expression levels between two or more different samples. Some of these require the incorporation of an isotopic label, either metabolically (SILAC), by chemical labeling (ICAT, ICPL), by enzymatic labeling (18O labeling), or with an isobaric tag (iTRAQ, TMT) (42). These methods have the following limitations: 1) metabolic labeling approaches require growth of cells in culture that preclude use with patient material; 2) problems arise in achieving high levels of labeling; and 3) greatly increased financial expense is associated with proprietary reagents. Moreover, uniform labeling strategies are not feasible in serial experiments that need to be done on samples acquired at different time points (months to years), as per serial clinical sample analysis in this study. An alternative to overcome these limitations is to use label-free methods. A quantitative label-free spectral counting is based on the principle that increased protein concentration gives a greater number of peptides and total MS scans per protein identification (24, 37). This approach is known to give highly correlative measures to protein abundance in a complex matrix (37). In addition, the method is highly sensitive to the detection of subtle changes in protein abundance (48). Analyzing samples by spectral counting requires prefractionation of a complex proteome. In addition, the fractionation method should be consistent throughout the experiments regardless of inherent differences in samples if any. To meet this requirement, we optimized a protocol using SDS-PAGE with DNA ladder as an internal marker that has been developed by Zhang et al. (71). The internal DNA marker guides us to cut the gels at the same lane, thereby overcoming the limitation of gel to gel reproducibility, and gives consistent fractionation on any day the sample is analyzed. This is very critical in label-free quantification approach, where samples are analyzed at different time points as in this study.
Previous studies on GBM protein biomarkers have used labeling techniques like iTRAQ and ICPL or gel-based proteomics strategies like 2DE or DIGE (2, 9, 10, 31, 55). They provided important insights into the molecular mechanisms in the disease. However, most of these reports utilized pooled sample analyses, where it would not be evident if the differential protein expression is from one or all of the patient samples without later verification by other techniques. Proteins that are the common mediators for a disease should be identified in all patient samples. Hence, in this study, we analyzed samples from each patient separately to identify the common regulators of GBM. Using a high-throughput novel proteomics workflow we will be able to identify biomarkers and reveal biological networks and signaling pathways that will allow for a better understanding of GBM pathophysiology. We have used SDS-PAGE with internal DNA marker for protein fractionation to reduce the complexity of the proteome. This is then followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis and label-free quantification to identify differential protein expression in GBM compared with nonneoplastic brain tissue from patients with intractable seizures (epilepsy). In addition to differentially expressed (up- or downregulated) proteins in GBM, we have identified and validated seven proteins that are significantly overexpressed in GBM: nestin (NEST), Ras GTPase-activating-like protein (IQGAP1), nicotinamide phosphoribosyltransferase (NAMPT), annexin A1 (ANXA1), peripherin (PERI), cathepsin B (CATB), and fetuin A (FETUA). These proteins are likely to be functionally significant to GBM as they are involved in tumor cell proliferation, survival, invasion, and angiogenesis. Therefore, the protein biomarkers identified in this study provide us with a better understanding of GBM pathophysiology and have the potential to serve as therapeutic targets with the hopes for better treatment options for GBM patients in the future.
MATERIALS AND METHODS
Materials.
Reagents and chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. Antibodies against IQGAP1, NAMPT, FETUA, CATB, and horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG were purchased from Abcam (Cambridge, MA). Anti-NEST was obtained from Novus Biologicals (Littleton, CO). Anti-PERI and HRP-conjugated donkey anti-goat IgG were purchased from Santa Cruz Biotechnology (Dallas, TX). Anti-ANXA1 antibody was supplied by Cell Signaling Technology, (Danvers, MA). Anti-β-actin was purchased from Sigma-Aldrich (St. Louis, MO). HRP-conjugated goat anti-mouse IgG was supplied by R&D Systems (Minneapolis, MN). SDS-PAGE and Western blotting materials were obtained from Life Technologies (Grand Island, NY). ZipTip pipette tips were supplied by EMD Millipore (Billerica, MA). Trypsin Gold was purchased from Promega (Madison, WI).
Sample collection.
Both GBM and epilepsy specimens were obtained from temporal region of the brain (5 right temporal and 5 left temporal in GBM and 6 right temporal and 4 left temporal from epilepsy patients). GBM tumor tissue and epileptic brain tissue specimens were collected at the time of tissue resection and snap-frozen in liquid nitrogen within 30 min of removal. The samples were stored at −80°C in the Brain and Spine Tissue Bank at the Medical College of Wisconsin (MCW) until used for sample analysis by MS.
A total of 10 GBM tumors and 10 epileptic brain tissue samples were analyzed in this study. GBM samples were resected from patients ranging in age from 48 to 67 yr (6 women and 4 men) and epileptic brain samples were from patients ranging in age from 21 to 61 yr (4 women and 6 men). All samples were collected after informed written consent was obtained from patients. The research protocol was approved by the Institutional Review Board at MCW. The GBM tumor tissues were evaluated by routine histologic, immunohistochemical, and angiogenic measurements. Each tissue biopsy sample was fixed in 10% buffered formalin, processed, embedded in paraffin, cut, and stained with hematoxylin and eosin and any other histochemical or immunohistochemical stains needed to fully evaluate the tissue. The diagnostic evaluation of each biopsy was performed in the Department of Pathology at MCW. Diagnosis of GBM was based on morphologic features that are considered histological hallmarks of GBM including high cellularity, nuclear hyperchromatism and pleomorphism, abundant mitoses, endothelial proliferation, and necrosis with or without pseudopalisades. Nonneoplastic brain tissues were obtained at craniotomy from epilepsy patients having surgery for an intractable seizure disorder.
Sample preparation.
GBM primary tumor and epileptic brain tissue samples were homogenized and powdered in liquid nitrogen using a mixer mill (Retsch). Samples were maintained at liquid N2 temperature throughout the process. Homogenized and powdered tissue samples were then resuspended in 5× volume of the weight of the tissue sample in a reducing buffer [125 mM Tris pH 6.8, 4% SDS (wt/vol), 10% glycerol (vol/vol), 5% 2-mercaptoethanol (vol/vol), Complete protease inhibitor (Roche), HALT phosphatase inhibitor (Thermo)]. Samples were then heated to 70°C with mixing at 1,400 rpm for 10 min, sonicated with a tip sonicator for 30 s at power level 3, and then centrifuged at 16,000 g for 10 min at room temperature. The supernatant was then collected and subjected to a Pierce 660 nm protein assay to determine protein concentration.
Gel-based sample fractionation.
Protein from tissue samples was fractionated by SDS-PAGE in a modification of the technique of Zhang et al. (71). In brief, 50 μg of protein for each sample was mixed with 5 μg of 1 kb Plus DNA Ladder (Life Technologies), sample buffer, and reducing agent and heated to 70°C for 10 min. Proteins from each sample were then separated based on molecular weight on a 7% Tris-acetate gel using Tris-acetate running buffer under reducing conditions by gel electrophoresis at 150 V in a cold room. Gels were fixed for 30 min with 7% acetic acid/40% ethanol and then stained with indoine blue (0.025% in 7% acetic acid/40% ethanol) for 25 min. Destaining with fixing buffer for 10 min followed by rehydration of the gel in MilliQ water overnight allowed for visualization of the DNA ladder marker mixed in with the sample. For each GBM tumor and epileptic brain tissue sample, gel lanes were then reproducibly fractionated by cutting at specific DNA ladder bands (0.85 kb and 0.3 kb bands). This approach resulted in the separation of sample proteins based on molecular weight resulting in three fractions: high-, medium-, and low-molecular-weight fractions (Fig. 1B).
Fig. 1.
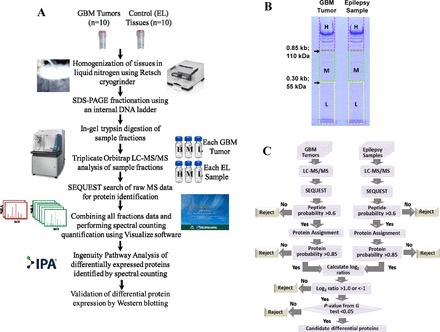
A: proteomics workflow for the identification of differential protein expression in glioblastoma (GBM) using label-free quantification proteomics: GBM and epileptic brain tissue (EL) samples were analyzed separately by SDS-PAGE with internal DNA markers followed by in-gel trypsin digestion and nano-liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis on a Thermo LTQ Orbitrap Velos instrument in triplicate. Protein identification was carried out by SEQUEST algorithm and spectral counting quantification by Visualize tool. The differentially expressed proteins were further characterized by Ingenuity Pathway Analysis (IPA) network analysis. Proteins significantly altered and associated with cancer and GBM were validated by Western blotting. B: SDS-PAGE separation of tumor proteome from GBM and control EL samples. An internal DNA marker was used for consistent fractionation of the in-gel separated proteins. Gels were cut at 110 and 55 kDa protein, which is equivalent to elution of DNA of 0.85 and 0.30 kb. Thus, each fraction is cut into high (H), medium (M), and low (L) mass fractions, thereby reducing the complexity of the proteome before MS analysis. C: flowchart for the identification of differentially expressed proteins in GBM vs. EL by label-free spectral counting quantification using Visualize tool.
Destaining and in-gel trypsin digestion of gel pieces.
Each sample fraction (i.e., high-, medium-, and low-molecular-weight fractions) was cut into 1 mm gel pieces. Gel pieces were washed three times 10 min in 25 mM NH4HCO3/50% methanol, three times 1 h in 5% acetic acid/50% methanol, and two times 20 min in HPLC grade water, shaking at room temperature. Gel pieces were then dehydrated by shaking in 100% acetonitrile at room temperature until the gel pieces were opaque white. Excess acetonitrile was removed and discarded, and sample gel pieces were dried in a Speed-Vac for 30 min at medium heat. Gels were rehydrated in a solution of 20 ng/μl Trypsin Gold (Promega) in 100 mM NH4HCO3 and digested overnight at 37°C. Peptides were extracted from the gel pieces in three volumes of 5% trifluoracetic acid/50% acetonitrile for 1 h and then with 0.1% trifluoracetic acid/75% acetonitrile for 1 h, shaking both times at room temperature. Supernatants from both extractions were combined and dried under vacuum at high heat (65°C) for 3–4 h. Samples were desalted with C18 resin-packed ZipTip pipette tips according to the manufacturer's instructions.
