

Abstract
Nephrin is required during kidney development for the maturation of podocytes and formation of the slit diaphragm junctional complex. Because nephrin expression is downregulated in acquired glomerular diseases, nephrin deficiency is considered a pathologic feature of glomerular injury. However, whether nephrin deficiency exacerbates glomerular injury in glomerular diseases has not been experimentally confirmed. Here, we generated mice with inducible RNA interference–mediated nephrin knockdown. Short-term nephrin knockdown (6 weeks), starting after the completion of kidney development at 5 weeks of age, did not affect glomerular structure or function. In contrast, mice with long-term nephrin knockdown (20 weeks) developed mild proteinuria, foot process effacement, filtration slit narrowing, mesangial hypercellularity and sclerosis, glomerular basement membrane thickening, subendothelial zone widening, and podocyte apoptosis. When subjected to an acquired glomerular insult induced by unilateral nephrectomy or doxorubicin, mice with short-term nephrin knockdown developed more severe glomerular injury compared with mice without nephrin knockdown. Additionally, nephrin-knockdown mice developed more exaggerated glomerular enlargement when subjected to unilateral nephrectomy and more podocyte apoptosis and depletion after doxorubicin challenge. AKT phosphorylation, which is a slit diaphragm–mediated and nephrin-dependent pathway in the podocyte, was markedly reduced in mice with long-term or short-term nephrin knockdown challenged with uninephrectomy or doxorubicin. Taken together, our data establish that under the basal condition and in acquired glomerular diseases, nephrin is required to maintain slit diaphragm integrity and slit diaphragm–mediated signaling to preserve glomerular function and podocyte viability in adult mice.
Keywords: nephrin, podocyte, glomerulopathy, slit diaphragm, kidney disease, animal model
Nephrin is considered the molecular building block of the slit diaphragm.1 Nephrin bridges the filtration slit to form a 35-nm-long globular cross-strand that contributes to a porous slit diagram scaffold.2 It also serves as a signaling hub3,4 by interacting with other slit diagram proteins.5,6 However, all ascribed functions of nephrin were derived from studies of cultured podocytes or mice with inherited nephrin deficiency.7,8 The function of nephrin and the effect of acquired nephrin loss in the adult mice are not known.
Nephrin is required for podocyte maturation during glomerular development.9 In humans and mice with inherited nephrin deficiency due to loss-of-function mutations of NPHS110 or Nphs1 deletion (Nphs1−/−),7,8 massive proteinuria develops in utero or soon after birth. A series of elegant studies from the laboratories of Holzman,11,12 Quaggin,13 and Holthofer14,15 highlighted the function of nephrin in rodents. In humans, nephrin is downregulated in many forms of acquired glomerular diseases, such as diabetic nephropathy,16–19 minimal-change disease,20–22 FSGS,23 and membranous nephropathy,21,23,24 and others.24,25 It has been suggested that nephrin deficiency causes podocyte injury in vivo, but this has not been directly confirmed. Development of glomerular dysfunction in rodents injected with an antinephrin antibody,26,27 as well as recurrence of nephropathy due to alloimmunization against nephrin after kidney transplantation in individuals with congenital nephropathy of the Finnish type,28,29 suggests that antinephrin antibody is detrimental to glomerular function. The mechanism of antinephrin antibody causing glomerular dysfunction in these cases, however, is not known.26
Here we produced several lines of genetically engineered mice with inducible nephrin knockdown mediated by short hairpin RNA (shRNA). We sought to directly characterize nephrin function by examining adult mice with acquired rather than inherited nephrin loss.
Results
Murine Model of Inducible Nephrin Knockdown
We generated transgenic mice with inducible shRNA-mediated Nphs1 knockdown using an in vivo RNA interference approach.30,31 Three different lines of mice encoding an miR30-based expression cassette under the transcriptional control of a doxycycline (DOX)-responsive promoter element were produced. Each line harbors a different 21-bp Nphs1 guide sequence. These three lines are called sh1401, sh1416, and sh3703. The DOX-responsive transgene, TGM, was configured with a GFP "spacer" between the tetracycline-responsive promoter element and the miR30-based expression cassette (Supplemental Figure 1A). Mice with a single copy of TGM were generated and then bred with mice that possess widespread expression of reverse tetracycline transactivator (rtTA), hereafter referred to as CAGs-rtTA mice,30 to produce double transgenic CAGs-rtTA/+; TGM/+ mice. These double transgenic mice were born at the expected Mendelian ratio and were indistinguishable from wild-type mice and singly transgenic mice harboring the CAGs-rtTA or TGM transgene.
Murine kidney development is completed between postnatal days 7 and 8.32 Induction of nephrin knockdown started at 5 weeks of age with DOX-supplemented chow (650 mg/kg). The nephrin protein and mRNA levels of 11-week-old mice induced with DOX for 6 weeks were significantly lower in all three lines of nephrin-knockdown mice than in control Luci.1309 mice, which express an shRNA targeting the firefly luciferase gene30 (Supplemental Figure 1, B and C). Two Western bands specific to nephrin with apparent molecular masses of 160 kD and 180 kD have been described.33 The 180-kD band corresponds to N-glycosylated nephrin.33 Green fluorescent protein (GFP) expression was similar between Luci.1309 and sh3703 mice. The sh3703 line demonstrated the most robust knockdown. Nephrin expression in the renal cortex of sh3703 mice was knocked down by approximately 80% compared with control Luci.1309 mice after 6 weeks of induction. Because sh3703 mice have the most robust knockdown, they were used in all subsequent experiments.
Robust knockdown was observed after 3 weeks of DOX induction and was sustainable for up to 20 weeks of induction (Figure 1A). Nephrin expression of age-matched sh3703 mice without DOX feeding (sh3703 No DOX_20w) did not differ from that of control Luci.1309 mice with and without DOX induction for 20 weeks (DOX_20w and No DOX_20w) and was markedly higher than that of sh3703 mice with DOX induction (Supplemental Figure 1D). sh3703 mice that were induced with a higher dose of DOX (650 mg/kg DOX food plus 2 mg/ml DOX in the drinking water [DOX food+H2O_20w]) had a degree of nephrin knockdown similar to that in those induced with only 650 mg/kg DOX food (DOX_20w) (Supplemental Figure 1D). The kinetics of nephrin knockdown followed a biphasic pattern of early, rapid decay followed by a progressive decline (Figure 1A). This biphasic pattern is consistent with the thought that nephrin is an integral transmembrane protein involved in stable trans interaction with adjacent foot processes and thus is not readily turned over under the basal condition. Maximal knockdown appeared to plateau at approximately 80%–90% for both nephrin protein and mRNA transcript, suggesting that the shRNA-mediated knockdown in our system is robust but not complete. Immunostaining of nephrin confirmed that glomerular nephrin expression was markedly reduced in GFP-positive glomerular cells of sh3703 mice but not Luci.1309 mice (Figure 1C). These GFP-positive glomerular cells are predominantly podocytes and not mesangial or endothelial cells.31
Figure 1.
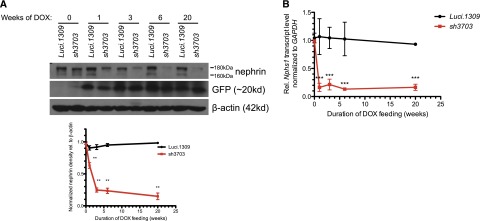

A murine model of inducible nephrin knockdown. An inducible knockdown of nephrin is robust and sustainable up to 20 weeks of induction. Inducible nephrin knockdown mice (sh3703) and control Luci.1309 mice with inducible overexpression of an shRNA against the firefly luciferase gene received DOX in the chow (625 mg/kg) starting at 5 weeks of age and continued for 20 weeks until 25 weeks of age. (A) Representative Western blots of nephrin in kidney protein lysates of sh3703 and Luci.1309 mice that received DOX for 0, 1, 3, 6, and 20 weeks. A bar graph of normalized ratios of Western band densities of nephrin/β-actin for the Western blots shown. (B) Nephrin mRNA transcript levels of sh3703 mice relative to age-matched, DOX-fed Luci.1309 mice in the kidney. Each point on the graph contains samples from at least three mice. ***P<0.001 compared with matched Luci.1309 mice. (C) Nephrin immunostaining and GFP fluorescence on frozen kidney sections of mice after 0, 1, 3, 6, and 20 weeks of induction. Scale bars, 50 μm. Error bars, SEM.
