

Abstract
Neurodegenerative diseases (NDs) collectively afflict more than 40 million people worldwide. The majority of these diseases lack therapies to slow or stop progression due in large part to the challenge of disentangling the simultaneous presentation of broad, multifaceted pathophysiologic changes. Present technologies and computational capabilities suggest an optimistic future for deconvolving these changes to identify novel mechanisms driving ND onset and progression. In particular, integration of highly multi-dimensional omic analytical techniques (e.g., microarray, mass spectrometry) with computational systems biology approaches provides a systematic methodology to elucidate new mechanisms driving NDs. In this review, we begin by summarizing the complex pathophysiology of NDs associated with protein aggregation, emphasizing the shared complex dysregulation found in all of these diseases, and discuss available experimental ND models. Next, we provide an overview of technological and computational techniques used in systems biology that are applicable to studying NDs. We conclude by reviewing prior studies that have applied these approaches to NDs and comment on the necessity of combining analysis from both human tissues and model systems to identify driving mechanisms. We envision that the integration of computational approaches with multiple omic analyses of human tissues, and mouse and in vitro models, will enable the discovery of new therapeutic strategies for these devastating diseases.
Graphical abstract
1 Introduction
Neurodegeneration refers to the progressive death and loss of neurons in the brain, a process that begins with dysregulation at the molecular level and leads to gross regional dysfunction and eventually clinical disability. Collectively, neurodegenerative disorders affect more than 40 million people worldwide.1–3 The greatest risk factor for many neurodegenerative diseases (NDs) is age,4–6 a fact that highlights the need to develop effective therapeutics for our aging global population. Commonly studied NDs include Alzheimer's disease (AD), frontotemporal dementia (FTD), amyotrophic lateral sclerosis (ALS), Parkinson's disease (PD), and Huntington's disease (HD). These diseases share two primary features that make them accessible to the research community. First, all are considered proteinopathies – that is, they are believed to stem from pathological protein aggregation (Table 1). Second, each disease has variants with known monogenic causes that can be used to model the disease in vitro and in vivo in an effort to understand the more common and complex sporadic (no family history) form of the disease (with the exception of HD, which is purely a monogenic disorder).
Table 1. Characteristics of protein-aggregating neurodegenerative diseases.
| Primary constituent of protein aggregation | Location of Aggregates | Commonly Studied Familial Genes | Familial Forms (% of total disease count) | Primary Region of Neuronal Loss | Average age at onset | |
|---|---|---|---|---|---|---|
| AD | Aβ, tau | extracellular | PSEN1, PSEN2, APP, APOE32 | < 5%32 | entorhinal cortex, hippocampus | 7033–35 |
| FTD | ubiquitin, TDP-43, FUS, tau | cytoplasmic (neuronal and glial), intranuclear (neuronal) | TARDBP, FUS, C9ORF72, PGRN VCP, MAPT36 | 25-50%32,37,38 | frontal and/or temporal lobe | 6039,40 |
| ALS | ubiquitin, SOD1, TDP-43, FUS | cytoplasmic | SOD1, C9ORF72, FUS, TARDBP, OPTN41–43 | 5-10%32,44 | motor cortex, brainstem, spinal cord | 6045 |
| PD | α-synuclein | cytoplasmic | SNCA, LRRK2, PINK1, PRKN, DJ132 | <10%46 | substantia nigra | 6033 |
| HD | htt | intranuclear and cytoplasmic | HTT47 | ∼90%32 | striatum | * |
Dependent on number of CAG repeats.48
Due to the pathological presentation of protein aggregates in the above NDs, the dominant paradigm for studying each of these diseases has traditionally focused on identification of mechanisms behind protein generation and aggregation, and the direct influence of these aggregates on neuronal function.7–11 However, therapies aimed at reducing protein processing and clearance for AD have been unsuccessful in clinical trials, and one study reported an acceleration of cognitive decline in treated patients.12–14 These results suggest that targeting protein aggregation alone may be insufficient to treat NDs.
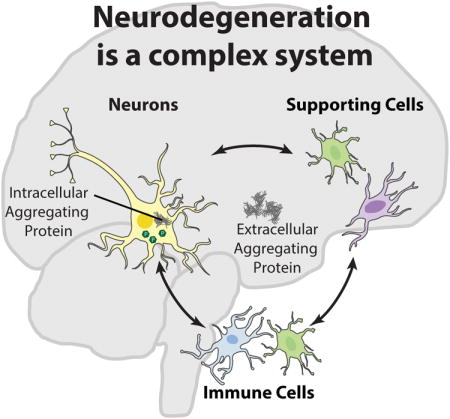
NDs are pathologically defined by the primary aggregating protein as well as the location of the aggregate and neuronal loss.15–18 Nevertheless, protein aggregation represents only one portion of a complex, integrated pathophysiology in each of these diseases and is commensurate with loss of homeostatic regulation, including immune response, metabolic changes, synaptic loss, and neuronal death. Moreover, neurodegeneration is associated with a break-down of the blood-brain barrier (BBB)19 and/or blood spinal cord barrier (BSCB)20 which enables peripheral immune cells21 to infiltrate the central nervous system (CNS) (Fig. 1), further contributing to homeostatic dysregulation in the affected tissues.
Figure 1.

NDs are associated with broad physiologic responses in the CNS. A primary feature common to many NDs is the neuronal expression of intra- and/or extra-cellular aggregating protein (AP). Intra-neuronal APs, including α-synuclein, tau, the huntingtin protein, and superoxide dismutates-1 (SOD1), harm neurons directly by interfering with cellular processes, including microtubule and synaptic function.7–11 Extracellular APs, including amyloid-β, α-synuclein, and tau not only affect neurons, but stimulate responses from supporting cells (e.g., astrocytes and oligodendrocytes), and immune cells (e.g., astrocytes, and microglia). The supportive and immune cell response leads to production of reactive oxygen species (ROS) that are directly deleterious to neuron survival.22 Cytokines and other factors may also be directly harmful to neurons23 and promote BBB/BSCB leakiness.19,20,22 Combined with enhanced cytokine production, the leaky BBB/BSCB enhances peripheral monocyte and leukocyte infiltration, contributing to homeostatic dysregulation.21,22 This figure was created using cell art adapted from Servier Medical Art (http://www.servier.com/Powerpoint-image-bank).
Due to the complexity of these diseases and the heterogeneity of onset and progression, it remains unclear whether neurodegeneration is triggered and driven by protein aggregation (causative pathophysiology) or whether protein nucleation and aggregation represent a physiological response to a pre-existing homeostatic dysregulation (responsive pathophysiology) whereby the aggregation event is possibly an attempt to attenuate the toxic effect of the misfolded protein.24–26 Contributing to the debate in AD, extracellular (amyloid beta; Aβ) and intracellular (tau) protein aggregation can occur in cognitively normal individuals at levels consistent with AD tissues, but without significant neuro-inflammation or neuronal death.27,28
Disentangling causative pathogenic events from responsive pathology is nearly impossible by analyzing postmortem tissue alone. First, pathological protein deposition begins years to decades prior to onset of clinical symptoms29,30 and decades before detailed bio-chemical measurements are collected from postmortem tissues.31 Second, a multitude of changes occur simultaneously during disease progression including cell death and inflammation. Each of these pathophysiological traits is expected to evolve throughout disease progression. To build temporal and spatial models of progression, researchers often use transgenic mouse models to access tissue at different phases of disease. The most commonly used models express genetic mutations identified by familial linkage studies in monogenic forms of the respective disease (Table 1).
Like in the human disease, mouse models elicit a multitude of simultaneous pathophysiologic changes. While in vivo models are essential for modeling the complexity of the human diseases, the central challenge of how to isolate causative mechanisms remains. The field of systems biology provides a broad collection of multivariate computational tools suitable for identifying causative mechanisms based on modern, highly multi-dimensional omics datasets. Together, with systems analysis-directed perturbation analysis and in vitro models, the field is poised to identify new mechanisms driving NDs.
In this article, we will begin by reviewing some of the key genetic and pathological elements of NDs and how these elements interact as a complex system. Second, we will review some of the computational analysis tools available for analyzing data from primary human tissues, mouse, and cell culture models. We will then review the current literature applying systems analysis approaches to NDs, and conclude by emphasizing the need for integrated systems analysis of human tissues together with perturbation analysis in model systems to identify mechanisms driving NDs.
2 Neurodegenerative Diseases Share Complex Pathophysiology
2.1 Alzheimer's Disease
AD is the most common form of dementia. Symptomatically, it begins with loss of short term memory, followed by disorientation and behavioral changes, and ultimately loss of language and motor function.4,49 Pathological onset is marked by the accumulation of extracellular Aβ plaques50 in the neocortex, entorhinal cortex, and hippocampus.51 Microglia and astrocytes are attracted to the plaque locations and express cytokines52,53 which likely amplify astrocyte recruitment to the location.54 Simultaneous with this immune response, neurons in the plaque vicinity show hyper-phosphorylation of microtubule-associated protein tau.55 Hyper-phosphorylated tau (hp-tau) aggregates to form intracellular neurofibrillary tangles (NFTs). It is hypothesized that aggregation of hp-tau depletes the cell of functional tau which normally serves to stabilize microtubules and is necessary for neurite outgrowth and synaptic function.56 Affected neurons become dystrophic and die.53 Due to the role of tau in AD pathogenesis, AD is considered to be a member of a larger family of tau aggregate-involving NDs called tauopathies.57
Extracellular deposition of Aβ plaques is the hallmark pathology of AD, leading to the hypothesis that the cellular processes that regulate Aβ production, synthesis, or accumulation drive pathogenic mechanisms in AD. This hypothesis is supported by the finding that certain mutations in the amyloid precursor protein (APP), which creates Aβ when cleaved, cause accelerated deposition of Aβ plaques and early-onset familial AD (FAD).58,59 Interestingly, a rare coding mutation in APP was recently identified to protect against AD and cognitive decline in the elderly.60 Additional FAD pathogenic mutations have been identified in presenilin 1 and presenilin 2, members of the γ-secretase complex which cleaves APP to form Aβ.59 The convergence of pathology and genetics on the theme of increased amyloid production has led to the prominent belief in the Aβ-hypothesis that has dominated the field for more than a decade. Nevertheless, a recent report has described sporadic AD “mismatch” cases that have pronounced Aβ pathology, yet no cognitive decline or neuronal death.28 Taken together, these results suggest that Aβ pathology may be a necessary but not sufficient condition for driving cognitive decline and neuronal death. Furthermore, monogenic forms of AD may be a more direct result of accumulation of amyloid while sporadic forms of the disease are possibly a result of a more complex interaction of pathogenic events.
