

Abstract
Ischemia-reperfusion (IR) injury is the most common cause of AKI. The susceptibility to develop AKI varies widely among patients. However, little is known about the genes involved. 20-Hydroxyeicosatetraenoic acid (20-HETE) has an important role in the regulation of renal tubular and vascular function and has been implicated in IR injury. In this study, we examined whether a deficiency in the renal formation of 20-HETE enhances the susceptibility of Dahl salt-sensitive (SS) rats to ischemic AKI. Transfer of chromosome 5 containing the CYP4A genes responsible for the formation of 20-HETE from the Brown Norway (BN) rat onto the SS genetic background increased renal 20-HETE levels after ischemia and reduced plasma creatinine levels (±SEM) 24 hours after IR from 3.7±0.1 to 2.0±0.2 mg/dl in an SS.5BN-consomic strain. Transfer of this chromosome also prevented the secondary decline in medullary blood flow and ischemia that develops 2 hours after IR in the susceptible SS strain. Blockade of the synthesis of 20-HETE with HET0016 reversed the renoprotective effects in SS.5BN rats. Similar results were observed in an SS.5Lew-congenic strain, in which a smaller region of chromosome 5 containing the CYP4A genes from a Lewis rat was introgressed onto the SS genetic background. These results indicate that 20-HETE has a protective role in renal IR injury by maintaining medullary blood flow and that a genetic deficiency in the formation of 20-HETE increases the susceptibility of SS rats to ischemic AKI.
Keywords: AKI, 20-HETE, genetics, intrarenal blood flow
AKI is a common condition that is associated with significant mortality.1 The incidence of AKI has increased by 10% per year over the last decade, and the mortality rate has more than doubled.2 Renal ischemia-reperfusion (IR) injury is the most frequent cause of AKI, and the incidence of AKI exceeds 50% after major cardiac, aortic, or transplant surgery.3,4 Unfortunately, there is no approved therapy for the treatment of AKI.5 The susceptibility to develop AKI also varies widely among patients6 and among different strains of rats. In this regard, Basile et al.7,8 previously reported that Brown Norway (BN) rats are more resistant to the development of IR injury than Sprague–Dawley or Dahl salt-sensitive (SS) rats. More recently, transfer of the X chromosome and chromosomes 3–8, 10, and 15 from BN rats has been reported to confer partial protection to renal IR injury in consomic strains of SS rats.8 However, little is known about the genes or mechanisms involved.
Previous studies have indicated that 20-hydroxyeicosatetraenoic acid (20-HETE) plays an important role in the regulation of renal tubular and vascular function, and a deficiency in the renal formation of 20-HETE has been linked to the development of hypertension in SS rats.9–11 Cytochromes P450 4A11 (CYP4A11) and 4F2 (CYP4F2) are the isoforms responsible for the synthesis of 20-HETE in the human kidney, and sequence variants in these genes have been linked to the development of hypertension.12–16 However, the role of these genes in AKI is unknown. Our group has reported that the genes encoding the CYP4A enzymes that catalyze the renal formation of 20-HETE are located on chromosome 5 in the rat17 and that the expression of CYP4A enzymes and the formation of 20-HETE are reduced in SS rats relative to other strains.10,11 More recently, we found that administration of a 20-HETE agonist can protect the kidney from IR injury.18 This was associated with prevention of the secondary fall in medullary blood flow (MBF) and prolonged tissue hypoxia that developed 2 hours after reperfusion.18,19 Therefore, this study examined whether a deficiency in the renal formation of 20-HETE contributes to the increased susceptibility of SS rats to renal IR injury by determining if transfer of the CYP4A genes on chromosome 5 from the BN or Lewis rat onto the SS genetic background would increase renal 20-HETE formation and promote resistance to renal IR injury.
Results
A comparison of baseline data of SS and SS.5BN rats is presented in Supplemental Table 1. There was no significant difference in body or kidney weight, systolic BP, urine flow, or urinary sodium excretion in SS and SS.5BN rats. Plasma 20-HETE levels were significantly higher in SS.5BN rats than in SS rats (0.46±0.04 and 0.28±0.03 ng/ml, respectively) (Supplemental Figure 1). We also compared platelet aggregation and bleeding time in SS and SS.5BN rats, because 20-HETE has been reported to alter clotting.20 The time for platelet aggregation and bleeding time were not significantly different in SS and SS.5BN rats. Moreover, administration of an inhibitor of the synthesis of 20-HETE, HET0016,18,21 3 hours before experiments did significantly not affect platelet function of SS.5BN rats, in which 20-HETE levels are elevated.
Effect of Renal IR on Tissue Levels of 20-HETE
A comparison of the effects of renal IR on the levels of 20-HETE in the renal cortex and outer medulla of SS, SS.5BN, and normal Sprague–Dawley rats is presented in Figure 1. Basal levels of 20-HETE in the renal cortex and outer medulla were significantly higher in SS.5BN rats than in Sprague–Dawley or SS rats. 20-HETE levels increased in the renal cortex and outer medulla to the same extent in Sprague–Dawley and SS.5BN rats after 30 minutes of ischemia. The levels rapidly returned to control in the renal cortex of Sprague–Dawley rats after reperfusion but remained elevated in the outer medulla of Sprague–Dawley and SS.5BN rats. In contrast, the rise in 20-HETE levels in both the renal cortex and outer medulla after IR was blunted in SS rats relative to that seen in the other strains. IR did not significantly alter the levels of any of the other metabolites of arachidonic acid (AA), including 5-, 12–15-HETE, prostaglandin E2 (PGE2), PGF2α, 6-keto-PGE1, thromboxane B2, epoxyeicosatrienoic acids, and dihydroxyeicosatrienoic acids, in the renal cortex or outer medulla (data not shown).
Figure 1.

