

Abstract
The transcription factor nuclear factor-κB (NF-κB) mediates inflammation and stress signals in cells. To test NF-κB in the control of hepatic insulin sensitivity, we inactivated NF-κB in the livers of C57BL/6 mice through deletion of the p65 gene, which was achieved by crossing floxed-p65 and Alb-cre mice to generate L-p65-knockout (KO) mice. KO mice did not exhibit any alterations in growth, reproduction, and body weight while on a chow diet. However, the mice on a high-fat diet (HFD) exhibited an improvement in systemic insulin sensitivity. Hepatic insulin sensitivity was enhanced as indicated by increased pyruvate tolerance, Akt phosphorylation, and decreased gene expression in hepatic gluconeogenesis. In the liver, a decrease in intracellular cAMP was observed with decreased CREB phosphorylation. Cyclic nucleotide phosphodiesterase-3B (PDE3B), a cAMP-degrading enzyme, was increased in mRNA and protein as a result of the absence of NF-κB activity. NF-κB was found to inhibit PDE3B transcription through three DNA-binding sites in the gene promoter in response to tumor necrosis factor-α. Body composition, food intake, energy expenditure, and systemic and hepatic inflammation were not significantly altered in KO mice on HFD. These data suggest that NF-κB inhibits hepatic insulin sensitivity by upregulating cAMP through suppression of PDE3B gene transcription.
Introduction
The transcription factor nuclear factor-κB (NF-κB) is a master regulator of inflammation. It is required for expression of proinflammatory cytokines, such as interleukin (IL)-1β and IL-6. In the cytosol, NF-κB is associated with the inhibitor protein κB inhibitor α (IκBα), which controls nuclear translocation of NF-κB. Degradation of IκBα leads to NF-κB activation for transcriptional regulation of gene expression. IκBα degradation is initiated by serine kinase IκB kinase-β (IKKβ), which phosphorylates IκBα at serine residues to induce ubiquitination-mediated degradation in proteasomes. The roles of IKKβ were studied in the pathogenesis of insulin resistance in global and tissue-specific transgenic mice. Those studies suggested that IKKβ deficiency (IKKβ+/−) protected mice from obesity-induced insulin resistance (1), although the same result was not observed in a subsequent study by a different group (2). Tissue-specific effects of IKKβ provide a mechanism for the discrepancy. The phenotypes of tissue-specific IKKβ transgenic mice suggest that IKKβ contributes to insulin sensitivity when it is activated in liver (3,4) or myeloid cells (3) but not in skeletal muscle (5) or adipose tissue (6). Although IKKβ has been studied extensively in various tissues in transgenic mice, the mechanism remains unknown for its action in insulin resistance.
In liver-specific studies, IKKβ overexpression was found to inhibit insulin sensitivity through induction of IL-6 expression (4), and IKKβ knockout (KO) was found to protect insulin sensitivity through inhibition of IL-1β expression (3). Although both studies suggested a role of transcriptional regulation by NF-κB in the mechanism of IKKβ action, the details remain unknown because the downstream genes were different in the two studies. In addition, IKKβ regulates insulin sensitivity through a transcription-independent mechanism of insulin receptor substrate-1 serine phosphorylation (7,8). The relative significance of transcription-dependent and -independent mechanisms remains unknown for the IKKβ activity. Inactivation of NF-κB is an approach to addressing this issue. NF-κB is a heterodimer protein formed by two subunits p65 (RelA) and p50 (NF-κB 1). The transcriptional activity of NF-κB is determined by the subunit p65, which contains an activation domain. Whole-body p65 inactivation leads to embryonic lethality (9), which does not allow phenotypic analysis. In this study, we inactivated p65 gene in liver (L-p65-KO) and examined insulin sensitivity in a comprehensive phenotypic study.
L-p65-KO mice were made by crossing floxed-p65 mice with Alb-cre mice. The phenotypic study included analysis of insulin sensitivity and energy balance in mice fed either a chow diet or high-fat diet (HFD). The mechanistic studies were conducted with a focus on cAMP/protein kinase A (PKA) pathways to understand the metabolic effects of NF-κB.
