

Abstract
Endoplasmic reticulum (ER) stress and reactive oxygen species (ROS) generation in the brain circumventricular subfornical organ (SFO) mediate the central hypertensive actions of Angiotensin II (ANG II). However, the downstream signaling events remain unclear. Here we tested the hypothesis that angiotensin type 1a receptors (AT1aR), ER stress, and ROS induce activation of the transcription factor nuclear factor-κB (NF-κB) during ANG II-dependent hypertension. To spatiotemporally track NF-κB activity in the SFO throughout the development of ANG II-dependent hypertension, we used SFO-targeted adenoviral delivery and longitudinal bioluminescence imaging in mice. During low-dose infusion of ANG II, bioluminescence imaging revealed a prehypertensive surge in NF-κB activity in the SFO at a time point prior to a significant rise in arterial blood pressure. SFO-targeted ablation of AT1aR, inhibition of ER stress, or adenoviral scavenging of ROS in the SFO prevented the ANG II-induced increase in SFO NF-κB. These findings highlight the utility of bioluminescence imaging to longitudinally track transcription factor activation during the development of ANG II-dependent hypertension and reveal an AT1aR-, ER stress-, and ROS-dependent prehypertensive surge in NF-κB activity in the SFO. Furthermore, the increase in NF-κB activity before a rise in arterial blood pressure suggests a causal role for SFO NF-κB in the development of ANG II-dependent hypertension.
Keywords: hypertension; central nervous system; angiotensin II; NF-κB, ER stress
the hormone angiotensin ii (ANG II) has profound effects on cardiovascular regulation by acting within the central nervous system (CNS) to alter volume homeostasis and stimulate the sympathetic nervous system (10). In this regard, neurocardiovascular dysfunction due to inappropriate ANG II signaling is now recognized as a key mechanism contributing to the development of hypertension (9, 36). Although many brain regions are sensitive to ANG II, the circumventricular organs, areas lacking a blood-brain barrier (BBB), serve as sensors to detect circulating ANG II, a peptide too large to cross the BBB (8, 10, 11). Neural pathways originating from circumventricular organs project into a wide cardiovascular regulatory network, including hypothalamic nuclei and brain stem regions crucial for the control of the autonomic nervous system (8, 10, 11). In particular, the subfornical organ (SFO), a circumventricular forebrain region dense with angiotensin type 1a receptors (AT1aR), is crucial in driving ANG II-mediated neurogenic hypertension (8, 10, 11).
The endoplasmic reticulum (ER) is a specialized organelle responsible for the synthesis, assembly, folding, and sorting of proteins. Acute and/or chronic stress that challenges ER function, such as disturbed calcium homeostasis, alterations in cellular redox status, and inflammation, leads to an accumulation of misfolded proteins (i.e., ER stress). In an attempt to reestablish ER homeostasis, cells activate the unfolded protein response (UPR), a set of conserved signaling pathways (30, 31, 33), involving attenuation of translation, upregulation of ER chaperones, increased protein degradation, transcriptional activation, and, under extreme stress, programmed cell death (30, 33). Long-term activation of the UPR has recently emerged as a major paradigm for many chronic diseases (17, 30, 33). In line with this, we have recently shown that UPR activation in the SFO plays a causal role in ANG II-dependent hypertension (38). Indeed, systemic infusion of ANG II in mice resulted in a robust and selective induction of the UPR in the SFO, and pharmacological or genetic inhibition of ER stress in the SFO prevented the development of ANG II hypertension (38).
In addition to ER stress, our laboratory and others have extensively demonstrated that reactive oxygen species (ROS) in the CNS are linked to the development and progression of cardiovascular disease (40, 41, 43). For example, ANG II induces robust ROS production in the SFO, and selective scavenging of superoxide in this brain region abolishes ANG II-dependent hypertension (40), highlighting the importance of oxidant signaling in the SFO in mediating neurogenic hypertension. Interestingly, our recent findings demonstrate that ANG II-induced ER stress is a source of ROS production in the SFO (38). However, the mechanisms by which ER and oxidant stress within the brain translate into sustained elevations in arterial blood pressure remain unclear.
Emerging evidence suggests that ANG II-mediated activation of inducible transcription factors and the ensuing alterations in gene profiles within the CNS may contribute to the development of cardiovascular diseases, such as neurogenic hypertension (1, 2, 35). In this regard, the transcription factor nuclear factor-κB (NF-κB) may be particularly relevant (22). Abundant evidence has shown that ANG II induces activation of NF-κB in the peripheral vasculature and the heart (2), and more recently in other central cardiovascular regions (6, 18). Interestingly, NF-κB is known to modulate signaling cascades in response to ER and oxidant stress, including within CNS neurons (26, 39). The downstream consequence of CNS NF-κB activation includes a complex array of gene changes that influence everything from synaptic plasticity to neuromodulatory/neurotrophic factor production to synaptic structural alterations. Whether ANG II-induced ER stress and ROS production mediate activation of NF-κB in the SFO remains unknown.
We hypothesized that ANG II would activate NF-κB in the SFO and activation of this transcription factor would be AT1aR, ER stress, and ROS dependent. We used SFO-targeted delivery of an adenoviral vector encoding luciferase downstream of the NF-κB consensus sequence, in conjunction with bioluminescence imaging (BLI), to longitudinally track SFO NF-κB activity during the development and maintenance of ANG II hypertension. Our findings reveal an ANG II-mediated prehypertensive surge in NF-κB activity in the SFO. SFO-targeted ablation of AT1aR, inhibition of ER stress, or adenoviral scavenging of ROS in the SFO abolished the ANG II-induced increase in SFO NF-κB.
METHODS
Animals.
Studies were performed in adult (8–10 wk old) male C57Bl/6 mice obtained from an in-house colony. AT1aRfl/fl mice were initially obtained from the colony of Dr. Thomas Coffman (13) and used to establish our own colony. Mice were fed standard chow and water ad libitum and were housed with a 12:12-h light-dark cycle. All procedures were approved by the Animal Care and Use Committees at Cornell University, and care of the mice met the standards set forth by the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Slow-pressor ANG II infusion.