Renal 20-HETE levels increase to a greater extent in Sprague–Dawley (SD) and SS.5BN consomic than SS rats after bilateral renal IR. Levels of 20-HETE were measured in the renal cortex and outer medulla by liquid chromatography/mass spectrometry/mass spectrometry after 30 minutes of ischemia and 1 or 24 hours of reperfusion (R). Mean values±SEMs are presented from four to six rats per time point. *P<0.05 from the corresponding control value measured within a strain. †P<0.05 from the corresponding value measured in SS rats.
Comparison of Renal IR Injury in Sprague–Dawley, SS, and SS.5BN Rats
Baseline plasma creatinine concentration (PCr) was similar in Sprague–Dawley, SS, and SS.5BN rats. PCr rose to 3.7±0.1 mg/dl 24 hours after bilateral renal ischemia in SS rats and was significantly greater than the levels seen in Sprague–Dawley or SS.5BN rats (1.7±0.4 and 2.0±0.2 mg/dl, respectively) (Figure 2). Blockade of the formation of 20-HETE with HET0016 exacerbated the degree of IR injury in both Sprague–Dawley and SS.5BN rats, and PCr rose to the same level as that seen in SS rats.
Figure 2.

Evidence for increased renal injury in SS versus Sprague–Dawley (SD) and SS.5BN consomic rats 24 hrs following 30 minutes of bilateral renal ischemia. Pretreatment with a 20-HETE inhibitor, HET0016 (5 mg/kg subcutaneously), abolished the resistance to IR injury in SD and SS.5BN rats. Bilaterally nephrectomized (Nx) rats served as the control for complete renal failure within each group. Mean values±SEMs from six rats per group are presented. *P<0.05 from the corresponding control value measured within a strain. †P<0.05 from the corresponding value measured in SS rats at a particular time point.
A comparison of the histologic appearance and degree of tubular injury of the renal cortex and medulla 24 hours after IR in SS and SS.5BN rats is presented in Figure 3A. Diffuse tubular necrosis, intratubular debris, and tubular casts were present in the corticomedullary region and outer medulla of SS rats 24 hours after IR. In contrast, the severity of renal tubular injury was significantly less in SS.5BN rats. Blockade of the formation of 20-HETE with HET0016 markedly increased the incidence of tubular necrosis in SS.5BN rats, but it had little effect in SS rats.
Figure 3.
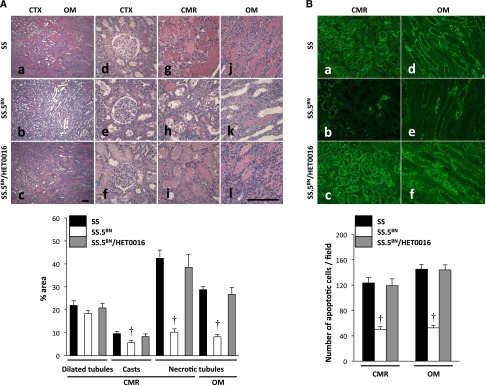
Renal tubular injury is greater in SS than SS.5BN rats 24 hours following 30 minutes of ischemia. (A) The severity of renal injury was less in SS.5BN consomic rats, with (b, e, h, and k) only focal areas of tubular necrosis or exfoliation of tubular cells (c, f, i, and l). Pretreatment of SS.5BN rats with the 20-HETE inhibitor, HET0016, increased the degree of necrotic tubular epithelium compared with the values seen in SS.5BN rats. Renal tubular injury was quantified using the eosin fluorescence method. The area of renal tubular injury was significantly reduced in the SS.5BN rats compared with the value seen in control SS or SS.5BN rats that received the 20-HETE inhibitor HET0016 (SS.5BN/HET0016). Original magnification, ×40 in a–c; ×200 in d–l. Scale bar, 100 μm. (B) The number of caspase 3-positive apoptotic cells (green) was significantly less in (b) the renal CMR and (e) OM of SS.5BN rats compared with the levels seen in (a and d) SS or (c and f) HET0016-pretreated SS.5BN rats (SS.5BN/HET0016). Quantitative analysis was performed on 10 random nonoverlapping fields at ×200 magnification in the renal CMR and OM. Mean values±SEMs from five to six rats per group are presented. CMR, corticomedullary region; CTX, cortex; OM, outer medulla. †P<0.05 from the corresponding value measured in SS rats.
The degree of renal tubular injury was determined by measuring the area of fluorescent necrotic tubular epithelial cells and the formation of tubular casts in hematoxylin and eosin (H&E)-stained sections as depicted in Supplemental Figure 2. The percentage of the area occupied by necrotic tubules and casts was significantly greater in the corticomedullary region and outer medulla of SS rats than that seen in SS.5BN rats (Figure 3A). Moreover, the number of caspase 3-positive apoptotic cells in the corticomedullary region and outer medulla of SS rats was significantly larger than that seen in SS.5BN rats (Figure 3B). Administration of N-Hydroxy-N′-(4-butyl-2-methylphenyl)-formamidine (HET0016) abolished the renoprotection observed in the SS.5BN rats, and the percentage of necrotic tubules and the number of apoptotic cells increased to the same level as those seen in SS rats.
The number of tubular cells undergoing autophagy identified as positive staining for autophagy-related protein 8 and CD68 antigen-positive macrophages in the corticomedullary region and outer medulla of SS rats was significantly larger than that seen in SS.5BN rats 24 hours after IR (Supplemental Figure 3, A and B). Administration of HET0016 increased the number of cells undergoing autophagy and macrophages in SS.5BN rats to the same level seen in SS rats after IR.
The urinary excretion of the renal injury biomarkers, including TNF-α, kidney injury molecule-1, neutrophil gelatinase–associated lipocalin, and heme oxygenase-1, was significantly greater in SS rats than that seen in SS.5BN rats 24 hours after IR (Supplemental Figure 4).
