

Abstract
Outcomes for lung transplantation are the worst of any solid organ, and ischemia-reperfusion injury (IRI) limits both short- and long-term outcomes. Presently no therapeutic agents are available to prevent IRI. Sphingosine 1-phosphate (S1P) modulates immune function through binding to a set of G protein-coupled receptors (S1PR1-5). Although S1P has been shown to attenuate lung IRI, the S1P receptors responsible for protection have not been defined. The present study tests the hypothesis that protection from lung IRI is primarily mediated through S1PR1 activation. Mice were treated with either vehicle, FTY720 (a nonselective S1P receptor agonist), or VPC01091 (a selective S1PR1 agonist and S1PR3 antagonist) before left lung IR. Function, vascular permeability, cytokine expression, neutrophil infiltration, and myeloperoxidase levels were measured in lungs. After IR, both FTY720 and VPC01091 significantly improved lung function (reduced pulmonary artery pressure and increased pulmonary compliance) vs. vehicle control. In addition, FTY720 and VPC01091 significantly reduced vascular permeability, expression of proinflammatory cytokines (IL-6, IL-17, IL-12/IL-23 p40, CC chemokine ligand-2, and TNF-α), myeloperoxidase levels, and neutrophil infiltration compared with control. No significant differences were observed between VPC01091 and FTY720 treatment groups. VPC01091 did not significantly affect elevated invariant natural killer T cell infiltration after IR, and administration of an S1PR1 antagonist reversed VPC01091-mediated protection after IR. In conclusion, VPC01091 and FTY720 provide comparable protection from lung injury and dysfunction after IR. These findings suggest that S1P-mediated protection from IRI is mediated by S1PR1 activation, independent of S1PR3, and that selective S1PR1 agonists may provide a novel therapeutic strategy to prevent lung IRI.
Keywords: lung transplantation, inflammation, primary graft dysfunction, barrier function, invariant natural killer T cells
outcomes for lung transplantation are the worst of any solid organ (6, 17). Despite significant advancements over the past decade in lung transplantation, outcomes remain poor, and both short- and long-term graft survival is limited by the inherent threat of ischemia-reperfusion injury (IRI). Mechanisms involved in lung IRI include oxidative stress, epithelial cell apoptosis, alveolar macrophage activation, T-cell activation, and neutrophil infiltration (7, 32, 37, 40, 48, 49). Presently no therapeutic agents are available to prevent lung IRI, and treatment strategies are limited to maintaining pulmonary mechanics and oxygenation capacity.
Sphingolipids are ubiquitous components of cell membranes, and their metabolites (e.g., ceramide, sphingosine, and sphingosine 1-phosphate) are established regulators of a vast number of cellular processes (24). Sphingosine kinase 1 and 2 phosphorylate sphingosine to generate sphingosine 1-phosphate (S1P), a biologically active lipid growth factor that binds to a family of five G protein-coupled receptors (S1PR1-5) to regulate a spectrum of biological functions including proliferation, cell survival, angiogenesis, extracellular matrix assembly, endothelial cell barrier integrity, and immune cell trafficking and function (5, 14, 19, 33). Okazaki et al. (30) have demonstrated that S1P increases oxygenation capacity following experimental lung transplantation while decreasing proinflammatory cytokine production, endothelial cell apoptosis, and neutrophil numbers. Other studies have shown that S1P or FTY720, a nonspecific agonist for S1PR1 and S1PR3-5 (3, 25), provides significant protection in various models of acute lung injury (8, 28, 31). Although these studies serve as a foundation for potential S1P receptor-targeted therapies in lung injury and transplantation, pharmacological S1P analogs with differential receptor subtype affinities have yet to be examined in the setting of lung IRI or transplantation. Thus the purpose of this study was to evaluate the potential protective advantages afforded by VPC01091, a novel sphingosine analog, which is a selective S1PR1 agonist and S1PR3 antagonist (50), on lung IRI. In addition, we sought to concurrently evaluate FTY720 to determine the differential effects of S1PR3 targeting on lung IRI. We approached this study with the hypothesis that selective S1PR1 agonism will attenuate lung IRI and that S1PR3 antagonism would provide an additional protective advantage, as S1PR3 has been implicated as a biomarker of acute lung injury (41).
MATERIALS AND METHODS
Study design and animals.
Wild-type mice (C57BL/6, 8–12 wk; The Jackson Laboratory, Bar Harbor, ME) were randomly assigned to six groups. All animals were treated with either vehicle or test compound via intraperitoneal injection 30 min before surgery. Two groups (sham and IR) were treated with 0.2 ml vehicle (3% fatty acid-free BSA/PBS solution; Sigma, St. Louis, MO). Two groups (sham and IR) were treated with FTY720 [2-amino-2-(4-octylphenethyl)propane-1,3-diol, 0.3 mg/kg; Novartis, Basel, Switzerland], and two groups (sham and IR) were treated with VPC01091 [([1R,3S]-1-amino-3-[4-octylphenyl]cyclopentyl)methanol, 1.5 mg/kg, gift from Abbott Laboratories in Worcester, MA]. A separate IR group entailed the coadministration of VPC01091 with VPC44116 [(R)-(3-amino-4-[(3-octylphenyl)amino]-4-oxobutyl)phosphonic acid, 10 mg/kg, gift from Dr. Frank Foss at the University of Texas Arlington], a selective S1PR1 antagonist (12). Mice then underwent sham surgery or lung IR 30 min after intraperitoneal treatment, as this time frame has been established to achieve maximal drug effectiveness (2). A prior time-course study has also demonstrated that FTY720-mediated barrier enhancement is maximized at 30–60 min following administration (9). The present study conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institute of Health and was approved by the University of Virginia Institutional Animal Care and Use Committee.
Murine lung IRI.