Human ANG II (Sigma-Aldrich, St. Louis, MO) was diluted in 0.1% bovine serum albumin (BSA; Sigma-Aldrich) and was loaded into 2-wk osmotic minipumps (ALZET; Durect, Cupertino, CA) at the desired concentration (600 ng−kg−1·min−1) for infusion. BSA was used as a vehicle control. Minipumps were primed overnight at 37°C and were then implanted subcutaneously as previously described (5, 38, 40).
Arterial blood pressure measurements.
Mice were anesthetized (ketamine, 150 mg/kg + xylazine, 15 mg/kg ip) and instrumented with radiotelemetry probes (PA-C10; Data Sciences International) as previously described (5, 38, 40). The radiotelemeter catheter was implanted in the thoracic aorta via the left common carotid artery. The body of the probe was placed in a subcutaneous pocket on the right flank, and the wound was closed and sutured. Mice remained undisturbed in their home cages for a minimum of 7 days to achieve full recovery of normal circadian rhythm and cardiovascular parameters before obtaining recordings of arterial blood pressure (4).
Adenoviral targeting of the SFO and lateral ventricle cannulation.
Recombinant adenoviral vectors encoding an NF-κB luciferase reporter construct [AdNF-κB-luc, 1.0 × 1011 plaque-forming units (pfu)/ml], AP-1 luciferase reporter construct (AdAP-1-luc, 1.0 × 1011 pfu/ml), human cytoplasmic superoxide dismutase (AdCuZnSOD, 1.3 × 1012 pfu/ml), Cre-recombinase (AdCre, 4 × 1010 pfu/ml), and titer-matched AdLacZ were obtained from the Iowa Gene Transfer Vector Core. Construction and characterization of these viral vectors has been described previously (27, 40). Targeting of adenoviral vectors to the SFO was performed as previously described in detail by our laboratory (5, 27, 38, 40). Briefly, mice were anesthetized and placed in a stereotaxic device. The surface of the skull was visualized under a dissecting microscope and leveled between lambda and bregma. AdNF-κB-luc was targeted to the SFO via a custom pressure injection system using the following coordinates: 3.2 mm ventral from the dorsal surface of the skull at 0.3 mm rostral and 1.0 mm lateral to bregma. For certain experiments viral vectors were delivered in a 1:1 mixture of AdNF-κB-luc/AdLacZ, AdNF-κB-luc/AdCre, and AdNF-κB-luc/AdCuZnSOD. For experiments involving lateral cerebroventricle injections (ICV), mice were subsequently instrumented with an indwelling ICV cannula (PlasticsOne) as previously described (38, 40).
ICV injections.
The ER stress inducer thapsigargin (TG; EMD Chemicals, Gibbstown, NJ) was dissolved in dimethyl sulfoxide (DMSO) and diluted with saline to the desired concentration. The ER stress inhibitor tauroursodeoxycholic acid (TUDCA; EMD Chemicals) was diluted in artificial cerebrospinal fluid (aCSF) to the final concentration. TG was administered as a single ICV injection at a dose (1 μg) that has been confirmed extensively to induce ER stress in vivo (29, 38). Daily ICV TUDCA administration (5 μg) began on the day of osmotic minipump implantation and continued throughout the 2-wk infusion period (38). The appropriate vehicle control was used for TG (DMSO) and TUDCA (aCSF).
In vivo BLI.
To spatiotemporally track NF-κB and activator protein-1 (AP-1) activation in the SFO in vivo, an adenoviral vector containing a luciferase cassette downstream of the NF-κB or AP-1 consensus-binding sequence was delivered to the SFO (see above) in conjunction with in vivo BLI as described (3, 27). Mice with SFO-targeted AdNF-κB-luc and AdAP-1-luc were imaged 24 h after adenoviral delivery to confirm expression of the reporter construct and were then allowed 2 wk for recovery from surgery-induced elevations in transcription factor activity. Following recovery and baseline imaging over 3 days, osmotic minipumps were implanted for continuous infusion of ANG II, and daily in vivo bioluminescence images were acquired with an IVIS 200 (Caliper Life Sciences, Alameda, CA), following injection of the substrate d-luciferin (150 mg/kg ip) and transfer of the mice to the light-sealed imaging cabinet. To ensure uniformity, each of the groups was imaged within the same daily session and at the same time each day. At the end of the study, systemic endotoxin-induced activation of AdNF-κB-luc and AdAP-1-luc was assessed by injection of lipopolysaccharide (LPS, 8 μg/g ip) to confirm functional expression of the luciferase reporter in the SFO. Data were analyzed with Xenogen Living Image software as described (3, 27).
ROS detection in the SFO.
ROS production in SFO tissue was assessed by dihydroethidium (DHE) microfluorography as described (5, 38, 40). Brains from mice that had undergone ANG II infusion for 3, 5, 7, and 14 days were rapidly removed and flash frozen. Coronal sections (20 μM) were collected onto chilled microscope slides, thawed at room temperature, and incubated for 5 min in the dark with DHE (1 μM) followed by a 2-min wash with PBS. DHE fluorescence was visualized by confocal microscopy (Zeiss LSM 510), with laser settings kept constant across all samples. ImageJ software was used to quantify fluorescence intensity, which was normalized to fluorescence levels observed in untreated samples as described (5, 38, 40).
Quantitative real-time PCR.
Neuro2A cells were cultured as previously described (38) and were treated with vehicle (BSA), ANG II (100 nM), or ANG II following 30 min pretreatment with the AT1R antagonist losartan (3 μM). After treatment (24 h), cells were collected, and quantitative real-time PCR (qPCR) analysis was performed. Total RNA was isolated by Trizol (Invitrogen) extraction and reverse transcribed using random hexamer primers, and samples of 25 ng were subjected in triplicate to qPCR (ABI 7500FAST system) using Power SYBR Green (Applied Biosystems). For each target gene, the average expressed isoform was expressed relative to the calibrator (β-actin), and the relative fold change compared with the calibrator was calculated using the comparative ΔΔCt method. Primer sequences derived from Mus musculus (National Center for Biotechnology Information GenBank) were as follows: CHOP forward 5′-ATATCTCATCCCCAGGAAACG-3′ and reverse 5′-TCTTCCTTGCTCTTCCTCCTC-3′; GRP78 forward 5′- TTCAGCTGTCACTCGGAGAAT-3′ and reverse 5′- ATATCTCATCCCCAGGAAACG −3′; and β-actin forward 5′-CATCCTCTTCCTCCCTGGAGAAGA-3′ and reverse 5′-ACAGGATTCCATACCCAAGAAGGAAGG-3′.