LC-MS/MS analysis.
Tryptic peptides from each sample fraction were injected onto a C18 capillary column (5 μm particle size, 100 Å pore size; Michrom Bioresources) on a nanoACQUITY UPLC system (Waters). Columns were packed in-house to 10 cm long in 75 μm inner-diameter tubing (Polymicro Technologies) with a laser-pulled tip. Peptides were eluted over a 150 min gradient from buffer A (2% acetonitrile, 98% H2O, 0.1% formic acid) to buffer B (98% acetonitrile, 2% H2O, 0.1% formic acid). The gradient program began with 2 min at 98% A, followed by a 3 min ramp to 95% A, a 115 min ramp to 60% A, a 15 min ramp to 2% A, 3 min at 2% A, 2 min ramp to 98% A, then a 10 min equilibration in 98% A. Eluted peptides underwent electrospray ionization followed by data acquisition in a LTQ-Orbitrap Velos mass spectrometer (Thermo Scientific). MS1 scans were detected in the FTMS section of the Orbitrap Velos in profile mode at a resolution of 30,000 (full width of peak at half-maximum at 400 m/z). The 10 most abundant parent ions from each MS1 scan were selected for fragmentation via collision induced dissociation. Ion intensity threshold was set to 1,000 with normalized collision energy of 35% for MS2 scans in the LTQ section of the instrument. Dynamic exclusion settings omitted any mass observed more than twice in a 30 s interval from selection for fragmentation. Each sample fraction was analyzed in three injection replicates, resulting in nine MS runs for each tissue sample.
Protein identification and quantification by spectral counting analysis.
MS RAW data files were uploaded into the MCW Biotechnology and Bioengineering Center Workflow (20) and extracted with msConvert.(5) Identification of extracted spectra was carried out with the SEQUEST search algorithm against the National Center for Biotechnology Information RefSeq database (release date May 16, 2013). Search parameters included the differential modifications of methionine oxidation and alkylation of cysteine, trypsin cleavage (K, R) with three missed cleavages allowed, and a precursor m/z tolerance of ± 0.2 Da. Each raw MS data file resulted in one ez2 data file.
Bioinformatics analysis.
The data obtained is based on 180 liquid chromatography-mass spectrometry (LC-MS) experiments, where 20 samples were subjected to three fractions each and then three replicate experiments of each fraction on LC-MS. For each sample, the ez2 data files for each of the three fractions and technical replicates of each fraction were combined with Visualize, a software tool developed in-house (21). Spectral counting quantification and statistical analyses comparing the GBM tumor samples with epileptic brain tissue samples were also performed with Visualize. Data from all patient samples were combined and compared with combined data from epilepsy samples. Visualize gives an output file with data for each protein identified in both the samples, with information on total peptides identified and total scan counts in each sample. From the scan counts, it calculates the scan ratio, normalized scan ratio, and finally normalized log2 ratio, which we are using to identify the differential protein expression in this study. Normalization is done with respect to the total number of identified scans for each sample. The algorithm also provides the statistical significance of the results by calculating the P values between sample means and standard deviation within sample groups, which we took into consideration while evaluating the data. Proteins with significant differential expression consisted of those with protein probability ≥ 0.85 with at least two scans between samples, and P value ≤ 0.05. The flowchart of protein quantification by label-free spectral counting approach using Visualize tool is shown in Fig. 1C. Differentially expressed proteins were further analyzed by Ingenuity Pathway Knowledge Database (Ingenuity Systems, Redwood City, CA) to determine their biological significance and relationship to known biochemical pathways. Fisher's exact test was used to calculate a P value to determine the significance of each network and function assessed. P values ≤ 0.05 were considered significant. Activation z score was calculated as a measure of functional and translational activation in disease and functional protein networks and upstream regulator analysis. A positive z score signifies activation and negative z score signifies inhibition, and the scores ≥ 2 or ≤ −2 were considered significant in this study.
Western blot analysis and quantification.
Equal amounts of protein from each of the 10 GBM tumor samples or the 10 epileptic brain tissue samples were pooled into one sample resulting in a pooled GBM tumor sample and a pooled epileptic brain tissue sample. SDS-PAGE and Western blotting were performed by standard methods. The detailed approach with gel composition, membrane type, blocking conditions, and antibody dilutions were varied depending on the protein of interest and are detailed in supplementary methods.1 The Molecular Imager Gel Doc XR+ System (Bio-Rad) was used to capture images of resulting exposed film. ImageQuant software was used to quantify Western blot images. β-Actin loading controls for each experiment were used to normalize the signals from the proteins of interest.
RESULTS
To identify GBM tumor tissue biomarkers, we used SDS-PAGE fractionation followed by tandem mass spectral analysis and spectral counting quantification of GBM and epileptic brain tissue samples. The overall workflow of this study is represented in Fig. 1A. Ten GBM tumors and 10 epileptic brain tissue samples were analyzed separately and reproducibly fractionated into three fractions based on molecular weight (high, medium, and low) (Fig. 1B). Peptides for each fraction were analyzed in triplicate by LC-MS/MS, resulting in nine MS runs per sample. SEQUEST algorithm was used for protein identification and Visualize (21), a bioinformatics software program developed in-house, was used to combine data and perform spectral counting quantitative analyses. On average, 6,402 ± 1,201 proteins were identified per GBM tumor and epileptic brain tissue samples. Only proteins identified with protein probability ≥ 0.85 with ≥ 2 scans between samples were further used for spectral counting quantification. On the basis of these criteria, we quantified a total of 4,576 proteins. Proteins that were identified in all 10 GBM tumor samples with P value ≤ 0.05 (601 proteins) were selected for Ingenuity Pathway Analysis (IPA) and further validation. Data represented were obtained from mean value from three technical replicate (n = 3) experiments. The reproducibility was measured across three replicate injections and observed a technical variation <10%. The proteins that were considered differentially expressed and used for IPA and validations are those that were identified in every GBM sample analyzed. Hence, irrespective of the tumor location in brain, these proteins are expressed in all GBM tumors and, hence, highly specific to the disease condition.
Spectral counting quantification with Visualize software.
Analysis of differentially expressed proteins using spectral counting quantification resulted in a distribution of 601 proteins with P value ≤ 0.05, which is represented in Fig. 2. Of these, 213 proteins were identified as overexpressed and common to all 10 GBM tumor samples compared with epileptic brain tissue samples with log2 ratio ≥ 1 (or ≥2-fold). Of the 601 proteins, 323 were within log2 ratio 1 to −1, which we considered as equal expression or no significant differential expression of proteins. Lastly, 65 proteins were downregulated with log2 ratio ≤ −1 and common to all 10 GBM tumor samples compared with scan counts of combined 10 epileptic brain tissue samples. The list of top 100 upregulated proteins is represented in Table 1. The list of all 601 differentially expressed proteins along with the total number of scans observed in GBM and epileptic brain tissue samples that were used for spectral counting quantification and their normalized log2 ratios are represented in Supplementary Table S1.
Fig. 2.

Ratio distribution of the 601 differentially expressed proteins in GBM tumors compared with control EL samples. Of these, 213 proteins were upregulated (log2 ratio ≥ 1), 323 equal expression (log2 ratio 1 to −1), and 65 downregulated (log2 ratio ≤ −1). This set of proteins was used for IPA analysis.
Table 1.