The pathology described above is commensurate with much broader dysregulation that may further contribute to neuronal death and pathological progression. Glia exposed to Aβ produce reactive oxygen species (ROS)61 that are toxic to neurons62 and certain cytokines have been reported to enhance Aβ production.63 In addition, the CNS vasculature becomes activated, resulting in endothelial secretion of inflammatory cytokines, matrix metalloproteinases, and other species that may be neurotoxic.64 Some research suggests that hypoperfusion due to dysfunctional cerebrovasculature may be a mechanism driving neuronal death.65 Increased vascular permeability66 may also contribute to bacteria67 and immune cell68 migration from the periphery into the CNS, further contributing to the homeostatic dysregulation of the affected tissues.
2.2 Frontotemporal Dementia
FTD encompasses a diverse spectrum of clinical deficits, including cognitive, behavioral, semantic, and motor dysfunction.69 FTD is the third most common form of dementia after AD and vascular dementia, accounting for approximately 5-10% of diagnosed dementia cases.3 FTD often presents with ALS, as discussed in the next section.
FTD is structurally defined by atrophy of the frontal and temporal lobes and reduced glucose metabolism.70 In addition to neuronal atrophy, FTD is pathologically defined by intracellular inclusions comprised of one of the following proteins: tau, TAR DNA binding protein-43 (TDP-43; encoded by the TARDBP gene),71 fused in sarcoma (FUS) protein72, or superoxide dismutates-1 (SOD1; primarily found in ALS, but also observed in FTD41). Pathological inclusions of each of these proteins are either highly phosphorylated (tau),73 or highly ubiquitinated (TDP-43,71 FUS,72,74 SOD175). Many FTD cases consist of hp-tau aggregates within neurons and glia,76 and are members of the tauopathy family of NDs.57 Tau negative cases present primarily with intraneuronal inclusions consisting of highly ubiquitinated TDP-4371 or FUS.72
Approximately 25-50% of FTD cases are inherited and are a result of mutations in a number of genes (Table 1). Aggregates of tau, TDP-43, and FUS are also found in sporadic forms of FTD which, like AD and PD, implicates common pathways of aggregation for proteins and genes known to cause monogenic forms of disease. The presence of intracellular inclusions in neurons from affected tissues suggest a cell autonomous processes. However, like in AD, affected tissues present activated microglia,77 gliosis early in disease,78 increased oxidative stress,79 and increased expression of pro-inflammatory cytokines.80 Interestingly, unlike in AD, apoptotic glial death has been reported to be correlated with pathological severity in FTD.81
2.3 Amyotrophic Lateral Sclerosis
ALS primarily affects neurons in the motor cortex and spinal cord, causing muscle stiffness, spasms, weakness, and eventually muscle wasting, typically leading to death within 5-8 years of symptomatic onset.45,82–86 In addition to neuronal atrophy, ALS is pathologically defined by intracellular, highly ubiquinated inclusions comprised of one of the following proteins, many of which are also found in FTD: TDP-43,87 FUS72, optineurin (encoded by OPTN),42 or SOD1.75,43 While ALS and FTD affect different regions of the brain, many patients present initially with one disease and progress to develop features of both diseases. Thus, these diseases may be referred to as FTD-ALS (or FTD with Motor Neuron Disease).88,89
Approximately 5-10% of ALS cases are inherited,44 and these familial forms are associated with mutations in many of the genes connected to familial FTD (Table 1). In contrast to FTD, ALS affects motor neurons in the motor cortex and spinal cord.90 A direct mechanism linking these aggregates to neuronal death remains absent and a non-cell autonomous mechanisms of neuronal death has been suspected.91 Affected tissues in ALS exhibit a strong inflammatory response, including an increased number of activated microglia,92,93 reactive astrocytes,94 and upregulated inflammatory cytokines.92,95,96 We direct the reader to the review by McGeer & McGeer97 for an overview of inflammation in ALS. Increased oxidative stress98 and widespread metabolic changes99 are believed to further contribute to disease pathogenesis. Additionally, a recent report found a breakdown in the blood-spinal cord barrier (BSCB) in ALS associated with a reduction in pericyte numbers, resulting in red blood cell invasion into the spinal cord.100
In addition to the role of inflammatory response, a prominent recent theory hypothesizes that ALS and FTD are caused by a dysfunctional RNA stress granule response, resulting in nuclear stress granule inclusions found in postmortem tissues and mouse models.101 A number of ALS/FTD genes (including TARDBP, FUS, OPTN) are functionally implicated in RNA binding proteins and stress granule formation, and it is thought that mutations in these genes alter the stability and ability of stress granules to respond to cellular stresses.101
2.4 Parkinson's Disease
PD causes motor impairment such as muscle rigidity, tremors and bradykinesia as a result of cell death in the substantia nigra. In particular, the degeneration of dopaminergic neurons in the substantia nigra over time decreases dopamine release, leading to decreased stimulation of the motor cortex by the basal ganglia.102 Pathologically, PD is characterised by cell death, gliosis, and the presence of intracellular proteinaceous inclusions called Lewy bodies. The development of cognitive impairment can often occur late in disease, although some patients manifest with early dementia alongside the onset of motor symptoms (often called PD with dementia or dementia with Lewy bodies, DLB).103–105 The primary constituent of Lewy bodies is α-synuclein; therefore these NDs are often referred to as synucleinopathies.57
Less than ten percent of PD cases are familial forms.46 Kindred studies of familial forms of PD have identified rare point mutations, as well as chromosomal multiplications that cause familial PD (Table 1).106,107 The most well characterized disease association is the SNCA gene locus which encodes the α-synuclein protein. In these cases, age of onset and disease severity correlates closely with the expression level of α-synuclein.107
The understanding that Lewy bodies are found within neurons and α-synuclein is expressed by neurons led researchers to believe that dopaminergic neuronal death is cell autonomous, with α-synuclein acting as an intrinsic trigger of cytotoxicity. As a consequence of this belief, research has primarily focused on understanding cellular pathways that influence α-synuclein aggregation within the neuron and what effect these aggregation events have on the health and normal function of neurons.108,109 The more recent discovery of α-synuclein as a secreted protein has challenged this neurocentric view of PD. Recent studies have demonstrated that α-synuclein is secreted and taken-up by neurons, both in culture and in mouse models of PD.110,111 Additionally, α-synuclein has been detected in microglia, astrocytes, and oligodendrocytes in a number of synucleinopathies. This suggests that α-synuclein is not just transmitted by neurons to other neurons through a traditional synaptic model, but that α-synuclein may be transmitted to glial cells as well.112–114 Given the role that glia play in homeostatic maintenance of neuronal health and signal transduction in response to extracellular signals, it is likely that glial dysfunction plays a significant role in dopaminergic neuron vulnerability in PD. Furthermore, since glia are an integral component of the BBB, glial dysfunction is also believed to be a key contributor to BBB disruption in PD.115
2.5 Huntington's Disease
Symptoms of HD typically begin with subtle changes in mood and personality, then progress to include impairment of motor function followed by executive functions. These symptoms are associated with severe degeneration in the caudate and putamen and, to a lesser extent, in other regions including the substantia nigra, cortex, and hippocampus. See the review by Walker for an extensive review of HD symptoms and pathology.116
HD differs from other NDs discussed so far in that it is an autosomal dominant monogenic disorder. It is caused by an expansion of the CAG triplet repeat in the N-terminal of the huntingtin gene47 from a non-disease level of less than 36 repeats to full penetrance with more than 42 repeats.117,118 These repeats result in increased protein oligomerization, which is enhanced as the number of repeats increases. The huntingtin protein (htt) is primarily expressed in neurons, and at a lower level in glia.119
While the genetic cause of HD is known, the specific mechanisms leading from protein aggregation to neuronal death remain unknown. Nuclear and cytoplasmic inclusions of aggregated htt are found in neurons from diseased tissues.120 As observed in other NDs, affected tissues experience neuroinflammation, including elevated expression of pro-inflammatory cytokines such as IL-6, CCL2, and TNF-α,121 an increased number of activated microglia that correlates with pathological severity,122 and reduced expression of brain derived neurotrophic factor (BDNF).123 While the BBB and cerebrovascular function has had limited examination in HD, a recent study showed increased vascular densities in human HD tissues, and increased cerebral blood volume in the R6/2 mouse model, which expresses human htt with greater than 200 CAG repeats under the control of the human HTT promoter.124 While this study did not find differences in BBB function in the model, it did demonstrate a role for the vasculature in HD.
HD exemplifies the challenge of finding effective therapies for NDs. While the mutation causing HD was discovered more than 20 years ago,47 clinically effective therapies remain elusive.125 Since HD is monogenic, one approach currently under evaluation is to reduce htt expression.125 However, multiple studies emphasize our poor understanding of the relationship between htt aggregation and neuronal death. One population of neurons that is susceptible to death in HD, cortical pyramidal neurons, present with many intra-nuclear inclusions, suggesting that protein aggregation may be neurotoxic. However, medium spiny neurons found in the striatum are also prone to death, but rarely present with inclusions,126 suggesting that htt can cause neuronal death in a non-cell autonomous fashion. Moreover, longitudinal MRI analysis reveals that while the CAG repeat length correlates with rate of atrophy in specific cortical regions, it does not explain rate of atrophy in the caudate.127 Taken together, these findings illustrate that the mechanism connecting the CAG repeat to neuronal death is complex.
3 Experimental Models of Neurodegernative Diseases
The broad simultaneous changes described for each of the NDs discussed above illustrate the difficulty in delineating causal mechanisms from responsive physiology based on terminal human tissues alone. As we will discuss further in the following sections, a central means of identifying causal mechanisms is to experimentally perturb the pathological physiology which is not possible in the analysis of human tissues. Therefore, the incorporation of mouse and culture model systems is essential to identifying novel mechanisms that will lead to new therapeutic strategies.