Research Design and Methods
Generation of L-p65-KO Mice
LoxP p65 mice were generated on a C57BL/6 gene background as described elsewhere (10). Alb-cre mice on the C57BL/6 genetic background (stock number 003574) were purchased from The Jackson Laboratory (Bar Harbor, ME). L-p65-KO (p65f/f Cre+/−) mice were generated by crossing the floxed-p65 mice with Alb-cre mice. Floxed-p65 littermates (p65f/f) were used as the wild-type (WT) control for KO mice (L-p65-KO). The study was conducted in male mice at the animal facility of the Pennington Biomedical Research Center. The mouse housing environment included a 12-h light-dark cycle, constant room temperature (22–24°C), and free access to water and diet. The mice were fed the chow diet (5% weight for weight or 11% calories from fat, 5001; LabDiet, St. Louis, MO) or HFD (36% weight for weight or 58% calories from fat, D12331; Research Diets, New Brunswick, NJ). HFD feeding was started at 8 weeks of age to generate a diet-induced obese model. All procedures were performed in accordance with the National Institutes of Health guidelines for the care and use of animals and were approved by the Institutional Animal Care and Use Committee at the Pennington Biomedical Research Center.
Cell Culture
The human hepatoma cell line HepG2 (HB-8065) was purchased from the American Type Culture Collection (Manassas, VA). Primary hepatocytes were made from mice at 6–10 weeks of age using a protocol described elsewhere (11). HepG2 cells and the primary hepatocytes were maintained in DMEM supplemented with 10% FBS at 37°C in a 5% CO2 incubator. The cells were treated with tumor necrosis factor-α (TNF-α) in serum-free DMEM containing 0.25% fatty acid–free BSA. An ssIκBα cell line was made through stable transfection of HepG2 cells with an ssIκBα-pBABE expression vector.
Body Weight and Composition
Body weight and composition were measured every 2 weeks. Body composition was measured using quantitative nuclear magnetic resonance (NMR) (Minispec Mn10 NMR scanner, Brucker, Milton, ON, Canada) as described previously (12). In the test, conscious and unrestrained mice were placed individually in a small tube and then placed in the NMR analyzer. Fat and lean mass were recorded within 1 min.
Food Intake
Food intake was determined manually for individually housed mice on HFD for 14 weeks. The average daily food intake was determined over 3 days by the net reduction in diet weight with exclusion of spilled food. Food intake is expressed in grams per mouse per day.
Energy Expenditure
Energy metabolism was monitored in mice after 4 weeks on HFD using the indirect calorimetry system (Comprehensive Laboratory Animal Monitoring System; Columbus Instruments, Columbus, OH). Mice were kept in the metabolic chamber for 6 days. VO2, VCO2, spontaneous physical activity, and food intake were recorded daily. Energy expenditure in kilocalories per kilogram per hour was calculated with day 5 data using the following equation: energy expenditure = [3.815 + 1.232 × VCO2/VO2] × VO2 × 0.001 (13). Energy expenditure data were normalized with body lean mass.
Insulin, Glucose, and Pyruvate Tolerance Testing
An intraperitoneal insulin tolerance test (ITT) was performed in mice (15 weeks on HFD) by insulin injection (0.75 units/kg body weight, I9278; Sigma-Aldrich) after 4 h of fasting. An intraperitoneal glucose tolerance test (GTT) was performed in mice (14 weeks on HFD) using glucose (2 g/kg body weight) after overnight fasting. An intraperitoneal pyruvate tolerance test (PTT) was performed in mice (24 weeks on HFD) using pyruvate (2 g/kg body weight) after overnight fasting. Blood glucose was measured in tail vein blood using the FreeStyle blood glucose monitoring system (Therasense, Phoenix, AZ) at 0, 30, 60, 120, and 180 min after injection. Data are expressed as blood glucose concentration (mg/dL).
Western Blotting
Livers were collected from mice after 16 weeks on HFD and examined for gene expression. Whole-cell lysates were prepared from liver tissue/cells and used in Western blotting according to protocols described elsewhere (12). Antibodies to NF-κB p65 (sc-8008), NF-κB p50 (sc-114 X), c-JUN (sc-1694 X), Sp1 (sc-59 X), and CREB (sc-7583 X) were purchased from Santa Cruz Biotechnology (Dallas, TX). Antibodies to p-Akt (threonine [T] 308, ab38449), p-Akt (serine [S] 473, ab66138), p-CREB (S133, ab32096), phosphodiesterase-3B (PDE3B) (ab125675), β-actin (ab6276), and tubulin (ab7291) were obtained from Abcam (Cambridge, MA). Tubulin and β-actin were used as internal controls.