Comparison of Intrarenal Blood Flow after IR in SS and SS.5BN Rats
A comparison of the time course of changes in cortical blood flow (CBF), MBF, and mean arterial pressure (MAP) of SS, SS.5BN, and HET0016-treated SS.5BN rats is presented in Figure 4A. Baseline CBF, MBF (Supplemental Figure 5), and MAP were similar in SS, SS.5BN, and HET0016-treated SS.5BN rats. CBF and MBF decreased markedly during the ischemic period and then rapidly returned to the control after ischemia in SS, SS.5BN, and HET0016-treated SS.5BN rats. However, MBF exhibited a secondary fall in flow to about 30% of control 3 hours after reperfusion in SS rats and HET0016-treated SS.5BN rats. In contrast, this secondary fall in MBF did not occur in SS.5BN rats. MAP was stable after IR and did not differ at any time during the experiment in SS, SS.5BN, and HET0016-treated SS.5BN rats.
Figure 4.

Transfer of BN chromosome 5 prevents the secondary fall in medullary blood flow and attenuates renal medullary ischemia following IR in SS rats. (A) CBF and MBF were measured in SS, SS.5BN, and HET0016-treated SS.5BN rats (SS.5BN/HET0016) during a control period and after 30 minutes of ischemia and 3 hours of reperfusion. Mean values±SEMs from five rats per group are presented. A two-way ANOVA with a Holm–Sidak test was used to compare corresponding values in control and HET0016-treated SS.5BN rats with those measured in SS rats at the same time points. (B) The area of renal hypoxia was determined by Hypoxyprobe staining. The percentage area of hypoxic tissue was significantly less in (e) the renal CMR and (h) OM of SS.5BN rats than that seen in (d and g) SS or (f and i) HET0016-treated SS.5BN rats (SS.5BN/HET0016). Quantitative analysis of the percentage of the area stained with Hypoxyprobe was performed on 10 random nonoverlapping fields at ×200 magnification in the renal CTX, CMR, and OM. Mean values±SEMs from five rats per group are presented. CMR, corticomedullary region; CTX, cortex; OM, outer medulla. †P<0.05 from the corresponding value measured in SS rats at a given time point.
A comparison of the appearance of H&E-stained sections of the kidney 3 hours after IR in SS, SS.5BN, and HET0016-treated SS.5BN rats is presented in Supplemental Figure 6. A large number of red blood cells indicative of vascular congestion was found in the juxtamedullary cortex and the outer medulla of SS rats. The degree of vascular congestion was significantly less in SS.5BN rats. Blockade of the synthesis of 20-HETE with HET0016 restored the degree of vasocongestion in SS.5BN rats to the same level as that seen in SS rats.
A comparison of the degree of hypoxia in the renal cortex and outer medulla of SS, SS.5BN, and HET0016-treated SS.5BN rats 24 hours after IR is presented in Figure 4B. Hypoxyprobe staining in the corticomedullary region and outer medulla of SS rats and SS.5BN rats treated with HET0016 was much greater than that seen in SS.5BN rats.
Additional studies were performed in which the inhibitor of the synthesis of 20-HETE was given 3 hours after reperfusion to determine the time frame over which 20-HETE exerts its renoprotective effect. The results presented in Supplemental Figure 7 indicate that pretreatment with HET0016 enhanced the degree of renal injury 24 hours after IR in SS.5BN rats. However, administration of HET0016 3 hours after reperfusion did not.
Comparison of 20-HETE Levels and Susceptibility to Renal IR Injury in SS.5Lew 4A+ and 4A− Congenic Strains
To further explore whether the renoprotective effects of transfer of chromosome 5 in SS.5BN rats were caused by a sequence variant in one of the CYP4A genes in SS rats, we compared the degree of IR injury in overlapping SS.5Lew congenic strains, in which a much smaller region of chromosome 5 that includes (4A+) or excludes (4A−) CYP4A genes from the Lewis rat was introgressed onto the SS genetic background. Transfer of the CYP4A alleles from the Lewis rat onto the SS genetic background has been reported to increase renal 20-HETE levels and protect against the development of hypertension and proteinuria in an SS.5Lew 4A+ congenic strain, similar to what has been reported in SS.5BN consomic rats.10,11 Basal levels of 20-HETE in the cortex and renal medulla were significantly higher in the SS.5Lew 4A+ congenic strain than the overlapping SS.5Lew 4A− control congenic strain. The concentration of 20-HETE in the renal cortex and outer medulla increased significantly after 30 minutes of ischemia in the 4A+ congenic strain but not in the control 4A− congenic strain (Figure 5A).
Figure 5.
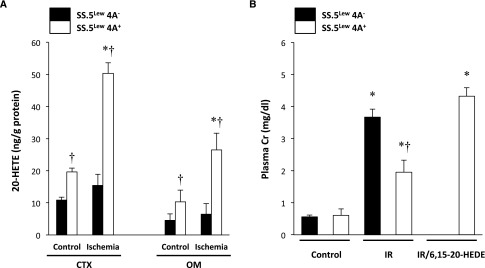
Transfer of a region of chromosome 5 containing CYP4A genes from the Lewis rat increases renal 20-HETE levels and reduces renal injury following IR in SS rats. (A) The concentration of free 20-HETE in the renal cortex and outer medulla of 4A+ and 4A− rats was measured by liquid chromatography/mass spectrometry/mass spectrometry. Baseline 20-HETE levels in the renal cortex and outer medulla were significantly higher in 4A+ rats compared with 4A− rats and increased after ischemia in 4A+ rats. (B) PCr was significantly less after IR in 4A+ rats compared with 4A− rats. Pretreatment with a 20-HETE antagonist (6,15–20-HEDE) abolished the resistance to IR injury in 4A+ rats. Mean values±SEMs from five to six rats per group are presented. CTX, cortex; OM, outer medulla. *P<0.05 from the corresponding control value measured within a strain. †P<0.05 from the corresponding value measured in 4A− control rats.