Western blotting using an antibody against the intracellular domain of nephrin revealed both of the nephrin-specific bands (180 kD and 160 kD) in the kidney cortex lysate. Consistent with previous reports, we also identified the 160-kDa nephrin band in extrarenal organs: heart, spleen, pancreas, and brain (Supplemental Figure 1E).7,34,35 The exclusive presence of the 180-kD band in the kidney lysate has been described previously.35 Because DOX does not cross the blood-brain barrier, GFP and nephrin expressions did not different between mice with and without induction. Histologic features of the myocardium did not appear to be different between sh3703 and Luci.1309 mice (Supplemental Figure 1F).
Long-term, but Not Short-term, Nephrin Knockdown in Adult Mice Promotes Proteinuria
sh3703 mice fed with DOX starting at 5 weeks of age developed more proteinuria than did control Luci.1309 mice after 24 weeks of induction (Figure 2A). In contrast, age-matched Luci.1309 mice with DOX induction did not develop marked proteinuria. The urinary albumin excretion in sh3703 mice was approximately 3-fold higher than that in Luci.1309 mice after 24 weeks of induction (mean±SD, 0.179±0.041 versus 0.052±0.014 μg of albumin per μg of creatinine; P<0.05). The serum creatinine of sh3703 and Luci.1309 mice did not differ before and after 20 weeks of induction (Figure 2B). The kidney-to-body weight ratio and serum glucose level were also not different (Supplemental Figure 2, A and B). The polyanionic charge of the glomerulus as assessed by Alcian blue staining also appeared to be similar after 6 and 20 weeks of knockdown (Figure 2C).
Figure 2.
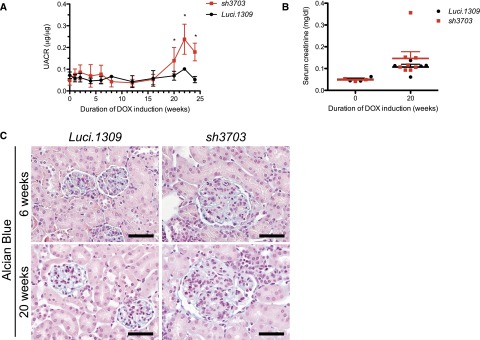
Long-term nephrin knockdown promotes mild proteinuria without effecting serum creatinine or glomerular polyanionic charge. Glomerular function and polyanionic charge of mice with and without nephrin knockdown. DOX-inducible nephrin knockdown started at 5 weeks of age and continued for 20 weeks. (A) Urinary albumin excretion as quantified by the ratio of urinary albumin to creatinine ratio (UACR, μg of albumin/μg of creatinine) after 0, 1, 2, 4, 6, 8, 12, 16, and 20 weeks of DOX induction. UACR of sh3703 mice was significantly higher than that of Luci.1309 mice (P=0.02) after 20 weeks of induction. *P<0.05 compared with sh3703 mice at 0 week of induction. (B) Serum creatinine of sh3703 and Luci.1309 mice was not statistically different at 20 weeks of induction. (C) Representative images of a kidney section stained with Alcian blue stain to assess the polyanionic charge of the glomerulus. Scale bars, 50 μm. Error bars, SEM.
Long-term Nephrin Depletion Disrupts All Components of the Glomerular Tuft
After 20 weeks of induction, the glomerular cross-sectional area of sh3703 mice was enlarged (Figure 3, A and B). In contrast, the glomerular structure and glomerular cross-sectional area of Luci.1309 kidney at up to 20 weeks of induction and sh3703 kidney at up to 6 weeks of induction appeared normal (Figure 3, A and B). Glomeruli of age-matched sh3703 mice without DOX induction appeared normal (Supplemental Figure 1G), thus ruling out any off-target effects or leakiness of shRNA as the cause of glomerular changes in DOX-induced sh3703 mice. Inspection of the glomerular tuft by transmission electron microscopy revealed diffuse podocyte effacement and foot process (FP) widening in more than half of the total glomerular basement membrane (GBM) surface area for sh3703 mice (Figure 3C). The GBM was irregularly thickened with a scalloped appearance (Figure 3C). The endothelial layer appeared disorganized with widening of the subendothelial zone, which was occupied by loose extracellular matrix (arrowheads in Figure 3C). To our surprise, slit diagram complexes were still present despite more than 80% nephrin knockdown (arrows in Figure 3C), albeit at a lower frequency because of diffuse FP effacement. Some of the slit diagram appeared apically displaced. The FP width in sh3703 mice was 4-fold greater than that in Luci.1309 mice after 20 weeks of induction (1582±139 nm versus 388±46 nm; P<0.01) (Figure 3D) but did not differ between sh3703 and Luci.1309 mice after 6 weeks of induction.
Figure 3.


Long-term nephrin depletion disrupts all components of the glomerular tuft. Glomerular histology and ultrastructure of mice with and without nephrin knockdown. (A) Representative images of period acid-Schiff–stained kidney sections of sh3703 and Luci.1309 mice after 0, 3, 6, and 20 weeks of DOX induction. (B) Graph of glomerular cross-sectional area (μm2) for Luci.1309 and sh3703 mice after 0, 3, 6, and 20 weeks of DOX feeding (5055±150 μm2 for Luci.1309_20w versus 7214±250 μm2 for sh3703_20w; **P<0.001). (C) Representative images of transmission electron microscopy for Luci.1309 and sh3703 after 6 and 20 weeks of induction. Slit diaphragms (arrows) are present in filtration slits of Luci.1309 and sh3703 mice after 6 and 20 weeks of induction. Widening of the subendothelial zone of the glomerular basement membrane (arrowheads) occupied by amorphous extracellular matrix material is observed. (D) Graph of the FP widths of indicated mice. **P<0.01; n=3 mice for Luci.1309 and sh3703 after 6 weeks of induction; n=3 mice for Luci.1309 after 6 weeks of induction; n=4 mice for sh3703 after 20 weeks of induction. (E) Relative glomerular expression of genes that encode for slit diaphragm components and podocyte-specific genes in sh3703 mice compared with Luci.1309 mice after 20 weeks of induction. Black squares, genes not statistically different between sh3703 and Luci.1309 mice. Red squares: genes statistically different between sh3703 and Luci.1309 mice. GOI, genes of interest. n=3 Luci.1309 mice and n=5 sh3703 mice. None of the genes examined were different by more than 2-fold. (F) Representative images of immunostaining for synaptopodin (Synpo) and podocin on frozen kidneys sections of mice after 20 weeks of induction. Scale bar, 50 μm. Error bars, SEM.
The glomerular transcript level of genes encoding for protein components of the slit diagram (e.g., Nphs2/podocin, Neph1, Fat1/proto-cadherin, Cdh3/P-cadherin, zona occludin 1, F11r/JAM-A, Ocln/occludin, and Cgn/cingulin) or involved in podocyte cytoskeleton organization (e.g., CD2AP, Inf2/inverted formin 2, Actn4/α-actinin 4, synaptopodin/Synpo and Wt1) was not different by more than 2-fold between sh3703 and Luci.1309 mice after 20 weeks of induction (Figure 3E). This lack of a difference in the glomerular expression of podocyte-specific genes is consistent with what has been described previously for Nphs1 knockout mice.9 Of note, the glomerular expression of Neph1 and Ocln in sh3703 mice was significantly less than in Luci.1309 mice, but the difference in expression levels was less than 2-fold. As expected, Nphs1 expression of sh3703 mice was reduced to less than 15% of that in Luci.1309 mice. Neph3 and Claudin3 are upregulated in nephrin-knockout mice.9,28 However, in sh3703 mice with acquired nephrin knockdown, glomerular Neph3 expression did not appear to be different and glomerular Claudin3 expression was actually lower than that in Luci.1309 mice. To confirm that the transcript levels reflect the protein levels, we immunostained for several slit diagram proteins: synaptopodin, podocin, and zona occludin 1. The intensity of immunostaining did not appear to be different (Figure 3F).