Quantitative Real-Time RT-PCR
Total mRNA was extracted from liver tissue or cells using TRIzol reagent following the manufacturer’s protocol (Invitrogen, Carlsbad, CA). TaqMan Universal PCR Master Mix (4304437; Applied Biosystems, Carlsbad, CA) was used to quantify gene mRNA in the RNA extracts using the ABI 7900 machine. Target mRNA was normalized to ribosomal 18S RNA, an endogenous control. Primers and probes were purchased from Applied Biosystems. These included IL-1β (Mm00434228_ml), IL-6 (Mm00446190_m1), MCP-1 (Mm00441242_m1), IκBα (Mm00477798_m1), PDE3B (Mm00691635_m1), PEPCK (Mm00440636_m1), and G6Pase (Mm00839363_m1). The sequence of SYBR Green primers for human PDE3B is 5′-AAATTCTGGAGGTGGAAATG-3′ (product number KSPQ12012G, H_PDE3B_1, NM_000922; Sigma-Aldrich). The primer was used with SYBR Green Master Mix (4309155; Applied Biosystems) for PDE3B mRNA in HepG2 cells.
Luciferase Reporter Assay
PDE3B-luciferase reporter (−5.1/−3.4 SX-luciferase PGL3) is described elsewhere (14). The vectors for p65-pcDNA, ssIκBα-pBABE, and corresponding control plasmids are also described elsewhere (15). The transfection was conducted in HepG2 cells using Lipofectamine 2000 (11668019; Life Technologies, Grand Island, NY). The luciferase assay was performed at 48 h using a 96-well luminometer with the dual-luciferase substrate system (E1960; Promega Corporation, Fitchburg, WI). SV40 (simian virus 40 Renilla luciferase) 0.1 μg/well was used as an internal control. Each experiment was repeated at least three times.
Electrophoretic Mobility Shift Assay
The nuclear extract was made from HepG2 cells, and the electrophoretic mobility shift assay (EMSA) was conducted as described elsewhere (15). The DNA probes for NF-κB binding sites in the mouse PDE3B gene promoter were synthesized according the following sequences: site a (5′-ACACTGGGGATTTGACCTCTA-3′), site b (5′-GCATTAGGGTCTTCCCATATA-3′), and site c (5′-TAAAGATGAGAGTCCCATGCC-3′). The authentic NF-κB (human IL-6-κB site) and Sp1 probes are described elsewhere (16,17). In oligonucleotide competition and antibody supershift experiments, a 50-fold excess of unlabeled oligonucleotide probe and 2 μg IgG were used, respectively.
Plasma Cytokine and Insulin
After overnight fasting, blood was collected from mice on HFD for 18 weeks through retro-optical bleeding. Plasma was isolated and used in cytokine assays for IL-1β, IL-6, and TNF-α using a multiplex kit (MCYTOMAG-70K; EMD Millipore). Plasma insulin was measured using a multiplex kit of mouse metabolic hormones (MMHMAG-44K; EMD Millipore).
Liver Function
Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were measured in the plasma using a standard enzymatic assay (TR71121, TR70121; Thermo Fisher Scientific Inc., Middletown, VA).
cAMP Assay
cAMP level was determined using an ELISA kit (ADI-900-067; Enzo Life Sciences, Inc.). cAMP concentrations were presented as fold change in nmol/g over WT control.
Statistical Analysis
Statistical analysis was performed using two-tailed, unpaired Student t test. P < 0.05 was considered significant. Results are presented as mean ± SEM.
Results
Decreased Inflammation in Liver of L-p65-KO Mice
The transcriptional activity of NF-κB depends on the p65 subunit. Inactivation of p65 is expected to decrease expression of inflammatory genes in hepatocytes. In this study, L-p65-KO mice (p65f/f, Alb-Cre+/−) were generated by crossing floxed-p65 mice with Alb-cre mice. Analysis of the phenotype was conducted in male L-p65-KO mice, and floxed-p65 littermates (p65f/f) were used as WT controls. The p65 protein was examined in the liver tissues to verify the gene KO; p65 was reduced by 90% in the liver tissue of KO mice relative to that of WT controls (Fig. 1A). Expression of NF-κB target genes (IκBα, IL-1β, and IL-6) was decreased in hepatocytes from KO mice (Fig. 1B). Primary hepatocytes were prepared and used in the gene expression studies to exclude the activities of macrophages (Kupffer cells). The results in Fig. 1 suggest that NF-κB function is inactivated in the hepatocytes of L-p65-KO mice.
Figure 1.

Decreased inflammation in liver of L-p65-KO mice on a chow diet. A: The p65 protein in liver tissue. Proteins were determined by Western blotting. B: Expression of mRNA of NF-κB target genes in primary hepatocytes. The primary hepatocytes were treated with TNF-α (20 ng/mL) for 2 h to induce expression of the indicated genes. Data are mean ± SEM (n = 3). **P < 0.001 by Student t test.