Transgenic mouse models enable the collection of longitudinal data that provide insight into early disease development, including mechanisms promoting protein aggregation, immune response, synaptic loss, and neuronal death. The ability to harvest tissue rapidly and in a tightly controlled experimental setting enables analysis of rapidly decaying proteins, such as phospho-proteins, which generally cannot be reliably analyzed in postmortem human tissue.128 However, mouse models present limitations which confound their use in isolating individual mechanisms of human disease. Specifically, mouse models i) incompletely reproduce the pathophysiology observed in the human disease, ii) simultaneously present multiple aspects of disease (e.g., protein aggregation, inflammatory response, and cell death), similar to the human disease, and iii) are mainly based on gene mutations found in familial cases, which may not reflect mechanisms driving sporadic disease cases.
Mouse models each have particular advantages and limitations in reflecting their associated human disease (Table 2). Therefore, certain models may be superior for reproducing the ND pathophysiology of interest. For example, until the discovery of TDP-43 mutations in FTD and ALS, ALS researchers were restricted to the use of SOD1 mice for modeling primarily spinal degeneration and dysfunction in ALS. The creation of various mutant TDP-43 lines expanded the affected tissue types from the spinal motor neurons affected in SOD1 mice to cortical regions, representing a more diverse range of ALS symptoms that are often regarded as more relevant to sporadic ALS.129 This model has limitations, however, because pure backcrossed lines present abnormalities in peripheral nervous system expression of TDP-43. 130
Table 2. Selected mouse models of neurodegenerative diseases.
| Model Name | Disease Applicability | Expressed Protein (Mutation) | Promotor | Pathological Features | Limitations |
|---|---|---|---|---|---|
| APPswe/PS1dE9143 | AD | APP (KM594/5NL), presenilin 1 (deletion of exon 9) | Mouse prion protein | Aβ plaques in cortex and hippocampus by 4mo144 | No NFTs, no neuronal death |
| Tg2576145 | AD | APP (K670/671L) | Hamster prion protein | Aβ plaques after 12mo146 | No NFTs, no neuronal death |
| 3xTg147 | AD | APP (K670/671L), presenilin 1 (M146V), tau (P301L) | Mouse Psen1 (presenilin 1), Mouse Thy1 (APP, tau) | Aβ plaques by 6mo, hippocampal then neocortical NFTs appear between 12-18mo | NFT formation may be independent of Aβ148 |
| rTg4510149 | AD, FTD | tau (P301L)* | Mouse prion protein (tau), mouse calmodulin-kinase II (transactivator) | Hippocampal NFTs and neuronal death by 4mo, cortical NFTS by 4mo150 | AD: no Aβ pathology FTD: Greatest NFT and neuronal death in hippocampus |
| Thy1-TDP-43151 | FTD, ALS | Human TDP-43 (wild-type over expressed) | Mouse Thy1 | Nuclear and cytoplasmic (ubiquitinated, phosphorylated) inclusions primarily in motor cortex and hippocampus by 6mo, neuronal death by 6mo | Subcellular localization of TDP-43 is not reported |
| Prnp-FUS152 | ALS | Human FUS (wild-type) | Mouse prion protein | Non-ubiquitinated inclusions in the motor and somatosensory cortices and spinal cord, 60% motor neuron death in spinal cord by 11 weeks old | No neuronal loss and little microglial or astrocytic response in brain |
| SOD1-G93A153 | ALS | SOD1 (G93A) | Endogenous human SOD1 promoter | 55% reduction in spinal cord neuron count by 123 days154 | Neuronal loss restricted to spinal cord |
| PDGF-β hα-syn (D-line)155 | PD (and DLB) | α-synuclein (wild-type) | Human PDGF-β | α- synuclein accumulation primarily in neocortex and limbic system, substantia nigra (rarely)156, motor deficits 9-12 months157 | α- synuclein aggregates not consistent with human Lewy bodies, no dopaminergic degeneration, glial expression of α- synuclein (protein and RNA) |
| mThy1 ha-syn158 | PD | α-synuclein (wild-type or A43T) | Mouse Thy1 | High α- synuclein expression in neocortex, limbic system, thalamus, some reports of spinal cord156 | No dopaminergic degeneration, few inclusions, mostly diffuse expression of α-synuclein |
| MPTP Neurotoxic lesion models159 | PD | Chemical lesion: MPTP | - | Dopaminergic degeneration | Acute models (not progressive models for onset), no α-synuclein inclusions in mice |
| YAC128160 | HD | htt (128 CAG repeats on exon 1) | Human HTT | Diffuse nuclear htt by 12mo, nuclear inclusions by 18mo, 15% reduction in striatal neuron count by 12mo | No cytoplasmic inclusions |
| HD94161 | HD | htt fragment (94 CAG repeats on exon 1)* | calmodulin-kinase IIα | Cytoplasmic and intranuclear inclusions in the striatum by 12 weeks old, striatal atrophy162 | No neuronal death |
Tetracycline-regulated transgene.
Cell culture-based ND models (e.g. human cell lines, primary neuron and glial cultures, and induced pluripotent stem cells; iPSCs) provide complementary capability to mouse models for isolating the roles of specific cell types in response to aggregating proteins, or genetic, pharmacologic, or other interventions. For example, primary neuron and glial cultures are valuable for assessing specific cell type responses to Aβ131,132 Human cell lines,133 and emerging iPSC models134–141 may provide further utility for replicating functional changes associated with genetic defects that cannot be exogenously applied to cultures. Additionally, human cell-derived cultures may not suffer from some of the limitations found in mice, such as the lack of endogenous mouse tau aggregation into NFTs.142
Mouse and cell culture models each provide particular value for reproducing specific aspects of disease pathophysiology. Integrating broad genomic and proteomic multi-dimensional data from these model systems will be central to identifying causative mechanisms driving NDs.
4 Systems Analysis Methodology
The NDs discussed in the previous sections present complex pathophysiology. This suggests that multivariate analysis of a broad collection of biological pathways will provide a substantial advantage in identifying mechanisms and pathways driving disease.
4.1 Choice of Analysis Methodology
The choice of computational analysis to analyze multivariate data depends on a variety of factors including a priori biological knowledge, the number of samples, the number of measurements, and the hypothesis or question explored. Systems analysis approaches lie along a spectrum that ranges from mechanistic (analysis which applies assumptions based on prior biological knowledge)163 to data-driven (analysis of experimental data without making specific mechanistic assumptions).164 Mechanistic approaches, such as differential reaction equations, require a detailed understanding of the underlying mechanisms and require a large number of experimental perturbations to reliably identify parameters.163 Since knowledge of detailed mechanisms is limited and a large number of perturbations are not always possible in vivo, mechanistic approaches are generally less suitable for in vivo studies.
For multi-dimensional omics analysis of in vivo or culture experiments, it is common to collect a large number of measurements (measurement variables) from a relatively small number of samples. For example, microarray datasets typically consist of gene expression measurements for tens of thousands of genes from tens to hundreds of samples.165 Mass spectrometry measures a similar number of proteins (total and phospho-proteins)166, while multiplexed xMAP® immunoassays (Luminex, Austin, TX) are able to measure 1-500 analytes (e.g., cytokines, total proteins, phospho-proteins), and flow cytometry is able to distinguish on the order of a dozen labels.167 See the review by Lyons et al.168 in this issue for a detailed review of technologies for systems biology applications. Key questions that can be asked when evaluating these datasets include:
Do the samples differ from one another based on experimental class or quantitative phenotype?
Which measurement variables most strongly discriminate between class or phenotype?
Does measurement variable A drive measurement variable B?
Data-driven approaches for analyzing multi-dimensional data begin with correlation approaches such as hierarchical clustering164,169 and multivariate projection approaches, including principal component analysis (PCA) and partial least squares (PLS) regression.170,171 Correlation approaches assess whether measurement variables are able to group samples into classes or along a spectrum based on a phenotypic Y-variable, and identify which measurement variables are correlated with class or phenotype. Prior knowledge pathway analysis approaches,172 such as gene set enrichment analysis (GSEA)173, extend correlation approaches by incorporating prior knowledge about gene co-regulation to identify clusters of genes that are differentially regulated between classes. Finally, Bayesian networks use prior knowledge together with current experimental data to construct a directed network graph that optimizes the probability that variable A causes B for every pair of measurement variables. The following sub-sections briefly review the methods and application of several prominent systems analysis techniques applied in the ND literature.
4.2 Multivariate Projection
Multivariate projection approaches are often used to find a “principal axis” separating samples based on either correlation between the measurement variables or correlation between the measurement variables and a qualitative or quantitative phenotype. These approaches are highly effective when the number of measurement variables is high relative to the number of samples.
Principal Component Analysis
A dataset consisting of N samples and M measurements variables can be thought of as N data points plotted in an M-dimensional space. PCA uses a singular value decomposition174 to identify directions in the data space where the M measurements are maximally co-varying across all data points.170 Re-plotting the data in terms of the first few maximally co-varying axes (principal components; PCs) often yields an enhanced clustering of samples associated with each class, or along a biologically meaningful spectrum, that was not immediately apparent from the representation in the original M-dimensional measurement space. This re-plotting of the data represents a new presentation of the data in a rotated coordinate frame. Each of the PCs represents a composite variable that is composed of a weighted sum of the M measurement variables. The weight of each of the M measurements in the PC indicates the relative importance of that measurement variable in distinguishing the data points. An important feature of PCA is that it can often separate meaningful biological variation in the data from uncorrelated variation which may be unrelated to the biological question of interest. Common sources of noise in in vivo samples include genetic and environmental differences, health or cause of death at the time of tissue collection, and inconsistencies in the tissue preparation and measurement assay.128
Partial Least Squares
Partial least squares (PLS) regression extends the concept of PCA by regressing the M measurement variables for each of the N samples against a phenotypic measurement (Y-variable). The Y-variable often represents a quantitative assessment of tissue state, pathology, or clinically-relevant endpoints. For example, it could represent pathological assessment of disease progression, such as Braak & Braak stage in AD,175 or a measurement of glial activation or neuronal density. PLS can also be used to regress against a discrete Y-variable that represents sample class, e.g., tissue samples from AD patients vs. samples from patients without AD.