Terminal deoxynucleotidyl transferase dUTP nick end labeling staining.
C57Bl/6 mice that had received systemic ANG II infusions were perfused transcardially with 37°C saline followed by ice-cold 4% paraformaldehyde at 14 days of infusion. Brains were removed and stored in 30% sucrose overnight. Cryosections (30 μm) were obtained and mounted directly onto glass slides and allowed to dry overnight. Slides were washed in 1× PBS and were then placed in a permeabilization solution of 0.1% Triton X-100 (Sigma-Aldrich) and 0.1% sodium citrate (Sigma-Aldrich) in ddH20 at 4°C for 5 min, followed by subsequent washing in 1× PBS. Tissue sections were incubated in working terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) solution (Roche, Indianapolis, IN) for 90 min. Slides were covered with cover slips with Aquamount mounting media with DAPI and imaged using a Zeiss Axio Imager.
Data analysis.
Data are expressed as means ± SE. A one-way ANOVA was used for comparison between multiple groups. For time course experiments, a two-way repeated-measures ANOVA was used. Post hoc comparisons were performed using a Tukey's test when appropriate. The alpha level was set at P < 0.05.
RESULTS
ANG II infusion evokes a prehypertensive surge of NF-κB in the SFO.
We used a mouse model in which 2-wk systemic infusion of ANG II causes slowly developing and persistent hypertension (38, 40). As shown in Fig. 1A and in line with our previous reports, low-dose infusion of ANG II causes an initial increase in arterial blood pressure at ∼8–10 days and a peak increase in blood pressure on days 14-16. To test if ANG II induces NF-κB activation in the SFO in this model, an NF-κB-dependent reporter virus, AdNF-κB-luc, was selectively targeted to the SFO, and BLI was used for real-time monitoring of NF-κB activation during the development of hypertension (27). Figure 1, B and C, illustrates original BLI images of SFO NF-κB activity (photon flux) and a summary of the temporal changes in NF-κB-dependent photon flux during ANG II-induced hypertension. Following implantation of osmotic minipumps for delivery of ANG II, SFO NF-κB activity remained at low levels over the first 4 days of infusion. However, at day 5 of ANG II infusion, NF-κB activity in the SFO surged (Fig. 1, B and C), a time point prior to a significant rise in arterial blood pressure. Interestingly, SFO NF-κB activation subsided within 2 days and remained at low levels for the remainder of the 2-wk ANG II infusion. NF-κB-dependent photon flux remained unchanged throughout the experimental protocol in vehicle-treated mice. The functional capacity of AdNF-κB-luc was tested at the end of the protocol using systemic administration of LPS (8 μg/g). In line with our previous findings (27), LPS induced profound increases in NF-κB activity in the SFO (Fig. 1C, right), in both vehicle- and ANG II-infused mice, confirming the functionality of AdNF-κB-luc throughout the course of the study. It is important to note that the luciferase construct was selectively targeted to the SFO. Therefore, although tissue scattering causes the light emission to appear widespread, the SFO-selective delivery of AdNF-κB-luc confirms that the NF-κB activation is specific for this brain region (3, 7, 27). Overall, these findings demonstrate that ANG II infusion evokes a prehypertensive surge in NF-κB activity in the SFO.
Fig. 1.
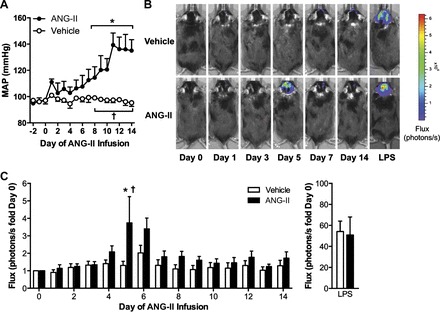
Slow-pressor angiotensin II (ANG II) infusion evokes a prehypertensive surge of nuclear factor-κB (NF-κB) in the subfornical organ (SFO). A: radiotelemetry measurements of mean arterial blood pressure (MAP) during infusion of vehicle (BSA) or ANG II. B: original bioluminescence images of NF-κB-dependent photon emission during infusion of vehicle or ANG II. The areas of high photon emission (flux) are displayed in red, and the areas of low photon emission are displayed in blue. C: summary of temporal changes in NF-κB activity. Changes in NF-κB activity in response to systemic lipopolysaccharide (LPS) at the end of the studies are shown on the right; note the different scale; n = 9–10/group. *P < 0.05 vs. day 0; †P < 0.05 vs. vehicle.
ANG II-induced SFO NF-κB activation is mediated by AT1aR.
To test if the ANG II-induced rise in NF-κB activity in the SFO is mediated by AT1aR, we used a mouse model in which exon 3 of Agtr1a gene is flanked by loxP sites (AT1aRfl/fl) (13). Targeted adenoviral delivery of Cre-recombinase (AdCre) to the SFO in these mice allows for a robust and efficient ablation of AT1aR in this brain region (37). Similar to the findings presented in Fig. 1, slow-pressor infusion of ANG II caused a prehypertensive and transient surge in SFO NF-κB activity at day 5 in control mice (Fig. 2A, left). Selective removal of AT1aR in the SFO abolished ANG II-induced NF-κB activation in the SFO such that NF-κB-dependent photon flux was not significantly different from baseline levels at any time point during ANG II infusion. Importantly, LPS-induced increases in SFO NF-κB activity were similar at the end of the study (4.39 ± 1.00 vs. 6.37 ± 1.87, AdLacZ vs. AdCre, P > 0.05), confirming that the attenuation of ANG II-induced NF-κB activation was the result of the removal of AT1aR and not the functionality of AdNF-κB-luc.