Enhanced Systemic Insulin Sensitivity in L-p65-KO Mice on HFD
Analysis of the phenotype was first performed in L-p65-KO mice on a chow diet. No phenotypic change was observed by ITT and GTT, body weight, body composition, food intake, energy expenditure, physical activity, and substrate use (data not shown). The data suggest that KO does not influence insulin sensitivity and energy balance in lean mice. In obese KO mice on HFD (58% calories from fat), insulin sensitivity was significantly improved as indicated by a 50% reduction in fasting insulin and enhanced tolerance to insulin and glucose (Fig. 2A–C). The data suggest that NF-κB inactivation in hepatocytes significantly improves systemic insulin sensitivity in L-p65-KO mice in obese but not lean conditions.
Figure 2.
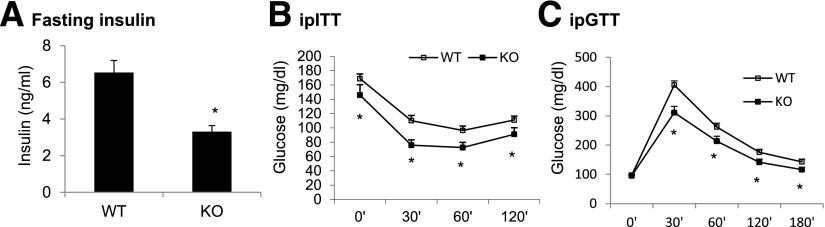
Systemic insulin sensitivity in L-p65-KO mice. A: Fasting insulin. Blood was collected from mice following overnight fasting after 18 weeks on HFD (n = 7–8). B: ITT. After 15 weeks on HFD, mice received insulin 0.75 units/kg i.p. following 4 h of fasting (n = 10–12). Blood glucose was determined at the indicated time points. C: GTT. After 14 weeks on HFD, mice received glucose 2 g/kg i.p. following overnight fasting (n = 10–12). Data are mean ± SEM. *P < 0.05. ip, intraperitoneal.
Enhanced Hepatic Insulin Sensitivity in L-p65-KO Mice on HFD
Liver insulin sensitivity was examined to understand the improved systemic insulin sensitivity in KO mice on HFD. Pyruvate tolerance was studied to determine hepatic gluconeogenesis, a process inhibited by insulin. Insulin sensitivity was inversely associated with glucose elevation in PTT. The tolerance was enhanced in L-p65-KO mice for a smaller increase in blood glucose (Fig. 3A). Phosphorylation of Akt was examined to determine insulin signaling activity in hepatocytes. Akt phosphorylation at T308 and S473 was enhanced in KO mice (Fig. 3B). Expression of gluconeogenic genes phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase) was examined in hepatocytes to determine the mechanism of reduced gluconeogenesis, and both were decreased in the KO liver (Fig. 3C). Liver weight was not significantly altered in KO mice on HFD (Fig. 3D), although plasma ALT was elevated in KO mice (Fig. 3E). These data suggest that hepatic insulin sensitivity is improved in L-p65-KO mice on HFD.
Figure 3.

Hepatic insulin action in L-p65-KO mice. A: PTT. After 24 weeks on HFD, mice received pyruvate 2 g/kg i.p. following overnight fasting (n = 8–10). B: Insulin signaling in hepatocytes. Primary hepatocytes were prepared from 8-week-old mice and cultured in vitro. After 15 min of insulin treatment, phosphorylation of Akt at T308 and S473 was examined in cell lysate by Western blotting. C: mRNA expression of gluconeogenic genes in hepatocytes of KO liver (n = 6). D: Liver weight. Livers were collected from mice on HFD for 16 weeks and used in the weight analysis (n = 10–12). E: Liver function. ALT and AST were tested using plasma collected from mice after 4 weeks on HFD (n = 7). Data are mean ± SEM. *P < 0.05.
Reduced cAMP Activity in Liver of L-p65-KO Mice
Insulin action is antagonized by effects of glucagon in the control of hepatic gluconeogenesis. Glucagon activity depends on activation of the cAMP/PKA pathway. In the cAMP/PKA pathway, cAMP elevation leads to activation of serine kinase PKA, which phosphorylates the transcription factor CREB on S133. After phosphorylation, CREB promotes gluconeogenesis through the induction of PEPCK and G6pase transcription. CREB phosphorylation was examined in the livers of mice after overnight fasting. The phosphorylation signal was significantly reduced in KO liver (Fig. 4A), and this reduction was associated with a decrease in intracellular cAMP (Fig. 4B) and an increase in PDE3B expression (Fig. 4C and D). PDE3B is an intracellular enzyme that decreases cAMP levels in hepatocytes through catalyzing cAMP hydrolysis. Because cAMP concentrations are determined by relative rates of synthesis and degradation in cells, the increased expression of PDE3B is most likely responsible for the decreased cAMP content and cAMP/PKA signaling in liver tissue of KO mice.