The top 100 upregulated proteins identified in GBM tumors using label-free quantification proteomics
| Symbol | Name | Total GBM Scans | Total EL Scans | Normalized Log2 Ratio | P Value |
|---|---|---|---|---|---|
| NEST | Nestin | 1,651 | 5 | 7.95 | 1.33E-35 |
| CD14 | Monocyte differentiation antigen CD14 | 191 | 2 | 6.16 | 6.68E-10 |
| LAC7 | Ig lambda-7 chain C region IGLC7 | 612 | 10 | 5.52 | 1.32E-09 |
| LAC6 | Ig lambda-6 chain C region IGLC6 | 598 | 10 | 5.49 | 1.01E-08 |
| ALS | Insulin-like growth factor-binding protein complex acid labile subunit IGFALS | 176 | 4 | 5.04 | 1.42E-41 |
| ITAM | Integrin alpha-M ITGAM | 352 | 9 | 4.87 | 1.85E-19 |
| VAT1 | Synaptic vesicle membrane protein VAT-1 homolog | 113 | 3 | 4.82 | 5.02E-18 |
| IQGA1 | Ras GTPase-activating-like protein IQGAP1 | 1,207 | 37 | 4.61 | 1.15E-06 |
| NAMPT | Nicotinamide phosphoribosyltransferase | 1,008 | 33 | 4.52 | 1.15E-06 |
| STT3A | Dolichyl-diphosphooligosaccharide-protein glycosyltransferase subunit | 86 | 3 | 4.43 | 1.17E-11 |
| SERPH | Serpin H1 SERPINH1 | 187 | 7 | 4.32 | 4.39E-11 |
| TAGL2 | Transgelin-2 TAGLN2 | 317 | 12 | 4.31 | 4.42E-06 |
| VTNC | Vitronectin VTN | 468 | 19 | 4.21 | 9.28E-14 |
| LAMA4 | Laminin subunit alpha-4 | 338 | 14 | 4.18 | 1.7E-05 |
| LAC1 | Ig lambda-1 chain C regions IGLC1 | 186 | 8 | 4.12 | 6.51E-13 |
| IGHG2 | Ig gamma-2 chain C region | 1,779 | 80 | 4.06 | 6.47E-05 |
| SAMH1 | SAM domain and HD domain-containing protein 1 SAMHD1 | 133 | 6 | 4.05 | 7.84E-19 |
| A2AP | Alpha-2-antiplasmin SERPINF2 | 688 | 34 | 3.92 | 0.000126 |
| ANXA1 | Annexin A1 | 1,388 | 69 | 3.91 | 3.12E-11 |
| F13A | Coagulation factor XIII A chain F13A1 | 637 | 33 | 3.85 | 3.12E-11 |
| CO5 | Complement C5 | 459 | 24 | 3.84 | 1.13E-07 |
| IGLL5 | Immunoglobulin lambda-like polypeptide 5 | 200 | 11 | 3.77 | 1.13E-07 |
| PERI | Peripherin PRPH | 140 | 8 | 3.71 | 0.000245 |
| LAMB1 | Laminin subunit beta-1 | 172 | 10 | 3.69 | 0.000245 |
| IGHG4 | Ig gamma-4 chain C region | 546 | 32 | 3.68 | 7.64E-10 |
| CO3A1 | Collagen alpha-1(III) chain COL3A1 | 136 | 8 | 3.67 | 7.46E-32 |
| FLNC | Filamin-C | 2,792 | 168 | 3.64 | 5.66E-15 |
| IGHG3 | Ig gamma-3 chain C region | 632 | 39 | 3.60 | 1.48E-06 |
| TENA | Tenascin TNC | 1,926 | 126 | 3.52 | 3.48E-11 |
| LUM | Lumican | 235 | 16 | 3.46 | 2.81E-06 |
| CLIC4 | Chloride intracellular channel protein 4 | 101 | 7 | 3.44 | 2.42E-13 |
| LAC3 | Ig lambda-3 chain C regions IGLC3 | 991 | 71 | 3.39 | 1.22E-10 |
| LAC2 | Ig lambda-2 chain C regions IGLC2 | 990 | 71 | 3.39 | 4.4E-57 |
| LAMP1 | Lysosome-associated membrane glycoprotein 1 | 152 | 11 | 3.37 | 4.23E-10 |
| NP1L1 | Nucleosome assembly protein 1-like 1 NAP1L1 | 151 | 11 | 3.36 | 4.23E-10 |
| APOE | Apolipoprotein E | 662 | 51 | 3.28 | 6.35E-08 |
| PSME2 | Proteasome activator complex subunit 2 | 227 | 18 | 3.24 | 6.35E-08 |
| CPNS1 | Calpain small subunit 1 CAPNS1 | 63 | 5 | 3.24 | 1E-05 |
| IGKC | Ig kappa chain C region | 2278 | 182 | 3.23 | 0.001792 |
| DPYD | Dihydropyrimidine dehydrogenase [NADP(+)] | 62 | 5 | 3.22 | 3.84E-29 |
| ANT3 | Antithrombin-III SERPINC1 | 859 | 70 | 3.20 | 5.24E-27 |
| LTBP1 | Latent-transforming growth factor beta-binding protein 1 | 85 | 7 | 3.19 | 5.37E-12 |
| LEG1 | Galectin-1 LGALS1 | 779 | 69 | 3.08 | 1.19E-07 |
| FINC | Fibronectin FN1 | 4,060 | 362 | 3.07 | 2.02E-25 |
| ROA1 | Heterogeneous nuclear ribonucleoprotein A1 HNRNPA1 | 163 | 15 | 3.03 | 2.02E-25 |
| COIA1 | Collagen alpha-1(XVIII) chain COL18A1 | 194 | 18 | 3.01 | 1.84E-11 |
| ITIH1 | Inter-alpha-trypsin inhibitor heavy chain H1 | 1,156 | 111 | 2.96 | 6.26E-11 |
| CLUS | Clusterin CLU | 983 | 95 | 2.96 | 5E-09 |
| PROS | Vitamin K-dependent protein S PROS1 | 81 | 8 | 2.92 | 3.56E-05 |
| HRG | Histidine-rich glycoprotein | 706 | 74 | 2.84 | 3.56E-05 |
| CATB | Cathepsin B CTSB | 180 | 19 | 2.83 | 0.003461 |
| ANXA2 | Annexin A2 | 790 | 86 | 2.78 | 9.24E-09 |
| AFAM | Afamin AFM | 211 | 23 | 2.78 | 1.99E-17 |
| FCGBP | IgGFc-binding protein | 100 | 11 | 2.77 | 1.7E-08 |
| CR2 | Complement receptor type 2 | 63 | 7 | 2.75 | 6.67E-05 |
| ITIH2 | Inter-alpha-trypsin inhibitor heavy chain H2 | 1,694 | 189 | 2.75 | 6.67E-05 |
| FETUA | Alpha-2-HS-glycoprotein AHSG | 133 | 15 | 2.73 | 1.94E-27 |
| FIBB | Fibrinogen beta chain FGB | 4,833 | 578 | 2.65 | 0.000125 |
| THRB | Prothrombin F2 | 165 | 20 | 2.63 | 0.006664 |
| ANGT | Angiotensinogen AGT | 488 | 60 | 2.61 | 0.006664 |
| AKP13 | A-kinase anchor protein 13 AKAP13 | 250 | 31 | 2.60 | 0.006664 |
| RPN2 | Dolichyl-diphosphooligosaccharide-protein glycosyltransferase subunit 2 | 298 | 38 | 2.56 | 1.05E-07 |
| AMPL | Cytosol aminopeptidase LAP3 | 405 | 53 | 2.52 | 4.81E-06 |
| AXA2L | Putative annexin A2-like protein ANXA2P2 | 508 | 67 | 2.51 | 4.34E-09 |
| VIME | Vimentin VIM | 9,652 | 1300 | 2.48 | 8.83E-06 |
| CO6A3 | Collagen alpha-3(VI) chain COL6A3 | 3,034 | 419 | 2.44 | 8.83E-06 |
| PDIA1 | Protein disulfide-isomerase P4HB | 724 | 100 | 2.44 | 2.71E-20 |
| IGHG1 | Ig gamma-1 chain C region | 2,335 | 325 | 2.43 | 1.16E-31 |
| RLA1 | 60S acidic ribosomal protein P1 RPLP1 | 100 | 14 | 2.42 | 4.05E-24 |
| GPX1 | Glutathione peroxidase 1 | 85 | 12 | 2.41 | 3.7E-54 |
| TSNAX | Translin-associated protein X | 63 | 9 | 2.39 | 6.39E-07 |
| SHOT1 | Shootin-1 KIAA1598 | 278 | 40 | 2.38 | 1.62E-05 |
| IC1 | Plasma protease C1 inhibitor SERPING1 | 582 | 84 | 2.38 | 0.00043 |
| ITIH4 | Inter-alpha-trypsin inhibitor heavy chain H4 | 567 | 88 | 2.27 | 0.012792 |
| IGHA2 | Ig alpha-2 chain C region | 219 | 34 | 2.27 | 0.012792 |
| RL6 | 60S ribosomal protein L6 RPL6 | 154 | 24 | 2.27 | 4.69E-08 |
| DYH5 | Dynein heavy chain 5, axonemal DNAH5 | 526 | 82 | 2.27 | 6.13E-12 |
| PGBM | Basement membrane-specific heparan sulfate proteoglycan core protein HSPG2 | 706 | 111 | 2.25 | 1.16E-06 |
| PTN13 | Tyrosine-protein phosphatase nonreceptor type 13 PTPN13 | 76 | 12 | 2.25 | 2.95E-05 |
| BAI2 | Brain-specific angiogenesis inhibitor 2 | 1,586 | 267 | 2.15 | 2.95E-05 |
| TLR1 | Toll-like receptor 1 | 59 | 10 | 2.14 | 2.73E-15 |
| CERU | Ceruloplasmin CP | 1,458 | 249 | 2.13 | 6.35E-14 |
| CLIP1 | CAP-Gly domain-containing linker protein 1 | 138 | 24 | 2.11 | 1.96E-11 |
| FLNB | Filamin-B | 681 | 123 | 2.05 | 2.1E-06 |
| SODM | Superoxide dismutase [Mn], mitochondrial SOD2 | 601 | 110 | 2.03 | 0.000795 |
| COPB2 | Coatomer subunit beta' | 152 | 28 | 2.02 | 3.23E-45 |
| AOFB | Amine oxidase [flavin-containing] B MAOB | 1,261 | 238 | 1.99 | 5.37E-05 |
| 1C18 | HLA class I histocompatibility antigen, Cw-18 alpha chain HLA-C | 90 | 17 | 1.99 | 2.73E-07 |
| MAGI3 | Membrane-associated guanylate kinase, WW and PDZ domain-containing protein 3 | 116 | 22 | 1.98 | 2E-08 |
| STOM | Erythrocyte band 7 integral membrane protein | 427 | 81 | 1.98 | 2E-08 |
| ZHX3 | Zinc fingers and homeoboxes protein 3 | 219 | 42 | 1.97 | 6.8E-06 |
| RBM27 | RNA-binding protein 27 | 67 | 13 | 1.95 | 0.001464 |
| AACT | Alpha-1-antichymotrypsin SERPINA3 | 1,288 | 254 | 1.93 | 0.001464 |
| FLNA | Filamin-A | 5,408 | 1,096 | 1.89 | 0.024456 |
| CFAH | Complement factor H CFH | 330 | 67 | 1.88 | 0.024456 |
| RAB5C | Ras-related protein Rab-5C | 123 | 25 | 1.88 | 0.024456 |
| APMAP | Adipocyte plasma membrane-associated protein | 170 | 35 | 1.86 | 0.024456 |
| IF5A1 | Eukaryotic translation initiation factor 5A-1 EIF5A | 165 | 34 | 1.86 | 2.08E-26 |
| BIEA | Biliverdin reductase A BLVRA | 82 | 17 | 1.85 | 8.16E-11 |
| FKBP5 | Peptidyl-prolyl cis-trans isomerase FKBP5 | 106 | 22 | 1.85 | 0.000176 |
The 7 proteins in boldface were validated by Western blotting analysis. GBM, glioblastoma; EL, epilepsy. Scans represent the number of mass spectra each of these proteins is identified from all 10 samples in each group. Data are presented as mean values; n = 3, P ≤ 0.05.