PLS approaches are often used in instances where it is not possible to obtain a unique solution by directly regressing a large number of independent variables (in this case, M measurement variables) against the Y-variable when the number of measurements is larger than the number of samples (M>N).176 PLS provides a solution by regressing against the first several “principal” components derived from finding the directions of maximum co-variance of the product of the measurement variables and the Y-variable. The form of this analysis resembles PCA as discussed above, except the “principal” components are referred to as latent variables (LVs). Unlike PCA, PLS analysis identifies variation in measurement variables that is correlated with the Y-variable. By directly accounting for co-variation between the Y- variable and measurement variables, PLS is capable of identifying biologically relevant signaling changes that are of a lower magnitude than other sources of noise in the system which are uncorrelated with the Y-variable.
Despite the advantage of PLS analysis for identifying Y-correlated changes in the measurement variables, it should be used with caution. A key concern with any regression analysis is whether the identified regression coefficients represent the underlying biology, or whether they are only fitting the small number of samples in the available dataset. This calls into question not only the separation of the samples in LV space, but also the weighted contribution of each of the M measurements in the LV. It is therefore important to test the predictive capability of the model against a validation dataset. For small sample sets, a leave-one-out cross validation is often used for validation, where a single sample is left out of the regression, then predicted based on the model derived from regressing the remaining data points. This process is repeated for all samples.
4.3 Prior Knowledge Pathway Analysis
Correlation approaches, such as PCA or PLS, are effective at determining which measurement variables can separate samples into classes. Given the large number of measurement variables produced by microarray analysis, mass spectrometry, etc., however, correlation analysis often identifies a very large number of differentially regulated genes or proteins that strongly distinguish between classes. A key challenge, therefore, is to extract biological meaning from the collection of differential measurement variables, many of which may not be mechanistically or functionally related. Alternatively, a univariate analysis may find correlation between a large number of related genes and a phenotype, but none of the correlation may reach the level of significance. Prior knowledge pathway analysis attempts to solve these problems by using curated prior knowledge of gene co-regulation to group measurement variables together.173,177 Prior knowledge is drawn from databases that relate genes based on mechanistic connections or functional relationships. The Kyoto Encyclopedia of Genes and Genomes (KEGG)178 and the Gene Ontology (GO) database179 are prominent databases curating mechanistic and functional relationships, respectively.
Many related algorithms have been proposed to incorporate prior knowledge into the pathway analysis.172 We briefly summarize two of these here which have been widely applied: Gene Set Enrichment Analysis (GSEA) and Association LIst Go AnnoTatOR (ALIGATOR). The GSEA algorithm computes an “enrichment” score for a group of genes based on how strongly each gene in the group is correlated with a particular class.173 A penalty is applied for strongly correlated genes that are not part of the group. By clustering genes into groups, GSEA has two major advantages in analyzing highly multi-dimensional datasets. First, it reduces a high-dimensional dataset into a smaller number of groups that are each composed of biologically related measurements. Second, because the GSEA score for each group is a composite value computed from the individual correlation value associated with a large group of functionally related genes, it rejects biological or sampling noise in any individual measurement variable. It is therefore able to more stably assess group/pathway activation across studies than individual measurement variables173. ALIGATOR180 is primarily targeted at genome wide association (GWA) studies. It first applies an arbitrary significance threshold to genes in the GWA study, and checks for overrepresentation of functionally related genes based on GO terms.
4.4 Bayesian Network Models
In contrast to correlation approaches and pathway analysis, which are directed at identifying how measurement variables or groups of measurement variables change with class, the primary goal of Bayesian network (BN) modeling is to identify causal relationships between measurement variables. BNs identify whether measurement variables are causally related (i.e., does variable A cause variable B?) in a dataset by explicitly integrating imperfect prior knowledge and data available from the current dataset.181– 183 In systems biology applications, BNs may be used to model relationships between expressed genes or proteins, or phosphorylation networks.181,184,185 Additionally, BNs can be extended to include temporal information, such as feedback loops.186 For example, these dynamic BNs may be particularly useful for analyzing time points drawn from conditioned culture models, or post-transgene activation in tetracycline-regulated mouse models (Table 2).
Bayesian networks attempt to identify causal relationships between variables by maximizing the a posteriori probability that local network interactions exist given a priori network information.182 A priori information may be drawn from mechanistic or ontology databases, such as the KEGG and GO databases. The Bayesian network is chosen to maximize the a posteriori likelihood that a collection of causal edges exist given imperfect prior knowledge about the network and given the dataset being modeled. A key advantage of Bayesian network models is that they are able to implicitly account for hidden intermediate variables that are not measured.181
Obtaining the network that maximizes the a posteriori probability function requires a search algorithm, which is computationally intensive (NP-complete)187 and is not tractable for datasets with a larger number of measurement variables. Therefore, heuristic search strategies are usually employed. An example heuristic strategy is a “greedy” search that starts with an initial collection of network edges and sequentially adds and removes edges until the posterior probability is locally maximized.188 The number of edges is exponential in the number of measurement variables, so highly multi-dimensional datasets (e.g., from microarray) present an intractable search space, even using a heuristic search. One means of reducing the search space is to initially identify a smaller collection of candidate edges based on a bi-variate statistic, such as correlation, since uncorrelated measurement variables will not be causally related.189
For analysis of datasets with a large number of variables, but a small number of samples, experimental conditions, or perturbations, a large collection of networks will similarly maximize the posterior probability. Many of these networks may share common edges, while other edges may appear only in a small subset of these networks. Therefore, an edge appearing in any single network may not reflect a true causal relationship. Edges that appear in a majority of networks are more likely to reflect a true causal connection.189 A Markov Chain Monte Carlo (MCMC) sampling procedure is the general approach to identifying all of the networks, but MCMC sampling is computationally intensive and is not tractable for large datasets.181 One proposed methodology, which is far more computationally tractable, is to employ a bootstrapping algorithm: a small numerical perturbation is applied to the dataset, the network learning procedure is applied, and the existence of the edge is checked for. This is repeated m times, and the “confidence” in the edge is computed as the fraction of times the edge appeared in the m perturbed networks.181 Of course, experimental perturbations are essential to verify that network-predicted edges are, in fact, causal.
4.5 Isolating Causative Factors
Correlative analysis tools, including clustering, multivariate projection, and pathway analysis are valuable for identifying which measurement variables or functional groups are most strongly correlated with each other, or with disease class or phenotypic measurements. They do not, however, provide any guarantee of a causal relationship between measurements, or between measurements and a phenotype. Even Bayesian networks, which maximize a probability function in an attempt to identify causative relationships based on prior information and the data, are not guaranteed to identify causal relationships. The question, then, is how do we systematically verify which of the hundreds to tens of thousands of measurement variables cause or modulate disease or phenotype?
Whether the initial dataset is from primary human tissues, or a model system, identification of causative variables requires perturbation analysis. Most often this will be practical in a mouse or culture model system. Initially suspected genes, proteins, phospho-proteins, etc., must be perturbed via pharmacologic, genetic, or other intervention, and all variables must be acquired under this new perturbation. The new data can then be integrated into the analysis to either verify the role of the node being perturbed, or suggest a new candidate causative node based on the new data. The identification of causal variables is therefore an iterative process.
5 A Review of Systems Studies of Neurodegenerative Diseases
5.1 Analysis of Pathway Enrichment
Systems analysis methodologies have been primarily applied to microarray analysis and GWA studies in postmortem human tissues due in part to the recent accessibility of these techniques, and the highly multi-dimensional datasets they produce. The key challenge with these datasets is identifying meaningful biological differences across the entire genome based on a relatively small number of samples. As discussed above, pathway analysis methodologies, (e.g. GSEA173 and GO179 enrichment) are valuable tools for analyzing composite changes in groups of genes based on prior knowledge of mechanistic or functional relationships.
Most basically, pathway analysis can be directly applied to a dataset with control and disease classes to identify differential pathway enrichment based on curated pathway databases. For example, Holmans et al. used ALIGATOR180 and GSEA applied to GWA study data to identify canonical pathways connected to leucocyte/lymphocyte activity and cytokine signaling associated with increased risk of PD.190 Similarly, Neueder & Bates applied GO enrichment to a gene expression dataset from HD and control brains, identifying increased activation of the complement system.191 It is important to note here that pathway analysis alone applied to a differential gene expression dataset is merely correlative, and does not provide evidence of causation.
Systems analysis can provide deeper insight when applied to both human tissues and a model system that can be perturbed to infer causal relationships. Wexler et al. used GSEA and GO enrichment analysis to identify Wnt1 signaling-associated changes in human neural progenitor (hNP) cells, and suggest a causal relationship between granulin haploinsufficiency and Wnt signaling in inherited FTD.192 The authors began by stimulating Wnt1 in hNP cells, finding differential regulation of genes involved in cell survival, metabolism, transcriptional and translational regulation, and KEGG pathways for AD, HD, and PD, and specifically reduced expression of granulin. They further found that Wnt1 signaling reduced progranulin expression in hNP cultures and that RNA interference hairpins against granulin up-regulated Wnt1 signaling, suggesting that Wnt1 and granulin are inversely regulated. Finally, the authors analyzed gene expression data from patients with granulin haploinsufficiency FTD, and found that a large number of canonical Wnt pathway genes were up-regulated. This study therefore combined analysis from culture and human tissues, together with perturbation in cell culture to suggest a causal connection between granulin haploinsufficiency FTD and Wnt signaling.
In addition to experimental perturbation, another means of enhancing confidence in the causality of a pathway is to identify pathways that are both differentially regulated based on gene expression and are implicated based on pathway enrichment of GWA data. A recent AD study by the International Genomics of Alzheimer's Disease Consortium used this approach to suggest potentially causative pathways in late onset AD.193 This study began by applying ALIGATOR and GSEA to GWA data to identify pathways involved in late onset AD. It then correlated the GWA pathway analysis with gene expression data to identify pathways derived from both modalities, including immune response, endocytosis, and cholesterol transport pathways.
To gain an understanding of epigenomic regulation in AD, a recent paper by Gjoneska et al.194 profiled gene expression (via RNA-seq) and chromatin binding (via ChIP-seq) in early vs. late pathology in the CK-p25 inducible mouse model. This model overexpresses the p25 subunit of CDK5R1 under the control of the CamKII promoter.195 The authors analyzed changes in hippocampal tissues of CK-p25 mice at 2 weeks and 6 weeks after transgene induction, representing early vs. late pathology, respectively. The authors combined their gene expression and chromatin mark analysis with GO enrichment to identify coordinated changes in gene expression and epigenomic regulation. They found that genes associated with synaptic plasticity showed consistently decreased expression at both time points and reduced chromatin enhancer and promoter marks in associated regulator regions. Similarly, genes associated with immune response showed up-regulated expression and increased enhancer and promoter marks in associated regulatory regions.