Fig. 2.
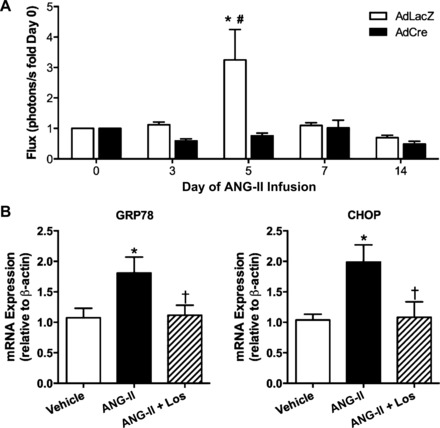
Angiotensin type 1a receptors (AT1aR) mediate ANG II-induced SFO NF-κB activation and endoplasmic reticulum (ER) stress. A: summary of temporal changes in NF-κB activity at key time points during ANG II infusion in AT1aRfl/fl mice with SFO-targeted adenoviral delivery of Cre-recombinase (AdCre) or control, AdLacZ; n = 5/group. *P < 0.05 vs. day 0; #P < 0.05 vs. AdCre. B: GRP78 (left) and CHOP (right) mRNA expression in Neuro2A cells treated with vehicle, ANG II, or ANG II plus losartan (Los). *P < 0.05 vs. vehicle; †P < 0.05 vs. ANG II.
AT1aR mediate ANG II-induced ER stress.
We have recently demonstrated that ANG II induces robust ER stress in the SFO, and this is functionally linked to the development of ANG II-dependent hypertension (38). However, the upstream mechanism(s) mediating ANG II-induced ER stress remain unknown. Here we tested if AT1R are involved in ANG II-evoked ER stress. Treatment of Neuro2A cells with ANG II resulted in an upregulation of mRNA expression of two key ER stress biomarkers, GRP78 and CHOP, whereas pretreatment of neurons with the AT1R antagonist losartan prevented the ANG II-induced increases in GRP78 and CHOP mRNA (Fig. 2B). These results build upon our recent findings (38) and demonstrate that ANG II-induced ER stress in neurons is mediated by AT1R.
ER stress mediates NF-κB activation in the SFO.
The findings presented in Figs. 1 and 2 demonstrate that ANG II evokes a prehypertensive surge in NF-κB in the SFO in an AT1aR-dependent manner. Given that ER stress and NF-κB activation are closely linked events (28, 39), and that AT1R mediate ANG II-induced ER stress (Fig. 2B), we tested if ER stress mediates ANG II-dependent SFO NF-κB activation. As a first step, we used ICV injections of TG, a chemical agent that induces robust ER stress in vivo (29, 38), in conjunction with BLI measurements of NF-κB-dependent photon flux in the SFO. As shown in Fig. 3A, C57Bl/6 mice treated with a single ICV injection of TG showed robust NF-κB activation in the SFO within 4 h, and this increase in NF-κB activity remained elevated at 24 h. No changes in SFO NF-κB-dependent photon flux were noted in mice that received ICV vehicle injection (Fig. 3A). These results illustrate that acute induction of ER stress in the brain evokes NF-κB activation in the SFO.
Fig. 3.
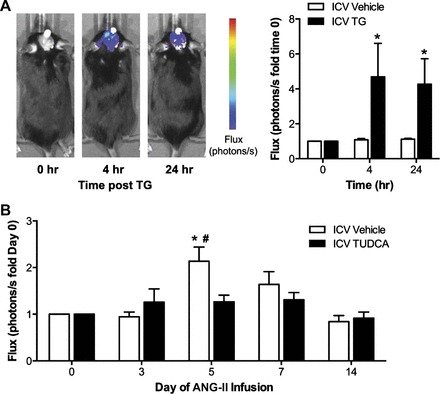
ER stress mediates ANG II-induced NF-κB activation in the SFO. A: original bioluminescence images of NF-κB-dependent photon emission during intracerebroventricular (icv) infusion of vehicle or the ER stress inducer thapsigargin (TG). Group summary data are shown on the right; n = 7–8/group. *P < 0.05 vs. time 0. B: summary of temporal changes in NF-κB activity during ANG II infusion in mice treated daily with icv vehicle or the ER stress inhibitor tauroursodeoxycholic acid (TUDCA); n = 7–8/group. *P < 0.05 vs. day 0; #P < 0.05 vs. ICV TUDCA.
We subsequently tested whether ER stress mediates SFO NF-κB activation during ANG II-dependent hypertension. As above, mice underwent SFO-targeted delivery of AdNF-κB-luc, and BLI was performed before and following implantation of osmotic minipumps for ANG II delivery. To prevent ANG II-induced ER stress in the SFO, mice were administered daily ICV injections of the chemical ER chaperone TUDCA, an approach that we have recently shown to prevent the development of ANG II-induced hypertension (25, 38). Infusion of ANG II in ICV vehicle-treated mice caused a robust surge in NF-κB activity in the SFO at day 5 that subsequently subsided and remained at low levels from day 7 to 14 (Fig. 3B, left). In contrast, mice that received daily ICV TUDCA showed no change in SFO NF-κB activity during the 2-wk ANG II infusion. LPS-induced NF-κB activation at the end of the experiment was similar between groups (11.91 ± 2.81 vs. 7.80 ± 2.49, ICV vehicle vs. ICV TUDCA, P > 0.05). Collectively, these results demonstrate that ER stress mediates NF-κB activation in the SFO during the development of ANG II-dependent hypertension.
Oxidative stress mediates ANG II-induced NF-κB activation in the SFO.