IPA.
To understand the biological significance of the differentially expressed proteins in GBM tumors, we used the IPA tool. This bioinformatics program identifies networks, diseases and disorders, molecular and cellular functions, physiological system development and functions, and canonical pathways that are most relevant to the input data. The number of proteins/molecules and significant P values from the data set are mapped to each network and pathway. The network analysis in the IPA system searches for significant molecular networks integrated from literature, gene expression, and annotation as represented in the commercial IPA knowledge base. We performed IPA analysis of upregulated (213 proteins), downregulated (65 proteins), and differentially expressed (601 proteins) to understand the biological significance of each of these datasets. A summary of the IPA analysis of differentially expressed proteins is given in Table 2, and those of up- and downregulated proteins individually in Supplementary Table S2. The top three diseases and disorders in the upregulated and differentially expressed proteins were cancer, neurological disease, and psychological disorders. Interestingly, neurological disease and psychological disorders have been listed as two of the top five diseases and disorders among downregulated proteins as can be seen in Table 2 and Supplementary Table S2. Of these lists, proteins associated with cancer and neurological diseases are of interest. Hence, we plotted these networks in IPA and those molecules involved in cancer and neurological disease from differentially expressed proteins are listed in Table 3. The table also shows the up- (by ↑ arrow) and downregulation (by ↓ arrow) of each protein enlisted. In each of these networks, 27 and 28 of the total 35 molecules, respectively, were identified to be differentially expressed. The interacting networks of these proteins are shown in Fig. 3.
Table 2.
Summary of the IPA analysis of differentially expressed proteins in GBM
| P Value | Molecules, n | ||
|---|---|---|---|
| Top Diseases and Biofunctions | |||
| Diseases and disorders | |||
| Cancer | 8.88E-23-3.66E-05 | 404 | |
| Neurological disease | 1.59E-21-3.04E-05 | 223 | |
| Psychological disorders | 8.54E-20-1.45E-05 | 169 | |
| Infectious disease | 6.97E-18-2.69E-05 | 136 | |
| Organismal injury and abnormalities | 2.52E-17-4.46E-05 | 242 | |
| Molecular and cellular functions | |||
| Cell death and survival | 8.75E-28-2.06E-05 | 255 | |
| Cellular assembly and organization | 2.20E-26-3.21E-05 | 186 | |
| Cellular function and maintenance | 2.20E-26-3.76E-05 | 237 | |
| Cellular movement | 1.86E-18-3.66E-05 | 170 | |
| Cell morphology | 9.55E-18-3.21E-05 | 209 | |
| Physiological system development and function | |||
| Tissue development | 1.88E-20-3.90E-05 | 162 | |
| Organismal survival | 5.16E-14-7.56E-08 | 174 | |
| Embryonic development | 1.08E-10-3.21E-05 | 77 | |
| Nervous system Development and function | 6.80E-10-3.88E-05 | 120 | |
| Immune cell trafficking | 9.98E-10-3.66E-05 | 76 | |
| P value | Ratio | ||
| Top canonical pathways | |||
| Remodeling of epithelial adherens junctions | 1.03E-30 | 32/70 (0.457) | |
| Epithelial adherens Junction signaling | 5.88E-24 | 37/154 (0.24) | |
| Signaling by Rho family GTPases | 2.72E-19 | 40/263 (0.152) | |
| 14-3-3-mediated signaling | 9.46E-19 | 29/121 (0.24) | |
| Actin cytoskeleton signaling | 6.56E-18 | 37/242 (0.153) | |
| z Score | P Value of Overlap | Predicted Activation State | |
| Top Upstream Regulators | |||
| SP1 | 3.845 | 1.90E-10 | Activated |
| STAT3 | 3.726 | 5.20E-08 | Activated |
| IL6 | 3.437 | 7.96E-09 | Activated |
| SRF | 3.054 | 4.88E-08 | Activated |
| CTNNa | −3.403 | 2.63E-05 | Inhibited |
IPA, Ingenuity Pathway Analysis.
Table 3.
Major networks and associated proteins obtained by IPA analysis of differentially expressed GBM proteins
| Top Diseases and Functions | Molecules in Network | Score | Focus Molecules |
|---|---|---|---|
| Neurological disease, cellular assembly and organization, cellular function and maintenance | 75 NGF, Alpha tubulin, AP-3, Beta Tubulin, ↑CLIP1, CRMP, ↓CRMP1, ↑CYB5R3, ↓DPYSL2, ↓DPYSL3, Dynein, ↑FNTA, ↑GFAP, ↓INA, ↓KIF5A, ↓KIF5C, ↑LAMP2, ↑LAP3, ↑MYO1E, ↓NEFH, ↓NEFL, ↓NEFM, NFkB (complex), ↑PRPH, ↓PTPLAD1, ↓RTN3, ↓RTN4, ↑S100B, ↑TANC1, ↑TUBA8, ↓TUBA1B, ↓TUBA3E, ↓TUBB1, ↓TUBB3, ↓TUBB2B | 42 | 28 |
| Cancer, endocrine system disorders, hematological disease | ↑ADH5, ALT, ↓CACNA1E, ↑CCT3, ↓CCT4, ↑CCT5, ↑CCT6A, ↑CFH, ↑CSNK1E, EIF3, ↑EIF3A, ↓EIF3B, ↑EIF4A1, Eif4 g, ↓EIF4G3, GOT, ↓GOT1, ↑HNRNPD, Ku, ↓MATR3, p70 S6k, ↑PCBP1, ↓PFKM, Pka, Rnr, ↓SLC1A3, ↑TAGLN2, ↓TUBA1A, ↓TUBA1C, ↓TUBA4A, ↓TUBB8, ↓TUBB2A, ↓TUBB4A, ↓TUBB4B, ↓VPS35 | 39 | 27 |
The boldfaced proteins are identified in our data, and their expression is shown by arrows: upregulation (↑) and downregulation (↓).
Fig. 3.
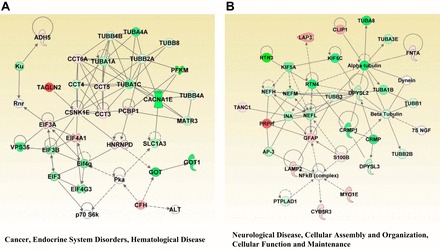
The top 2 diseases and disorders networks identified by IPA analysis. A: cancer, endocrine system disorders, hematological disease network; B: neurological disease, cellular assembly and organization, cellular function, and maintenance network. Red and green indicate up- and downregulation, respectively, of the proteins. Intensity of the color reflects the extent of differential expression.
The molecular and cellular functions associated with our lists of upregulated and differentially expressed proteins included cellular movement, cell death and survival, and cell morphology as top hits. In addition, cell assembly and organization and cell morphology were listed in downregulated as well as differentially expressed proteins. All these results point toward the significance of these proteins in cancer biology and specifically brain cancer biology as distinct from neurological disease. Further analysis of diseases and functions of the differentially expressed proteins gave very promising results. This function predicts the disease or functional activation based on the activation z score. The z score is calculated based on relationships in the molecular networks, which represent experimentally observed molecular expression and functional annotation data as derived from the information compiled in the IPA knowledge base. The activation z score ≥ 2 is considered significant, and we identified glioma as the only predicted increased activation in the list of cancer proteins with a z score of 2.236 with P value of 9.47E-08. The list of 41 proteins involved in the prediction of glioma annotation along with the log ratios is given in Table 4. The network annotation of the same is shown in Fig. 4. This demonstrates the efficiency of the experimental method and the bioinformatics analysis we used for biomarker identification.
Table 4.
Proteins involved in the prediction of glioma activation by IPA analysis of differentially expressed GBM proteins
| Genes/Proteins in Dataset | Prediction (based on expression direction) | Log2 Ratio | Findings |
|---|---|---|---|
| GFAP | increased | 1.406 | increases |
| LAMA4 | increased | 4.178 | increases |
| LAMB1 | increased | 3.689 | increases |
| SPTBN1 | increased | −0.655 | decreases |
| NES | increased | 7.951 | increases |
| KIAA1598 | affected | 2.381 | affects |
| COL1A1 | affected | 1.214 | affects |
| CDKL5 | affected | −0.465 | affects |
| GOT1 | affected | −1.244 | affects |
| IDH2 | affected | −0.411 | affects |
| KTN1 | affected | 0.371 | affects |
| PRKDC | affected | 1.466 | affects |
| FLNA | affected | 1.887 | affects |
| AKAP12 | affected | −0.907 | affects |
| MKI67 | affected | 0.809 | affects |
| COL3A1 | affected | 3.672 | affects |
| TF | affected | 1.759 | affects |
| FN1 | affected | 3.072 | affects |
| PZP | affected | 1.302 | affects |
| TTN | affected | 0.152 | affects |
| KIF5A | affected | −0.841 | affects |
| GSTM5 | affected | −0.672 | affects |
| PKM | affected | 0.596 | affects |
| COL4A2 | affected | 0.708 | affects |
| GNAO1 | affected | −0.397 | affects |
| HSPA2 | affected | −0.321 | affects |
| SPTBN4 | affected | −1.359 | affects |
| ALDOC | affected | −0.499 | affects |
| SRC | affected | 0.601 | affects |
| ACTN1 | affected | 0.127 | affects |
| TNC | affected | 3.518 | affects |
| CLU | affected | 2.955 | affects |
| HSPG2 | affected | 2.253 | affects |
| ASXL3 | affected | 0.469 | affects |
| ATR | affected | 1.184 | affects |
| MARCKS | affected | −0.697 | affects |
| PRKCB | affected | −1.273 | affects |
| HSPA5 | affected | 0.157 | affects |
| HSPA8 | affected | −0.278 | affects |
| COL1A2 | affected | 0.911 | affects |
| TRIP11 | affected | 0.862 | affects |
Glioma activation indicated by z score 2.236; overlap P value 9.47E-08.