Prenatal Nephrin Knockdown Causes Massive Proteinuria
To determine the effect of congenital nephrin knockdown, male mice that are homozygous for the CAGs-rtTA transgene and compound heterozygous for the Luci.1309 and sh3703 TGM transgenes (CAGs-rtTA/CAGs-rtTA; Luci.1309/sh3703) were crossed to wild-type C57BL6 female mice (+/+; +/+) (Figure 4A). Inseminated wild-type female mice with vaginal plugs were separated from the male mice and then fed DOX starting on E7. In three litters resulting from this breeding scheme, a total of 12 pups were born: Six (50%) were CAGs-rtTA/+; Luci.1309/+ and 6 (50%) were CAGs-rtTA/+; sh3703/+. All CAGs-rtTA/+; Luci.1309/+ pups survived to >6 weeks of age; all CAGs-rtTA/+; sh3703/+ pups died within 72 hours after birth. The amount of proteinuria in the 3 CAGs-rtTA/+; sh3703/+ pups that survived >24 hours was >90-fold higher than that of the 6 CAGs-rtTA/+; Luci.1309/+ pups (85.4±5.1 versus 0.99±0.3 μg albumin/μg of creatinine; P<0. 001) (Figure 4B). Nephrin expression was reduced in the CAGs-rtTA/+; sh3703/+ pups (Figure 4C). Tubular casts, tubular resorption droplets, and glomerular collapse with expanded Bowman capsule were observed in CAGs-rtTA/+; sh3703/+ kidneys but not CAGs-rtTA/+; Luci.1309/+ kidneys (Figure 4D). Glomerular capillary loops appeared distended, and the overlying glomerular basement membrane were denuded of visceral epithelial cell coverage. Because CAGs-rtTA/+; Luci.1309/+ and CAGs-rtTA/+; sh3703/+ pups were born at the expected Mendelian ratio, we concluded that nephrin is not essential for intrauterine survival. The phenotype of sh3703 mice with congenital nephrin knockdown is similar to what has been described for pups with Nphs1 inactivating mutations.
Figure 4.

Prenatal nephrin knockdown causes massive proteinuria. Prenatal DOX feeding for nephrin knockdown during development. (A) A schematic of the breeding scheme with the genotype of parents and pups specified. Inseminated female was fed DOX on day E7. The observed percentage of pups with a given genotype indicated within parentheses. (B) Urinary albumin excretion of pups with and without nephrin knockdown, sh3703 and Luci.1309, respectively. (C) Western blots of kidney lysates of sh3703 and Luci.1309 pups. Each lane is a lysate from a different mice. (D) Period acid-Schiff–stained kidney sections of Luci.1309 and sh3703 pups with prenatal DOX feeding. Yellow arrows in part c, dilated Bowman capule with collapsed glomeruli. Green arrow in part d, protein resorption droplets within tubular cells. Asterisk marks a tubular cast within the tubular lumen. Green arrowheads in part e, visceral epithelial glomerular cells. Green arrow in part e, segment of glomerular basement membrane without podocytes. Double-headed green arrow in part e, dilated capillary loop. Error bars, SEM.
Nephrin Reduction Promotes Hyperfiltration-Induced Glomerular Injury
Because glomerular hyperfiltration, which occurs in many pathologic conditions,29 transduces excessive hydraulic pressure on the filtration structure, we hypothesized that a nephrin knockdown could exacerbate hyperfiltration-induced glomerular injury. To test this hypothesis, we performed unilateral nephrectomy (UNPX) to induce glomerular hyperfiltration in the remnant kidney of mice with nephrin knockdown. Induction of nephrin knockdown started at 5 weeks of age and continued until 27 weeks of age (Figure 5A). UNPX was performed at 11 weeks of age. UNPX sh3703 mice developed a progressive increase in albuminuria starting as early as 19 weeks of age (8w after UNPX). In contrast, albuminuria in Luci.1309 mice with UNPX did not change markedly (Figure 5B). The urinary albumin-to-creatinine ratio of sh3703 mice was 63-fold higher than that of Luci.1309 mice at 27 weeks of age (5.369±2.804 μg/μg versus 0.085±0.034 μg/μg; P<0.001). Albumin was the predominant protein excreted in the urine as determined by Coomasie blue staining of urinary proteins resolved by gel electrophoresis (Figure 5C). The serum creatinine level of sh3703 mice was also significantly higher than that of Luci.1309 mice (0.2128±0.034 mg/dl versus 0.1216±0.011 mg/dl; P=0.05) (Figure 5D). The remnant kidney of UNPX sh3703 mice developed marked diffuse mesangial hypercellularity, glomerulomegaly, and tubular protein casts, but not in the remnant kidney of UNPX Luci.1309 mice (Figure 5E, Table 1). Variable involvement of segmental and global sclerosis, tubular protein casts, and interstitial inflammation was observed in the sh3703 group (Table 1). In contrast, none of the Luci.1309 mice developed glomerulosclerosis, tubular protein casts, or interstitial inflammation. The kidney-to-body weight ratio of the remnant kidney of sh3703 mice, however, was not significantly different from that of Luci.1309 mice (Figure 5F). The glomerular cross-sectional area of UNPX sh3703 mice was 72% larger than that of the UNPX Luci.1309 mice (8957±261.1 versus 4975±104 μm2; P<0.05; n=6 NPX sh3703 mice; n=4 NPX Luci.1309 mice; 20 glomeruli per mouse) (Figure 5G). The glomerular cross-sectional area of UNPX sh3703 mice was also significantly larger than that of matched sh3703 with the same duration of DOX feeding (sh3703_22w) (8957±261.1 versus 5814±264.4 μm2; P<0.05; n=6 NPX sh3703 mice; n=2 sh3703_22w mice; 20 glomeruli per mouse) (Figure 5G). Electron microscopy of the glomeruli demonstrated FP effacement. In some of the filtrations slits examined, the slit diagram appeared to be absent (Figure 5H).
Figure 5.


Nephrin reduction promotes hyperfiltration-induced glomerular injury in uninephrectomized mice. Glomerular function and structure of nephrin knockdown mice with UNPX. (A) Schema of study design. Five-week-old sh3703 and Luci.1309 mice were induced with DOX, and UNPX was performed at 11 weeks of age. Mice were euthanized at age 27 weeks. DOX induction was continuous from ages 5 to 27 weeks. (B) Urinary albumin excretion as quantified by the urinary albumin-to-creatinine ratio (UACR). P<0.05 compared with the sh3703 group at 15 weeks of age. P<0.001 compared with the Luci.1309 group at 27 weeks of age. (C) Urine (7.5 μl per lane) from each of the indicated mice obtained at 15 and 27 weeks of age was analyzed on a 10% denaturing polyacrylamide gel together with 15, 7.5, 1.5, and 0.75 μg BSA and then stained with Coomassie brilliant blue R250. (D) Bar graph of serum creatinine levels of mice at 27 weeks of age as determined by an HPLC-based measurement. *P<0.05. (E) Graph of kidney-to-body weight ratios for the remnant kidney for the indicated mice at the end of the experiment. (F) Representative histologic sections of mice at age 27 weeks. (G) Graph of the cross-sectional area of the glomerular tuft for mice at age 27 weeks. **P<0.01. Scale bar, 50 μm. (H) Representative images from transmission electron microscopy for sh3703 and Luci.1309 mice 16 weeks after UNPX. Glomerular basement membrane of sh3703 mice exhibits irregular thickening. Black arrows, slit diaphragms. Red arrows, filtration slits without visible slit diaphragms. Scale bar, 1μm. Error bars, SEM.
Table 1.