Figure 4.

Reduced cAMP content and signaling in liver of L-p65-KO mice. Livers were collected from mice on HFD for 16 weeks. A: pCREB. The phosphorylation was determined through Western blotting. B: cAMP levels in liver tissue. C: PDE3B protein in liver tissue. D: PDE3B mRNA expression in liver of HFD-fed mice (n = 5). Data are mean ± SEM. **P < 0.01.
Regulation of PDE3B by NF-κB
The data suggest that PDE3B plays an important role in the molecular mechanisms underlying improved insulin sensitivity in L-p65-KO mice. The PDE3 family contains two members encoded by distinct genes: PDE3A and PDE3B (18). Isoform PDE3A is relatively highly expressed in the cardiovascular system, and isoform PDE3B is relatively highly expressed in hepatocytes and adipocytes. mRNA data suggest that PDE3B gene transcription is upregulated after NF-κB inactivation. To test this possibility, NF-κB was activated in HepG2 cells by TNF-α treatment. PDE3B mRNA was examined in the system at multiple time points, and a decrease was observed after TNF-α treatment for 0.5 h (Fig. 5A). The TNF effect was blocked when NF-κB activity was inhibited by IκBα (Fig. 5B). The experiment was performed in an IκBα cell line made in HepG2 cells through stable transfection with an ssIκBα expression vector. In the stable cell line, the basal level of PDE3B mRNA was increased, and TNF-α could not inhibit PDE3B expression (Fig. 5B). The TNF-α activity was tested in the primary hepatocytes of L-p65-KO mice. The inhibition was observed in WT cells but not in KO cells (Fig. 5C), suggesting that NF-κB is required for TNF-α activity. These data suggest that NF-κB inhibits PDE3B expression in hepatocytes in the TNF-α signaling pathway.
Figure 5.
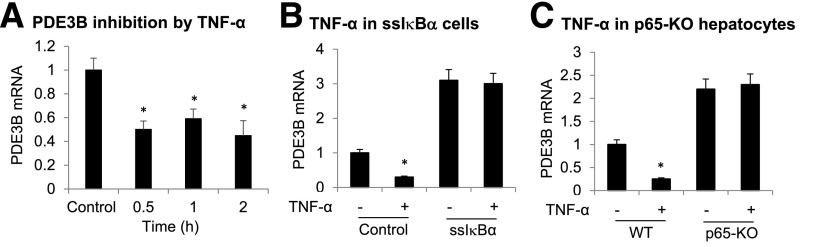
Inhibition of PDE3B expression by NF-κB. A: PDE3B inhibition after NF-κB activation by TNF-α treatment. PDE3B mRNA was examined in HepG2 cells after TNF-α treatment (20 ng/mL) (n = 3). B: Inhibition of TNF-α activity by IκBα. PDE3B expression was determined after TNF-α treatment of HepG2 cells that were stably transfected by an ssIκBα expression vector. C: TNF-α effect in p65-KO cells. PDE3B mRNA was examined in primary hepatocytes of L-p65-KO mice following TNF-α treatment for 30 min in vitro. Data are mean ± SEM (n = 3). *P < 0.05.
Inhibition of PDE3B Gene Promoter by NF-κB
As a transcription factor, NF-κB regulates gene expression through direct interaction with the gene promoter DNA. Potential NF-κB binding sites were searched in the PDE3B gene promoter through analysis of the nucleotide sequence. Three sites were identified in the mouse PDE3B gene promoter (Fig. 6A). Interaction of those sites with NF-κB was tested with EMSA through a competition against a classical NF-κB probe (Fig. 6B). All three sites competed with the classical probe, but with different efficiency. Site b exhibited the strongest competition, suggesting that it has the highest binding affinity to NF-κB protein. A radiolabeled probe was made from site b and further tested for NF-κB interaction in a supershift assay. The DNA-protein complex was shifted by the antibody to the NF-κB p50 subunit but not by the antibodies to c-JUN or Sp1 (Fig. 6B), suggesting that the probe specifically interacted with the NF-κB protein. The function of NF-κB sites was tested in the gene promoter of the luciferase reporter assay. The PDE3B promoter was inhibited by p65 in cotransfection, and the inhibition was blocked by IκBα in cotransfection (Fig. 6C). These data suggest that NF-κB inhibits PDE3B transcription through DNA-binding sites in the gene promoter.
Figure 6.