An established murine model of lung IRI was utilized as previously described by our laboratory (37, 48). Inhalational isoflurane anesthesia and orotracheal intubation permitted mechanical ventilation at 120 strokes/min with room air. Heparin was administered via the right external jugular vein (20 U/kg), and the left pulmonary hilum was exposed through an anterolateral thoracotomy at the third intercostal space. A 6-0 Prolene suture was passed around the left pulmonary hilum, and the two suture ends were passed through PE-60 tubing to permit hilar occlusion via tightening of suture and surgical clip application. Analgesia was administered (buprenorphine, 0.2 mg/kg) by intraperitoneal injection, and animals were returned to their cage during the 1 h of left-lung ischemia. Mice then underwent repeat anesthesia and intubation, and the hilar occlusion was released to begin reperfusion. Animals were then returned to their cages, whereupon reperfusion was continued for 2 h before functional evaluation, bronchoalveolar lavage, and histological analysis. Sham groups were identical to IR groups except that the left hilum was not occluded.
Pulmonary function.
Pulmonary function at the end of reperfusion was measured using an isolated, buffer-perfused lung apparatus (Hugo Sachs Elektronik, March-Huggstetten, Germany) as previously described (47). Mice were anesthetized and maintained on intratracheal ventilation (tidal volume = 7 μl/g body wt, rate = 100 strokes/min, positive end-expiratory pressure = 2 cmH2O) before exsanguination by inferior vena caval transection. The pulmonary artery was cannulated, and a left ventriculotomy permitted perfusate drainage. Lungs were perfused at a flow rate of 60 μl/g body wt per min with Krebs-Henseleit buffer. Following a 5-min period of equilibration, functional data (pulmonary artery pressure and pulmonary compliance) were recorded using PULMODYN data acquisition software (Hugo Sachs Elektronik) over an additional 5 min.
Bronchoalveolar lavage.
Following measurement of lung function, the left lung was isolated via ligation of the right pulmonary hilum with a surgical clip. An anterior tracheotomy was then performed and permitted intratracheal placement of a 20-G angiocatheter. A circumferential suture was secured around the trachea to limit pericatheter drainage. Two consecutive aspirates of 0.4 ml of 0.9% sodium chloride were then performed through the intratracheal cannula. Left lung bronchoalveolar lavage (BAL) fluid was immediately centrifuged at 4°C (1,500 revolution/min for 6 min), and supernatant was stored at −80°C.
Cytokine and myeloperoxidase measurements.
As previously described (36), cytokines were quantified in BAL fluid using a multiplex ELISA cytokine panel (Bio-Rad Laboratories, Hercules, CA), and myeloperoxidase (MPO) concentration in BAL fluid was measured by ELISA (Hycult Biotech, Uden, Netherlands).
Immunohistochemistry and neutrophil counting.
With the use of separate groups of animals, lungs were inflation fixed at 10 cmH2O with formalin at 4°C for 24 h before placement in 70% ethanol for paraffin embedding. Lung sections were immunostained for neutrophils using the Vectastain ABC kit (Vector Laboratories, Burlingame, CA) as previously described (47). Rat anti-mouse neutrophil antibody (AbD Serotec, Raleigh, NC) and alkaline phosphatase-conjugated anti-rat IgG (Sigma) secondary antibody were utilized. The signals were detected using Fast-Red (Sigma). The negative control utilized purified normal rat IgG (eBioscience, San Diego, CA). Neutrophil counting was performed by a blinded investigator. Three semistandardized fields per lung were counted at ×20 magnification, and the mean of these three values established the final neutrophil count per high-powered field for each mouse.
Pulmonary vascular permeability.
Vascular permeability in lungs was estimated using the Evans blue dye extravasation technique, which is an index of change in protein permeability, as previously described (22). With the use of separate groups of animals (n = 5/group), Evans blue (20 mg/kg, Sigma) was injected intravenously via the tail vein 30 min before euthanasia. The pulmonary vasculature was then perfused for 10 min with PBS to remove intravascular dye. Lungs were then homogenized in PBS to extract the Evans blue and centrifuged. The absorption of Evans blue was measured in the supernatant at 620 nm and corrected for the presence of heme pigments: A620corrected = A620 − (1.426 × A740 + 0.030). The concentration of Evans blue was determined according to a standard curve and expressed as micrograms/gram wet lung weight.
Flow cytometry.
Quantification of invariant natural killer T (iNKT) cells via flow cytometry was performed as previously described (37). Left lungs from mice were minced and incubated for 15 min at 37°C with collagenase type 1A (Sigma) in Dulbecco's PBS with 0.5% BSA and 2 mM EDTA. The lung tissue suspension was then passed through a 40-μm cell strainer (BD Falcon, Bedford, MA) and centrifuged at 1,000 revolution/min for 10 min. After treatment with red blood cell lysis buffer, the cell pellet was washed and resuspended in fluorescence-activated cell sorting (FACS) buffer (0.1% BSA, 0.01% sodium azide in PBS). Cells were stained with 7-aminoactinomycin (Invitrogen, Carlsbad, CA), PerCP-Cy5.5-labeled CD45 (eBioscience), PE-labeled CD1d tetramer loaded with PBS57 (1:500), an analog of α-galactosylceramide (αGalCer) (NIH Tetramer Facility, Emory University, Atlanta, GA), and FITC-labeled CD4 (eBioscience). For cell counting, 100 μl of cell suspension was mixed thoroughly with 100 μl of Caltag Counting Beads (Life Technologies, Frederick, MD) before acquisition by FACS. At least 1,000 bead events were acquired to ensure the accuracy of the assay. The absolute number of any gated cell population was calculated as follows: CD45+ cell absolute count (per lung) = [events of CD45+ cells counted/total number of beads counted (A + B) × input bead number]/lung. The total number of iNKT cells (per lung) was calculated by multiplying the CD45+ cell number and the percentage of the CD4+CD1d tetramer+ subset. For example, the CD4+CD1d tetramer+ cell number (per lung) = (total CD45+ cell number) × (percent of CD4+CD1d tetramer+ cells gated on the CD45+ cell population).
Statistics.