We have previously shown that superoxide production in the SFO mediates ANG II-dependent hypertension (40) and more recently have demonstrated that ANG II-induced ER stress is upstream of oxidant stress in this brain region (38). Having established that ER stress mediates SFO NF-κB activation during ANG II hypertension (Fig. 3B), we tested whether oxidant stress is linked to NF-κB activation. We first examined the temporal induction of SFO oxidative stress during low-dose ANG II infusion using DHE fluorescence measurements. Minimal ROS production within the SFO was found at 0 and 3 days of ANG II infusion (Fig. 4A). However, at day 5 of ANG II infusion a surge in ROS production occurred that remained elevated at days 7 and 14 (Fig. 4A). These findings indicate concomitant prehypertensive elevations in oxidative stress and NF-κB activation in the SFO during ANG II-dependent hypertension. To test the link between these two events, mice underwent SFO-targeted microinjections of AdNF-κB-luc plus an adenovirus encoding cytoplasmic superoxide dismutase (AdCuznSOD) or control vector. Following recovery, baseline BLI was performed, and osmotic minipumps were then implanted for infusion of ANG II. In line with our findings in Figs. 1, 2, and 3, ANG II caused a robust, prehypertensive increase in NF-κB activity in the SFO (Fig. 4B). Scavenging of superoxide in the SFO abolished the ANG II-induced rise in NF-κB activity at day 5. SFO NF-κB activity remained unchanged during vehicle infusion in control (AdLacZ) and AdCuZnSOD-treated mice (data not shown), and LPS-induced NF-κB activation was similar between groups at the commencement of the study (6.05 ± 1.22 vs. 5.45 ± 1.27, AdLacZ vs. AdCuZnSOD, P > 0.05). These results demonstrate that SFO ROS mediate prehypertensive NF-κB activation during ANG II hypertension.
Fig. 4.
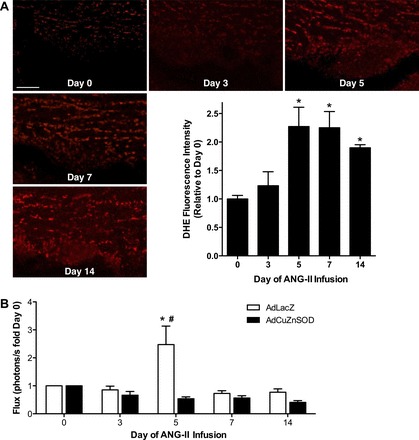
ANG II-induced activation of NF-κB in the SFO is mediated by oxidative stress. A: representative dihydroethidium (DHE) fluorescence images of the SFO from untreated mice and at key time points during ANG II infusion. Scale bar = 50 μm. B: summary of temporal changes in NF-κB activity at key time points during ANG II infusion in mice with SFO-targeted adenoviral delivery of cytoplasmic superoxide dismutase (AdCuZnSOD) or control, AdLacZ; n = 8–10/group. *P < 0.05 vs. day 0; #P < 0.05 vs. AdCuZnSOD.
ANG II does not cause cell death in the SFO.
Prolonged ER stress, oxidant stress, and subsequent transcription factor activation have been linked to apoptotic cell death (33). Our findings indicate that ANG II-induced ER stress and ROS production mediate NF-κB activation, and we therefore tested whether ANG II infusion leads to cell death in the SFO. In untreated mice, minimal evidence of cell death was seen in the SFO, as determined using TUNEL staining (Fig. 5A). In addition, administration of ANG II for 14 days did not significantly alter the number of positive TUNEL-labeled cells in the SFO (Fig. 5A). These findings therefore suggest that ANG II-mediated ER stress, oxidant stress, and NF-κB activation in the SFO do not lead to apoptotic cell death in this model of hypertension.
Fig. 5.
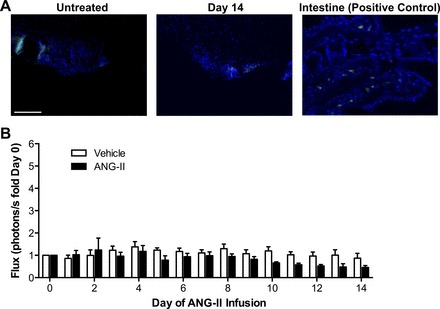
ANG II does not cause cell death or activate activator protein-1 (AP-1) in the SFO. A: representative terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining (green) in SFO sections from untreated (day 0) and ANG II-infused mice (day 14). A positive control section from the intestine is shown on the right. DAPI staining is shown in blue; n = 3. Scale bar = 100 μm. B: summary of temporal changes in SFO AP-1-dependent photon emission during ANG II infusion; n = 6–8/group.
The transcription factor AP-1 is not activated in the SFO during ANG II-dependent hypertension.
As a final step, we tested whether systemic elevations in ANG II result in transcription factor activation, in addition to NF-κB. The redox-sensitive transcription factor, AP-1, has been shown to be activated by ANG II, and we have previously demonstrated that AP-1 is activated in the CNS during the development of renovascular hypertension (3). Mice that underwent SFO-selective targeting of an AP-1-dependent reporter virus, AdAP-1-luc, were allowed to recover and underwent osmotic minipump implantation for infusion of ANG II or vehicle. No change in SFO AP-1 transcriptional activity was seen in ANG II- or vehicle-infused mice (Fig. 5B). Importantly, LPS induced robust activation of AP-1 at the end of the study, confirming the functionality of AdAP-1-luc (19.49 ± 3.74 vs. 16.12 ± 4.81, vehicle vs. ANG II, P > 0.05). These findings demonstrate that ANG II induces activation of NF-κB, but not AP-1, in the SFO during the development of ANG II-dependent hypertension.
DISCUSSION
By longitudinally monitoring transcription factor activation in vivo, the current findings demonstrate robust, prehypertensive NF-κB activation in the SFO during the development of ANG II-mediated hypertension. Our findings also demonstrate that ANG II-induced SFO NF-κB activation is mediated by AT1aR, ER stress, and redox-sensitive mechanisms. Collectively, these data highlight a complex, yet integrated, signaling pathway that drives ANG II-mediated NF-κB activation in the SFO (Fig. 6). Furthermore, the increase in NF-κB activity before a rise in arterial blood pressure suggests a causal role for SFO NF-κB in the development of ANG II-dependent hypertension.
Fig. 6.