Inhibition of PDE3B gene promoter by NF-κB. A: NF-κB binding sites in PDE3B gene promoter. The sites were identified by sequence analysis. B: Characterization of the NF-κB sites a, b, and c in gel shift assays. NF-κB was activated in hepatocytes by TNF-α, and the nuclear extracts were used in EMSA. In the competition assay, cold probes of NF-κB sites were used to compete with the radiolabeled authentic NF-κB probe. NF-κB site b of PDE3B was radiolabeled and used as a probe in the supershift assay with antibodies to p50 (α p50), c-JUN (α Jun), and Sp1 (α Sp1). C: Inhibition of PDE3B gene promoter by p65. The PDE3B-luciferase reporter was cotransfected with a p65 or an ssIκBα expression vector (μg) in HepG2 cells. The luciferase activity was normalized with the internal control (Renilla luciferase). Data are mean ± SEM (n = 3). **P < 0.01. ATG, adenine thymine guanine; bp, base pair; luc, luciferase.
Energy Balance and Systemic Inflammation
Energy balance was monitored in KO mice by checking body weight, body composition, food intake, and metabolic rate. Those parameters were not altered in L-p65-KO mice on the HFD (Fig. 7A–E) or chow diet (Fig. 7F). Systemic inflammatory status was examined by measuring plasma proinflammatory cytokines in mice on HFD, including TNF-α, IL-6, IL-1β, IL-10, IL-15, and vascular endothelial growth factor (VEGF). The cytokine levels were not altered in KO mice in nonfasted or fasted conditions (Fig. 7G and H). These data suggest that p65 inactivation in liver does not alter the energy balance and systemic inflammatory status in mice.
Figure 7.

Energy balance and inflammatory cytokines. Energy balance was determined by analysis of body weight, body composition, energy metabolism, and food intake in the mice. A: Body weight. B: Body fat. C: Body lean mass. D: Food intake. Data represent average food intake over 3 days in the metabolic cage at 14 weeks on HFD. E: Energy expenditure. The test was performed after 4 weeks on HFD, and the data were normalized with body lean mass. F: Body weight on chow diet. G: Plasma cytokines in nonfasted mice. H: Plasma cytokines in overnight-fasted mice. Data are mean ± SEM (n = 9–12).
Discussion
This study provides evidence that systemic insulin sensitivity is enhanced by liver-selective NF-κB inactivation in diet-induced obese mice, suggesting a role of hepatic NF-κB in the regulation of insulin sensitivity. The observation is consistent with that reported for hepatic IKKβ activities in overexpression and KO studies (3,4); however, the molecular mechanism of NF-κB action is different from those reported for IKKβ. IKKβ was reported to inhibit insulin sensitivity through induction of IL-1β (3) or IL-6 expression (4). The relative significance of the two cytokines is unknown in the IKKβ model. In addition, the cytokines are controversial in the pathogenesis of insulin resistance because their activities are not observed in the induction of insulin resistance in all studies (19). Although NF-κB was reported to mediate IKKβ activity, the role of PDE3B was not found downstream of NF-κB in those early studies (3,4). A novel finding of the current study is that PDE3B is a target gene of NF-κB in the regulation of insulin sensitivity. The data suggest that increased PDE3B after NF-κB inactivation contributes to insulin sensitivity in L-p65-KO mice, which involves a reduction in cAMP activity.
This study demonstrates that NF-κB is a transcriptional repressor of the PDE3B gene. PDE3B belongs to a large and complex superfamily of 11 phosphodiesterase (PDE1–11) gene families. PDE3B is one of the genes that mediates the cross talk of insulin and glucagon in the regulation of energy metabolism (18,20). PDE3B inhibits glucagon activity through downregulation of cAMP. Glucagon stimulates glucose production in liver through activation of the cAMP/PKA pathway, and insulin activates PDE3B to inhibit the glucagon activity. Little information exists about PDE3B in humans, but in mice, global inactivation of PDE3B increases energy expenditure through an effect in adipose tissue (21), and this inactivation leads to insulin resistance in the KO mice due to increased cAMP activity in liver (22). Inhibition of PDE3B by a chemical inhibitor (cilostazol) was reported to reduce obesity and improve insulin sensitivity in db/db mice (23). In 3T3-L1 adipocytes, TNF-α inhibits PDE3B expression to increase the activity of the cAMP/PKA pathway (24). However, the molecular mechanism was unknown for the PDE3B inhibition before the present report. NIK (NF-κB–inducing kinase) and IKKε are two serine kinases in the signaling pathway of TNF-α (25). Although both are activated by TNF-α, they were reported to have opposite activities in the regulation of the cAMP signaling pathway. NIK upregulated the pathway by direct phosphorylation of the CREB protein in a study of glucagon signaling in liver (26). IKKε downregulated the pathway through induction of PDE3B activity in a study of IKKε in adipocytes (27) in which IKKε was found to phosphorylate the PDE3B protein. The current study suggests that TNF-α enhances the pathway activity through inhibition of PDE3B expression by activation of NF-κB. We observed that NF-κB inhibited the gene promoter activity of PDE3B and found that NF-κB activity was required for TNF-α inhibition of PDE3B expression. NF-κB inhibition by ssIκBα overexpression or p65 gene inactivation eliminated the TNF-α activity. PDE3B expression was increased in liver under NF-κB inactivation in the L-p65-KO mice. NF-κB binding sites were identified in the PDE3B gene promoter using oligonucleotide competition and antibody-mediated supershift assays in EMSA. The inhibitory activity of NF-κB was proven in the reporter assay of the PDE3B gene promoter. The detail mechanism of NF-κB action remains unknown after binding to the PDE3B promoter DNA, but NF-κB activity may involve recruitment of a corepressor to the gene promoter or sequestration of coactivators from other activators in the gene promoter. Other activators in the gene promoter include CREB in the PDE3B gene promoter (14). Inhibition of PDE3B by NF-κB leads to increased cAMP activity, which provides a new mechanism for TNF-α induction of the cAMP/PKA pathway.