Fig. 4.
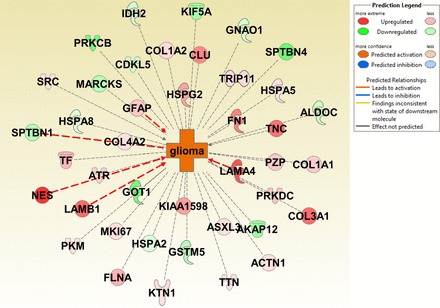
Network annotation of glioma activation identified by IPA. Proteins involved in the prediction of glioma activation are radially arranged where 5 proteins (NEST, SPTBN1, LAMB1, LAMA4, and GFAP) lead to activation, and the others have a correlative relationship with the disease. Color is used to indicate both the direction (red, upregulation; green, downregulation) and the magnitude (color intensity) of the expression change of the target proteins.
The canonical pathways along with up- and downregulated proteins in each pathway were represented in stack-bar chart of the differentially expressed proteins. The top three canonical pathways include remodeling of epithelial adherens junctions, epithelial adherens junctions signaling, and signaling by Rho family GTPases. The complete list of canonical pathways with −log P value of 7 is shown in Supplemental Fig. S1. Fisher's exact test P value method was used for scoring in this analysis. The overlapping canonical pathways from this list with filter values of a maximum of 25 pathways displayed with 12 common molecules overlapping these pathways are displayed in Supplemental Fig. S2. This represents the interaction of the pathways and their role in cancer biology, specific to GBM in our study.
Another interesting analysis that we performed with IPA is upstream regulator analysis. This tool predicts which upstream regulators are activated or inhibited based on the protein expression changes observed in the experimental data. This information helps us understand the biological activities occurring in the GBM tumor tissues. Our data predict increased activation for upstream regulators SP1 (z score 3.845), STAT3 (z score 3.726), and several other growth factors, whereas alpha-catenin (CTNNα) (z score −3.403) was inhibited among the top regulators with P value of overlap ≤ 0.05. The network analyses of these three upstream regulators and the interacting molecules that were used for their prediction are shown in Fig. 5.
Fig. 5.
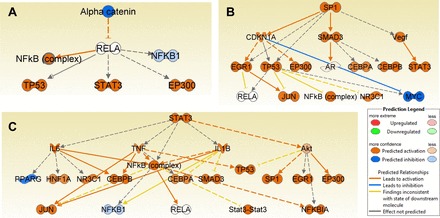
Upstream regulator analysis of differentially expressed proteins in GBM. α-Catenin (CTNNα) was predicted to be inhibited (A); SP1 and STAT3 (B and C) were predicted to be activated as determined by IPA.
Verification of protein biomarkers by Western blotting.
To further validate the results of our MS-based protein expression analysis, we selected seven protein candidates that were significantly overexpressed in GBM for Western blotting experiments. These proteins were selected based on functional relevance to GBM and cancer. As seen in Table 1, these are among the top 25% of the upregulated proteins in all GBM tumor samples as measured by our spectral counting quantitative analysis. The seven candidate proteins chosen for validation included: NEST, IQGAP1, NAMPT, ANXA1, PERI, CATB, and FETUA. To demonstrate the extent of differential expression, total MS scan numbers for each of these proteins in GBM and epileptic brain tissue samples and their log2 ratios are given in Table 1. Western blotting results and quantification of signal intensities of these seven proteins in GBM and epileptic brain tissue samples are shown in Fig. 6.
Fig. 6.
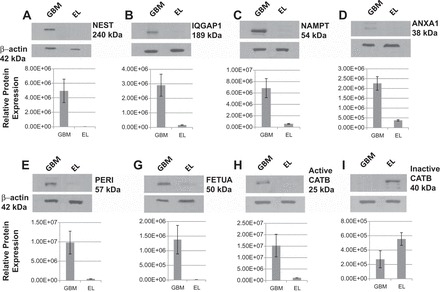
Western blot analysis of nestin (NEST) (A) Ras GTPase-activating-like protein (IQGAP1) (B), nicotinamide phosphoribosyltransferase (NAMPT) (C), annexin A1 (ANXA1) (D) peripherin (PERI) (E), fetuin A (FETUA) (F), active cathepsin B (CATB) (G), and inactive CATB (H) in GBM and control EL tissues. Quantification of band intensities was carried out by ImageQuant software.
As shown in Table 1 and Fig. 6A, NEST is highly expressed in GBM compared with control epileptic brain tissue samples analyzed. The log2 ratio for NEST in GBM is 7.95 relative to the control. Due to its extensively higher expression in GBM and known existence as a neural stem cell marker, it is our first choice for validation. The Western blot image showed a band at 240 kDa for NEST in the GBM sample and negligible signal in the control sample at the same molecular weight range. Densitometry showed an approximately sevenfold increase in intensity in GBM compared with control (Fig. 6A). Similarly, IQGAP1, NAMPT, ANXA1, PERI, CATB, and FETUA were overexpressed with log2 ratios 4.61, 4.52, 3.91, 3.71, 2.83, and 2.73, respectively. The Western blot images complement the MS spectral counting results as demonstrated in Fig. 6 and Table 1. Interestingly, the 25 kDa active form of CATB (log2 ratio 2.83) was identified in GBM sample only, whereas the 40 kDa inactive form of CATB is observed in the control sample.
DISCUSSION
This study aimed to identify novel biomarkers in GBM. Due to the highly invasive and diffuse nature of GBM, seemingly healthy tissues surrounding the tumor cannot reliably be used as controls. Hence, nonneoplastic and nonmalignant epileptic brain tissue is commonly used as a control for GBM in biomarker studies (62a, 59). In addition, tissue specimens from both GBM and controls should be procured in the same way to keep the degree of variation minimal due to sample handling. Difference in sample collection changes the proteome of a given sample. Hence, we found epilepsy specimens to be the best available controls for GBM, which are nonneoplastic brain tissues and procured in the same manner as GBM tissues from live individuals during surgical resection of biopsy with patient's consent.
Proteins differentially expressed with high significance in GBM tumors compared with epileptic brain tissue are often considered to be GBM-specific biomarkers that could be used as potential therapeutic targets. To identify biomarkers in GBM, we used gel-based fractionation by SDS-PAGE followed by tandem mass spectrometry and spectral counting quantification analysis. The gel-based fractionation used in this study is a high-throughput technique that allows for consistent and reproducible fractionation, which is crucial in label-free quantification. In addition, our label-free spectral counting quantification approach eliminated the limitations of previously reported 2D gel-based and labeling quantification techniques. The outcome also suggests that the workflow we used is very efficient for quantitative proteomics aimed at biomarker identification. This workflow has been used for the analysis of each individual GBM and epileptic brain tissue sample, unlike most of the previous studies where pooled sample analysis has been used for differential expression studies in GBM (31). To our knowledge, this is the first comprehensive study to analyze tumor biopsies in large scale from each patient individually, where high-throughput protein identification and quantification by MS has been used for GBM biomarker identification.
Many of the differentially expressed proteins identified in this study have been previously reported in GBM biomarker studies using other proteomic methods including 2-DE, 2D-DIGE, and other MS-based techniques (12, 31, 46). In addition, we identified several new proteins that were not reported before in the context of GBM (Supplementary Table S1). The proteins that we identified to be differentially expressed have significant biological relevance to GBM molecular biology and pathophysiology as determined by IPA. Our findings of cancer and neurological disease as the top disease networks and cancer-associated pathways as the main cellular functions in the IPA analysis of differentially expressed GBM proteins validate the efficacy of our technique for the identification of GBM biomarkers. Moreover, the top canonical pathways identified in our analysis include remodeling of epithelial adherens junctions, epithelial adherens junctions signaling, and signaling by Rho family GTPases. Adherens junction signaling and Rho family GTPases are known to be associated with GBM invasion and tumor progression (15, 52). Proteins associated with the canonical pathways acute phase response signaling, caveolar-mediated endocytosis signaling, intrinsic and extrinsic prothrombin activation pathways, and coagulation systems are all upregulated as seen in Supplemental Fig. S1. Acute phase response is an inflammatory response triggered either during injury, infection, or malignant growth. Caveolar-mediated endocytosis in associated with cancer progression. These two pathways have also been reported as the top canonical pathways among upregulated proteins in a previous study (55). The involvement of the hemostatic system in angiogenesis and tumor malignancy is known, which explains the identification of upregulation of the associated proteins (49, 68).
The top networks related to the differentially expressed proteins point to several interesting regulatory proteins including STAT3, SP1, and CTNNα. STAT3 is a transcription factor activated by many cytokines and growth factors, and its activity is required for tumor growth as it promotes angiogenesis, migration, invasion, cell cycle progression, antiapoptosis, and cell survival. The role of STAT3 in tumor growth has been reported in many types of cancer, including GBM (16, 38). We identified STAT3 as an upstream regulator with activation z score 3.726, indicating increased activity in the GBM samples studied. Another transcription factor, SP1 (z score 3.845), is shown to be activated in our data. SP1 is known to regulate several cellular processes including cell growth, differentiation, and apoptosis. It is also known to regulate cancer-associated molecule CD147 in lung cancer (33). Multiple studies have reported that the inhibition of SP1 suppresses several different cancers, including GBM (62, 72). In addition, we identified several cytokines (IL6, IL4, OSM, EDN1, and IL5), growth factors (FGF2, ANGPT2, NRG1, and the PDGF family), and other kinases involved in cancer progression with activation z scores ≥ 2. Similarly, among the inhibited regulators, CTNNα is identified with activation z-score −3.403. CTNNα is involved in cell adhesion through association with cadherins. Thus, inhibition of CTNNα expression may lead to decreased adhesion, thereby increasing metastasis and invasion in cancer. The role of CTNNα in invasiveness and metastatic potential in several cancers has already been reported (56). Our data suggest that inhibition of CTNNα might lead to increased invasion in GBM. Thus, this set of proteins provides further understanding of GBM molecular biology and pathophysiology as well as the potential for new therapeutic targets for the treatment of GBM.