Statistical analyses were performed using one-way ANOVA with a post-hoc Tukey's multiple-comparisons correction. A P value of <0.05 represented statistical significance. Results are presented as the means ± SE.
RESULTS
Pulmonary function after IR is improved by VPC01091 and FTY720 treatment.
To determine whether S1PR1 agonism improves lung function after IR, mice were treated with VPC01091 or FTY720 before ischemia or sham surgery. Vehicle-treated mice demonstrated significant lung dysfunction following IR as shown by increased pulmonary artery pressure and decreased pulmonary compliance (Fig. 1). Compared with vehicle, both FTY720 and VPC01091 significantly decreased pulmonary artery pressure and increased pulmonary compliance after IR. No significant differences were observed between sham groups treated with vehicle, VPC01091, or FTY720 (Fig. 1). These results demonstrate that selective agonism of S1PR1 by VPC01091 improves lung function after IR to the same level as FTY720.
Fig. 1.
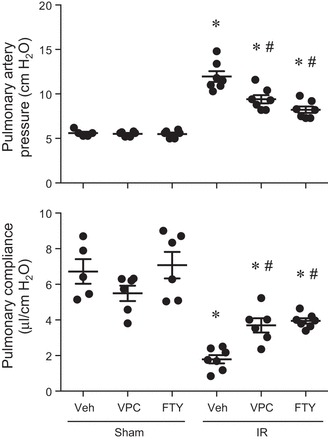
VPC01091 and FTY720 treatment improves lung function after ischemia-reperfusion (IR). Mice were pretreated with vehicle (Veh), VPC01091 (VPC), or FTY720 (FTY) 30 min before sham surgery or ischemia. Pulmonary artery pressure and pulmonary compliance were measured after 2 h of reperfusion. Results are presented as means ± SE. *P < 0.05 vs. vehicle sham, #P < 0.05 vs. vehicle IR, n = 5–7/group.
VPC01091 and FTY720 reduce pulmonary vascular permeability after IR.
To investigate the extent to which VPC01091 or FTY720 affects vascular permeability after IR, pulmonary vascular leak was assessed using the Evans blue dye extravasation technique. IR significantly increased vascular permeability, which was significantly attenuated by VPC01091 or FTY720 (Fig. 2). These results demonstrate that FTY720-mediated attenuation of vascular permeability after IR can be reproduced by selective S1PR1 agonism by VPC01091.
Fig. 2.
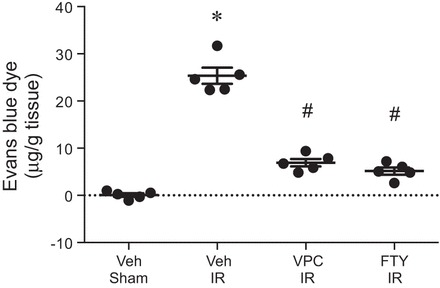
VPC01091 and FTY720 treatment attenuates pulmonary vascular permeability after IR. Mice were pretreated with vehicle, VPC01091, or FTY720 30 min before sham surgery or ischemia. Vascular permeability was assessed after 2 h of reperfusion by measuring Evans blue dye content in the lung (μg/g lung tissue) as described in materials and methods. Results are presented as means ± SE. *P < 0.05 vs. vehicle sham, #P < 0.05 vs. vehicle IR, n = 5/group.
VPC01091 and FTY720 attenuate proinflammatory cytokine production following IR.
Pulmonary inflammation after IR was assessed by measuring levels of proinflammatory cytokines in BAL fluid. IR significantly increased expression of IL-6, IL-17, IL-12 p70, IL-12/IL-23 p40, CC chemokine ligand-2 (CCL2), and TNF-α in vehicle-treated animals (Fig. 3). VPC01091 treatment resulted in significantly decreased levels of IL-6, IL-17, IL-12 p70, IL-12/IL-23 p40, CCL2, and TNF-α following IR vs. vehicle treatment. Similarly, FTY720 treatment significantly decreased levels of IL-6, IL-17, IL-12/IL-23 p40, CCL2, and TNF-α after IR vs. vehicle treatment. Cytokine levels were similar among sham animals treated with vehicle, FTY720, or VPC01091 (Fig. 3). These results demonstrate that FTY720 and VPC01091 have similar inhibitory effects on proinflammatory cytokine expression after IR.
Fig. 3.
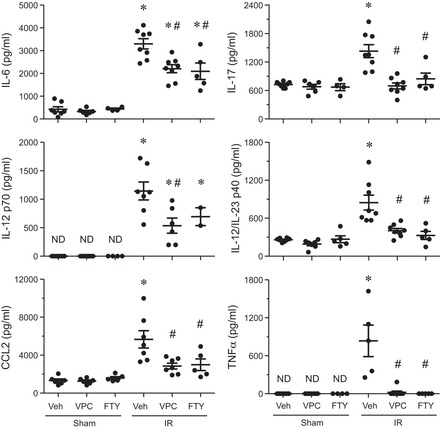
VPC01091 and FTY720 treatment before IR results in a significant decrease in proinflammatory cytokines vs. vehicle-treated IR control. Results are presented as means ± SE. *P < 0.05 vs. vehicle sham, #P < 0.05 vs. vehicle IR; n = 5–8/group. ND, not detectable, CCL2, CC chemokine ligand-2.
Neutrophil activation and infiltration after IR are decreased by VPC01091 and FTY720 treatment.
Neutrophil infiltration is a hallmark of lung inflammation after IR, and thus neutrophil numbers and activation status were assessed in lungs. Immunostaining of lung sections demonstrated that elevated neutrophil infiltration after IR was significantly attenuated by both VPC01091 and FTY720 treatments (Fig. 4, A and B). The concentration of MPO, a peroxidase enzyme abundantly present in neutrophil granulocytes and released upon activation, in BAL fluid was significantly increased after IR vs. sham in vehicle-treated animals (Fig. 4C). Both VPC01091 and FTY720 treatments resulted in significantly decreased MPO concentration vs. vehicle-treated IR. There were no significant differences in neutrophil counts or MPO levels between sham animals treated with vehicle, VPC01091, or FTY720 (data not shown). These data suggest that FTY720-mediated effects on neutrophil infiltration and activation after IR are reproduced by selective S1PR1 agonism by VPC01091.