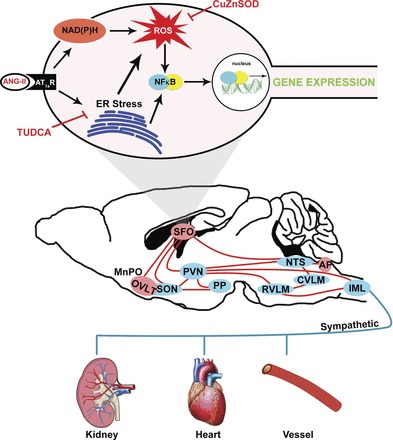
Schematic illustrating the central mechanisms within the SFO that may mediate ANG II-dependent hypertension. ANG II, via AT1aR, induces ER stress and NADPH oxidase-dependent reactive oxygen species (ROS) production in the SFO. ER stress and oxidant signaling lead to activation of NF-κB, subsequent changes in gene expression, and downstream stimulation of sympathoexcitatory circuits. MnPO, median preoptic nucleus; OVLT, organum vasculosum lamina terminalis; SON, supraoptic nucleus; PVN, paraventricular nucleus; PP, posterior pituitary; RVLM, rostral ventral lateral medulla; CVLM, caudal ventral lateral medulla; NTS, nucleus tractus solitarii; AP, area postrema; IML, intermediolateral column of the spinal cord.
A major mechanism by which extracellular signals cause long-lasting alterations in cellular function is through the activation of transcription factors and subsequent regulation of gene expression (1, 21, 22). In the CNS, DNA binding of transcription factors ultimately leads to changes in the level of proteins required for synthesis of neurotransmitters, receptors, ion channels, or cytoskeletal structures (21, 22). Indeed, chronic alterations in CNS function in the setting of both physiological (e.g., long-term memory) and pathophysiological (e.g., neurodegeneration) processes are due to changes in gene expression (21, 22). Similar to other CNS diseases, the concept that ANG II-mediated neurocardiovascular diseases are mediated by changes in neuronal gene expression is gaining strength (6, 18, 35). However, evaluating transcriptional activation in the brain is challenging because of the technical difficulties posed, particularly when evaluating genetic mechanisms throughout the development of disease. Conventional methods for monitoring NF-κB activation over time (e.g., electrophoretic mobility shift assays and nuclear localization) require the death of many animals, at numerous time points, to have sufficient tissue for assays (32). In addition, recent findings suggest that in vitro assays are not entirely informative because DNA binding and nuclear localization alone cannot distinguish between transcriptionally active and inactive transcription factors (32). To overcome these difficulties, in the current study we employed BLI for noninvasive in vivo longitudinal monitoring of transactivation of NF-κB selectively in the SFO (27). Our findings demonstrate that NF-κB transcriptional activation in the SFO was minimal over the first 4 days of ANG II infusion. This transitioned into a robust surge in SFO NF-κB activity at 5 days of infusion, a time point at which arterial blood pressure is not yet elevated (Fig. 1A). Interestingly, the increase in NF-κB activation was transient and returned to basal levels within 24 h and remained at low levels for the duration of the study. These findings are in line with the seminal work of Hoffman et al. (16) who revealed that NF-κB can undergo several different forms of activation, including sustained expression, pulsatile changes, or transient increases. Moreover, the dynamics of NF-κB activation determine the classes of genes that are expressed; some genes require persistent NF-κB activation, whereas others do not, and even short transient surges in NF-κB activation result in transcriptional activation of genes (14, 16). While downstream NF-κB-dependent gene changes in the SFO remain yet to be explored, these data highlight the utility of BLI to longitudinally track transcription factor activation during the development of ANG II-dependent hypertension in vivo. Indeed, if conventional methods were used, the robust and transient surge in NF-κB activation in the SFO may have been missed.
Our findings also build upon an emerging body of evidence that ANG II-induced activation of transcription factors may underlie neurocardiovascular diseases. For example, the current results are in line with our previous work demonstrating a robust surge in transcriptional activity in the CNS during the development of renovascular hypertension, a disease also associated with activation of the renin-angiotensin system (3). In addition, abundant evidence has demonstrated that ANG II induces activation of AP-1 family members in cultured neurons (35). More recently Mitra et al. (23) have shown that ANG II treatment resulted in upregulation of NF-κB subunits and DNA binding in CATH.a neurons, a response that was AT1R dependent. The current finding that selective ablation of AT1aR from the SFO prevents ANG II-induced NF-κB activation supports and extends this work to the in vivo setting. Interestingly, we did not find an increase in AP-1 transcriptional activity in the SFO during ANG II-induced hypertension, suggesting that in this model of hypertension and within the SFO, transcriptional activation may be selective to NF-κB.
Recent findings from Francis and colleagues have demonstrated upregulation of NF-κB subunits in the paraventricular nucleus of the hypothalamus (PVN) at 14 days of ANG II infusion in rats. Moreover, inhibition of NF-κB in the PVN prevented the development of ANG II-mediated hypertension (6, 18). Importantly, ANG II is too large to cross the BBB and signals to cardiovascular brain centers through sensory circumventricular organs, including the SFO, which contains extensive efferent connections to hypothalamic nuclei (8, 10). Our finding that NF-κB transcriptional activity in the SFO occurred at day 5, a prehypertensive time point, suggests that upstream SFO NF-κB-mediated activation may occur before PVN NF-κB activation. Therefore, targeted inhibition of NF-κB in the SFO may represent an alternative treatment strategy for ANG II-induced hypertension, although future studies are warranted. Similarly, further investigation into the cell type(s) responsible for ANG II-mediated NF-κB activation is necessary, given that adenoviral vector administration within the brain results in delivery of the viral construct to both neuronal and glial cell types (34), and that NF-κB activation has been shown to occur in both cell types (21, 22), as well as recent findings that distinct neural populations in the SFO mediate physiological responses (e.g., thirst) (24).