Liver damage may occur in L-p65-KO mice as indicated by increased ALT levels. ALT is found mainly in the liver but is also found in smaller amounts in the kidneys, heart, muscles, and pancreas. Low levels of ALT are found in the blood under normal conditions. When the liver is damaged, it releases ALT into the bloodstream; thus ALT level was measured to determine liver damage in L-p65-KO mice. Because NF-κB is known to inhibit cell apoptosis, inactivation of NF-κB should cause hepatic apoptosis to increase and result in liver damage, as indicated by ALT elevation.
In summary, this study provides evidence that NF-κB inactivation in hepatocytes improves hepatic insulin sensitivity and downregulates cAMP/PKA signaling. The decreased cAMP levels were observed with the enhanced PDE3B activity in the hepatocytes of L-p65-KO mice, which is most likely a result of elevated gene transcription in the absence of NF-κB activity. NF-κB was found to inhibit the PDE3B gene promoter through three DNA binding sites. The study suggests a new molecular mechanism for inflammation in the pathogenesis of insulin resistance in which NF-κB promotes cAMP signaling activity through downregulation of PDE3B transcription in hepatocytes. The study also suggests that glucagon signaling activity may be reduced in L-p65-KO mice, a possibility that remains to be tested.
Article Information
Acknowledgments. The authors thank Hanguan Liu, Pulmonary Cardiovascular Branch, National Heart, Lung, and Blood Institute/National Institutes of Health, for generating the PDE3B reporter vectors.
Funding. This study was supported by the National Natural Science Foundation of China (grant number 81370915 to Z.G.). V.M. is supported by the National Heart, Lung, and Blood Institute Intramural Research Program. This study was also supported by the National Key Basic Research Program of China (2012CB526700) and Guangzhou City International Cooperation (2012J5100017) (to B.W.) and the National Institutes of Health (NIH) (DK085495 and DK068036 to J.Y.). The quantitative real-time RT-PCR test, metabolic phenotyping, and imaging studies were conducted in the genomic core, phenotyping core, and imaging core, which are supported by NIH grants P30-DK-072476 and P20-RR-021945.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. B.K., Z.Z., X.Y., and Z.G. conducted the experiments and read and approved the manuscript. V.M., B.W., and J.Y. designed the study and prepared, read, and approved the manuscript. J.Y. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
References
- 1.Yuan M, Konstantopoulos N, Lee J, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 2001;293:1673–1677 [DOI] [PubMed] [Google Scholar]
- 2.Röhl M, Pasparakis M, Baudler S, et al. Conditional disruption of IkappaB kinase 2 fails to prevent obesity-induced insulin resistance. J Clin Invest 2004;113:474–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arkan MC, Hevener AL, Greten FR, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med 2005;11:191–198 [DOI] [PubMed] [Google Scholar]
- 4.Cai D, Yuan M, Frantz DF, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med 2005;11:183–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cai D, Frantz JD, Tawa NE Jr, et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 2004;119:285–298 [DOI] [PubMed] [Google Scholar]
- 6.Jiao P, Feng B, Ma J, et al. Constitutive activation of IKKβ in adipose tissue prevents diet-induced obesity in mice. Endocrinology 2012;153:154–165 [DOI] [PubMed] [Google Scholar]
- 7.Zhang J, Gao Z, Yin J, Quon MJ, Ye J. S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-(α) signaling through IKK2. J Biol Chem 2008;283:35375–35382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao Z, Hwang D, Bataille F, et al. Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J Biol Chem 2002;277:48115–48121 [DOI] [PubMed] [Google Scholar]
- 9.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature 1995;376:167–170 [DOI] [PubMed] [Google Scholar]