A comparison of the degree of renal IR injury in the 4A+ and 4A− strains is presented in Figure 5B. Baseline PCr was similar in SS, 4A+, and 4A− rats. After 30 minutes of bilateral renal ischemia and 24 hours of reperfusion, PCr rose to 3.7±0.1 mg/dl in SS rats and 3.7±0.3 mg/dl in the 4A− congenic strain. PCr was significantly lower in the 4A+ congenic strain (1.9±0.4 mg/dl). Administration of a 20-HETE antagonist, 6, 15–20-hydroxydienoic acid (6, 15–20-HEDE), abolished the resistance to renal IR injury in 4A+ rats, and PCr rose to 4.3±0.3 mg/dl.
A comparison of the appearance of the renal cortex and outer medulla after renal IR in the 4A+ and 4A− congenic strains is presented in Supplemental Figure 8. Diffuse tubular necrosis, intratubular debris, and tubular casts were present 24 hours after ischemia in the kidney of the control 4A− congenic strain. In contrast, the severity of renal injury in the 4A+ congenic strain was significantly less, with only a few focal areas of tubular necrosis and exfoliation of tubular cells. Pretreatment of 4A+ rats with a 20-HETE antagonist (6, 15–20-HEDE) restored the degree of tubular necrosis to the same level as that seen in SS and 4A− control rats.
Discussion
AKI remains a major health problem with few therapeutic options. The susceptibility to develop AKI varies widely among patients6 and in different strains of rats. However, little is known about the genes involved. Previous studies have indicated that BN rats are more resistant to the development of renal injury than Sprague–Dawley and SS rats after 45 minutes of ischemia.7,8 More recently, Basile et al.8 reported that transfer of several chromosomes, including chromosome 5, from BN to SS rats imparted partial resistance to renal IR injury. Because the CYP4A genes that produce 20-HETE are located on chromosome 5 and because the expression of CYP4A protein and the production of 20-HETE are reduced in SS rats relative to other strains,17 this study examined whether strain differences in the production of 20-HETE contribute to the renoprotective effect of transfer of BN chromosome 5 on ischemic AKI.
We first compared the levels of 20-HETE after renal IR in Sprague–Dawley, SS, SS.5BN consomic, and SS.5Lew congenic strains. The results indicate that renal ischemia increases the formation and/or release of 20-HETE in the renal cortex and outer medulla, whereas the levels of other eicosanoids are not affected. This finding is consistent with the idea that ischemia raises intracellular calcium levels to activate cytosolic phospholipase A2 and increase the release of AA from membrane phospholipids that can be metabolized to 20-HETE.9,23,24 Our data also reveal that cortical levels of 20-HETE return to control within 1 hour of reperfusion but that the levels in the renal medulla remain elevated for a more prolonged period of time.
The results of this study also indicate that 20-HETE levels increase to a much greater extent after IR in the renal outer medulla in the Sprague–Dawley, SS.5BN consomic, and SS.5Lew 4A+ congenic strains that are resistant to renal injury than in SS rats or the SS.5Lew 4A− control congenic strain, which are more susceptible to injury. Although the mechanism of action remains to be determined, previous studies have indicated that IR injury is associated with vasocongestion and prolonged hypoxia in the renal outer medulla.25–28 In this regard, we found that SS rats developed a secondary fall in MBF and pronounced vasocongestion 2–3 hours after renal IR. In contrast, transfer of chromosome 5 in the SS.5BN strain prevented the secondary fall in MBF, renal hypoxia, and vasocongestion, and this was associated with renoprotection. Pretreatment of SS.5BN rats with an inhibitor of the synthesis of 20-HETE reversed the renoprotective effects associated with transfer of chromosome 5. Similar renoprotection was seen in the SS.5Lew 4A+ congenic strain, in which a much smaller region of chromosome 5 containing just the CYP4A region from the Lewis rat was transferred onto the SS genetic background. Taken together, these findings indicate that increased release of 20-HETE after renal ischemia opposes renal injury, in part, by preserving MBF and preventing the secondary medullary hypoxia that develops 2–3 hours after IR. These results are consistent with our previous findings that administration of a 20-HETE agonist protects against the development of bilateral renal IR injury in Sprague–Dawley rats.18 However, Hoff et al.23 reported that pretreatment of rats with a 20-HETE inhibitor or antagonist protected against renal ischemia in a uninephrectomized model. The reason for the difference seems to be related to the different model systems (bilateral versus unilateral ischemia), because a follow-up study by Roman et al.29 confirmed that inhibition of 20-HETE was protective in the unilateral ischemic kidney model but enhanced injury in the bilateral model.
The results of this study also indicate that SS rats are more susceptible to renal IR injury than Sprague–Dawley rats after 30 minutes of ischemia. This finding differs from previous reports indicating that renal IR injury is similar in SS and Sprague–Dawley rats after 45 minutes of ischemia.7,8 The difference seems to be because of the longer ischemic period that produced more severe injury in the previous study.
The mechanism underlying the renoprotective effect of 20-HETE in bilateral IR injury remains to be determined. 20-HETE constricts the afferent arteriole,9 and the elevation in renal 20-HETE levels after IR would be expected to lower renal blood flow and aggravate IR injury. However, 20-HETE has been reported to increase rather than decrease MBF in normal rats.30 The vasodilator effects of 20-HETE on MBF were blocked by indomethacin but not Nω-nitro-L-arginine methyl ester (L-NAME), suggesting that 20-HETE is metabolized by cyclooxygenase to a vasodilatory metabolite.31 We also considered that 20-HETE might inhibit platelet aggregation, leukocyte adhesion, and plugging of vasa recta capillaries. However, in this study, 20-HETE failed to alter platelet aggregation or bleeding time in SS.5BN rats, although plasma levels of 20-HETE were elevated relative to SS rats. 20-HETE inhibits sodium transport in proximal tubule and thick ascending loop of Henle.9 Thus, it might attenuate renal medullary hypoxia by decreasing oxygen consumption and may prevent the secondary fall in MBF by reducing cell swelling, tubular necrosis, and physical occlusion of the adjacent vasa recta capillaries after renal IR.32 Regardless of the mechanism involved, these results suggest that the 20-HETE level is elevated immediately after reperfusion and that it protects against renal IR injury. Moreover, a deficiency in the renal production of 20-HETE contributes to the enhanced IR injury seen in SS rats relative to SS.5BN, SS.5Lew 4A+, and Sprague–Dawley rats. The critical time window is early in the reperfusion phase, because pretreatment with a 20-HETE inhibitor before IR reversed the renoprotection in SS.5BN rats but was ineffective when given 3 hours after reperfusion.