Histologic features of mice with glomerular hyperfiltration
| Mouse Model | Glomerular Compartment | Tubular Compartment | ||
|---|---|---|---|---|
| Glomeruli with Sclerosis (%) | Mesangial Expansion (1–3+) | Tubular Area with Protein Casts (%) | Interstitial Inflammation (1–3+) | |
| Luci.1309 | ||||
| 1 | 0 | 0 | 0 | 0 |
| 2 | 0 | 1 | 0 | 0 |
| 3 | 0 | 0 | 0 | 0 |
| sh3703 | ||||
| 1 | <10 | 3 | 20 | 1 |
| 2 | <10 | 3 | 20 | 0 |
| 3 | 0 | 1 | 0 | 0 |
| 4 | 0 | 3 | 0 | 0 |
| 5 | <10 | 3 | 20 | 1 |
Nephrin Knockdown Accelerates Doxorubicin-Induced Glomerulosclerosis
Next, we evaluated the development of doxorubicin-induced glomerulosclerosis in mice with nephrin knockdown. Most inbred mouse strains, including C57BL6 and FVBN, are resistant against doxorubicin-induced renal injury.36 Luci.1309 and sh3703 mice are on a mixed genetic background with contribution from C57BL6 and 129sv strains. We first evaluated the dose-dependent response of our mouse strain to doxorubicin-induced nephropathy. Mice that have been induced with DOX for 6 weeks were injected with one of three different doses of doxorubicin: 25 mg/kg, 18.8 mg/kg, and 15 mg/mkg. At doxorubicin doses of 25 mg/kg and 18.8 mg/kg, all sh3703 mice died within 10 days after doxorubicin injection (n=2 mice at 25 mg/kg and n=6 mice at 18.8 mg/kg). In contrast, all Luci.1309 mice injected with doxorubicin 25 mg/kg or 18.8 mg/kg survived. When doxorubicin 15 mg/kg was given, 80% of the sh3703 mice (n=5) and 100% of the Luci.1309 mice were viable 4 weeks after doxorubicin injection. The renal findings of mice that received doxorubicin 15 mg/kg were characterized.
Four weeks after doxorubicin injection, sh3703 mice developed significantly more weight loss, albuminuria, and serum creatinine elevation compared with Luci.1309 mice (Figure 6, A–E). Semi-quantitative histologic evaluation of the Luci.1309 kidneys revealed that tubular casts were present in <10% of the tubular compartment and glomerular abnormality was absent (Figure 6F, panel a and b, Table 2). In contrast, kidneys of sh3703 mice that received doxorubicin had tubular protein resorption droplets, tubular protein casts involving >20% of the tubular area (range, 20%–80%), and glomerulosclerosis involving 20%–60% of the glomeruli (Figure 6F, panels c–h, Table 2). Prominent interstitial inflammation was observed in 5 of the 7 mice in the sh3703 group (Figure 6F, panel f, Table 2). Mesangial expansion with increased mesangial cell number and mesangial matrix deposition were present in all the sh3703 mice but in only 1 of 3 Luci.1309 mice. Marked glomerular epithelial cell hyperplasia was present in all the sh3703 mice (Figure 6F, panel g, arrows), suggesting a compensatory response to podocyte depletion. Consistent with the possibility of podocyte depletion in sh3703 mice, segments of denuded glomerular basement membrane without podocyte coverage (Figure 6F, panel h, arrows) and dilated capillary loops with capsular adhesion of the glomerular tuft (Figure 6F, panel h, arrowhead) were observed in the sh3703 kidneys, but not the Luci.1309 kidneys. Focal areas of denuded GBM without any FP coverage were noted (Figure 6G). Diffuse FP effacement and irregularly thickened GBM were also present in doxorubicin-injected sh3703 kidneys (Figure 6G). Electron-dense junctional complex bridging the FPs appeared to be absent in some filtration slits (Figure 6G).
Figure 6.


Nephrin deficiency accelerates doxorubicin-induced glomerulosclerosis. Nephrin knockdown promotes doxorubicin-induced disruption of glomerular function and structure. (A) Schema of study design. Five-week-old sh3703 and Luci.1309 mice were induced with DOX until 15 weeks of age. At 11 weeks of age, both groups of mice were injected with doxorubicin (15 mg/kg body weight) via the tail vein and followed for 4 more weeks. (B) Graph of normalized body weights of sh3703 mice at 11, 12, 13, 14, and 15 weeks of age relative to their body weights at 11 weeks of age. *P<0.05 for a comparison of 15-week-old Luci.1309 versus sh3703 mice. (C) Graph of urinary albumin-to-creatinine ratios (UACR) of mice at 11, 13, and 15 weeks of age. P<0.05 and P<0.001 for comparisons of sh3703 mice at 13 and 15 weeks of age to matched sh3703 mice at 11 weeks of age. *P<0.05 for a comparison of 15-week-old Luci.1309 versus sh3703 mice. (D) Urine (7.5 μl per lane) from each of the indicated mice before (11 weeks of age) and after (15 weeks of age) doxorubicin injection was analyzed on a 10% denaturing polyacrylamide gel together with 15, 7.5, 1.5, and 0.75 μg BSA and then stained with Coomassie brilliant blue R250. (E) Graph of serum creatinine levels of the indicated mice before and after doxorubicin injection as determined by an HPLC-based measurement. P<0.001 for comparisons of sh3703 mice at 13 and 15 weeks of age to matched sh3703 mice at 11 weeks of age. *P<0.05 for a comparison of 15-week-old Luci.1309 versus 15-week-old sh3703 mice. (F) Representative histologic sections of mice at 4 weeks after doxorubicin injection. #, tubular protein casts. Normal-appearing histologic sections of doxorubicin-injected Luci.1309 mice 4 weeks after doxorubicin injection (panels a and b). Doxorubicin-injected sh3703 mice developed tubular protein casts (c), glomerulosclerosis (d), tubular protein resorption droplets (e), tubulointerstitial inflammation (f), glomerular epithelial cell proliferation (arrows in part g), dilated capillary loops with denuded basement membrane (arrows in part h), and adhesion of the capillary tuft to the Bowman capsule (arrowhead in part h). Scale bar, 50 μm. (G) Representative electron microscopy images. Doxorubicin-injected Luci.1309 mice display focal FP effacement (part a, magnified view in part b, and another magnified view in part c), but GBM is always covered by FP. Slit diaphragms are present in the filtration slits of Luci.1309 mice (arrows in part d). In contrast, denuded GBM without any FP coverage is observed in doxorubicin-injected sh3703 mice (arrowheads in part e and magnified view in f). Diffuse effacement of FP (part g) and occasional absence of slit diaphragm from filtration slits (arrowheads in part h) are present in doxorubicin-injected sh3703 mice. E, endothelial cell; P, podocytes. Error bars, SEM.
Table 2.
Histology of mice with doxorubicin nephropathy
| Mouse Model | Glomerular Compartment | Tubular Compartment | |||
|---|---|---|---|---|---|
| Glomeruli with Sclerosis (%) | Mesangial Expansion (1–3+) | Severity of Glomerular Epithelial Cell Hyperplasia (1–3) | Tubular Area with Protein Casts (%) | Interstitial Inflammation (1–3+) | |
| Luci.1309 | |||||
| 1 | 0 | 0 | 0 | 20 | 1 |
| 2 | 0 | 1 | 0 | 0 | 0 |
| 3 | 0 | 0 | 0 | 0 | 0 |
| sh3703 | |||||
| 1 | 40 | 2 | 2 | 60 | 3 |
| 2 | 60 | 3 | 2 | 20 | 3 |
| 3 | 60 | 3 | 2 | 80 | 3 |
| 4 | 20 | 1 | 1 | 20 | 1 |
| 5 | 60 | 3 | 2 | 80 | 3 |
| 6 | 60 | 3 | 2 | 20 | 3 |
| 7 | 20 | 3 | 1 | 40 | 2 |
AKT Signaling and Podocyte Viability Are Impaired in Nephrin Knockdown Mice
Nephrin is a major molecular component of the slit diagram, which is a key signaling hub of the podocyte. Nephrin stimulates PI3K-dependent AKT signaling and prevents Bad-mediated apoptosis of podocytes in culture.37 In our model, long-term nephrin knockdown (i.e., 20 weeks) was associated with reduced glomerular AKT phosphorylation (Figure 7A). Glomerular AKT phosphorylation was significantly lower in Luci.1309 kidneys with UNPX and doxorubicin nephropathy compared with Luci.1309 mice without kidney disease. AKT phosphorylation of sh3703 glomeruli was less than Luci.1309 glomeruli in mice without glomerular injury (i.e., DOX 20w). In mice with glomerular injury (UNPX or doxorubicin), AKT phosphorylation was also less in sh3703 mice compared with Luci.1309 mice. Because AKT phosphorylation prevents podocyte apoptosis in vitro,37 we assessed podocyte apoptosis by transferase deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick end labeling assay (TUNEL) staining and Wilms’ tumor-1 (WT-1) coimmunolabeling. TUNEL and WT-1 double-positive cells were detected in glomeruli of sh3703 mice after 20 weeks of nephrin knockdown as well as sh3703 mice injected with doxorubicin (Figure 7B), but not in glomeruli of Luci.1309 mice, sh3703 mice with UNPX, or sh3703 mice after 6 weeks of DOX induction (data not shown). Podocyte density as quantified by the number of WT-1–positive cells per glomerular cross-sectional was lower in the sh3703 group than the Luci.1309 group after 20 weeks of DOX induction, as well as in the doxorubicin model of acquired glomerular injury (Figure 7C). The glomerular density of WT-1–positive nuclei in the doxorubicin-treated sh3703 kidney was 56.6% of the Luci.1309 kidney (2.009±0.16 versus 3.549±0.21 nuclei per 1000 μm2 of glomerular cross-sectional area; P<0.001). In contrast, the density of WT-1–positive cells in glomeruli of sh3703 mice did not differ from that of Luci.1309 mice after 6 weeks of DOX induction or after UNPX. These findings suggest that nephrin reduction in adult mice impairs AKT phosphorylation, which is a nephrin-dependent, slit diagram–mediated signaling event in the podocyte. This event is associated with loss of podocyte viability and a reduction in podocyte number, both under the basal condition as well as in the doxorubicin model of acquired podocyte injury.