While these unbiased, transcriptome- and genome-wide approaches have confirmed previous hypotheses and highlighted new areas of research, the integration of prior knowledge is still biased by what is ‘known’ about the function of certain proteins in certain pathways. Researchers should therefore be cautious not to over interpret findings based solely on pathway analysis. For example, it is highly likely that inflammation plays a key role in neurodegeneration but prior knowledge of canonical inflammatory pathways is driven by the experimental history of these proteins in peripheral diseases. The multiple functions of proteins and cell-specific functions of proteins may be altered in CNS cell types or within the different environment of the CNS.
5.2 Identification of Novel Pathways
Network analysis methodologies, such as BN analysis discussed above, provide a framework for identifying correlation and potentially causal relationships between genes and proteins. In contrast to pathway analysis, these approaches provide methods for identifying novel mechanisms or functional relationships between genes, proteins, etc.
A network analysis-based study by Miller et al. analyzed microarray data from AD tissues and age-stratified healthy hippocampal tissues to identify gene expression changes associated with aging and changes specific to AD.196 This work relied on a weighted gene co-expression analysis (a network analysis based on correlation197) together with hierarchical clustering to identify “modules” of highly correlated genes. The analysis revealed PSEN1 expression as a central node that is highly co-expressed with myelin expressing genes. This relationship was associated with AD, but not normal aging, and suggests that PSEN1 may affect oligodendrocyte function. The analysis also found that YWHAZ, which encodes a protein involved in signaling and regulation of cell cycle,198 was a central node in both AD and normal aging. YWHAZ was previously not connected to either AD or normal aging.
Using a PCA of microarray data from the frontal cortex of late-onset AD patients and age-matched controls, a more recent study by Zhang et al.199 identified a PC composed of genes associated with microglial activation that was highly correlated with multiple measures of neuropathology. Starting with this subset of genes, the authors derived a microglial Bayesian network and generated cis-eQTLs (expression quantitative trait loci) as prior knowledge to identify causal driving nodes within the proposed network. This network model identified microglial activation submodules associated with the complement cascade, toll-like receptor signaling, cytokine signaling, histocompatibility complex, and the Fc-receptor system. This network analysis identified TYROBP as a central gene regulated in AD. The authors then validated their model by overexpressing TYROBP in a mouse microglial cell line and demonstrated that the microglial gene expression (measured via RNA-seq) network shifted to be more consistent with the network found in human AD samples.
A temporal study of HD by Xu et al. used microarray analysis of a tetracycline-regulated HD94 Huntington's mouse model (Table 2) at multiple points after transgene induction together with a temporal regression approach to find genes that were differentially regulated post-induction.200 The authors separately regressed gene expression values for time points from the HD94 model and transactivator controls against a temporal regression model to identify genes that had the most significantly different temporal profiles. Interestingly, some genes showed strong differential expression during the first two-weeks post-induction, but not later; others showed differential expression 6 weeks-post induction, but not earlier. These findings provide evidence for the temporal complexity of NDs and highlight the need to gather temporal understanding of molecular pathology as this understanding may alter future therapeutic approaches.
There is clear value to combining multiple omics modalities (e.g., GWA, gene expression, protein expression, etc.) to identify relationships that are not identifiable based on a single modality. For example, one article by Chen et al.201 combined genetic defect information from the Online Mendelian Inheritance in Man database (OMIM)202 database with genetic defect information from the Online Predicated Human Interaction Database203 to identify proteins, including β-catenin, that were connected with multiple AD-associated genes. Another report, by Krauthammer et al., mined published articles to build a molecular interaction network based on proteins, genes, mRNA, and other molecules.204 The authors validated the approach by using the network to identify proteins associated with four known AD genes, APOE, APP, PSEN1, PSEN2, and predicting other AD-associated genes including MAPT. Finally, Caberlotto et al.205 formulated a network analysis by combining gene expression, SNP, molecular drug targets, and protein-protein interaction data to identify genes that are strongly associated with multiple known AD genes. The authors identified AMP-activated protein kinase as being strongly connected to multiple known AD risk factor genes.
Due to the large number of interactions examined using genome-wide databases, literature surveys, and analytical techniques, there is potential for false-positive associations to be drawn from the network analyses. There are multiple ways to address this limitation. As we will discuss later, experimental perturbation in model disease systems is essential for verifying whether computation-predicted associations hold in the context of the ND under study. However, to determine whether protein interactions can physically occur, it is possible to incorporate direct protein-protein interaction data from yeast 2-hybrid (Y2H)206 screens. For example, Liu et al.,207 combined gene co-expression, co-citation data mining, Y2H data and other data sources to build a Bayesian network. The authors used the network to predict genes associated with APOE, APP, PSEN1 and PSEN2, identifying multiple previously known AD-associated genes, and some previously unknown genes. Similarly, Soler-López et al.208 built a protein-protein interaction network based on multiple protein interaction databases, including Human Protein Reference Database (HPRD), and a gene co-expression database to identify proteins connected to multiple causative AD genes. The authors then used a Y2H protein interaction screen to test network predictions, eliminating some false positives, and identified programmed cell death 4 (PDCD4) as being connected with PSEN2 and APOE.
5.3 Integration of Multi-ND Data
As discussed previously, the pathophysiologic overlap between the various NDs is substantial. In addition to protein aggregation, each ND shares similarities that include microglial and astrocyte activation, cytokine elevation, increased ROS, and broad metabolic changes. The BBB/BSCB loses integrity, and peripheral immune cells may migrate into the affected CNS tissues. Given the broad similarities among NDs, despite different sources and mechanisms of pathological protein aggregation, it is conceivable that different APs trigger a similar mechanism driving neuronal vulnerability. These common mechanisms could exist between diseases, despite specific genes and pathways being associated with each disease, because of different regulatory networks in specific brain regions (i.e., substantia nigra vs entorhinal cortex). If a common mechanism is shared between some or all of these diseases, it could be associated with loss of homeostatic regulation, increase of neurotoxic regulation, or even a signaling event that leads to apoptotic or autophagic neuronal death.
While multi-ND analyses have been few, several groups have analyzed data to investigate pathway activation and identify novel genes and proteins connected with multiple NDs. For example, a report by Ramanan & Saykin209 reviewed AD and PD GWA study data, identifying that specificity protein 1 (encoded by SP1) and Jun/Fos transcription factors were commonly modified in both diseases. A report relying on protein interaction data by Limviphuvadh, et al.210 used a literature survey to create protein-protein interaction networks for AD, PD, ALS, HD, detatorubral-pallidoluysian atrophy, and prion disease. The authors identified common proteins involved in apoptosis and mitogen-activated protein kinase (MAPK) signaling. Combining genetic and protein data, a report by Nguyen et al.211 integrated genetic defect information from the OMIM database202 with an interaction network built from the Interologous Interaction Database (i2d).203 The authors used these data together with GO annotations to compare biological pathways connected to gene mutations in nine NDs, including all of the NDs described in this review, identifying the Toll-like receptor pathway as being commonly connected to multiple NDs.
Finally, a recent report by Caberlotto et al. has suggested a role for autophagy in neurodegenerative disease by combining data from a broad collection of NDs.212 The authors created a protein interaction network based on known genetic defects in multiple NDs, and the known and predicted targets of existing drugs from every stage of the drug discovery pipeline. They built a network by combining genetic defect information from OMIM202 with protein interaction information from the i2d. The network combined data for AD, FTD, HD, Lewy body dementia, progressive supranuclear palsy, corticobasal dementia, Pick's disease, Prion disease, and ALS with Parkinsonism to identify a collection of proteins that may be affected in many of these diseases. The result was identification of autophagy-associated proteins, including Akt, BCL2, ATG5 that were mutated in AD, FTD, and ALS with Parkinsonism. This finding supports a growing interest in the role of autophagy in NDs.213 Therapeutic modulation of autophagy has been proposed for a number of NDs and is currently under evaluation in a number of labs and clinical trials.214 While most research has focused on the ability of autophagy to enhance clearance of aggregate-prone proteins, recent findings linking autophagy to healthy immune regulation could enhance the therapeutic value of autophagy modulation for chronic degenerative disorders.215
The above studies demonstrate the value of systems analysis for broadly characterizing similarities between multiple NDs and suggest that multiple mechanisms may be shared among NDs. The development of tools to easily analyze data from multiple NDs will make the exploration and analysis of multi-ND data more accessible to the community. One such tool is the NeuroDNet216 platform which provides a toolset for analyzing proteins that interact with 300 genes connected to 12 NDs based on data from multiple protein-protein interaction databases.
6 Model Systems and Perturbations are Necessary to Identify Causal Relationships
A critical consideration for each disease is deciding how to deconvolve the multitude of simultaneous changes to identify the driving neurodegenerative mechanisms. Systems analysis of primary human tissues yields many novel insights, as discussed above. However, analysis of terminal tissues alone provides only correlations, or at best suggests causative relationships, between measurement variables or between measurement variables and disease phenotypes. Even looking early in disease onset in a mouse model, a multivariate analysis may identify a large number of correlates with disease, making it difficult to isolate potentially causative pathways and mechanisms.
The power of systems analysis for identifying driving mechanisms is unlocked when we integrate multiple physiologic perturbations171,217 – a methodology which is not feasible to apply in humans. Therefore, it is essential to rely on an integrated systems analysis of data from human tissues, mouse models, and culture models to isolate causative factors. The work by Wexler et al.192 discussed above, employed perturbations in culture, together with a systems analysis, to identify pathway activation downstream of Wnt signaling in FTD. Culture based systems analysis is valuable for isolating many aspects of disease. However, culture models lack many aspects of ND pathology, and will be insufficient to elucidate the complete disease when used alone.
Perturbations have rarely been applied to inform systems analysis of in vivo models. One study by Lau et al. employed pharmacologic perturbations in a mouse model to inform a systems analysis that identified the role of immune cells in TNF-α induced apoptosis in the intestinal epithelium.218 This work illustrates the efficacy of combining in vivo perturbation with systems analysis to identify physiologic mechanisms. A similar application to mouse models of NDs will result in computational models that more accurately reflect the underlying biology driving disease in these models. Mouse models possessing human genes that cause ND are available for each of the diseases discussed here (Table 2). Models possessing inducible transgenes provide particular functionality for observing kinetics of gene/protein expression during disease onset in adult animals without sampling changes related to development. These models may therefore prove more useful for understanding sporadic forms of disease.