There is considerable evidence indicating that the mutations in CYP4A11 and CYP4F2 are common in patients and that they are associated with the development of hypertension.12–16 However, the role of these genes in determining genetic susceptibility to the development of AKI has not been considered previously. Fibrates upregulate the expression of CYP4A33,34 and CYP2C11 and enhance the formation of both 20-HETE and epoxyeicosatrienoic acids35 in the kidney. Moreover, clofibrate has been reported to reduce renal injury after IR.36 The results of this study suggest that the renoprotective effects of fibrates might be associated with upregulation of the CYP4A pathway, and a clinical trial might be warranted. This is not to say that alterations in CYP4A/20-HETE are the only pathway that contributes to the strain difference in SS and BN rats to renal IR injury. Indeed, the chromosomal substitution study by Basile et al.8 clearly indicated that the strain difference in the susceptibility to renal IR injury is polygenic and that BN chromosomes 3, 4, 6–8, 10, and 15 and the X chromosome all partially protect from renal IR injury in consomic strains of SS rats. These chromosomes undoubtedly harbor sequence variants in genes that affect IR injury by not only altering renal MBF but likely, affecting other mechanisms, such as tubular cell survival, blood clotting, and immune response, all of which are known to contribute to the severity of renal IR injury.37
In summary, this study indicates that renal IR injury is enhanced in SS rats that are deficient in the renal formation of 20-HETE and that increasing 20-HETE levels by transfer of the CYP4A genes on chromosome 5 in SS.5BN consomic or SS.5Lew 4A+ congenic rats partially protects against renal IR injury. The increase in 20-HETE levels in the renal outer medulla after IR prevents the secondary fall in MBF and the prolonged hypoxia that develops in the corticomedullary region and outer medulla of the kidney and contributes to tubular necrosis. These results suggest that patients with mutations in CYP4A11 or CYP4F2 who have reduced formation of 20-HETE might also be more susceptible to ischemic AKI, and therapeutic approaches that target the formation of 20-HETE might be renoprotective.
Concise Methods
General
Experiments were performed on 9-week-old male Sprague–Dawley, SS, SS.5BN, 4A+, and 4A− rats. The Sprague–Dawley rats were purchased from Charles River Laboratories (Wilmington, MA). The SS, SS.5BN, 4A+, and 4A− rats were obtained from inbred colonies maintained at the University of Mississippi Medical Center as previously described.38–40 The SS, SS.5BN, 4A+, and 4A− rats were maintained on a 0.3% NaCl diet (Harlan Teklad 7034; Harlan Laboratories, Madison, WI) that opposes the development of hypertension. All protocols were approved by the Institutional Animal Care and Use Committee of the University of Mississippi Medical Center.
Renal IR Injury Model
The rats were anesthetized with 3.0% isoflurane and placed on a heated surgical table to maintain body temperature at 37°C. A midline abdominal incision was made to expose the kidneys, and the renal arteries and veins were bilaterally occluded using microvascular clamps for 30 minutes. The clamps were then removed, the abdominal incision was closed, and the rats were allowed to recover. Sham-operated control rats underwent the same procedure without clamping of the renal vessels. Additional rats underwent bilateral nephrectomy and served as a renal failure control group. Twenty-four hours after IR, the rats were anesthetized with isoflurane, and a blood sample was collected from the aorta for measurement of the PCr using a creatinine assay kit (Wako Pure Chemical, Osaka, Japan). Both kidneys were collected for biochemical and histologic analyses. In some studies, the SS.5BN rats received vehicle (11% sulfobutyl-β-cyclodextrin in 165 mM mannitol solution) or HET0016 (5 mg/kg subcutaneously), a selective inhibitor of 20-HETE formation.18,21 Some of the SS.5Lew 4A+ congenic rats received vehicle or a 20-HETE antagonist (20-hydroxyeicosa-6(Z), 15(Z)-dienoic acid [6, 15-20-HEDE]; 10 mg/kg subcutaneously)22,23 30 minutes before clamping of the renal vessels.
Measurement of CYP450 Eicosanoids in the Kidney
Samples of the renal cortex and outer medulla (approximately 0.3 g) were homogenized in 3 mL 10 mmol/L KPO4 buffer containing 250 mmol/L sucrose and 1 mmol/L EDTA (pH 7.7). The homogenate was centrifuged at 4000×g for 5 minutes. The supernatant was extracted two times with 3 mL ethyl acetate after the addition of 2 ng internal standard (20-HETE-d6), and the organic phase was dried under nitrogen. Samples were reconstituted in methanol, and the metabolites of AA produced were measured using an ABI-Sciex 4000 Q-Trap liquid chromatography/mass spectrometry/mass spectrometry as previously described.41–43 Values are expressed as the amount of the eicosanoids per gram of the tissue protein.
Histopathologic Analysis of Renal Injury
The kidneys were fixed in 10% formalin, and paraffin sections (3 μm) were prepared and stained with H&E to detect eosin autofluorescence in necrotic tubular cells.44 The sections were examined using a Nikon Eclipse 55i microscope equipped with a 540-nm excitation filter and a 590-nm emission filter and a Nikon DS-Fi1 color camera (Nikon Instruments Inc., Melville, NY). Ten randomly chosen corticomedullary and outer medullary fields were photographed (×200 total magnification). After background thresholding, the percentage of the area of the image containing fluorescent necrotic tubular epithelium was quantified using the NIS-Elements D 3.0 software (Nikon Instruments Inc.). All morphometric analyses of the kidney samples were performed in blinded manner.