Figure 7.


AKT signaling and podocyte viability are impaired in nephrin knockdown mice. AKT phosphorylation, apoptosis, and glomerular density of podocytes in mice with nephrin knockdown. (A) Representative Western blots of glomerular lysates from indicated mice for phospho-AKT (Ser473), total AKT, nephrin, and β-actin. After 20 weeks of DOX induction (DOX 20w), AKT phosphorylation was markedly reduced in sh3703 mice compared with Luci.1309, but not after 6 weeks of DOX induction (DOX 6w). AKT phosphorylation was markedly reduced in the remnant kidney of Luci.1309 mice with UNPX and further reduced in the remnant kidney of sh3703 mice with UNPX compared with Luci.1309 mice without UNPX. AKT phosphorylation was also reduced in doxorubicin-treated Luci.1309 mice and further reduced in doxorubicin-treated sh3703 mice. Bar graph shows the normalized ratio of phospho-to-total AKT band density plotted for each of the indicated group of mice. (B) Representative images of kidney sections from the indicated mice labeled with an antibody against WT-1 to identify podocytes as well as TUNEL for apoptotic nuclei. Glomerular cells with colabeling of WT-1 and TUNEL were detected in kidneys of sh3703 mice after 20 weeks of DOX induction (DOX 20w) and doxorubicin-injected sh3703 mice induced with DOX. Arrows indicate cells with colocalization of the Hoescht nuclear stain, TUNEL, and WT-1. Scale bar, 50 μm. (C) Representative images of kidney sections stained with WT-1. Bar graphs below represent quantification of the density of WT-1–labeled cells per glomerular tuft area (cells/1000 μm2). The density of WT-1 cells was significantly reduced for sh3703 mice after 20 weeks of DOX induction (DOX 20w) as well as doxorubicin-injected sh3703 compared with their corresponding matched control Luci.1309 mice. ***P<0.001. Error bars, SEM. NS, not significant.
Discussion
Here we observed that acquired, long-term nephrin knockdown affects all components of the glomerulus—podocytes, mesangial cells and matrix, GBM, and endothelial cells—which had not been seen previously in other models of nephrin deficiency. Prior studies have delineated cell-cell and cell-matrix cross-talks within the glomerular compartment.38 In particular, podocyte-specific injury causes mesangial cell proliferation,39 and disrupted VEGF-mediated podocyte-endothelial crosstalk leads to endothelial injury40 and mesangial expansion.41 Because nephrin acts in concert with other slit diagram proteins to regulate the organization of membrane lipid rafts, we reasoned that acquired nephrin deficiency disrupts clustering of membrane raft microdomains to prevent nephrin-mediated signaling (i.e., AKT phosphorylation), which could directly affect the podocytes or other glomerular cells that interact with podocytes.
It remains to be determined why proteinuria, FP effacement, and podocyte dropout occurred with long-term but not short-term nephrin knockdown even though robust nephrin depletion was achieved after the third week of knockdown. Others have questioned the central role of nephrin in guiding, forming, and maintaining the slit diagram on the basis of the observation that birds lack an ortholog of NPHS1 but still form slit diagram within their kidneys and can retain serum albumin in their blood.3,42,43 So perhaps, as previously suggested by others,3,42 it is not nephrin per se that is critical for preventing proteinuria but the slit diagram itself that is required to maintain the glomerular ultrafilter. Consistent with this hypothesis, slit diagrams were observed in mice with >80% nephrin depletion.
It appears that the degree of knockdown was greater for the 160-kD than for the 180-kD nephrin band. Because shRNA-mediated nephrin knockdown depletes the total nephrin pool, one would not expect preferential targeting of the nephrin with and without N-glycosylation. We speculate the reason that 160-kD nephrin band is depleted to a greater extent is because the N-glycosylated (180-kD) nephrin participates in stable intercellular nephrin-to-nephrin interactions and has a slower turnover compared with nonglycoslyated (160-kD) nephrin, which makes the 180-kD nephrin more resistant to shRNA-mediated depletion. This supposition is consistent with our observation that both nephrin bands were equally reduced in nephrin knockdown mice with doxorubicin-induced podocyte injury, which is a pathologic condition known to increase nephrin turnover.
Because nephrin expression is reduced in glomerular diseases, reduction of nephrin is considered to cause glomerular and podocyte injury either directly or indirectly. Hauser et al. proposed that endothelin-1–mediated shedding of nephrin from podocytes is a key determinant of urinary protein loss in the context of endothelial injury, such as pre-eclampsia, hypertension, and diabetes.44 Others have postulated that nephrin is a critical determinant of insulin responsiveness in podocytes and nephrin loss could contribute to the development of diabetic nephropathy.1,4 In addition, pharmacologic blockade of angiotensin II activity prevents the reduction of nephrin in experimental models of diabetic nephropathy45 and passive Heymann nephritis46 and clearly mitigates the progression of renal disease in those models. However, the causal relationship between nephrin reduction and glomerular injury has never been demonstrated. Here we provide the first direct experimental demonstration that nephrin loss causes podocyte and glomerular injury under basal conditions as well as in experimental glomerular injury models.
Our findings suggest that nephrin is critical for glomerular function under basal conditions as well as in glomerular diseases. Thus, preservation of nephrin expression is a potential therapeutic approach that should be explored in patients with glomerular disease to mitigate podocyte loss and glomerular injury.
Concise Methods
Transgenic Mice and Experimental Models of Glomerular Disease
Genotyping
The genotyping for the hairpins was done using the ColA1-TGM primers: ColA1-For 5′-AAT CAT CCC AGG TGC ACA GCA TTG CGG-3′, ColA1-Rev 5′- ACT TGA GGG CTC ATG AAC CTC CCA GG-3′, SAdpa-Rev 5′-ATC AAG GAA ACC CTG GAC TAC TGC G-3′ (amplicon size wild-ype 220-bp and mutant 295-bp). Thermo profile for the PCR reaction was as follows: 95°C for 6 minutes followed by 35 cycles at 95°C for 40 seconds, 62°C for 45 seconds, and 72°C for 1 minute, then 72°C for 10 minutes. The genotyping for the CAGs-rtTA mice was done using the following primers: Ch8 For1 5′- TGC CTA TCA TGT TGT CAA A-3′. SApA For1 5′- CTG CTG TCC ATT CCT TAT TC-3′, Ch8 Rev2 5′- CGA AAC TCT GGT TGA CAT G-3′ (amplicon size wild-type 363-bp and mutant 330-bp). Thermo profile for the PCR reaction was as follows: 95°C for 2 minutes followed by 35 cycles at 95°C for 30 seconds, 56°C for 30 seconds, and 72°C for 45 seconds, then 72°C for 5 minutes. All animal studies were performed in accordance with the guidelines of and approved by the Institutional Animal Care and Use Committee at the Icahn School of Medicine at Mount Sinai.