To further validate the efficiency of our approach and data analysis, we did IPA network analysis of proteins that were identified in all 10 epileptic brain tissue samples in this study. In addition to the neurological disease and nervous system development and function as the top most disease and function, we identified seizure disorder (z score 4.22), seizures (z score 4.21), movement disorders (z score 3.95), and ataxia (z score 3.55) to be increased, which are all relevant to epilepsy. This is what we would expect from an analysis of epileptic brain tissues, and it increases our confidence in conclusions drawn based on IPA analysis of the GBM dataset.
In addition to the identification of differentially expressed proteins, we have also validated potential GBM biomarkers by Western blotting. We chose seven potential biomarkers for validation based on both the extent of differential expression and the known functional significance of the protein in either GBM or cancer. Of these, NEST, IQGAP1, NAMPT, ANXA1, CATB, and FETUA were upregulated in GBM, common to all GBM tumors we analyzed, and have all been previously identified to be related to GBM by other methods. We also validated the overexpression observed by MS for a seventh protein, PERI, which has never been reported in the context of adult GBM.
NEST is the most highly overexpressed of all the proteins identified. It is a type VI intermediate filament protein that is transiently expressed in neural stem/progenitor cells during the development of the central nervous system but does not normally persist into adulthood. Recent studies have shown a link between NEST expression and carcinogenesis with a role in tumor angiogenesis, glioma stem cell proliferation, and tumor cell infiltration (7, 41). NEST has been identified in one other GBM proteomics study using a MS-based technique, but these results were based on analysis of only one GBM tumor sample (66). In addition to NEST, other intermediate filament proteins already known to be associated with GBM that were found in our study to be significantly overexpressed and identified in all GBM tumor samples include vimentin (log2 ratio 2.47), desmin (log2 ratio 1.68), and glial fibrillary acidic protein (log2 ratio 1.40).
IQGAP1 is a ubiquitously expressed scaffolding protein involved in regulating various cellular processes including actin cytoskeleton organization, cell proliferation, cell migration, cell adhesion, cell cycle regulation, and angiogenesis. Overexpression of IQGAP1 has been associated with several different cancers, including GBM (1, 29, 67). Interestingly, the expression of IQGAP1 specifies a subpopulation of amplifying nestin+ cancer cells in a rat model of glioma (1). Furthermore, it has been shown that IQGAP1 regulates cell signaling through the RAF/MEK/ERK pathway in RAS-MAP kinase-driven tumors (27, 58). To date, IQGAP1 has not been identified by a proteomics study as a GBM biomarker. In addition, the conjunctive unique expression of IQGAP1 and Nestin in human GBM tumor samples has not been observed in any previous proteomics studies.
NAMPT catalyzes the condensation of nicotinamide with 5-phosphoribosyl-1-pyrophosphate to yield nicotinamide mononucleotide, the rate-limiting intermediate in the biosynthesis of NAD+. NAMPT is a pleiotropic protein that functions not only as an enzyme, but also as an adipocytokine, a growth factor, and a cytokine. This protein plays a role in many cellular processes including DNA damage and repair mechanisms, metabolism, cell proliferation, angiogenesis, inflammation, and resistance to cell death. Overexpression of NAMPT has been linked to several different cancers resulting in the development of therapeutic targeting NAMPT (4, 64). In addition to other cancers, previous studies have also shown an association of NAMPT in GBM patient serum (4, 64).
Several annexins (II, IV, V, and A5) have been previously identified in proteomic studies to be differentially overexpressed in GBM tumors (8, 23, 40, 61). Annexins are a family of calcium- and phospholipid-binding proteins, which have diverse functions including vesicle trafficking, cell division, apoptosis, calcium signaling, and growth regulation and have been identified to be associated with several cancers (13). We have identified a number of annexins in our study to be overexpressed in GBM tumors including ANXA1 (log2 ratio 3.91), ANXA2 (log2 ratio 2.78), and ANXA5 (log2 ratio 1.35). Previous studies have shown that ANXA2 and ANXA5 promote glioma cell invasion and tumor progression and are shown to be overexpressed in angiogenic compared with invasive GBM tumors (57, 70). ANXA1, the annexin with the greatest magnitude of GBM overexpression in our study, has been found to be a major component in necrotic tumor cell-derived stimulants of the growth of GBM via the activation of formyl peptide receptor (FPR1) (69). One other proteomics study has identified and validated ANXA1 as an overexpressed protein in a single GBM tumor sample (66).
CATB is a lysosomal cysteine protease that is associated with increased tumor cell invasion by proteolytic activation and degradation of extracellular matrix components. Its overexpression has been identified in various cancers including lung, prostate, breast, colorectal, and GBM (43, 54). This protein is secreted as an inactive proenzyme (∼43 kDa) and is activated via proteolytic cleavage (∼25–30 kDa) (44). Interestingly, in our Western blot assay, we observed a band at 25 kDa, corresponding to active CATB, in the GBM tumor samples and not in epileptic brain tissue samples. Conversely, we observed a more prominent CATB band at 40 kDa, the inactive CATB, in epileptic brain tissue samples, but not in GBM. Previous proteomics studies have identified cathepsins D and L in GBM, but not CATB (22, 23, 73). Thus our results further corroborate the potential for CATB as a GBM therapeutic target.
Fetuins are proteins that are made in the liver and secreted into the bloodstream. These proteins bind to carrier proteins such as albumin and mediate their transport and availability. In addition, fetuins inhibit soft tissue calcification. A previous study has shown that serum FETUA levels are associated with survival of GBM patients. In that study, serum FETUA correlated inversely with glioma grade and GBM patients with normal FETUA serum levels survived longer than those patients with low FETUA serum levels (53). Though we have not correlated the expression level of FETUA with survival of GBM patients, we did observe overexpression of FETUA in GBM tumors. FETUA binds to toll-like receptor 4 (50), and therefore it might be involved in signaling events in GBM tumor cells. Hence, FETUA is overexpressed in tumor tissues as observed in our study.
In addition to the six above-mentioned proteins, we also identified a new GBM biomarker, which was found to be common to all GBM tumor samples analyzed. We identified and validated the overexpression of PERI in GBM tumors compared with control epileptic brain tissue. PERI is a type III intermediate filament expressed in peripheral neurons and known to be overexpressed in neuroblastoma cells (51). Previous studies have shown that roles played by intermediate filaments in the progression of different cancers include epithelial-to-mesenchymal transition, regulation of major signaling pathways, cell migration and invasion, and cell-cell and cell-substrate adhesions (14, 51). Thus, PERI is presumed to have a significant role in GBM tumor progression.
Conclusions
The findings from this study suggest that using a high-throughput novel proteomics strategy with SDS-PAGE with internal DNA ladder followed by tandem mass spectrometry and spectral counting quantification, we could identify differential protein expression in GBM. These proteins are highly specific to GBM irrespective of their location in brain, thus providing important insights into the disease pathology as has been demonstrated by the validated proteins and their involvement in various events of tumorigenesis. Our analysis also provided several other upregulated proteins, like SERPH, PDIA1, CERU, TENA, VTNC, APOE, LEG1, HRG, and FKBP5, that are known to be involved in tumor progression, aggressiveness, and invasion in GBM (3, 17, 18, 28, 30, 32, 39, 45, 47). In addition to known GBM-associated proteins, novel potential biomarkers identified in this study include CLIC4, NP1L1, IGKC, TAGL2, and YES (Supplementary Table S1). These proteins have been identified to be involved in processes promoting cancer progression but have never been previously associated with GBM (35, 36, 60, 63, 65). With further validations, these proteins may serve as novel therapeutic targets in GBM. Thus, our data provide a unique wealth of knowledge for understanding GBM pathophysiology. These insights suggest potential therapeutic targets, which could provide new and better treatment options in GBM.
GRANTS
This work was funded by The James C. Benjamin Fund for Brain Tumor Research, a Froedtert Foundation Grant, and an Advancing a Healthier Wisconsin - Clinical and Translational Science Institute Pilot Research Grant.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.S.H., M.A.C., W.M.M., S.D.R., E.J.C., P.S.L., K.M.S., and S.P.M. performed experiments; M.S.H., M.A.C., B.D.H., M.A.-G., and S.P.M. analyzed data; M.S.H., M.A.C., J.M.C., W.M.M., E.J.C., P.S.L., and S.P.M. interpreted results of experiments; M.S.H., M.A.C., and S.P.M. prepared figures; M.S.H., M.A.C., M.A.-G., and S.P.M. drafted manuscript; M.S.H., M.A.C., B.D.H., M.A.-G., S.D.R., P.S.L., M.G.M., K.M.S., and S.P.M. edited and revised manuscript; M.S.H., M.A.C., B.D.H., M.A.-G., J.M.C., W.M.M., S.D.R., E.J.C., P.S.L., M.G.M., K.M.S., and S.P.M. approved final version of manuscript; J.M.C., M.G.M., K.M.S., and S.P.M. conception and design of research.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Innovation Center Mass Spectrometry Facility for MS instrumentation support and the MCW Brain and Spine Tissue Bank for providing the tumor samples used in this study.
Footnotes
The online version of this article contains supplemental material.