Fig. 4.
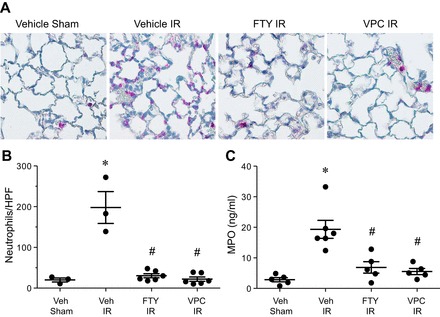
Neutrophil infiltration and activation after IR is significantly decreased by VPC01091 or FTY720 treatment compared with vehicle-treated mice. A: representative immunostaining of neutrophils (pink staining) within indicated groups; ×40 magnification. B: quantification of neutrophils per high-powered field (HPF) for each group. C: myeloperoxidase (MPO) levels in bronchoalveolar lavage fluid after IR were significantly reduced by VPC01091 and FTY720 treatment vs. vehicle treatment. No significant differences in neutrophil counts or MPO levels between vehicle, VPC01091, and FTY720 treatment of sham animals were observed (data not shown). Results are presented as means ± SE. *P < 0.05 vs. vehicle sham; #P < 0.05 vs. vehicle IR, n = 3–6/group.
S1PR1 agonism is essential for VPC01091-mediated attenuation of lung IRI.
To determine whether the S1PR3 antagonist functionality of VPC01091 potentially contributes to VPC01091-mediated protection after IR, an S1PR1 antagonist, VPC44116, was coadministered with VPC01091. Coadministration of VPC01091 and VPC44116 resulted in a reversal of protection from lung dysfunction after IR (Fig. 5). VPC1091/VPC44116 combined treatment significantly reversed the VPC1091-mediated decrease in pulmonary artery pressure after IR. Similarly, VPC1091/VPC44116 combined treatment also reversed VPC01091-mediated improvement in pulmonary compliance although this did not reach significance. These results confirm S1PR1 as the principle protective mediator of lung IRI by S1P analogs such as VPC01091.
Fig. 5.
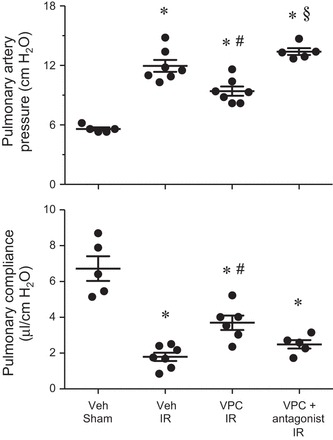
VPC01091-mediated functional protection after IR is reversed with coadministration of an S1PR1 antagonist (VPC44116). The VPC + antagonist group is shown compared with relevant groups shown in Fig. 1. Results are presented as means ± SE. *P < 0.05 vs. vehicle sham; #P < 0.05 vs. vehicle IR, §P < 0.05 vs. VPC IR, n = 5–7/group.
VPC01091 does not affect infiltration of iNKT cells after lung IR.
To identify iNKT cells, a PE-labeled CD1d tetramer loaded with an analog of αGalCer was utilized as previously described (37). The total number of iNKT cells (CD4+ CD1d tetramer+) was increased over fourfold in the left lung after IR (71,706 ± 8,086 iNKT cells) compared with sham (16,977 ± 3,357 iNKT cells) in vehicle-treated mice (Fig. 6). Treatment with VPC01091 did not significantly affect iNKT cell numbers after IR (90,357 ± 10,429 iNKT cells) vs. vehicle (Fig. 6).
Fig. 6.
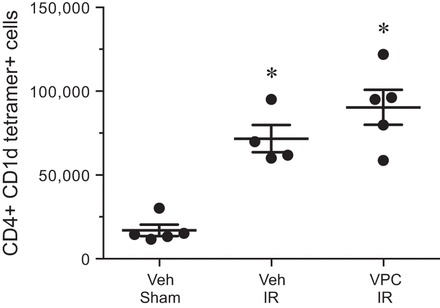
VPC01091 does not affect invariant natural killer T (iNKT) cell infiltration after IR. Total iNKT cell numbers (CD4+ CD1d tetramer+ cells) were counted in left lungs by flow cytometry as described in materials and methods. Mice pretreated with vehicle or VPC01091 demonstrated similar and significant elevations in iNKT cell numbers after IR vs. sham. Results are presented as means ± SE. *P < 0.05 vs. vehicle sham, n = 4–5/group.
DISCUSSION
Use of pharmacological S1P receptor-targeted drugs have demonstrated promise in the regulation of immune-mediated disease through the inhibition of lymphocyte egress from lymphoid organs (46). This strategy has been adopted to attenuate acute injury and graft rejection in kidney and liver transplantation with a proposed protective mechanism through S1PR1 binding (1, 2, 42). Several studies have supported the application of S1P and FTY720 in the reduction of injury in experimental lung transplantation models (18, 30). Although these studies support a protective role for S1P and S1P analogs in IRI, further understanding of the role of specific S1P receptor subtypes in lung IRI is needed. S1PR3 has been linked to decreased epithelial integrity within the lung in addition to promoting airway hyperreactivity, systemic hypertension, coronary artery vasoconstriction, and profibrotic responses within the lung (13, 15, 23, 29, 39, 43). Additionally, the nitrated form of S1PR3 is increased in the plasma of mice and humans with sepsis-induced acute lung injury, and reduced S1PR3 expression is associated with an attenuation of increased vascular permeability during acute lung injury (41). Thus the design of agents with differential activities at S1P receptor subtypes may help provide optimal protection from lung IRI with reduced negative side effects.