Doxorubicin Murine Model
In the doxorubicin murine model, the Luci.1309 mice and sh3703 mice (both 5 weeks old) were fed with DOX (650 mg/kg) for 6 weeks and received intravenous doxorubicin by tail-vein injection.36 Urine was collected weekly to assess for proteinuria, and serum was collected to assess for serum creatinine. Mice were euthanized 4 weeks after doxorubicin injection.Unilateral NephrectomyMice were fed with DOX (650 mg/kg) for 6 weeks and then the right kidney was removed through a flank surgical approach, as previously described.47 Each mouse was anesthetized and a skin incision was made parallel to the last rib beginning midway between that rib and the iliac crest. The renal vessels and the ureter were ligated and severed, and the kidney was withdrawn through the incision.
Mice Breeding
First, we generated male mice that were homozygous for the CAGs-rtTA transgene and compound heterozygous for the Luci.1309 and sh3703 ColTGM transgene (CAGs-rtTA/CAGs-rtTA; Luci1309/sh3703) by breeding a female CAGs-rtTA/+; Luci1309/+ mouse with a male CAGs-rtTA/+; sh3703/+ mouse. Male CAGs-rtTA/CAGs-rtTA; Luci1309/sh3703 mice were then crossed with wild-type C57BL6 female mice (+/+; +/+). Impregnated wild-type C57BL6 female mice (+/+; +/+) were then separated from the male mice and fed with DOX (625 mg/kg chow) starting on E7. The day of conception was determined by checking the vaginal plug.
Antibodies
A rabbit antibody against the intracellular domain of nephrin was produced by Lawrence Holzman (University of Pennsylvania, Philadelphia, PA), and a mouse antibody to synaptopodin was a gift from Dr. Peter Mundel (Massachusetts General Hospital, Boston, MA). We also used rabbit antibody to podocin (Sigma-Aldrich, St. Louis, MO), rabbit antibody to WT-1 (Santa Cruz Technology, Santa Cruz, CA), mouse antibody to GFP (Santa Cruz Technology), and rabbit antibody to phospho-Serine 473-AKT and total-AKT (Cell Signaling Technology, Beverly, MA).
Glomeruli Isolation
Mouse glomeruli were isolated as described elsewhere.48 Briefly, animals were perfused with Hank's buffered salt solution containing 2.5 mg/ml iron oxide and 1% BSA. At the end of perfusion, kidneys were removed, decapsulated, minced into 1-mm3 pieces, and digested in Hank's buffered salt solution containing 1 mg/ml collagenase A and 100 units/ml deoxyribonuclease I. Digested tissue was then passed through a 100-μm cell strainer and collected by centrifugation. The tissue was resuspended in 2 ml of Hank's buffered salt solution, and glomeruli were collected using a magnet. Total RNA was isolated using TRIzol (Invitrogen).
Measurements of Serum Creatinine
Serum creatinine was measured by an HPLC-based method as described previously.49 Five microliters of serum per mouse was processed as described and then assayed using a Shimadzu Prominence HPLC unit (Columbia, MD) connected with a cation exchange column (PRP-X200; Hamilton Co., Reno, NV).
Histopathology and Immunohistochemistry Staining of Kidney Tissue
Kidney samples were fixed in 10% formalin, embedded in paraffin, and sectioned to 4-μm thickness. Periodic acid-Schiff–stained sections were used to assess kidney histologic features. Renal histologic abnormalities were scored as previously described.50 The glomerular parameters scored included the percentage of glomeruli exhibiting segmental or global sclerosis, the severity of mesangial expansion (graded on a semiquantitative scale from 0 to 3+), and the severity of podocyte hyperplasia (graded on a semiquantitative scale from 0 to 3+). For the latter parameter, podocyte hyperplasia was graded as 0 (absent), 1+ (involving 1%–25% of all glomeruli sampled), 2+ (involving 26%–50% of glomeruli), and 3+ (involving >50% of glomeruli). For the evaluation of the tubulointerstitial compartment, we assessed the percentage of total renal parenchyma occupied by tubular casts and/or microcysts as well as the severity of interstitial inflammation (graded on a semiquantitative scale from 0 to 3+). For the latter parameter, interstitial inflammation was graded as 0 (absent), 1+ (involving 1%–25% of the cortical parenchyma), 2+ (involving 25%–50%), and 3+ (involving > 50%).
Frozen sections were used for immunofluorescence staining for nephrin, podocin, synaptopodin, and WT-1, and images were taken by using a Zeiss Axioplan 2 IE microscope.
Alcian Blue Staining
Deparaffinized tissue sections were stained in 1% Alcian blue (Sigma-Aldrich) solution and counterstained with 0.1% fast red (Vector) as described previously.51
TUNEL Staining
TUNEL was performed using TUNEL labeling mix (Promega Corp., Fitchburg, WI), according to the manufacturer’s instructions. At least 30 glomeruli from each mouse were photographed at 400× magnification. The number of WT-1–positive nuclei were counted and the cross-sectional area of the glomerular tuft encompassing the WT-1–positive nuclei was measured using ImageJ software, version 1.47. The number of WT-1–positive nuclei per 1000 μm2 of glomerular tuft area assessed was calculated.
Transmission Electron Microscopy
Mice were perfused with phosphate-buffered saline and then immediately fixed in glutaraldehyde. Sections were mounted on a copper grid and photographed under a Hitachi H7650 microscope.
Urine Albumin and Creatinine
Urine creatinine was quantified using commercial kits from BioAssay Systems (Hayward, CA). Urine albumin was determined using a commercial assay from Bethyl Laboratory Inc. (Houston, TX). Urine albumin excretion was expressed as the ratio of urine albumin to creatinine.
Coomasie Blue Staining of Urinary Proteins
Urine was denatured with Laemmli sample buffer and then resolved on 10% SDS-PAGE. Proteins on the gel were detected by Coomassie blue staining. Each lane contains 7.5 µl of urine.
Quantitative RT-PCR
One microgram of total RNA extracted from glomerular extracts was reverse transcribed to complementary DNA, which was used for quantitative RT-PCR. Primers for RT-PCR were designed by using Primer-Blast (National Center for Biotechnology Information) (see Table 3). Gene expression was normalized to Gapdh and fold change in expression relative to the control group was calculated using the 2-∆∆Ct method. Two technical replicates per gene were used.
Table 3.
Primers for RT-PCR
| Gene | Forward | Reverse |
|---|---|---|
| Gapdh | 5′-GCCATCAACGACCCCTTCAT | 5′-ATGATGACCCGTTTGGCTCC |
| Nphs1 | 5′-GTGCCCTGAAGGACCCTACT | 5′-CCTGTGGATCCCTTTGACAT |
| Nphs2 | 5′-CTTGGCACATCGATCCCTCA | 5′-CGCACTTTGGCCTGTCTTTG |
| Cldn3 | 5′-GTCCGTCAGTTTTCGAAGGG | 5′-AGGATGGACACCACGATGAG |
| Synpo | 5′-CTTTGGGGAAGAGGCCGATTG | 5′-GTTTTCGGTGAAGCTTGTGC |
| Neph1 | 5′-TCACAAGCAGGGCTTTAGGA | 5′-CTGGCTGAAGCGAGTCTGAG |
| Neph3 | 5′-ACATTTCCGGAACCCAGGAG | 5′-CCACAATCCGGACAGTAGGC |
| CD2AP | 5′-TGGTGGAGTGGAACCCTGAA | 5′-GTGAGGTAGGGCCAGTCAAA |
| P-cadherin | 5′-GATTTTGAGGCTCAGGACCA | 5′-GACCTTGGAAGGTGGAACAA |
| Inf2 | 5′-CACCATCTCTCGGCTGTACC | 5′-CGAGTAGTTGACCACCGAGG |
| Fat1 | 5′-CCAGGGAAGTCCGTTCACAA | 5′-TTGACTCTGGGTGGATTGCC |
| Actn4 | 5′-TTCGACAACAAGCACACCAA | 5′-CCAGTGCCCCGCCAT |
| Wt1 | 5′-GAGAGCCAGCCTACCATCC | 5′-GGGTCCTCGTGTTTGAAGGAA |
| JAM-A | 5′-CGCGTCGGGATTGTAACTGT | 5′-CACCGAACCCTTGCCTTGTA |
| Occludin | 5′-AGGTGAGCACCTTGGGATTC | 5′-TTCAAAAGGCCTCACGGACA |
| Cingulin | 5′-TGTGAATCCGGGAGCACTGA | 5′-TAGCACCCTCTGGCTCTGTA |
| ZO-1 | 5′-TAGCACGGACAGTAGACACA | 5′-ATGGAAGTTGGGGTTCATAG |
Western Blot Analysis
Western blotting was performed using specific antibodies for proteins of interest as described previously.52
Statistical Analyses
Data are expressed as mean±SEM. For comparison of means between three or more groups, two-way ANOVA with Bonferroni post-test was applied. For comparisons of means between two groups, two-tailed, unpaired t tests were performed. Prism 5 (GraphPad Software, La Jolla, CA) was used for statistical analyses.