REFERENCES
- 1.Balenci L, Clarke ID, Dirks PB, Assard N, Ducray F, Jouvet A, Belin MF, Honnorat J, Baudier J. IQGAP1 protein specifies amplifying cancer cells in glioblastoma multiforme. Cancer Res 66: 9074–9082, 2006. [DOI] [PubMed] [Google Scholar]
- 2.Banerjee HN, Mahaffey K, Riddick E, Banerjee A, Bhowmik N, Patra M. Search for a diagnostic/prognostic biomarker for the brain cancer glioblastoma multiforme by 2D-DIGE-MS technique. Mol Cell Biochem 367: 59–63, 2012. [DOI] [PubMed] [Google Scholar]
- 3.Behrem S, Zarkovic K, Eskinja N, Jonjic N. Distribution pattern of tenascin-C in glioblastoma: correlation with angiogenesis and tumor cell proliferation. Pathol Oncol Res 11: 229–235, 2005. [DOI] [PubMed] [Google Scholar]
- 4.Bi TQ, Che XM. Nampt/PBEF/visfatin and cancer. Cancer Biol Ther 10: 119–125, 2010. [DOI] [PubMed] [Google Scholar]
- 5.Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, Hoff K, Kessner D, Tasman N, Shulman N, Frewen B, Baker TA, Brusniak MY, Paulse C, Creasy D, Flashner L, Kani K, Moulding C, Seymour SL, Nuwaysir LM, Lefebvre B, Kuhlmann F, Roark J, Rainer P, Detlev S, Hemenway T, Huhmer A, Langridge J, Connolly B, Chadick T, Holly K, Eckels J, Deutsch EW, Moritz RL, Katz JE, Agus DB, MacCoss M, Tabb DL, Mallick P. A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotechnol 30: 918–920, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang BY, Kim SA, Malla B, Kim SY. The effect of selective estrogen receptor modulators (SERMs) on the tamoxifen resistant breast cancer cells. Toxicol Res 27: 85–93, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chinnaiyan P, Wang M, Rojiani AM, Tofilon PJ, Chakravarti A, Ang KK, Zhang HZ, Hammond E, Curran W, Jr, Mehta MP. The prognostic value of nestin expression in newly diagnosed glioblastoma: report from the Radiation Therapy Oncology Group. Radiat Oncol 3: 32, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chumbalkar VC, Subhashini C, Dhople VM, Sundaram CS, Jagannadham MV, Kumar KN, Srinivas PN, Mythili R, Rao MK, Kulkarni MJ, Hegde S, Hegde AS, Samual C, Santosh V, Singh L, Sirdeshmukh R. Differential protein expression in human gliomas and molecular insights. Proteomics 5: 1167–1177, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Collet B, Guitton N, Saikali S, Avril T, Pineau C, Hamlat A, Mosser J, Quillien V. Differential analysis of glioblastoma multiforme proteome by a 2D-DIGE approach. Proteome Sci 9: 16, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Com E, Clavreul A, Lagarrigue M, Michalak S, Menei P, Pineau C. Quantitative proteomic Isotope-Coded Protein Label (ICPL) analysis reveals alteration of several functional processes in the glioblastoma. J Proteom 75: 3898–3913, 2012. [DOI] [PubMed] [Google Scholar]
- 11.D'Alessandris QG, Montano N, Cenci T, Martini M, Lauretti L, Bianchi F, Larocca LM, Maira G, Fernandez E, Pallini R. Targeted therapy with bevacizumab and erlotinib tailored to the molecular profile of patients with recurrent glioblastoma. Preliminary experience. Acta Neurochirurg 155: 33–40, 2013. [DOI] [PubMed] [Google Scholar]
- 12.Deighton RF, McGregor R, Kemp J, McCulloch J, Whittle IR. Glioma pathophysiology: insights emerging from proteomics. Brain Pathol 20: 691–703, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fatimathas L, Moss SE. Annexins as disease modifiers. Histol Histopathol 25: 527–532, 2010. [DOI] [PubMed] [Google Scholar]
- 14.Foley J, Witte D, Chiu FC, Parysek LM. Expression of the neural intermediate filament proteins peripherin and neurofilament-66/alpha-internexin in neuroblastoma. Lab Invest 71: 193–199, 1994. [PubMed] [Google Scholar]
- 15.Fortin Ensign SP, Mathews IT, Symons MH, Berens ME, Tran NL. Implications of Rho GTPase signaling in glioma cell invasion and tumor progression. Front Oncol 3: 241, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh MK, Sharma P, Harbor PC, Rahaman SO, Haque SJ. PI3K-AKT pathway negatively controls EGFR-dependent DNA-binding activity of Stat3 in glioblastoma multiforme cells. Oncogene 24: 7290–7300, 2005. [DOI] [PubMed] [Google Scholar]
- 17.Gladson CL, Cheresh DA. Glioblastoma expression of vitronectin and the alpha v beta 3 integrin. Adhesion mechanism for transformed glial cells. J Clin Invest 88: 1924–1932, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goplen D, Wang J, Enger PO, Tysnes BB, Terzis AJ, Laerum OD, Bjerkvig R. Protein disulfide isomerase expression is related to the invasive properties of malignant glioma. Cancer Res 66: 9895–9902, 2006. [DOI] [PubMed] [Google Scholar]
- 19.Grossman SA, Batara JF. Current management of glioblastoma multiforme. Semin Oncol 31: 635–644, 2004. [DOI] [PubMed] [Google Scholar]
- 20.Halligan BD, Geiger JF, Vallejos AK, Greene AS, Twigger SN. Low cost, scalable proteomics data analysis using Amazon's cloud computing services and open source search algorithms. J Proteome Res 8: 3148–3153, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Halligan BD, Greene AS. Visualize: a free and open source multifunction tool for proteomics data analysis. Proteomics 11: 1058–1063, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hill JJ, Moreno MJ, Lam JC, Haqqani AS, Kelly JF. Identification of secreted proteins regulated by cAMP in glioblastoma cells using glycopeptide capture and label-free quantification. Proteomics 9: 535–549, 2009. [DOI] [PubMed] [Google Scholar]
- 23.Hiratsuka M, Inoue T, Toda T, Kimura N, Shirayoshi Y, Kamitani H, Watanabe T, Ohama E, Tahimic CG, Kurimasa A, Oshimura M. Proteomics-based identification of differentially expressed genes in human gliomas: down-regulation of SIRT2 gene. Biochem Biophys Res Commun 309: 558–566, 2003. [DOI] [PubMed] [Google Scholar]
- 24.Hoehenwarter W, Wienkoop S. Spectral counting robust on high mass accuracy mass spectrometers. Rapid Commun Mass Spectrom 24: 3609–3614, 2010. [DOI] [PubMed] [Google Scholar]
- 25.Hou LC, Veeravagu A, Hsu AR, Tse VC. Recurrent glioblastoma multiforme: a review of natural history and management options. Neurosurg Focus 20: E5, 2006. [DOI] [PubMed] [Google Scholar]
- 27.Jameson KL, Mazur PK, Zehnder AM, Zhang J, Zarnegar B, Sage J, Khavari PA. IQGAP1 scaffold-kinase interaction blockade selectively targets RAS-MAP kinase-driven tumors. Nat Med 19: 626–630, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang W, Cazacu S, Xiang C, Zenklusen JC, Fine HA, Berens M, Armstrong B, Brodie C, Mikkelsen T. FK506 binding protein mediates glioma cell growth and sensitivity to rapamycin treatment by regulating NF-kappaB signaling pathway. Neoplasia 10: 235–243, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson M, Sharma M, Henderson BR. IQGAP1 regulation and roles in cancer. Cell Signal 21: 1471–1478, 2009. [DOI] [PubMed] [Google Scholar]
- 30.Jung TY, Jung S, Ryu HH, Jeong YI, Jin YH, Jin SG, Kim IY, Kang SS, Kim HS. Role of galectin-1 in migration and invasion of human glioblastoma multiforme cell lines. J Neurosurg 109: 273–284, 2008. [DOI] [PubMed] [Google Scholar]
- 31.Kalinina J, Peng J, Ritchie JC, Van Meir EG. Proteomics of gliomas: initial biomarker discovery and evolution of technology. Neuro-oncology 13: 926–942, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karrlander M, Lindberg N, Olofsson T, Kastemar M, Olsson AK, Uhrbom L. Histidine-rich glycoprotein can prevent development of mouse experimental glioblastoma. PLoS One 4: e8536, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kong LM, Liao CG, Fei F, Guo X, Xing JL, Chen ZN. Transcription factor Sp1 regulates expression of cancer-associated molecule CD147 in human lung cancer. Cancer Sci 101: 1463–1470, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krex D, Klink B, Hartmann C, von Deimling A, Pietsch T, Simon M, Sabel M, Steinbach JP, Heese O, Reifenberger G, Weller M, Schackert G. Long-term survival with glioblastoma multiforme. Brain 130: 2596–2606, 2007. [DOI] [PubMed] [Google Scholar]
- 35.Lee EK, Han GY, Park HW, Song YJ, Kim CW. Transgelin promotes migration and invasion of cancer stem cells. J Proteome Res 9: 5108–5117, 2010. [DOI] [PubMed] [Google Scholar]
- 36.Lee JH, Pyon JK, Kim DW, Lee SH, Nam HS, Kim CH, Kang SG, Lee YJ, Park MY, Jeong DJ, Cho MK. Elevated c-Src and c-Yes expression in malignant skin cancers. J Exp Clin Cancer Res 29: 116, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu H, Sadygov RG, Yates JR., 3rd A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Analyt Chem 76: 4193–4201, 2004. [DOI] [PubMed] [Google Scholar]
- 38.Luwor RB, Stylli SS, Kaye AH. The role of Stat3 in glioblastoma multiforme. J Clin Neurosci 20: 907–911, 2013. [DOI] [PubMed] [Google Scholar]
- 39.Manjula S, Aroor AR, Raja A, Rao SN, Rao A. Elevation of serum ceruloplasmin levels in brain tumours. Acta Neurol Scand 86: 156–158, 1992. [DOI] [PubMed] [Google Scholar]