Immunohistochemistry
Formalin-fixed, paraffin-embedded kidney samples were sectioned at 3-μm thickness and mounted on slides. The slides were deparaffinized in xylene, rehydrated through a decreasing ethanol gradient, and rinsed in PBS. The slides were pretreated with proteinase K (Dako, Carpinteria, CA) for 10 minutes and exposed to a blocking solution (Dako) for 30 minutes at room temperature. They were rinsed in PBS, incubated with primary antibodies caspase 3, autophagy-related protein 8 (1:100; Abcam, Inc., Cambridge, MA), or a monoclonal CD68 macrophage antigen antibody (ED-1) (1:100; AbD Serotec, Raleigh, NC) overnight at 4°C, rinsed in PBS, and then incubated in secondary antibodies conjugated with Alexa Fluor 488 (1:200; Jackson ImmunoResearch, West Grove, PA) for 1 hour. After three rinses in PBS, they were then counterstained with 0.001% Evans Blue (Sigma-Aldrich, St. Louis, MO), rinsed in distilled water, and mounted with fluorescent mounting medium with 4,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA). Images were generated using a Nikon Eclipse 55i microscope equipped with a 540-nm excitation filter and a 590-nm emission filter and a Nikon DS-Fi1 color camera.
Assessment of Intrarenal Hemodynamics
Rats were anesthetized with 1.0%–2.0% isoflurane. Catheters were placed in the femoral artery for measurement of arterial pressure. The left kidney was exposed by a midline incision, and two single-mode optical fibers were implanted 4 mm into the kidney for measurement of MBF by laser Doppler flowmetry with a Laser Doppler Flowmeter (PeriFlux System 5000; Perimed Inc., Ardmore, PA) as previously described.18,45 CBF was measured using additional laser Doppler probes held in static position by two micromanipulators above the renal cortex. After surgery, CBF and MBF were recorded every 5 minutes during a 1-hour control period. Then, the blood supply to the left kidney was occluded for 30 minutes. CBF and MBF were recorded during the ischemic period and for 3 hours after reperfusion. The values measured in perfusion units were expressed as a percentage of the control value.
Assessment of Renal Hypoxia
Intrarenal hypoxia was assessed using the hypoxia-sensitive marker 2-pimonidizole Hypoxyprobe-1 Plus (Hypoxyprobe Inc., Burlington, MA) as previously described.46,47 In brief, conscious rats subjected to 30 minutes of ischemia and 24 hours of reperfusion were injected with a 60-mg/kg intravenous bolus dose of Hypoxyprobe-1 through the tail vein. They were then anesthetized with isoflurane, and a 0.5 ml sample of blood was collected from the aorta. The kidneys were then quickly collected, hemisected, and immediately placed in ice-cold 10% buffered formalin in <30 seconds to minimize warm ischemic time and the generation of a hypoxic signal. The kidneys were embedded in paraffin, sectioned, and stained with an FITC-conjugated Hypoxyprobe-1 mAb antibody, and they were examined using a fluorescence microscope.
Statistical Analyses
Mean values±SEMs are presented. The significance of differences in mean values between two groups was determined using an unpaired t test. The significance of difference in mean values between multiple groups was determined using one-way ANOVA or two-way repeated measures ANOVA for time course data followed by the Holm–Sidak test for preplanned comparisons back to the control value within a group or the corresponding value measured in SS rats at a given time point. A P value <0.05 using a two-tailed test was considered to be statistically significant.
Disclosures
None.
Supplementary Material
Acknowledgments
We thank Ms. Christine A. Purser for assistance with the liquid chromatography/mass spectrometry/mass spectrometry analysis.
This study was supported, in part, by National Institutes of Health Grants HL-36279 (to R.J.R.) and DK104184 (to R.J.R.); core facilities were supported by National Institutes of Health Grant P01-GM104357 and Robert A. Welch Foundation Grant I-0011 (to J.R.F.).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2014090868/-/DCSupplemental.