With this understanding, the present study evaluated the efficacy of a novel sphingosine analog, VPC01091, that serves as a selective S1PR1 agonist and S1PR3 antagonist (50). Comparison of VPC01091 to the nonselective FTY720 agonist demonstrated an equivalent level of protection, suggesting that that S1P- and FTY720-mediated protection is achieved primarily through S1PR1 activation. Although FTY720 also acts on S1PR4 and S1PR5 receptors, S1PR5 is exclusively expressed in the brain and skin (27). The biological activities of S1PR4 are not well established; however, a recent study has demonstrated that S1PR4 deficiency results in impaired dendritic cell migration, cytokine secretion, and Th-17 cell differentiation (35). This supports a potential mechanistic role for S1PR4 in IRI pathogenesis (21, 33). The present data demonstrate that antagonism at S1PR3 does not result in altered therapeutic efficacy of VPC01091. Although superiority of VPC01091 over FTY720 in protection was not demonstrated in the present study, the use of VPC01091 may be advantageous to avoid potential long-term effects of S1PR3 receptor agonism by S1P, FTY720, or similar nonspecific agonists.
Important to the advancement of pharmacological S1P receptor agonist therapy is the timing of delivery, as S1P receptor expression at the cell membrane can vary according to the cell activation state (26). Graeler et al. (16) demonstrated that activation of CD4+ T cells results in decreased S1P receptor expression, suppressing the potentially beneficial effects of S1P administration after onset of injury (16). However, pretreatment before T-cell activation results in a significant inhibition of chemokine-directed migration. These prior findings provided rationale for the use a pretreatment strategy in the present study and support the translation of this therapy to the treatment of the donor lung or transplant recipient before transplantation.
Debate persists regarding the mechanisms of protection afforded by S1P or FTY720 administration, supporting our application of VPC01091 as a novel agent for prevention of lung IRI. FTY720 has been demonstrated to have more potent effects than S1P on immune cell trafficking and recruitment to secondary lymphoid organs, as it is not metabolized as efficiently as the natural ligand (19). These findings introduce potential limitations to S1P delivery for prevention of IRI, supporting the design and utilization of selective S1P receptor agonists that are optimized for maximal biological effect. FTY720 treatment was found to downregulate S1PR1 expression, creating a temporary pharmacological S1PR1-null state in lymphocytes (26). This strategy has been adopted in models of renal IRI, with antagonist studies suggesting the mechanism of activity to be at S1PR1 (2). The coadministration of VPC01091 with a selective S1PR1 antagonist (VPC44116) in the present study supports a similar mechanism of protection from lung IRI through S1PR1 agonism.
A multitude of studies suggests a central role for S1P signaling in the maintenance of endothelial barrier function (44). S1P and FTY720 have been suggested to sustain the endothelial cell barrier during a state of inflammation through S1PR1 activation and induction of hepatocyte growth factor, acting through a Gi-coupled receptor, tyrosine kinases, and lipid rafts (4, 10, 11). Parallel studies with S1P and FTY720 in the setting of reduced S1PR1 expression (via siRNA) have demonstrated an absence of effect on pulmonary endothelial barrier enhancement, suggesting that the protective effects are dependent on the S1PR1 (9). Although these mechanisms of protective actions remain unclear, the present study importantly demonstrates that both FTY720 and VPC01091 provide similar protection from lung IRI, including potent preservation of endothelial barrier function. The reduction in vascular permeability by FTY720 and VPC01091 after IR (Fig. 2) could, however, result from direct effects on endothelial cells, indirect effects of reduced proinflammatory cytokines/chemokines, or both. Furthermore, while validating prior studies that support the efficacy of FTY720 in attenuation of IRI, our utilization of VPC01091 demonstrates that antagonistic activity at the S1PR3 receptor may not limit therapeutic potential and suggests VPC01091 as a more strategic therapeutic approach for prevention of lung IRI through S1PR1 agonism (18).
Prior study has demonstrated that S1P analogs reduce inflammation through the negative regulation of IL-12p70 following LPS administration (34). Furthermore, S1P is decreased in patients with cystic fibrosis, and supplementation has demonstrated the potential to rescue major histocompatibility complex-II and CD40 expression on lung dendritic cells (45). These data are supported by our finding that protection from lung IRI by FTY720 and VPC01091 was associated with decreased IL-12 (p70) expression as well as IL-12/IL-23 (p40) expression (p40 is a subunit of both IL-12 and IL-23), suggesting mechanistic effects on the dendritic cell-T-cell axis during lung IRI. The reduction in IL-12/IL-23 by FTY720 and VPC01091 likely contributed to the observed decrease in IL-17 production after IR, which confirms our previous results that demonstrate a principle role for IL-17 production by iNKT cells in lung IRI (37). Thus it is possible that S1PR1 agonism attenuates lung IRI, at least in part, by dampening the IL-23/IL-17 axis. This becomes more relevant because we observed that VPC01091 did not affect iNKT cell trafficking into the lung after IR (Fig. 6), which supports findings by Hwang et al. (20) showing that S1PR1 agonism affects NKT cells largely by inhibiting cytokine production rather than affecting migration (20). Thus results of the present study suggest that S1PR1 agonism potently attenuates lung IRI by modulating both endothelial barrier function and iNKT cell activation.
Although the present study provides important insights into the potential for S1P receptor-targeted therapies in prevention of lung IRI, inherent limitations exist. First, the lung IRI model entails warm ischemia and reperfusion of the lung but does not involve transplantation. Although this model has been validated by prior study and is an accepted model for mechanistic studies in IRI, immunoregulatory effects of these agents on donor-recipient cell interactions cannot be concluded from the present study. Second, the present study involves a 2-h period of reperfusion and thus focuses on acute IRI. Although acute injury and chronic rejection pathologies have causal and associative linkage in lung transplantation, no conclusions regarding the effects of these compounds on chronic graft function can be made from the present study. Importantly, however, we predict that VPC01091, through prevention of IRI, would benefit long-term immunoregulation and graft function. Thus it is possible that S1PR1 agonism and paired S1PR3 antagonism may provide optimal S1P receptor-targeted prevention of rejection pathology after lung transplant while limiting the profibrotic activity that has been associated with S1PR3 receptor activation (5). This hypothesis is based on a prior study in a bleomycin model of lung injury that demonstrated an exacerbation of lung injury with diffuse alveolar damage and more significant hyaline membrane deposition with repeat FTY720 administration (38).