The upstream mechanism(s) by which ANG II, through AT1aR, mediate NF-κB activation in the CNS remains unclear. We have recently shown that ER stress in the SFO is a causative factor in the development of ANG II-mediated hypertension (38). Building upon these findings, the current results demonstrate that ANG II-induced neuronal ER stress is mediated by AT1R receptors and that acute pharmacological induction of ER stress in the CNS evokes NF-κB activation in the SFO. Moreover, chronic inhibition of ER stress prevented SFO NF-κB activation during the development of ANG II hypertension, highlighting ANG II-induced ER stress as a key upstream mechanism mediating this transcription factor activation (Fig. 6). These findings are in line with an emerging body of literature that ER stress signaling pathways converge with NF-κB activation in a variety of tissues, including the CNS (26, 39). For example, blockade of ER stress in the mediobasal hypothalamus of the brain during high-fat diet feeding in mice prevented NF-κB activation and the subsequent development of metabolic dysfunction (39). While our findings clearly demonstrate that ER stress mediates NF-κB activation in the SFO during ANG II hypertension, the specific arms of the UPR that are implicated remain unknown. Indeed, the three ER sensors, double-stranded RNA-dependent protein kinase-like ER kinase, inositol-requiring 1α, and activating transcription factor 6, have all been linked to NF-κB activation (30), although their role in mediating ANG II-induced NF-κB activation in the CNS remains uninvestigated.
In addition to ER stress, free radicals are crucial in integrating NF-κB-dependent responses (2, 15, 22). In this regard, our laboratory and others have demonstrated that ROS production mediates neurocardiovascular diseases, such as ANG II-mediated hypertension. Indeed, the current data demonstrate a prehypertensive surge (day 5) in ROS generation in the SFO during low-dose ANG II infusion, building upon previous observations of SFO oxidative stress at mid (7–8 days) and established (day 14) hypertension (38, 40). As such, we sought to determine if transcription factor activation in the SFO, due to ANG II, is mediated by redox-dependent mechanisms. In addition to the parallel elevations in ROS and NF-κB activation at day 5, we found that selective adenoviral scavenging of cytoplasmic superoxide prevented ANG II-mediated activation of NF-κB in the SFO. These findings are in line with our previous work showing that selective overexpression of AdCuZnSOD in the SFO prevents ANG II hypertension (40) and demonstrate NF-κB as a downstream target of ROS production in this brain region (Fig. 6).
Perspectives
Hypertension is an epidemic and is mediated, in part, due to neurogenic mechanisms (12, 19). In this regard, ANG II is a primary culprit, acting through circumventricular organs, such as the SFO (9, 10, 38, 40). With the use of longitudinal BLI, the current findings provide the first evidence for activation of the transcription factor NF-κB in the SFO during ANG II-dependent hypertension. Furthermore, our findings illustrate a complex, yet interconnected, pathway within the SFO by which ANG II signaling leads to NF-κB activation (Fig. 6). ANG II, via AT1R, induced ER stress may directly lead to SFO NF-κB transcriptional activation. In addition, NADPH oxidase-derived oxidant production (20, 42) provides an alternative direct mechanism by which ANG II induces NF-κB activation in the SFO. However, we have recently shown that ER stress is a source of ROS in the SFO during ANG II hypertension. Indeed, selective inhibition of ER stress in the SFO prevented ANG II-induced increases in ROS in this region (38). Therefore, while ER and oxidant stress may independently activate NF-κB in the SFO, it is plausible that ER stress induces ROS production, which results in subsequent activation of NF-κB. Although theoretical, ANG II-induced free radical accumulation may also evoke ER stress and subsequently NF-κB activation, an area that is currently being actively investigated. Overall, these findings further our understanding of the potential mechanisms underlying ANG II-mediated neurogenic hypertension.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-063887 and HL-084207. C. N. Young was supported by an American Physiological Society Fellowship, AHA13POST14410020 and K99HL116776.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: C.N.Y., C.G.C., and R.L.D. conception and design of research; C.N.Y., A.L., F.N.D., J.A.H., and C.G.C. performed experiments; C.N.Y., A.L., F.N.D., J.A.H., and C.G.C. analyzed data; C.N.Y., A.L., F.N.D., J.A.H., C.G.C., and R.L.D. interpreted results of experiments; C.N.Y., A.L., F.N.D., J.A.H., and C.G.C. prepared figures; C.N.Y. drafted manuscript; C.N.Y., A.L., F.N.D., J.A.H., C.G.C., and R.L.D. edited and revised manuscript; C.N.Y., A.L., F.N.D., J.A.H., C.G.C., and R.L.D. approved final version of manuscript.
REFERENCES
- 1.Blume A, Herdegen T, Unger T. Angiotensin peptides and inducible transcription factors. J Mol Med 77: 339–357, 1999. [DOI] [PubMed] [Google Scholar]
- 2.Brasier AR, Jamaluddin M, Han Y, Patterson C, Runge MS. Angiotensin II induces gene transcription through cell-type-dependent effects on the nuclear factor-kB (NF-kB) transcription factor. 212: 155–169, 2000. [PubMed] [Google Scholar]
- 3.Burmeister MA, Young CN, Braga VA, Butler SD, Sharma RV, Davisson RL. In vivo bioluminescence imaging reveals redox-regulated activator protein-1 activation in paraventricular nucleus of mice with renovascular hypertension. Hypertension 57: 289–297, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Butz GM, Davisson RL. Chronic blood pressure recording in pregnant mice by radiotelemetry. Physiol Genomics 2001. [Google Scholar]
- 5.Cao X, Peterson JR, Wang G, Anrather J, Young CN, Guruju MR, Burmeister MA, Iadecola C, Davisson RL. Angiotensin II-dependent hypertension requires cyclooxygenase 1-derived prostaglandin E2 and EP1 receptor signaling in the subfornical organ of the brain. Hypertension 59: 869–876, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cardinale JP, Sriramula S, Mariappan N, Agarwal D, Francis J. Angiotensin II-induced hypertension is modulated by nuclear factor-kappaBin the paraventricular nucleus. Hypertension 59: 113–121, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Contag CH. Molecular imaging using visible light to reveal biological changes in the brain. Neuroimaging Clin N Am 16: 633–654, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Cottrell GT, Ferguson AV. Sensory circumventricular organs: central roles in integrated autonomic regulation. Regul Pept 117: 11–23, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Davisson RL. Physiological genomic analysis of the brain renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol 285: R498–R511, 2003. [DOI] [PubMed] [Google Scholar]