References
- 1.Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW: Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol 16: 3365–3370, 2005 [DOI] [PubMed] [Google Scholar]
- 2.Hsu RK, McCulloch CE, Dudley RA, Lo LJ, Hsu CY: Temporal changes in incidence of dialysis-requiring AKI. J Am Soc Nephrol 24: 37–42, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thadhani R, Pascual M, Bonventre JV: Acute renal failure. N Engl J Med 334: 1448–1460, 1996 [DOI] [PubMed] [Google Scholar]
- 4.Aydin Z, van Zonneveld AJ, de Fijter JW, Rabelink TJ: New horizons in prevention and treatment of ischaemic injury to kidney transplants. Nephrol Dial Transplant 22: 342–346, 2007 [DOI] [PubMed] [Google Scholar]
- 5.Jo SK, Rosner MH, Okusa MD: Pharmacologic treatment of acute kidney injury: Why drugs haven’t worked and what is on the horizon. Clin J Am Soc Nephrol 2: 356–365, 2007 [DOI] [PubMed] [Google Scholar]
- 6.Sreedharan R, Devarajan P, Van Why SK: Pathogenesis of acute renal failure. In: Pediatric Nephrology, 6th Ed., edited by Avner E, Harmon W, Niaudet P, Yoshikawa N, Heidelberg, Germany, Springer-Verlag, 2009, pp 1579–1602 [Google Scholar]
- 7.Basile DP, Donohoe D, Cao X, Van Why SK: Resistance to ischemic acute renal failure in the Brown Norway rat: A new model to study cytoprotection. Kidney Int 65: 2201–2211, 2004 [DOI] [PubMed] [Google Scholar]
- 8.Basile DP, Dwinell MR, Wang SJ, Shames BD, Donohoe DL, Chen S, Sreedharan R, Van Why SK: Chromosome substitution modulates resistance to ischemia reperfusion injury in Brown Norway rats. Kidney Int 83: 242–250, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roman RJ: P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev 82: 131–185, 2002 [DOI] [PubMed] [Google Scholar]
- 10.Williams JM, Sarkis A, Hoagland KM, Fredrich K, Ryan RP, Moreno C, Lopez B, Lazar J, Fenoy FJ, Sharma M, Garrett MR, Jacob HJ, Roman RJ: Transfer of the CYP4A region of chromosome 5 from Lewis to Dahl S rats attenuates renal injury. Am J Physiol Renal Physiol 295: F1764–F1777, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williams JM, Fan F, Murphy S, Schreck C, Lazar J, Jacob HJ, Roman RJ: Role of 20-HETE in the antihypertensive effect of transfer of chromosome 5 from Brown Norway to Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 302: R1209–R1218, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gainer JV, Bellamine A, Dawson EP, Womble KE, Grant SW, Wang Y, Cupples LA, Guo CY, Demissie S, O’Donnell CJ, Brown NJ, Waterman MR, Capdevila JH: Functional variant of CYP4A11 20-hydroxyeicosatetraenoic acid synthase is associated with essential hypertension. Circulation 111: 63–69, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Mayer B, Lieb W, Götz A, König IR, Aherrahrou Z, Thiemig A, Holmer S, Hengstenberg C, Doering A, Loewel H, Hense HW, Schunkert H, Erdmann J: Association of the T8590C polymorphism of CYP4A11 with hypertension in the MONICA Augsburg echocardiographic substudy. Hypertension 46: 766–771, 2005 [DOI] [PubMed] [Google Scholar]
- 14.Fu Z, Nakayama T, Sato N, Izumi Y, Kasamaki Y, Shindo A, Ohta M, Soma M, Aoi N, Sato M, Matsumoto K, Ozawa Y, Ma Y: Haplotype-based case study of human CYP4A11 gene and cerebral infarction in Japanese subject. Endocrine 33: 215–222, 2008 [DOI] [PubMed] [Google Scholar]
- 15.Laffer CL, Gainer JV, Waterman MR, Capdevila JH, Laniado-Schwartzman M, Nasjletti A, Brown NJ, Elijovich F: The T8590C polymorphism of CYP4A11 and 20-hydroxyeicosatetraenoic acid in essential hypertension. Hypertension 51: 767–772, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu H, Zhao Y, Nie D, Shi J, Fu L, Li Y, Yu D, Lu J: Association of a functional cytochrome P450 4F2 haplotype with urinary 20-HETE and hypertension. J Am Soc Nephrol 19: 714–721, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stec DE, Deng AY, Rapp JP, Roman RJ: Cytochrome P4504A genotype cosegregates with hypertension in Dahl S rats. Hypertension 27: 564–568, 1996 [DOI] [PubMed] [Google Scholar]
- 18.Regner KR, Zuk A, Van Why SK, Shames BD, Ryan RP, Falck JR, Manthati VL, McMullen ME, Ledbetter SR, Roman RJ: Protective effect of 20-HETE analogues in experimental renal ischemia reperfusion injury. Kidney Int 75: 511–517, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Regner KR, Roman RJ: Role of medullary blood flow in the pathogenesis of renal ischemia-reperfusion injury. Curr Opin Nephrol Hypertens 21: 33–38, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu JY, Li N, Yang J, Li N, Qiu H, Ai D, Chiamvimonvat N, Zhu Y, Hammock BD: Metabolic profiling of murine plasma reveals an unexpected biomarker in rofecoxib-mediated cardiovascular events. Proc Natl Acad Sci U S A 107: 17017–17022, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyata N, Taniguchi K, Seki T, Ishimoto T, Sato-Watanabe M, Yasuda Y, Doi M, Kametani S, Tomishima Y, Ueki T, Sato M, Kameo K: HET0016, a potent and selective inhibitor of 20-HETE synthesizing enzyme. Br J Pharmacol 133: 325–329, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ge Y, Murphy SR, Lu Y, Falck J, Liu R, Roman RJ: Endogenously produced 20-HETE modulates myogenic and TGF response in microperfused afferent arterioles. Prostaglandins Other Lipid Mediat 102-103: 42–48, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoff U, Lukitsch I, Chaykovska L, Ladwig M, Arnold C, Manthati VL, Fuller TF, Schneider W, Gollasch M, Muller DN, Flemming B, Seeliger E, Luft FC, Falck JR, Dragun D, Schunck WH: Inhibition of 20-HETE synthesis and action protects the kidney from ischemia/reperfusion injury. Kidney Int 79: 57–65, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chien KR, Han A, Sen A, Buja LM, Willerson JT: Accumulation of unesterified arachidonic acid in ischemic canine myocardium. Relationship to a phosphatidylcholine deacylation-reacylation cycle and the depletion of membrane phospholipids. Circ Res 54: 313–322, 1984 [DOI] [PubMed] [Google Scholar]