In conclusion, the present study demonstrates that S1P analogs provide a promising modality for the prevention of lung IRI. The finding that VPC01091 affords equal protection to FTY720 in lung IRI demonstrates that the protective mechanisms, such as preservation of endothelial barrier function or modulation of iNKT cell activation, are primarily dependent on S1PR1 agonism. Use of VPC01091 may, therefore, be a more effective approach to S1P receptor-targeted therapy, as it avoids the limited potency of S1P alone while also avoiding potential deleterious effects of S1P3-mediated profibrotic processes after lung injury.
GRANTS
This study was funded by NIH grants T32HL007849 (I. Kron), R01HL077301 (V. Laubach), and R01GM067958 (K. Lynch).
DISCLOSURES
VPC01091 is claimed in an issued U.S. Patent owned by the University of Virginia (with Kevin R. Lynch as an inventor) and is currently unlicensed. There are no other conflicts of interest declared by the remaining authors.
AUTHOR CONTRIBUTIONS
Author contributions: M.L.S., A.K.S., I.L.K., K.R.L., and V.E.L. conception and design of research; M.L.S., A.K.S., Y.Z., E.J.C., and W.F.J. performed experiments; M.L.S., A.K.S., Y.Z., E.J.C., and W.F.J. analyzed data; M.L.S., A.K.S., I.L.K., K.R.L., and V.E.L. interpreted results of experiments; M.L.S., Y.Z., and V.E.L. prepared figures; M.L.S. drafted manuscript; M.L.S., A.K.S., K.R.L., and V.E.L. approved final version of manuscript; K.R.L. and V.E.L. edited and revised manuscript.
REFERENCES
- 1.Anselmo DM, Amersi FF, Shen XD, Gao F, Katori M, Lassman C, Ke B, Coito AJ, Ma J, Brinkmann V, Busuttil RW, Kupiec-Weglinski JW, Farmer DG. FTY720 pretreatment reduces warm hepatic ischemia reperfusion injury through inhibition of T-lymphocyte infiltration. Am J Transplant 2: 843–849, 2002. [DOI] [PubMed] [Google Scholar]
- 2.Awad AS, Ye H, Huang L, Li L, Foss FW Jr, Macdonald TL, Lynch KR, Okusa MD. Selective sphingosine 1-phosphate 1 receptor activation reduces ischemia-reperfusion injury in mouse kidney. Am J Physiol Renal Physiol 290: F1516–F1524, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Brinkmann V, Davis MD, Heise CE, Albert R, Cottens S, Hof R, Bruns C, Prieschl E, Baumruker T, Hiestand P, Foster CA, Zollinger M, Lynch KR. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem 277: 21453–21457, 2002. [DOI] [PubMed] [Google Scholar]
- 4.Camp SM, Bittman R, Chiang ET, Moreno-Vinasco L, Mirzapoiazova T, Sammani S, Lu X, Sun C, Harbeck M, Roe M, Natarajan V, Garcia JG, Dudek SM. Synthetic analogs of FTY720 [2-amino-2-(2-[4-octylphenyl]ethyl)-1,3-propanediol] differentially regulate pulmonary vascular permeability in vivo and in vitro. J Pharmacol Exp Ther 331: 54–64, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiyo M, Iwata T, Webb TJ, Vasko MR, Thompson EL, Heidler KM, Cummings OW, Yoshida S, Fujisawa T, Brand DD, Wilkes DS. Silencing S1P1 receptors regulates collagen-V reactive lymphocyte-mediated immunobiology in the transplanted lung. Am J Transplant 8: 537–546, 2008. [DOI] [PubMed] [Google Scholar]
- 6.Christie JD, Edwards LB, Kucheryavaya AY, Aurora P, Dobbels F, Kirk R, Rahmel AO, Stehlik J, Hertz MI. The Registry of the International Society for Heart and Lung Transplantation: twenty-seventh official adult lung and heart-lung transplant report–2010. J Heart Lung Transplant 29: 1104–1118, 2010. [DOI] [PubMed] [Google Scholar]
- 7.de Perrot M, Young K, Imai Y, Liu M, Waddell TK, Fischer S, Zhang L, Keshavjee S. Recipient T cells mediate reperfusion injury after lung transplantation in the rat. J Immunol 171: 4995–5002, 2003. [DOI] [PubMed] [Google Scholar]
- 8.Ding R, Han J, Tian Y, Guo R, Ma X. Sphingosine-1-phosphate attenuates lung injury induced by intestinal ischemia/reperfusion in mice: role of inducible nitric-oxide synthase. Inflammation 35: 158–166, 2012. [DOI] [PubMed] [Google Scholar]
- 9.Dudek SM, Camp SM, Chiang ET, Singleton PA, Usatyuk PV, Zhao Y, Natarajan V, Garcia JG. Pulmonary endothelial cell barrier enhancement by FTY720 does not require the S1P1 receptor. Cell Signal 19: 1754–1764, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ephstein Y, Singleton PA, Chen W, Wang L, Salgia R, Kanteti P, Dudek SM, Garcia JG, Jacobson JR. Critical role of S1PR1 and integrin beta4 in HGF/c-Met-mediated increases in vascular integrity. J Biol Chem 288: 2191–2200, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finigan JH, Dudek SM, Singleton PA, Chiang ET, Jacobson JR, Camp SM, Ye SQ, Garcia JG. Activated protein C mediates novel lung endothelial barrier enhancement: role of sphingosine 1-phosphate receptor transactivation. J Biol Chem 280: 17286–17293, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Foss FW Jr, Snyder AH, Davis MD, Rouse M, Okusa MD, Lynch KR, Macdonald TL. Synthesis and biological evaluation of gamma-aminophosphonates as potent, subtype-selective sphingosine 1-phosphate receptor agonists and antagonists. Bioorg Med Chem 15: 663–677, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fryer