- 10.Ferguson AV. Angiotensinergic regulation of autonomic and neuroendocrine outputs: critical roles for the subfornical organ and paraventricular nucleus. Neuroendocrinology 89: 370–376, 2009. [DOI] [PubMed] [Google Scholar]
- 11.Ferguson AV. The subfornical organ: a central integrator in the control of neurohypophysial hormone secretion. 99: 435–455, 1987. [Google Scholar]
- 12.Fisher JP, Young CN, Fadel PJ. Central sympathetic overactivity: maladies and mechanisms. Auton Neurosci 148: 5–15, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gurley SB, Riquier-Brison AD, Schnermann J, Sparks MA, Allen AM, Haase VH, Snouwaert JN, Le TH, McDonough AA, Koller BH, Coffman TM. AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab 13: 469–475, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hager GL, McNally JG, Misteli T. Transcription dynamics. Mol Cell 35: 741–753, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev 18: 2195–2224, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science 298: 1241–1245, 2002. [DOI] [PubMed] [Google Scholar]
- 17.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140: 900–917, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang YM, Ma Y, Zheng JP, Elks C, Sriramula S, Yang ZM, Francis J. Brain nuclear factor-kappa B activation contributes to neurohumoral excitation in angiotensin II-induced hypertension. Cardiovasc Res 82: 503–512, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J. Global burden of hypertension: analysis of worldwide data. Lancet 365: 217–223, 2005. [DOI] [PubMed] [Google Scholar]
- 20.Lob HE, Schultz D, Marvar PJ, Davisson RL, Harrison DG. Role of the NADPH oxidases in the subfornical organ in angiotensin II-induced hypertension (Abstract). Hypertension 61: 382, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mattson MP, Camandola S. NF-kappaB in neuronal plasticity and neurodegenerative disorders. 107: 247–54, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meffert MK, Baltimore D. Physiological functions for brain NF-kappaB. 28: 37–43, 2005. [DOI] [PubMed] [Google Scholar]
- 23.Mitra AK, Gao L, Zucker IH. Angiotensin II-induced upregulation of AT1 receptor expression: sequential activation of NF-κB and Elk-1 in neurons. Am J Physiol Cell Physiol 299: C561–C569, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Oka Y, Ye M, Zuker CS. Thirst driving and suppressing signals encoded by distinct neural populations in the brain. Nature In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313: 1137–1140, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park SH, Choi HJ, Yang H, Do KH, Kim J, Lee DW, Moon Y. Endoplasmic reticulum stress-activated C/EBP homologous protein enhances nuclear factor-κB signals via repression of peroxisome proliferator-activated receptor γ. J Biol Chem 285: 35330–35339, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peterson JR, Infanger DW, Braga VA, Zhang Y, Sharma RV, Engelhardt JF, Davisson RL. Longitudinal noninvasive monitoring of transcription factor activation in cardiovascular regulatory nuclei using bioluminescence imaging. Physiol Genomics 33: 292–299, 2008. [DOI] [PubMed] [Google Scholar]
- 28.Purkayastha S, Zhang G, Cai D. Uncoupling the mechanisms of obesity and hypertension by targeting hypothalamic IKK-beta and NF-kappaB. Nat Med 17: 883–887, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Purkayastha S, Zhang H, Zhang G, Ahmed Z, Wang Y, Cai D. Neural dysregulation of peripheral insulin action and blood pressure by brain endoplasmic reticulum stress. Proc Natl Acad Sci USA 108: 2939–2944, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8: 519–529, 2007. [DOI] [PubMed] [Google Scholar]
- 31.Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocr Rev 29: 317–333, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmitz ML, Mattioli I, Buss H, Kracht M. NF-kappaB: a multifaceted transcription factor regulated at several levels. Chembiochem 5: 1348–1358, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Ann Rev Biochem 74: 739–789, 2005. [DOI] [PubMed] [Google Scholar]
- 34.Sinnayah P, Lindley TE, Staber PD, Cassell MD, Davidson BL, Davisson RL. Selective gene transfer to key cardiovascular regions of the brain: comparison of two viral vector systems. Hypertension 39: 603–608, 2002. [DOI] [PubMed] [Google Scholar]
- 35.Sumners C, Fleegal MA, Zhu M. Angiotensin AT1 receptor signalling pathways in neurons. Clin Exp Pharmacol Physiol 29: 483–490, 2002. [DOI] [PubMed] [Google Scholar]
- 36.Veerasingham SJ, Raizada MK. Brain renin-angiotensin system dysfunction in hypertension: recent advances and perspectives. Br J Pharmacol 139: 191–202, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Young CN, Butler SD, Li A, Parsons KK, Coffman TM, Mark AL, Davisson RL. Angiotensin type 1a receptors (AT1a) in the subfornical organ contribute to the weight-reducing effects of leptin (Abstract). 58: e42, 2011. [Google Scholar]
- 38.Young CN, Cao X, Guruju MR, Pierce JP, Morgan DA, Wang G, Iadecola C, Mark AL, Davisson RL. ER stress in the brain subfornical organ mediates angiotensin-dependent hypertension. J Clin Invest 122: 3960–3964, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 135: 61–73, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zimmerman MC, Lazartigues E, Sharma RV, Davisson RL. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res 95: 210–216, 2004. [DOI] [PubMed] [Google Scholar]
- 41.Zimmerman MC, Davisson RL. Redox signaling in central neural regulation of cardiovascular function. Prog Biophys Mol Biol 84: 125–149, 2004. [DOI] [PubMed] [Google Scholar]
- 42.Zimmerman MC, Dunlay RP, Lazartigues E, Zhang Y, Sharma RV, Engelhardt JF, Davisson RL. Requirement for rac1-dependent NADPH oxidase in the cardiovascular and dipsogenic actions of angiotensin II in the brain. Circ Res 95: 532–539, 2004. [DOI] [PubMed] [Google Scholar]
- 43.Zimmerman MC, Sharma RV, Davisson RL. Superoxide mediates angiotensin II-induced influx of extracellular calcium in neural cells. Hypertension 45: 717–723, 2005. [DOI] [PubMed] [Google Scholar]