Disclosures
P.K.P. is the chief executive officer of Mirimus, Inc., which specializes in engineering customized mouse models with reversible gene silencing.
Supplementary Material
Acknowledgments
We thank Dr. Peter Mundel (Massachusetts General Hospital, Boston, MA) for sharing the synaptopodin antibody.
The study was supported by National Institutes of Health grants 5K08-DK082760, 1R01-DK098126, and 1R01-DK078897; Chinese 973 fund grant 2012CB517601; and Veterans Affairs Merit Review Award 1I01-BX000345.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2014040405/-/DCSupplemental.
References
- 1.Welsh GI, Saleem MA: Nephrin-signature molecule of the glomerular podocyte? J Pathol 220: 328–337, 2010 [DOI] [PubMed] [Google Scholar]
- 2.Wartiovaara J, Ofverstedt LG, Khoshnoodi J, Zhang J, Mäkelä E, Sandin S, Ruotsalainen V, Cheng RH, Jalanko H, Skoglund U, Tryggvason K: Nephrin strands contribute to a porous slit diaphragm scaffold as revealed by electron tomography. J Clin Invest 114: 1475–1483, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grahammer F, Schell C, Huber TB: The podocyte slit diaphragm—from a thin grey line to a complex signalling hub. Nat Rev Nephrol 9: 587–598, 2013 [DOI] [PubMed] [Google Scholar]
- 4.George B, Holzman LB: Signaling from the podocyte intercellular junction to the actin cytoskeleton. Semin Nephrol 32: 307–318, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barletta GM, Kovari IA, Verma RK, Kerjaschki D, Holzman LB: Nephrin and Neph1 co-localize at the podocyte foot process intercellular junction and form cis hetero-oligomers. J Biol Chem 278: 19266–19271, 2003 [DOI] [PubMed] [Google Scholar]
- 6.Gerke P, Huber TB, Sellin L, Benzing T, Walz G: Homodimerization and heterodimerization of the glomerular podocyte proteins nephrin and NEPH1. J Am Soc Nephrol 14: 918–926, 2003 [DOI] [PubMed] [Google Scholar]
- 7.Putaala H, Soininen R, Kilpeläinen P, Wartiovaara J, Tryggvason K: The murine nephrin gene is specifically expressed in kidney, brain and pancreas: Inactivation of the gene leads to massive proteinuria and neonatal death. Hum Mol Genet 10: 1–8, 2001 [DOI] [PubMed] [Google Scholar]
- 8.Rantanen M, Palmén T, Pätäri A, Ahola H, Lehtonen S, Aström E, Floss T, Vauti F, Wurst W, Ruiz P, Kerjaschki D, Holthöfer H: Nephrin TRAP mice lack slit diaphragms and show fibrotic glomeruli and cystic tubular lesions. J Am Soc Nephrol 13: 1586–1594, 2002 [DOI] [PubMed] [Google Scholar]
- 9.Doné SC, Takemoto M, He L, Sun Y, Hultenby K, Betsholtz C, Tryggvason K: Nephrin is involved in podocyte maturation but not survival during glomerular development. Kidney Int 73: 697–704, 2008 [DOI] [PubMed] [Google Scholar]
- 10.Kestilä M, Lenkkeri U, Männikkö M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T, Nissinen M, Herva R, Kashtan CE, Peltonen L, Holmberg C, Olsen A, Tryggvason K: Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Mol Cell 1: 575–582, 1998 [DOI] [PubMed] [Google Scholar]
- 11.Moeller MJ, Kovari IA, Holzman LB: Evaluation of a new tool for exploring podocyte biology: Mouse Nphs1 5′ flanking region drives LacZ expression in podocytes. J Am Soc Nephrol 11: 2306–2314, 2000 [DOI] [PubMed] [Google Scholar]
- 12.Moeller MJ, Sanden SK, Soofi A, Wiggins RC, Holzman LB: Two gene fragments that direct podocyte-specific expression in transgenic mice. J Am Soc Nephrol 13: 1561–1567, 2002 [DOI] [PubMed] [Google Scholar]
- 13.Ly JP, Onay T, Quaggin SE: Mouse models to study kidney development, function and disease. Curr Opin Nephrol Hypertens 20: 382–390, 2011 [DOI] [PubMed] [Google Scholar]
- 14.Juhila J, Lassila M, Roozendaal R, Lehtonen E, Messing M, Langer B, Kerjaschki D, Verbeek JS, Holthofer H: Inducible nephrin transgene expression in podocytes rescues nephrin-deficient mice from perinatal death. Am J Pathol 176: 51–63, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Juhila J, Roozendaal R, Lassila M, Verbeek SJ, Holthofer H: Podocyte cell-specific expression of doxycycline inducible Cre recombinase in mice. J Am Soc Nephrol 17: 648–654, 2006 [DOI] [PubMed] [Google Scholar]
- 16.Doublier S, Salvidio G, Lupia E, Ruotsalainen V, Verzola D, Deferrari G, Camussi G: Nephrin expression is reduced in human diabetic nephropathy: Evidence for a distinct role for glycated albumin and angiotensin II. Diabetes 52: 1023–1030, 2003 [DOI] [PubMed] [Google Scholar]
- 17.Toyoda M, Suzuki D, Umezono T, Uehara G, Maruyama M, Honma M, Sakai T, Sakai H: Expression of human nephrin mRNA in diabetic nephropathy. Nephrol Dial Transplant 19: 380–385, 2004 [DOI] [PubMed] [Google Scholar]
- 18.Cooper ME, Mundel P, Boner G: Role of nephrin in renal disease including diabetic nephropathy. Semin Nephrol 22: 393–398, 2002 [DOI] [PubMed] [Google Scholar]
- 19.Jim B, Ghanta M, Qipo A, Fan Y, Chuang PY, Cohen HW, Abadi M, Thomas DB, He JC: Dysregulated nephrin in diabetic nephropathy of type 2 diabetes: A cross sectional study. PLoS ONE 7: e36041, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wernerson A, Dunér F, Pettersson E, Widholm SM, Berg U, Ruotsalainen V, Tryggvason K, Hultenby K, Söderberg M: Altered ultrastructural distribution of nephrin in minimal change nephrotic syndrome. Nephrol Dial Transplant 18: 70–76, 2003 [DOI] [PubMed] [Google Scholar]
- 21.Furness PN, Hall LL, Shaw JA, Pringle JH: Glomerular expression of nephrin is decreased in acquired human nephrotic syndrome. Nephrol Dial Transplant 14: 1234–1237, 1999 [DOI] [PubMed] [Google Scholar]
- 22.Srivastava T, Whiting JM, Garola RE, Dasouki MJ, Ruotsalainen V, Tryggvason K, Hamed R, Alon US: Podocyte proteins in Galloway-Mowat syndrome. Pediatr Nephrol 16: 1022–1029, 2001 [DOI] [PubMed] [Google Scholar]
- 23.Kim BK, Hong HK, Kim JH, Lee HS: Differential expression of nephrin in acquired human proteinuric diseases. Am J Kidney Dis 40: 964–973, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Wang SX, Rastaldi MP, Pätäri A, Ahola H, Heikkilä E, Holthöfer H: Patterns of nephrin and a new proteinuria-associated protein expression in human renal diseases. Kidney Int 61: 141–147, 2002 [DOI] [PubMed] [Google Scholar]
- 25.Huh W, Kim DJ, Kim MK, Kim YG, Oh HY, Ruotsalainen V, Tryggvason K: Expression of nephrin in acquired human glomerular disease. Nephrol Dial Transplant 17: 478–484, 2002 [DOI] [PubMed] [Google Scholar]