RM, Muthukumarana A, Harrison PC, Nodop Mazurek S, Chen RR, Harrington KE, Dinallo RM, Horan JC, Patnaude L, Modis LK, Reinhart GA. The clinically-tested S1P receptor agonists, FTY720 and BAF312, demonstrate subtype-specific bradycardia (S1P1) and hypertension (S1P3) in rat. PLoS One 7: e52985, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, Bamberg JR, English D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest 108: 689–701, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gon Y, Wood MR, Kiosses WB, Jo E, Sanna MG, Chun J, Rosen H. S1P3 receptor-induced reorganization of epithelial tight junctions compromises lung barrier integrity and is potentiated by TNF. Proc Natl Acad Sci USA 102: 9270–9275, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Graeler M, Shankar G, Goetzl EJ. Cutting edge: suppression of T cell chemotaxis by sphingosine 1-phosphate. J Immunol 169: 4084–4087, 2002. [DOI] [PubMed] [Google Scholar]
- 17.Granton J. Update of early respiratory failure in the lung transplant recipient. Curr Opin Crit Care 12: 19–24, 2006. [DOI] [PubMed] [Google Scholar]
- 18.Hirt SW, von Suesskind-Schwendi M, Puehler T, Schmid C, Lehle K. Early administration of FTY720 prevents chronic airway as well as vascular destruction in experimental rat lung transplantation. Transplant Proc 45: 783–786, 2013. [DOI] [PubMed] [Google Scholar]
- 19.Hla T. Signaling and biological actions of sphingosine 1-phosphate. Pharmacol Res 47: 401–407, 2003. [DOI] [PubMed] [Google Scholar]
- 20.Hwang SJ, Kim JH, Kim HY, Kim S, Chung DH. FTY720, a sphingosine 1-phosphate receptor modulator, inhibits CD1d-restricted NKT cells by suppressing cytokine production but not migration. Lab Invest 90: 9–19, 2010. [DOI] [PubMed] [Google Scholar]
- 21.Kreisel D, Sugimoto S, Zhu J, Nava R, Li W, Okazaki M, Yamamoto S, Ibrahim M, Huang HJ, Toth KA, Ritter JH, Krupnick AS, Miller MJ, Gelman AE. Emergency granulopoiesis promotes neutrophil-dendritic cell encounters that prevent mouse lung allograft acceptance. Blood 118: 6172–6182, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lapar DJ, Hajzus VA, Zhao Y, Lau CL, French BA, Kron IL, Sharma AK, Laubach VE. Acute hyperglycemic exacerbation of lung ischemia-reperfusion injury is mediated by receptor for advanced glycation end-products signaling. Am J Respir Cell Mol Biol 46: 299–305, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levkau B, Hermann S, Theilmeier G, van der Giet M, Chun J, Schober O, Schafers M. High-density lipoprotein stimulates myocardial perfusion in vivo. Circulation 110: 3355–3359, 2004. [DOI] [PubMed] [Google Scholar]
- 24.Maceyka M, Spiegel S. Sphingolipid metabolites in inflammatory disease. Nature 510: 58–67, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mandala S, Hajdu R, Bergstrom J, Quackenbush E, Xie J, Milligan J, Thornton R, Shei GJ, Card D, Keohane C, Rosenbach M, Hale J, Lynch CL, Rupprecht K, Parsons W, Rosen H. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 296: 346–349, 2002. [DOI] [PubMed] [Google Scholar]
- 26.Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, Allende ML, Proia RL, Cyster JG. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 427: 355–360, 2004. [DOI] [PubMed] [Google Scholar]
- 27.Melendez AJ. Sphingosine kinase signalling in immune cells: potential as novel therapeutic targets. Biochim Biophys Acta 1784: 66–75, 2008. [DOI] [PubMed] [Google Scholar]
- 28.Muller HC, Hocke AC, Hellwig K, Gutbier B, Peters H, Schonrock SM, Tschernig T, Schmiedl A, Hippenstiel S, N'Guessan PD, Rosseau S, Suttorp N, Witzenrath M. The sphingosine-1 phosphate receptor agonist FTY720 dose dependently affected endothelial integrity in vitro and aggravated ventilator-induced lung injury in mice. Pulm Pharmacol Ther 24: 377–385, 2011. [DOI] [PubMed] [Google Scholar]
- 29.Murakami A, Takasugi H, Ohnuma S, Koide Y, Sakurai A, Takeda S, Hasegawa T, Sasamori J, Konno T, Hayashi K, Watanabe Y, Mori K, Sato Y, Takahashi A, Mochizuki N, Takakura N. Sphingosine 1-phosphate (S1P) regulates vascular contraction via S1P3 receptor: investigation based on a new S1P3 receptor antagonist. Mol Pharmacol 77: 704–713, 2010. [DOI] [PubMed] [Google Scholar]
- 30.Okazaki M, Kreisel F, Richardson SB, Kreisel D, Krupnick AS, Patterson GA, Gelman AE. Sphingosine 1-phosphate inhibits ischemia reperfusion injury following experimental lung transplantation. Am J Transplant 7: 751–758, 2007. [DOI] [PubMed] [Google Scholar]
- 31.Peng X, Hassoun PM, Sammani S, McVerry BJ, Burne MJ, Rabb H, Pearse D, Tuder RM, Garcia JG. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med 169: 1245–1251, 2004. [DOI] [PubMed] [Google Scholar]
- 32.Ross SD, Tribble CG, Gaughen JR Jr, Shockey KS, Parrino PE, Kron IL. Reduced neutrophil infiltration protects against lung reperfusion injury after transplantation. Ann Thorac Surg 67: 1428–1433, 1999. [DOI] [PubMed] [Google Scholar]