As discussed previously, mouse models do have limitations. They are characterized in terms of specific mirrored disease pathology, as summarized in Table 2, but fail to capture the entire human disease. Systems analysis of highly multi-dimensional omics techniques provides the capability to further characterize these models, not just in terms of pathological indicators, but in terms of broader network changes. Comparison of these network changes with analysis of human tissues will enable us to isolate portions of the network (e.g., signaling, metabolism, etc.), that shift similarly in the mouse and human disease. Finally, mouse models are well complemented by human-derived culture models, and can be used together to elucidate complex human disease.
7 Conclusion
Application of systems analysis methodology to NDs is a fledgling field and is, to date, represented by a limited number of studies. Given the simultaneous onset and presentation of multiple aspects of ND pathophysiology, the structured computational methodologies available from systems biology will surely be an essential asset in deciphering the mechanisms driving these diseases. Integrated analysis of human tissues together with mouse and culture models holds the prospect of, first, identifying model limitations in reflecting the human disease at the systems level. Second, systematic integration of perturbation and time point analysis from models provides the ability to identify causative pathways and mechanisms that drive disease at the systems level. Finally, the striking similarities in pathophysiologic presentation among NDs, despite differences in the specific AP, aggregate localization, and regional degeneration, suggest that comparative analysis among diseases may identify common changes responsible for neuronal death. Ultimately, new understandings derived through these experimental and analytical approaches may in turn lead to novel therapeutic strategies for these devastating diseases.
Insight, Innovation, and Integration.
Effective therapeutic strategies remain absent for a broad collection of neurodegenerative diseases (NDs) associated with pathological protein aggregation. In this review, we first provide insight into the pathophysiology of these NDs viewed as complex, dysregulated systems. We emphasize that many pathophysiological features are shared between NDs, despite differences in the location and identity of protein aggregates. We then review tools available from the field of systems biology for isolating mechanisms driving ND onset and progression. Finally, we comment on current literature applying systems biology methodologies to elucidate NDs, and remark on importance of integrating data and analysis from human tissues and model systems to identify new therapeutic targets.
Acknowledgments
This review was supported in part by the National Institutes of Health under grant number 5R01AG040530. S.D.S was supported by the National Science Foundation Graduate Research Fellowship under grant number DGE-1122374 and the Barbara J. Weedon Fellowship.
Footnotes
Author Information: L.B.W., A.R.W., and S.D.S. researched the literature and wrote the manuscript. The authors declare no competing financial interests. A.R.W. is an employee and shareholder of Pfizer, Inc.
References
- 1.Prince M, Prina M, Guerchet M. World Alzheimer Report 2013 Journey of Caring: An Analysis of Long-Term Care for Dementia. 2013 [Google Scholar]
- 2.Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, Marshall FJ, Ravina BM, Schifitto G, Siderowf A, Tanner CM. Neurology. 2007;68:384–386. doi: 10.1212/01.wnl.0000247740.47667.03. [DOI] [PubMed] [Google Scholar]
- 3.Abbott A. Nature. 2011;475:S2–S4. doi: 10.1038/475S2a. [DOI] [PubMed] [Google Scholar]
- 4.Fargo K, Bleiler L. Alzheimers Dement. 2014;10:e47–92. [Google Scholar]
- 5.Niccoli T, Partridge L. Curr Biol. 2012;22:741–752. doi: 10.1016/j.cub.2012.07.024. [DOI] [PubMed] [Google Scholar]
- 6.Reeve A, Simcox E, Turnbull D. Ageing Res Rev. 2014;14:19–30. doi: 10.1016/j.arr.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tanzi RE, Bertram L. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 8.Igaz LM, Kwong LK, Lee EB, Chen-Plotkin A, Swanson E, Unger T, Malunda J, Xu Y, Winton MJ, Trojanowski JQ, Lee VMY. J Clin Invest. 2011;121:726–738. doi: 10.1172/JCI44867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Periquet M, Fulga T, Myllykangas L, Schlossmacher MG, Feany MB. J Neurosci. 2007;27:3338–3346. doi: 10.1523/JNEUROSCI.0285-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Landles C, Bates GP. EMBO Rep. 2004;5:958–963. doi: 10.1038/sj.embor.7400250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spires-Jones TL, Stoothoff WH, de Calignon A, Jones PB, Hyman BT. Trends Neurosci. 2009;32:150–9. doi: 10.1016/j.tins.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 12.Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG, Aisen PS, Siemers E, Sethuraman G, Mohs R. N Engl J Med. 2013;369:341–50. doi: 10.1056/NEJMoa1210951. [DOI] [PubMed] [Google Scholar]
- 13.Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Ferris S, Reichert M, Ketter N, Nejadnik B, Guenzler V, Miloslavsky M, Wang D, Lu Y, Lull J, Tudor IC, Liu E, Grundman M, Yuen E, Black R, Brashear HR. N Engl J Med. 2014;370:322–33. doi: 10.1056/NEJMoa1304839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, Raman R, Sun X, Aisen PS, Siemers E, Liu-Seifert H, Mohs R. N Engl J Med. 2014;370:311–21. doi: 10.1056/NEJMoa1312889. [DOI] [PubMed] [Google Scholar]
- 15.Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Trojanowski JQ, Vinters HV, Hyman BT. Acta Neuropathol. 2012;123:1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cairns NJ, Bigio EH, Mackenzie IRA, Neumann M, Lee VMY, Hatanpaa KJ, White CL, Schneider JA, Grinberg LT, Halliday G, Duyckaerts C, Lowe JS, Holm IE, Tolnay M, Okamoto K, Yokoo H, Murayama S, Woulfe J, Munoz DG, Dickson DW, Ince PG, Trojanowski JQ, Mann DMA. Acta Neuropathol. 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, Halliday GM, Hardy J, Leverenz JB, Del Tredici K, Wszolek ZK, Litvan I. Lancet Neurol. 2009;8:1150–1157. doi: 10.1016/S1474-4422(09)70238-8. [DOI] [PubMed] [Google Scholar]
- 18.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 19.Desai BS, Monahan AJ, Carvey PM, Hendey B. Cell Transplantation. 2007;16:285–299. doi: 10.3727/000000007783464731. [DOI] [PubMed] [Google Scholar]
- 20.Garbuzova-Davis S, Rodrigues MCO, Hernandez-Ontiveros DG, Louis MK, Willing AE, Borlongan CV, Sanberg PR. Brain Res. 2011;1398:113–125. doi: 10.1016/j.brainres.2011.04.049. [DOI] [PubMed] [Google Scholar]
- 21.Rezai-Zadeh K, Gate D, Town T. J Neuroimmune Pharmacol. 2009;4:462–475. doi: 10.1007/s11481-009-9166-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zlokovic BV. Nat Rev Neurosci. 2011;12:723–739. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schulz J. Neurodegenerative Disease: Neurobiology, Pathogenesis and Therapeutics. 2005:80–93. [Google Scholar]
- 24.Lee H, Zhu X, Nunomura A, Perry G, Smith MA. Curr Alzheimer Res. 2006;3:75–80. doi: 10.2174/156720506775697124. [DOI] [PubMed] [Google Scholar]
- 25.Dewey CM, Cenik B, Sephton CF, Johnson BA, Herz J, Yu G. Brain Res. 2012;1462:16–25. doi: 10.1016/j.brainres.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quilty MC, King AE, Gai WP, Pountney DL, West AK, Vickers JC, Dickson TC. Exp Neurol. 2006;199:249–256. doi: 10.1016/j.expneurol.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 27.Price JL, Morris JC. Ann Neurol. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 28.Perez-Nievas BG, Stein TD, Tai HC, Dols-Icardo O, Scotton TC, Barroeta-Espar I, Fernandez-Carballo L, De Munain EL, Perez J, Marquie M, Serrano-Pozo A, Frosch MP, Lowe V, Parisi JE, Petersen RC, Ikonomovic MD, López OL, Klunk W, Hyman BT, Gómez-Isla T. Brain. 2013;136:2510–2526. doi: 10.1093/brain/awt171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bateman RJ, Xiong C, Benzinger TLS, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jack CR, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, Vemuri P, Wiste HJ, Weigand SD, Lesnick TG, Pankratz VS, Donohue MC, Trojanowski JQ. Lancet Neurol. 2013;12:207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sperling Ra, Aisen PS, Beckett La, Bennett Da, Craft S, Fagan AM, Iwatsubo T, Jack CR, Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH. Alzheimer's Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bertram L, Tanzi RE. J Clin Invest. 2005;115:1449–1457. doi: 10.1172/JCI24761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li YJ, Scott WK, Hedges DJ, Zhang F, Gaskell PC, Nance Ma, Watts RL, Hubble JP, Koller WC, Pahwa R, Stern MB, Hiner BC, Jankovic J, Allen Fa, Goetz CG, Mastaglia F, Stajich JM, Gibson Ra, Middleton LT, Saunders AM, Scott BL, Small GW, Nicodemus KK, Reed AD, Schmechel DE, Welsh-Bohmer Ka, Conneally PM, Roses AD, Gilbert JR, Vance JM, Haines JL, Pericak-Vance Ma. Am J Hum Genet. 2002;70:985–993. doi: 10.1086/339815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamboh MI, Barmada MM, Demirci FY, Minster RL, Carrasquillo MM, Pankratz VS, Younkin SG, Saykin aJ, Sweet Ra, Feingold E, DeKosky ST, Lopez OL. Mol Psychiatry. 2012;17:1340–6. doi: 10.1038/mp.2011.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore Pa, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith aD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schürmann B, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frölich L, Hampel H, Hüll M, Rujescu D, Goate AM, Kauwe JSK, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Mühleisen TW, Nöthen MM, Moebus S, Jöckel KH, Klopp N, Wichmann HE, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans Pa, O'Donovan M, Owen MJ, Williams J. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen L, Bagyinszky E, Youn YC, An SSA, Kim S. Toxicol Environ Health Sci. 2013;5:113–130. [Google Scholar]