- 40.Maruo T, Ichikawa T, Kanzaki H, Inoue S, Kurozumi K, Onishi M, Yoshida K, Kambara H, Ouchida M, Shimizu K, Tamaru S, Chiocca EA, Date I. Proteomics-based analysis of invasion-related proteins in malignant gliomas. Neuropathology 33: 264–275, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsuda Y, Hagio M, Ishiwata T. Nestin: a novel angiogenesis marker and possible target for tumor angiogenesis. World J Gastroenterol 19: 42–48, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mirza SP. Quantitative mass spectrometry-based approaches in cardiovascular research. Circ Cardiovasc Genet 5: 477, 2012. [DOI] [PubMed] [Google Scholar]
- 43.Mohanam S, Jasti SL, Kondraganti SR, Chandrasekar N, Lakka SS, Kin Y, Fuller GN, Yung AW, Kyritsis AP, Dinh DH, Olivero WC, Gujrati M, Ali-Osman F, Rao JS. Down-regulation of cathepsin B expression impairs the invasive and tumorigenic potential of human glioblastoma cells. Oncogene 20: 3665–3673, 2001. [DOI] [PubMed] [Google Scholar]
- 44.Moin K, Day NA, Sameni M, Hasnain S, Hirama T, Sloane BF. Human tumour cathepsin B. Comparison with normal liver cathepsin B. Biochem J 285: 427–434, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mustafa DA, Sieuwerts AM, Zheng PP, Kros JM. Overexpression of colligin 2 in glioma vasculature is associated with overexpression of heat shock factor 2. Gene Regul Syst Biol 4: 103–107, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Niclou SP, Fack F, Rajcevic U. Glioma proteomics: status and perspectives. J Proteom 73: 1823–1838, 2010. [DOI] [PubMed] [Google Scholar]
- 47.Nicoll JA, Zunarelli E, Rampling R, Murray LS, Papanastassiou V, Stewart J. Involvement of apolipoprotein E in glioblastoma: immunohistochemistry and clinical outcome. Neuroreport 14: 1923–1926, 2003. [DOI] [PubMed] [Google Scholar]
- 48.Old WM, Meyer-Arendt K, Aveline-Wolf L, Pierce KG, Mendoza A, Sevinsky JR, Resing KA, Ahn NG. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol Cell Proteom 4: 1487–1502, 2005. [DOI] [PubMed] [Google Scholar]
- 49.Ornstein DL, Meehan KR, Zacharski LR. The coagulation system as a target for the treatment of human gliomas. Sem Thrombos Hemostas 28: 19–28, 2002. [DOI] [PubMed] [Google Scholar]
- 50.Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, Ray S, Majumdar SS, Bhattacharya S. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med 18: 1279–1285, 2012. [DOI] [PubMed] [Google Scholar]
- 51.Pedersen WA, Becker LE, Yeger H. Expression and distribution of peripherin protein in human neuroblastoma cell lines. Int J Cancer 53: 463–470, 1993. [DOI] [PubMed] [Google Scholar]
- 52.Perego C, Vanoni C, Massari S, Raimondi A, Pola S, Cattaneo MG, Francolini M, Vicentini LM, Pietrini G. Invasive behaviour of glioblastoma cell lines is associated with altered organisation of the cadherin-catenin adhesion system. J Cell Sci 115: 3331–3340, 2002. [DOI] [PubMed] [Google Scholar]
- 53.Petrik V, Saadoun S, Loosemore A, Hobbs J, Opstad KS, Sheldon J, Tarelli E, Howe FA, Bell BA, Papadopoulos MC. Serum alpha 2-HS glycoprotein predicts survival in patients with glioblastoma. Clin Chem 54: 713–722, 2008. [DOI] [PubMed] [Google Scholar]
- 54.Podgorski I, Sloane BF. Cathepsin B and its role(s) in cancer progression. Biochem Soc Sympos 263–276, 2003. [DOI] [PubMed] [Google Scholar]
- 55.Polisetty RV, Gautam P, Sharma R, Harsha HC, Nair SC, Gupta MK, Uppin MS, Challa S, Puligopu AK, Ankathi P, Purohit AK, Chandak GR, Pandey A, Sirdeshmukh R. LC-MS/MS analysis of differentially expressed glioblastoma membrane proteome reveals altered calcium signaling and other protein groups of regulatory functions. Mol Cell Proteom 11: M111 013565, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Raftopoulos I, Davaris P, Karatzas G, Karayannacos P, Kouraklis G. Level of alpha-catenin expression in colorectal cancer correlates with invasiveness, metastatic potential, and survival. J Surg Oncol 68: 92–99, 1998. [DOI] [PubMed] [Google Scholar]
- 57.Rajcevic U, Petersen K, Knol JC, Loos M, Bougnaud S, Klychnikov O, Li KW, Pham TV, Wang J, Miletic H, Peng Z, Bjerkvig R, Jimenez CR, Niclou SP. iTRAQ-based proteomics profiling reveals increased metabolic activity and cellular cross-talk in angiogenic compared with invasive glioblastoma phenotype. Mol Cell Proteom 8: 2595–2612, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sanchez-Laorden B, Viros A, Marais R. Mind the IQGAP. Cancer Cell 23: 715–717, 2013. [DOI] [PubMed] [Google Scholar]
- 59.Santosh V, Arivazhagan A, Sreekanthreddy P, Srinivasan H, Thota B, Srividya MR, Vrinda M, Sridevi S, Shailaja BC, Samuel C, Prasanna KV, Thennarasu K, Balasubramaniam A, Chandramouli BA, Hegde AS, Somasundaram K, Kondaiah P, Rao MR. Grade-specific expression of insulin-like growth factor-binding proteins-2, -3, and -5 in astrocytomas: IGFBP-3 emerges as a strong predictor of survival in patients with newly diagnosed glioblastoma. Cancer Epidemiol Biomark Prevent 19: 1399–1408, 2010. [DOI] [PubMed] [Google Scholar]
- 60.Schmidt M, Micke P, Hengstler JG. IGKC and prognosis in breast cancer. Clin Cancer Res 19: 304, 2013. [DOI] [PubMed] [Google Scholar]
- 61.Schwartz SA, Weil RJ, Johnson MD, Toms SA, Caprioli RM. Protein profiling in brain tumors using mass spectrometry: feasibility of a new technique for the analysis of protein expression. Clin Cancer Res 10: 981–987, 2004. [DOI] [PubMed] [Google Scholar]
- 62.Seznec J, Silkenstedt B, Naumann U. Therapeutic effects of the Sp1 inhibitor mithramycin A in glioblastoma. J Neuro-oncol 101: 365–377, 2011. [DOI] [PubMed] [Google Scholar]
- 62a.Shonka N. http://www.clinicaltrials.gov/ct2/show/NCT01493219, U.S. National Institutes of Health. [PubMed]
- 63.Simon HU, Mills GB, Kozlowski M, Hogg D, Branch D, Ishimi Y, Siminovitch KA. Molecular characterization of hNRP, a cDNA encoding a human nucleosome-assembly-protein-I-related gene product involved in the induction of cell proliferation. Biochem J 297: 389–397, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tan B, Young DA, Lu ZH, Wang T, Meier TI, Shepard RL, Roth K, Zhai Y, Huss K, Kuo MS, Gillig J, Parthasarathy S, Burkholder TP, Smith MC, Geeganage S, Zhao G. Pharmacological inhibition of nicotinamide phosphoribosyltransferase (NAMPT), an enzyme essential for NAD+ biosynthesis, in human cancer cells: metabolic basis and potential clinical implications. J Biol Chem 288: 3500–3511, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tung JJ, Hobert O, Berryman M, Kitajewski J. Chloride intracellular channel 4 is involved in endothelial proliferation and morphogenesis in vitro. Angiogenesis 12: 209–220, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang Y, Rudnick PA, Evans EL, Li J, Zhuang Z, Devoe DL, Lee CS, Balgley BM. Proteome analysis of microdissected tumor tissue using a capillary isoelectric focusing-based multidimensional separation platform coupled with ESI-tandem MS. Analyt Chem 77: 6549–6556, 2005. [DOI] [PubMed] [Google Scholar]
- 67.White CD, Brown MD, Sacks DB. IQGAPs in cancer: a family of scaffold proteins underlying tumorigenesis. FEBS Lett 583: 1817–1824, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wojtukiewicz MZ, Sierko E, Klement P, Rak J. The hemostatic system and angiogenesis in malignancy. Neoplasia 3: 371–384, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang Y, Liu Y, Yao X, Ping Y, Jiang T, Liu Q, Xu S, Huang J, Mou H, Gong W, Chen K, Bian X, Wang JM. Annexin 1 released by necrotic human glioblastoma cells stimulates tumor cell growth through the formyl peptide receptor 1. Am J Pathol 179: 1504–1512, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhai H, Acharya S, Gravanis I, Mehmood S, Seidman RJ, Shroyer KR, Hajjar KA, Tsirka SE. Annexin A2 promotes glioma cell invasion and tumor progression. J Neurosci 31: 14346–14360, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang G, Fenyo D, Neubert TA. Use of DNA ladders for reproducible protein fractionation by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) for quantitative proteomics. J Proteome Res 7: 678–686, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhao Y, Zhang W, Guo Z, Ma F, Wu Y, Bai Y, Gong W, Chen Y, Cheng T, Zhi F, Zhang Y, Wang J, Jiang B. Inhibition of the transcription factor Sp1 suppresses colon cancer stem cell growth and induces apoptosis in vitro and in nude mouse xenografts. Oncol Rep 30: 1782–1792, 2013. [DOI] [PubMed] [Google Scholar]
- 73.Zhou L, Wang Y, Zhang YT, Geng YP, Si LS, Wang YL. Proteomics-based analysis of a pair of glioma cell lines with different tumor forming characteristics. Neurosci Lett 401: 59–64, 2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.