- 25.Yamamoto K, Wilson DR, Baumal R: Outer medullary circulatory defect in ischemic acute renal failure. Am J Pathol 116: 253–261, 1984 [PMC free article] [PubMed] [Google Scholar]
- 26.Chujo K, Ueno M, Asaga T, Sakamoto H, Shirakami G, Ueki M: Atrial natriuretic peptide enhances recovery from ischemia/reperfusion-induced renal injury in rats. J Biosci Bioeng 109: 526–530, 2010 [DOI] [PubMed] [Google Scholar]
- 27.Salom MG, Cerón SN, Rodriguez F, Lopez B, Hernández I, Martínez JG, Losa AM, Fenoy FJ: Heme oxygenase-1 induction improves ischemic renal failure: Role of nitric oxide and peroxynitrite. Am J Physiol Heart Circ Physiol 293: H3542–H3549, 2007 [DOI] [PubMed] [Google Scholar]
- 28.Conesa EL, Valero F, Nadal JC, Fenoy FJ, López B, Arregui B, Salom MG: N-acetyl-L-cysteine improves renal medullary hypoperfusion in acute renal failure. Am J Physiol Regul Integr Comp Physiol 281: R730–R737, 2001 [DOI] [PubMed] [Google Scholar]
- 29.Roman RJ, Akbulut T, Park F, Regner KR: 20-HETE in acute kidney injury. Kidney Int 79: 10–13, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oyekan AO: Differential effects of 20-hydroxyeicosatetraenoic acid on intrarenal blood flow in the rat. J Pharmacol Exp Ther 313: 1289–1295, 2005 [DOI] [PubMed] [Google Scholar]
- 31.Cheng MK, McGiff JC, Carroll MA: Renal arterial 20-hydroxyeicosatetraenoic acid levels: Regulation by cyclooxygenase. Am J Physiol Renal Physiol 284: F474–F479, 2003 [DOI] [PubMed] [Google Scholar]
- 32.Linkermann A, Chen G, Dong G, Kunzendorf U, Krautwald S, Dong Z: Regulated cell death in AKI. J Am Soc Nephrol 25: 2689–2701, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson TW, Alonso-Galicia M, Roman RJ: Effects of lipid-lowering agents in the Dahl salt-sensitive rat. Hypertension 31: 225–231, 1998 [DOI] [PubMed] [Google Scholar]
- 34.Ishizuka T, Ito O, Tan L, Ogawa S, Kohzuki M, Omata K, Takeuchi K, Ito S: Regulation of cytochrome P-450 4A activity by peroxisome proliferator-activated receptors in the rat kidney. Hypertens Res 26: 929–936, 2003 [DOI] [PubMed] [Google Scholar]
- 35.Huang H, Morisseau C, Wang J, Yang T, Falck JR, Hammock BD, Wang MH: Increasing or stabilizing renal epoxyeicosatrienoic acid production attenuates abnormal renal function and hypertension in obese rats. Am J Physiol Renal Physiol 293: F342–F349, 2007 [DOI] [PubMed] [Google Scholar]
- 36.Portilla D, Dai G, Peters JM, Gonzalez FJ, Crew MD, Proia AD: Etomoxir-induced PPARalpha-modulated enzymes protect during acute renal failure. Am J Physiol Renal Physiol 278: F667–F675, 2000 [DOI] [PubMed] [Google Scholar]
- 37.Devarajan P: Update on mechanisms of ischemic acute kidney injury. J Am Soc Nephrol 17: 1503–1520, 2006 [DOI] [PubMed] [Google Scholar]
- 38.Garrett MR, Rapp JP: Two closely linked interactive blood pressure QTL on rat chromosome 5 defined using congenic Dahl rats. Physiol Genomics 8: 81–86, 2002 [DOI] [PubMed] [Google Scholar]
- 39.Mattson DL, Dwinell MR, Greene AS, Kwitek AE, Roman RJ, Jacob HJ, Cowley AW, Jr.: Chromosome substitution reveals the genetic basis of Dahl salt-sensitive hypertension and renal disease. Am J Physiol Renal Physiol 295: F837–F842, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Onwochei MO, Rapp JP: Hyposecretion of atrial natriuretic factor by prehypertensive Dahl salt-sensitive rat. Hypertension 13: 440–448, 1989 [DOI] [PubMed] [Google Scholar]
- 41.Williams JM, Sarkis A, Lopez B, Ryan RP, Flasch AK, Roman RJ: Elevations in renal interstitial hydrostatic pressure and 20-hydroxyeicosatetraenoic acid contribute to pressure natriuresis. Hypertension 49: 687–694, 2007 [DOI] [PubMed] [Google Scholar]
- 42.Ito O, Roman RJ: Role of 20-HETE in elevating chloride transport in the thick ascending limb of Dahl SS/Jr rats. Hypertension 33: 419–423, 1999 [DOI] [PubMed] [Google Scholar]
- 43.Fan F, Sun CW, Maier KG, Williams JM, Pabbidi MR, Didion SP, Falck JR, Zhuo J, Roman RJ: 20-Hydroxyeicosatetraenoic acid contributes to the inhibition of K+ channel activity and vasoconstrictor response to angiotensin II in rat renal microvessels. PLoS ONE 8: e82482, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.von Overbeck J, Saraga P, Gardiol D: An autofluorescence method for the diagnosis of early ischaemic myocardial lesions. A systematic study on 732 autopsies, including 182 cases of sudden death. Virchows Arch A Pathol Anat Histopathol 409: 535–542, 1986 [DOI] [PubMed] [Google Scholar]
- 45.Zou AP, Muirhead EE, Cowley AW, Mattson DL, Falck JR, Jiang J, Roman RJ: Role of changes in renal hemodynamics and P-450 metabolites of arachidonic acid in the reversal of one-kidney, one clip hypertension. J Hypertens 13: 557–566, 1995 [DOI] [PubMed] [Google Scholar]
- 46.Zhong Z, Arteel GE, Connor HD, Yin M, Frankenberg MV, Stachlewitz RF, Raleigh JA, Mason RP, Thurman RG: Cyclosporin A increases hypoxia and free radical production in rat kidneys: Prevention by dietary glycine. Am J Physiol 275: F595–F604, 1998 [DOI] [PubMed] [Google Scholar]
- 47.Efrati S, Berman S, Hamad RA, Siman-Tov Y, Ilgiyaev E, Maslyakov I, Weissgarten J: Effect of captopril treatment on recuperation from ischemia/reperfusion-induced acute renal injury. Nephrol Dial Transplant 27: 136–145, 2012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.