- 10.Gao Z, Zhang J, Henagan TM, et al. P65 inactivation in adipocytes and macrophages attenuates adipose inflammatory response in lean but not in obese mice. Am J Physiol Endocrinol Metab 2015;308:E496–E505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao Z, Yin J, Zhang J, He Q, McGuinness OP, Ye J. Inactivation of NF-kappaB p50 Leads to insulin sensitization in liver through post-translational inhibition of p70S6K. J Biol Chem 2009;284:18368–18376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang T, Zhang J, Yin J, et al. Uncoupling of inflammation and insulin resistance by NF-kappaB in transgenic mice through elevated energy expenditure. J Biol Chem 2010;285:4637–4644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hofmann SM, Zhou L, Perez-Tilve D, et al. Adipocyte LDL receptor-related protein-1 expression modulates postprandial lipid transport and glucose homeostasis in mice. J Clin Invest 2007;117:3271–3282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu H, Tang JR, Choi YH, et al. Importance of cAMP-response element-binding protein in regulation of expression of the murine cyclic nucleotide phosphodiesterase 3B (Pde3b) gene in differentiating 3T3-L1 preadipocytes. J Biol Chem 2006;281:21096–21113 [DOI] [PubMed] [Google Scholar]
- 15.Gao Z, He Q, Peng B, Chiao PJ, Ye J. Regulation of nuclear translocation of HDAC3 by IkappaBalpha is required for tumor necrosis factor inhibition of peroxisome proliferator-activated receptor gamma function. J Biol Chem 2006;281:4540–4547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young SH, Ye J, Frazer DG, Shi X, Castranova V. Molecular mechanism of tumor necrosis factor-alpha production in 1→3-beta-glucan (zymosan)-activated macrophages. J Biol Chem 2001;276:20781–20787 [DOI] [PubMed] [Google Scholar]
- 17.Ye J, Zhang X, Dong Z. Characterization of the human granulocyte-macrophage colony-stimulating factor gene promoter: an AP1 complex and an Sp1-related complex transactivate the promoter activity that is suppressed by a YY1 complex. Mol Cell Biol 1996;16:157–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Degerman E, Ahmad F, Chung YW, et al. From PDE3B to the regulation of energy homeostasis. Curr Opin Pharmacol 2011;11:676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye J, McGuinness OP. Inflammation during obesity is not all bad: evidence from animal and human studies. Am J Physiol Endocrinol Metab 2013;304:E466–E477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem 2007;76:481–511 [DOI] [PubMed] [Google Scholar]
- 21.Guirguis E, Hockman S, Chung YW, et al. A role for phosphodiesterase 3B in acquisition of brown fat characteristics by white adipose tissue in male mice. Endocrinology 2013;154:3152–3167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi YH, Park S, Hockman S, et al. Alterations in regulation of energy homeostasis in cyclic nucleotide phosphodiesterase 3B-null mice. J Clin Invest 2006;116:3240–3251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wada T, Onogi Y, Kimura Y, et al. Cilostazol ameliorates systemic insulin resistance in diabetic db/db mice by suppressing chronic inflammation in adipose tissue via modulation of both adipocyte and macrophage functions. Eur J Pharmacol 2013;707:120–129 [DOI] [PubMed] [Google Scholar]
- 24.Rahn Landström T, Mei J, Karlsson M, Manganiello V, Degerman E. Down-regulation of cyclic-nucleotide phosphodiesterase 3B in 3T3-L1 adipocytes induced by tumour necrosis factor alpha and cAMP. Biochem J 2000;346:337–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Razani B, Reichardt AD, Cheng G. Non-canonical NF-κB signaling activation and regulation: principles and perspectives. Immunol Rev 2011;244:44–54 [DOI] [PubMed] [Google Scholar]
- 26.Sheng L, Zhou Y, Chen Z, et al. NF-κB–inducing kinase (NIK) promotes hyperglycemia and glucose intolerance in obesity by augmenting glucagon action. Nat Med 2012;18:943–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mowers J, Uhm M, Reilly SM, et al. Inflammation produces catecholamine resistance in obesity via activation of PDE3B by the protein kinases IKKε and TBK1. eLife 2013;2:e01119. [DOI] [PMC free article] [PubMed] [Google Scholar]