- 33.Sawicka E, Zuany-Amorim C, Manlius C, Trifilieff A, Brinkmann V, Kemeny DM, Walker C. Inhibition of Th1- and Th2-mediated airway inflammation by the sphingosine 1-phosphate receptor agonist FTY720. J Immunol 171: 6206–6214, 2003. [DOI] [PubMed] [Google Scholar]
- 34.Schroder M, Richter C, Juan MH, Maltusch K, Giegold O, Quintini G, Pfeilschifter JM, Huwiler A, Radeke HH. The sphingosine kinase 1 and S1P1 axis specifically counteracts LPS-induced IL-12p70 production in immune cells of the spleen. Mol Immunol 48: 1139–1148, 2011. [DOI] [PubMed] [Google Scholar]
- 35.Schulze T, Golfier S, Tabeling C, Rabel K, Graler MH, Witzenrath M, Lipp M. Sphingosine-1-phosphate receptor 4 (S1P4) deficiency profoundly affects dendritic cell function and TH17-cell differentiation in a murine model. FASEB J 25: 4024–4036, 2011. [DOI] [PubMed] [Google Scholar]
- 36.Sharma AK, LaPar DJ, Stone ML, Zhao Y, Kron IL, Laubach VE. Receptor for advanced glycation end products (RAGE) on iNKT cells mediates lung ischemia-reperfusion injury. Am J Transplant 13: 2255–2267, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma AK, LaPar DJ, Zhao Y, Li L, Lau CL, Kron IL, Iwakura Y, Okusa MD, Laubach VE. Natural killer T cell-derived IL-17 mediates lung ischemia-reperfusion injury. Am J Respir Crit Care Med 183: 1539–1549, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shea BS, Brooks SF, Fontaine BA, Chun J, Luster AD, Tager AM. Prolonged exposure to sphingosine 1-phosphate receptor-1 agonists exacerbates vascular leak, fibrosis, and mortality after lung injury. Am J Respir Cell Mol Biol 43: 662–673, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sobel K, Menyhart K, Killer N, Renault B, Bauer Y, Studer R, Steiner B, Bolli MH, Nayler O, Gatfield J. Sphingosine-1-phosphate (S1P) receptor agonists mediate pro-fibrotic responses in normal human lung fibroblasts via S1P2 and S1P3 receptors and Smad-independent signaling. J Biol Chem 288: 14839–14851, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stammberger U, Gaspert A, Hillinger S, Vogt P, Odermatt B, Weder W, Schmid RA. Apoptosis induced by ischemia and reperfusion in experimental lung transplantation. Ann Thorac Surg 69: 1532–1536, 2000. [DOI] [PubMed] [Google Scholar]
- 41.Sun X, Singleton PA, Letsiou E, Zhao J, Belvitch P, Sammani S, Chiang ET, Moreno-Vinasco L, Wade MS, Zhou T, Liu B, Parastatidis I, Thomson L, Ischiropoulos H, Natarajan V, Jacobson JR, Machado RF, Dudek SM, Garcia JG. Sphingosine-1-phosphate receptor-3 is a novel biomarker in acute lung injury. Am J Respir Cell Mol Biol 47: 628–636, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tedesco-Silva H, Mourad G, Kahan BD, Boira JG, Weimar W, Mulgaonkar S, Nashan B, Madsen S, Charpentier B, Pellet P, Vanrenterghem Y. FTY720, a novel immunomodulator: efficacy and safety results from the first phase 2A study in de novo renal transplantation. Transplantation 79: 1553–1560, 2005. [PubMed] [Google Scholar]
- 43.Trifilieff A, Fozard JR. Sphingosine-1-phosphate-induced airway hyper-reactivity in rodents is mediated by the sphingosine-1-phosphate type 3 receptor. J Pharmacol Exp Ther 342: 399–406, 2012. [DOI] [PubMed] [Google Scholar]
- 44.Wilkerson BA, Argraves KM. The role of sphingosine-1-phosphate in endothelial barrier function. Biochim Biophys Acta 1841: 1403–1412, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu Y, Krause A, Limberis M, Worgall TS, Worgall S. Low sphingosine-1-phosphate impairs lung dendritic cells in cystic fibrosis. Am J Respir Cell Mol Biol 48: 250–257, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yanagawa Y, Sugahara K, Kataoka H, Kawaguchi T, Masubuchi Y, Chiba K. FTY720, a novel immunosuppressant, induces sequestration of circulating mature lymphocytes by acceleration of lymphocyte homing in rats. II. FTY720 prolongs skin allograft survival by decreasing T cell infiltration into grafts but not cytokine production in vivo. J Immunol 160: 5493–5499, 1998. [PubMed] [Google Scholar]
- 47.Yang Z, Sharma AK, Linden J, Kron IL, Laubach VE. CD4+ T lymphocytes mediate acute pulmonary ischemia-reperfusion injury. J Thorac Cardiovasc Surg 137: 695–702; discussion 702, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang Z, Sharma AK, Marshall M, Kron IL, Laubach VE. NADPH oxidase in bone marrow-derived cells mediates pulmonary ischemia-reperfusion injury. Am J Respir Cell Mol Biol 40: 375–381, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao M, Fernandez LG, Doctor A, Sharma AK, Zarbock A, Tribble CG, Kron IL, Laubach VE. Alveolar macrophage activation is a key initiation signal for acute lung ischemia-reperfusion injury. Am J Physiol Lung Cell Mol Physiol 291: L1018–L1026, 2006. [DOI] [PubMed] [Google Scholar]
- 50.Zhu R, Snyder AH, Kharel Y, Schaffter L, Sun Q, Kennedy PC, Lynch KR, Macdonald TL. Asymmetric synthesis of conformationally constrained fingolimod analogs—discovery of an orally active sphingosine 1-phosphate receptor type-1 agonist and receptor type-3 antagonist. J Med Chem 50: 6428–6435, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]