- 26.Topham PS, Kawachi H, Haydar SA, Chugh S, Addona TA, Charron KB, Holzman LB, Shia M, Shimizu F, Salant DJ: Nephritogenic mAb 5-1-6 is directed at the extracellular domain of rat nephrin. J Clin Invest 104: 1559–1566, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Orikasa M, Matsui K, Oite T, Shimizu F: Massive proteinuria induced in rats by a single intravenous injection of a monoclonal antibody. J Immunol 141: 807–814, 1988 [PubMed] [Google Scholar]
- 28.Wang SX, Ahola H, Palment T, Solin ML, Luimula P, Holthöfer H: Recurrence of nephrotic syndrome after transplantation in CNF is due to autoantibodies to nephrin. Exp Nephrol 9: 327–331, 2001 [DOI] [PubMed] [Google Scholar]
- 29.Patrakka J, Ruotsalainen V, Reponen P, Qvist E, Laine J, Holmberg C, Tryggvason K, Jalanko H: Recurrence of nephrotic syndrome in kidney grafts of patients with congenital nephrotic syndrome of the Finnish type: role of nephrin. Transplantation 73: 394–403, 2002 [DOI] [PubMed] [Google Scholar]
- 30.Premsrirut PK, Dow LE, Kim SY, Camiolo M, Malone CD, Miething C, Scuoppo C, Zuber J, Dickins RA, Kogan SC, Shroyer KR, Sordella R, Hannon GJ, Lowe SW: A rapid and scalable system for studying gene function in mice using conditional RNA interference. Cell 145: 145–158, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chuang PY, Xu J, Dai Y, Jia F, Mallipattu SK, Yacoub R, Gu L, Premsrirut PK, He JC: In vivo RNA interference models of inducible and reversible Sirt1 knockdown in kidney cells. Am J Pathol 184: 1940–1956, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernhardt WM, Schmitt R, Rosenberger C, Münchenhagen PM, Gröne HJ, Frei U, Warnecke C, Bachmann S, Wiesener MS, Willam C, Eckardt KU: Expression of hypoxia-inducible transcription factors in developing human and rat kidneys. Kidney Int 69: 114–122, 2006 [DOI] [PubMed] [Google Scholar]
- 33.Holzman LB, St John PL, Kovari IA, Verma R, Holthofer H, Abrahamson DR: Nephrin localizes to the slit pore of the glomerular epithelial cell. Kidney Int 56: 1481–1491, 1999 [DOI] [PubMed] [Google Scholar]
- 34.Wagner N, Morrison H, Pagnotta S, Michiels JF, Schwab Y, Tryggvason K, Schedl A, Wagner KD: The podocyte protein nephrin is required for cardiac vessel formation. Hum Mol Genet 20: 2182–2194, 2011 [DOI] [PubMed] [Google Scholar]
- 35.Aström E, Rinta-Valkama J, Gylling M, Ahola H, Miettinen A, Timonen T, Holthöfer H: Nephrin in human lymphoid tissues. Cell Mol Life Sci 63: 498–504, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee VW, Harris DC: Adriamycin nephropathy: A model of focal segmental glomerulosclerosis. Nephrology (Carlton) 16: 30–38, 2011 [DOI] [PubMed] [Google Scholar]
- 37.Huber TB, Hartleben B, Kim J, Schmidts M, Schermer B, Keil A, Egger L, Lecha RL, Borner C, Pavenstädt H, Shaw AS, Walz G, Benzing T: Nephrin and CD2AP associate with phosphoinositide 3-OH kinase and stimulate AKT-dependent signaling. Mol Cell Biol 23: 4917–4928, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Menon MC, Chuang PY, He CJ: The glomerular filtration barrier: components and crosstalk. Int J Nephrol 2012: 749010, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, Saunders TL, Dysko RC, Kohno K, Holzman LB, Wiggins RC: Podocyte depletion causes glomerulosclerosis: Diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol 16: 2941–2952, 2005 [DOI] [PubMed] [Google Scholar]
- 40.Sison K, Eremina V, Baelde H, Min W, Hirashima M, Fantus IG, Quaggin SE: Glomerular structure and function require paracrine, not autocrine, VEGF-VEGFR-2 signaling. J Am Soc Nephrol 21: 1691–1701, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Makris A, Thornton C, Thompson J, Thomson S, Martin R, Ogle R, Waugh R, McKenzie P, Kirwan P, Hennessy A: Uteroplacental ischemia results in proteinuric hypertension and elevated sFLT-1. Kidney Int 71: 977–984, 2007 [DOI] [PubMed] [Google Scholar]
- 42.Miner JH: Life without nephrin: It’s for the birds. J Am Soc Nephrol 23: 369–371, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Völker LA, Petry M, Abdelsabour-Khalaf M, Schweizer H, Yusuf F, Busch T, Schermer B, Benzing T, Brand-Saberi B, Kretz O, Höhne M, Kispert A: Comparative analysis of Neph gene expression in mouse and chicken development. Histochem Cell Biol 137: 355–366, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hauser PV, Collino F, Bussolati B, Camussi G: Nephrin and endothelial injury. Curr Opin Nephrol Hypertens 18: 3–8, 2009 [DOI] [PubMed] [Google Scholar]
- 45.Blanco S, Bonet J, López D, Casas I, Romero R: ACE inhibitors improve nephrin expression in Zucker rats with glomerulosclerosis. Kidney Int Suppl (93, Suppl): S10–S14, 2005 [DOI] [PubMed] [Google Scholar]
- 46.Benigni A, Tomasoni S, Gagliardini E, Zoja C, Grunkemeyer JA, Kalluri R, Remuzzi G: Blocking angiotensin II synthesis/activity preserves glomerular nephrin in rats with severe nephrosis. J Am Soc Nephrol 12: 941–948, 2001 [DOI] [PubMed] [Google Scholar]
- 47.Otsuka M, Meistrich ML: Acceleration of late radiation damage of the kidney by unilateral nephrectomy. Int J Radiat Oncol Biol Phys 22: 71–78, 1992 [DOI] [PubMed] [Google Scholar]
- 48.Takemoto M, Asker N, Gerhardt H, Lundkvist A, Johansson BR, Saito Y, Betsholtz C: A new method for large scale isolation of kidney glomeruli from mice. Am J Pathol 161: 799–805, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yuen PS, Dunn SR, Miyaji T, Yasuda H, Sharma K, Star RA: A simplified method for HPLC determination of creatinine in mouse serum. Am J Physiol Renal Physiol 286: F1116–F1119, 2004 [DOI] [PubMed] [Google Scholar]
- 50.Ratnam KK, Feng X, Chuang PY, Verma V, Lu TC, Wang J, Jin Y, Farias EF, Napoli JL, Chen N, Kaufman L, Takano T, D’Agati VD, Klotman PE, He JC: Role of the retinoic acid receptor-α in HIV-associated nephropathy. Kidney Int 79: 624–634, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Michael AF, Blau E, Vernier RL: Glomerular polyanion. Alteration in aminonucleoside nephrosis. Lab Invest 23: 649–657, 1970 [PubMed] [Google Scholar]
- 52.Chuang PY, Dai Y, Liu R, He H, Kretzler M, Jim B, Cohen CD, He JC: Alteration of forkhead box O (foxo4) acetylation mediates apoptosis of podocytes in diabetes mellitus. PLoS ONE 6: e23566, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.