- 37.Neary D, Snowden J, Mann D. Lancet Neurol. 2005;4:771–780. doi: 10.1016/S1474-4422(05)70223-4. [DOI] [PubMed] [Google Scholar]
- 38.Rosso SM, Donker Kaat L, Baks T, Joosse M, de Koning I, Pijnenburg Y, de Jong D, Dooijes D, Kamphorst W, Ravid R, Niermeijer MF, Verheij F, Kremer HP, Scheltens P, van Duijn CM, Heutink P, van Swieten JC. Brain. 2003;126:2016–22. doi: 10.1093/brain/awg204. [DOI] [PubMed] [Google Scholar]
- 39.Hodges JR, Davies R, Xuereb J, Kril J, Halliday G. Neurology. 2003;61:349–354. doi: 10.1212/01.wnl.0000078928.20107.52. [DOI] [PubMed] [Google Scholar]
- 40.Chow TW, Miller BL, Hayashi VN, Geschwind DH. Arch Neurol. 1999;56:817–822. doi: 10.1001/archneur.56.7.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Katz JS, Katzberg HD, Woolley SC, Marklund SL, Andersen PM. Amyotroph Lateral Scler. 2012;13:567–569. doi: 10.3109/17482968.2012.678365. [DOI] [PubMed] [Google Scholar]
- 42.Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, Kinoshita Y, Kamada M, Nodera H, Suzuki H, Komure O, Matsuura S, Kobatake K, Morimoto N, Abe K, Suzuki N, Aoki M, Kawata A, Hirai T, Kato T, Ogasawara K, Hirano A, Takumi T, Kusaka H, Hagiwara K, Kaji R, Kawakami H. Nature. 2010;465:223–226. doi: 10.1038/nature08971. [DOI] [PubMed] [Google Scholar]
- 43.Chen S, Sayana P, Zhang X, Le W. Mol Neurodegener. 2013;8:28. doi: 10.1186/1750-1326-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC. Lancet. 2011;377:942–955. doi: 10.1016/S0140-6736(10)61156-7. [DOI] [PubMed] [Google Scholar]
- 45.Howard R, Orrell R. Postgrad Med J. 2002;78:736–741. doi: 10.1136/pmj.78.926.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thomas B, Flint Beal M. Hum Mol Genet. 2007;16:183–194. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]
- 47.Macdonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, Barnes G, Taylor SA, James M, Groot N, Macfarlane H, Jenkins B, Anderson MA, Wexler NS, Gusella JF, Bates GP, Baxendale S, Hummerich H, Kirby S, North M, Youngman S, Mott R, Zehetner G, Sedlacek Z, Poustka A, Frischauf AM, Lehrach H, Buckler AJ, Church D, Doucettestamm L, Odonovan MC, Ribaramirez L, Shah M, Stanton VP, Strobel SA, Draths KM, Wales JL, Dervan P, Housman DE, Altherr M, Shiang R, Thompson L, Fielder T, Wasmuth JJ, Tagle D, Valdes J, Elmer L, Allard M, Castilla L, Swaroop M, Blanchard K, Collins FS, Snell R, Holloway T, Gillespie K, Datson N, Shaw D, Harper PS. Cell. 1993;72:971–983. [Google Scholar]
- 48.Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR. Clin Genet. 2004;65:267–277. doi: 10.1111/j.1399-0004.2004.00241.x. [DOI] [PubMed] [Google Scholar]
- 49.Holtzman DM, Morris JC, Goate AM. Sci Transl Med. 2011;3:77sr1. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Proc Natl Acad Sci U S A. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thal DR, Rüb U, Orantes M, Braak H. Neurology. 2002;58:1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 52.Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dickson DW. J Neuropathol Exp Neurol. 1997;56:321–339. doi: 10.1097/00005072-199704000-00001. [DOI] [PubMed] [Google Scholar]
- 54.Dorf ME, Berman MA, Tanabe S, Heesen M, Luo Y. J Neuroimmunol. 2000;111:109–121. doi: 10.1016/s0165-5728(00)00371-4. [DOI] [PubMed] [Google Scholar]
- 55.Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, Irizarry MC. Neurology. 2004;62:925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 56.Johnson GVW, Stoothoff WH. J Cell Sci. 2004;117:5721–5729. doi: 10.1242/jcs.01558. [DOI] [PubMed] [Google Scholar]
- 57.Bayer TA. Eur Neuropsychopharmacol. 2015;25:713–724. doi: 10.1016/j.euroneuro.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 58.Murrell J, Farlow M, Ghetti B, Benson MD. Science. 1991;254:97–99. doi: 10.1126/science.1925564. 80. [DOI] [PubMed] [Google Scholar]
- 59.Zekanowski C, Styczyńska M, Pepłońska B, Gabryelewicz T, Religa D, Ilkowski J, Kijanowska-Haładyna B, Kotapka-Minc S, Mikkelsen S, Pfeffer A, Barczak A, Łuczywek E, Wasiak B, Chodakowska-Zebrowska M, Gustaw K, Łaczkowski J, Sobów T, Kuźnicki J, Barcikowska M. Exp Neurol. 2003;184:991–6. doi: 10.1016/S0014-4886(03)00384-4. [DOI] [PubMed] [Google Scholar]
- 60.Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, Hoyte K, Gustafson A, Liu Y, Lu Y, Bhangale T, Graham RR, Huttenlocher J, Bjornsdottir G, Andreassen OA, Jönsson EG, Palotie A, Behrens TW, Magnusson OT, Kong A, Thorsteinsdottir U, Watts RJ, Stefansson K. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 61.Block ML, Zecca L, Hong JS. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 62.Brown GC, Bal-Price A. Mol Neurobiol. 2003;27:325–355. doi: 10.1385/MN:27:3:325. [DOI] [PubMed] [Google Scholar]
- 63.Ringheim GE, Szczepanik AM, Petko W, Burgher KL, Zhu SZ, Chao CC. Mol Brain Res. 1998;55:35–44. doi: 10.1016/s0169-328x(97)00356-2. [DOI] [PubMed] [Google Scholar]
- 64.Grammas P, Sanchez A, Tripathy D, Luo E, Martinez J. Cleve Clin J Med. 2011;78:S50–S53. doi: 10.3949/ccjm.78.s1.09. [DOI] [PubMed] [Google Scholar]
- 65.De La Torre JC. Lancet Neurol. 2004;3:184–190. doi: 10.1016/S1474-4422(04)00683-0. [DOI] [PubMed] [Google Scholar]
- 66.Biron KE, Dickstein DL, Gopaul R, Jefferies WA. PLoS One. 2011;6 doi: 10.1371/journal.pone.0023789. Article no: e23789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Balin BJ, Appelt DM. J Am Osteopat Assoc. 2001;101:S1–6. [PubMed] [Google Scholar]
- 68.Gate D, Rezai-Zadeh K, Jodry D, Rentsendorj A, Town T. J Neural Transm. 2010;117:961–970. doi: 10.1007/s00702-010-0422-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ. Arch Neurol. 2001;58:1803. doi: 10.1001/archneur.58.11.1803. [DOI] [PubMed] [Google Scholar]
- 70.Foster NL, Heidebrink JL, Clark CM, Jagust WJ, Arnold SE, Barbas NR, DeCarli CS, Turner RS, Koeppe RA, Higdon R, Minoshima S. Brain. 2007;130:2616–2635. doi: 10.1093/brain/awm177. [DOI] [PubMed] [Google Scholar]
- 71.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, Mccluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 72.Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar Ha, Mackenzie IRa. Brain. 2009;132:2922–31. doi: 10.1093/brain/awp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alonso ADC, Mederlyova A, Novak M, Grundke-Iqbal I, Iqbal K. J Biol Chem. 2004;279:34873–34881. doi: 10.1074/jbc.M405131200. [DOI] [PubMed] [Google Scholar]
- 74.Deng HX, Zhai H, Bigio EH, Yan J, Fecto F, Ajroud K, Mishra M, Ajroud-Driss S, Heller S, Sufit R, Siddique N, Mugnaini E, Siddique T. Ann Neurol. 2010;67:739–748. doi: 10.1002/ana.22051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cheroni C, Peviani M, Cascio P, DeBiasi S, Monti C, Bendotti C. Neurobiol Dis. 2005;18:509–522. doi: 10.1016/j.nbd.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 76.Lee VMY, Goedert M, Trojanowski JQ. Annu Rev Neurosci. 2001;24:1121–159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 77.Cagnin A, Rossor M, Sampson EL, MacKinnon T, Banati RB. Ann Neurol. 2004;56:894–897. doi: 10.1002/ana.20332. [DOI] [PubMed] [Google Scholar]
- 78.Kersaitis C, Halliday GM, Kril JJ. Acta Neuropathol. 2004;108:515–523. doi: 10.1007/s00401-004-0917-0. [DOI] [PubMed] [Google Scholar]
- 79.Castellani R, Smith MA, Richey PL, Kalaria R, Gambetti P, Perry G. Brain Res. 1995;696:268–271. doi: 10.1016/0006-8993(95)00535-x. [DOI] [PubMed] [Google Scholar]
- 80.Sjögren M, Folkesson S, Blennow K, Tarkowski E. J Neurol Neurosurg Psychiatry. 2004;75:1107–1111. doi: 10.1136/jnnp.2003.019422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Broe M, Kril J, Halliday GM. Brain. 2004;127:2214–2220. doi: 10.1093/brain/awh250. [DOI] [PubMed] [Google Scholar]
- 82.Ghatak NR, Campbell WW, Lippman RH, Hadfield MG. Anterior horn changes of motor neuron disease associated with demyelinating radiculopathy. 1986;45 doi: 10.1097/00005072-198607000-00001. [DOI] [PubMed] [Google Scholar]
- 83.Hughes JT. Adv Neurol. 1982;36:61–74. [PubMed] [Google Scholar]
- 84.Kawamata T, Akiyama H, Yamada T, McGeer PL. Am J Pathol. 1992;140:691–707. [PMC free article] [PubMed] [Google Scholar]
- 85.Ekblom J, Jossan SS, Oreland L, Walum E, Aquilonius SM. J Neural Transm Suppl. 1994;41:253–258. doi: 10.1007/978-3-7091-9324-2_33. [DOI] [PubMed] [Google Scholar]
- 86.Murayama S, Inoue K, Kawakami H, Bouldin TW, Suzuki K. Acta Neuropathol. 1991;82:456–461. doi: 10.1007/BF00293379. [DOI] [PubMed] [Google Scholar]
- 87.Neumann M, Sampathu D, Kwong L. Science. 2006;314:130–133. doi: 10.1126/science.1134108. 80. [DOI] [PubMed] [Google Scholar]
- 88.Lomen-Hoerth C, Anderson T, Miller B. Neurology. 2002;59:1077–1079. doi: 10.1212/wnl.59.7.1077. [DOI] [PubMed] [Google Scholar]
- 89.Achi EY, Rudnicki Sa. Neurol Res Int. 2012;2012 doi: 10.1155/2012/806306. Article no: 806306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pasinelli P, Brown RH. Nat Rev Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- 91.Ilieva H, Polymenidou M, Cleveland DW. J Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Henkel JS, Engelhardt JI, Siklós L, Simpson EP, Kim SH, Pan T, Goodman JC, Siddique T, Beers DR, Appel SH. Ann Neurol. 2004;55:221–235. doi: 10.1002/ana.10805. [DOI] [PubMed] [Google Scholar]
- 93.Turner MR, Cagnin A, Turkheimer FE, Miller CCJ, Shaw CE, Brooks DJ, Leigh PN, Banati RB. Neurobiol Dis. 2004;15:601–609. doi: 10.1016/j.nbd.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 94.Barbeito LH, Pehar M, Cassina P, Vargas MR, Peluffo H, Viera L, Estévez AG, Beckman JS. Brain Res Brain Res Rev. 2004;47:263–274. doi: 10.1016/j.brainresrev.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 95.Rentzos M, Nikolaou C, Rombos A, Boufidou F, Zoga M, Dimitrakopoulos A, Tsoutsou A, Vassilopoulos D. Amyotroph Lateral Scler. 2007;8:283–287. doi: 10.1080/17482960701419232. [DOI] [PubMed] [Google Scholar]
- 96.Elliott JL. Brain Res Mol Brain Res. 2001;95:172–178. doi: 10.1016/s0169-328x(01)00242-x. [DOI] [PubMed] [Google Scholar]
- 97.McGeer PL, McGeer EG. Muscle Nerve. 2002;26:459–70. doi: 10.1002/mus.10191. [DOI] [PubMed] [Google Scholar]
- 98.Barber SC, Mead RJ, Shaw PJ. Biochim Biophys Acta - Mol Basis Dis. 2006;1762:1051–1067. doi: 10.1016/j.bbadis.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 99.Dodge JC, Treleaven CM, Fidler Ja, Tamsett TJ, Bao C, Searles M, Taksir TV, Misra K, Sidman RL, Cheng SH, Shihabuddin LS. Proc Natl Acad Sci U S A. 2013;110:10812–7. doi: 10.1073/pnas.1308421110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Winkler EA, Sengillo JD, Sullivan JS, Henkel JS, Appel SH, Zlokovic BV. Acta Neuropathol. 2013;125:111–120. doi: 10.1007/s00401-012-1039-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li YR, King OD, Shorter J, Gitler AD. J Cell Biol. 2013;201:361–372. doi: 10.1083/jcb.201302044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Purves D, Augustine GJ, Fitzpatrick D, Hall WC, LaMantia AS, McNamara JO, White LE. Neuroscience. 4th. Sinaur Associates, Inc.; Sunderland, MA: 2008. [Google Scholar]
- 103.Emre M, Aarsland D, Brown R, Burn DJ, Duyckaerts C, Mizuno Y, Broe GA, Cummings J, Dickson DW, Gauthier S, Goldman J, Goetz C, Korczyn A, Lees A, Levy R, Litvan I, McKeith I, Olanow W, Poewe W, Quinn N, Sampaio C, Tolosa E, Dubois B. Mov Disord. 2007;22:1689–1707. doi: 10.1002/mds.21507. [DOI] [PubMed] [Google Scholar]
- 104.Ballard C, Ziabreva I, Perry R, Larsen JP, O'Brien J, McKeith I, Perry E, Aarsland D. Neurology. 2006;67:1931–1934. doi: 10.1212/01.wnl.0000249130.63615.cc. [DOI] [PubMed] [Google Scholar]
- 105.Apaydin H, Ahlskog JE, Parisi JE, Boeve BF, Dickson DW. Arch Neurol. 2002;59:102–112. doi: 10.1001/archneur.59.1.102. [DOI] [PubMed] [Google Scholar]
- 106.Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destée A. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- 107.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 108.Waxman EA, Giasson BI. Biochim Biophys Acta - Mol Basis Dis. 2009;1792:616–624. doi: 10.1016/j.bbadis.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL, Reports E, Lavedant C, Leroyt E, Papapetropoulos T, Di G, Golbe L. Science. 1997;276:2045–2048. doi: 10.1126/science.276.5321.2045. 80. [DOI] [PubMed] [Google Scholar]
- 110.Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K. J Neurosci. 2010;30:6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L, Vanderburg CR, McLean PJ. Mol Neurodegener. 2012;7:42. doi: 10.1186/1750-1326-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Papp MI, Kahn JE, Lantos PL. J Neurol Sci. 1989;94:79–100. doi: 10.1016/0022-510x(89)90219-0. [DOI] [PubMed] [Google Scholar]
- 113.Hishikawa N, Hashizume Y, Yoshida M, Sobue G. Neuropathol Appl Neurobiol. 2001;27:362–372. doi: 10.1046/j.1365-2990.2001.00345.x. [DOI] [PubMed] [Google Scholar]
- 114.Braak H, Sastre M, Del Tredici K. Acta Neuropathol. 2007;114:231–241. doi: 10.1007/s00401-007-0244-3. [DOI] [PubMed] [Google Scholar]
- 115.Cabezas R, Avila M, Gonzalez J, El-Bachá RS, Báez E, García-Segura LM, Jurado Coronel JC, Capani F, Capani F, Cardona-Gomez GP, Barreto GE. Front Cell Neurosci. 2014;8:211. doi: 10.3389/fncel.2014.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Walker FO. Lancet. 2007;369:218–28. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- 117.Rubinsztein DC, Leggo J, Coles R, Almqvist E, Biancalana V, Cassiman JJ, Chotai K, Connarty M, Crauford D, Curtis A, Curtis D, Davidson MJ, Differ AM, Dode C, Dodge A, Frontali M, Ranen NG, Stine OC, Sherr M, Abbott MH, Franz ML, Graham CA, Harper PS, Hedreen JC, Hayden MR. Am J Hum Genet. 1996;59:16–22. [PMC free article] [PubMed] [Google Scholar]
- 118.Brinkman RR, Mezei MM, Theilmann J, Almqvist E, Hayden MR. Am J Hum Genet. 1997;60:1202–1210. [PMC free article] [PubMed] [Google Scholar]
- 119.Singhrao SK, Thomas P, Wood JD, MacMillan JC, Neal JW, Harper PS, Jones AL. Exp Neurol. 1998;150:213–222. doi: 10.1006/exnr.1998.6778. [DOI] [PubMed] [Google Scholar]
- 120.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 121.Silvestroni A, Faull RLM, Strand AD, Möller T. Neuroreport. 2009;20:1098–1103. doi: 10.1097/WNR.0b013e32832e34ee. [DOI] [PubMed] [Google Scholar]
- 122.Pavese N, Gerhard A, Tai YF, Ho AK, Turkheimer F, Barker RA, Brooks DJ, Piccini P. Neurology. 2006;66:1638–1643. doi: 10.1212/01.wnl.0000222734.56412.17. [DOI] [PubMed] [Google Scholar]
- 123.Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- 124.Lin CY, Hsu YH, Lin MH, Yang TH, Chen HM, Chen YC, Hsiao HY, Chen CC, Chern Y, Chang C. Exp Neurol. 2013;250:20–30. doi: 10.1016/j.expneurol.2013.08.019. [DOI] [PubMed] [Google Scholar]
- 125.Sampaio C, Borowsky B, Reilmann R. Mov Disord. 2014;29:1419–1428. doi: 10.1002/mds.26021. [DOI] [PubMed] [Google Scholar]
- 126.Sieradzan KA, Mann DM. Neuropathol Appl Neurobiol. 2001;27:1–21. doi: 10.1046/j.0305-1846.2001.00299.x. [DOI] [PubMed] [Google Scholar]
- 127.Rosas HD, Reuter M, Doros G, Lee SY, Triggs T, Malarick K, Fischl B, Salat DH, Hersch SM. Mov Disord. 2011;26:1691–1697. doi: 10.1002/mds.23762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Espina V, Mueller C, Edmiston K, Sciro M, Petricoin EF, Liotta LA. Proteomics - Clin Appl. 2009;3:874–882. doi: 10.1002/prca.200800001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH. Proc Natl Acad Sci U S A. 2009;106:18809–18814. doi: 10.1073/pnas.0908767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hatzipetros T, Bogdanik LP, Tassinari VR, Kidd JD, Moreno AJ, Davis C, Osborne M, Austin A, Vieira FG, Lutz C, Perrin S. Brain Res. 2014;1584:59–72. doi: 10.1016/j.brainres.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 131.Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, Spires-Jones T, Xie H, Arbel-Ornath M, Grosskreutz CL, Bacskai BJ, Hyman BT. J Neurosci. 2010;30:2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Johnstone M, Gearing AJ, Miller KM. J Neuroimmunol. 1999;93:182–193. doi: 10.1016/s0165-5728(98)00226-4. [DOI] [PubMed] [Google Scholar]
- 133.Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D/'Avanzo C, Chen H, Hooli B, Asselin C, Muffat J, Klee JB, Zhang C, Wainger BJ, Peitz M, Kovacs DM, Woolf CJ, Wagner SL, Tanzi RE, Kim DY. Nature. 2014;515:274–278. doi: 10.1038/nature13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhang D, Pekkanen-Mattila M, Shahsavani M, Falk A, Teixeira AI, Herland A. Biomaterials. 2014;35:1420–1428. doi: 10.1016/j.biomaterials.2013.11.028. [DOI] [PubMed] [Google Scholar]
- 135.Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, Carson CT, Laurent LC, Marsala M, Gage FH, Remes AM, Koo EH, Goldstein LSB. Nature. 2012;482:216–20. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Muratore CR, Rice HC, Srikanth P, Callahan DG, Shin T, Benjamin LNP, Walsh DM, Selkoe DJ, Young-Pearse TL. Hum Mol Genet. 2014;23:3523–3536. doi: 10.1093/hmg/ddu064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bilican B, Serio A, Barmada SJ, Nishimura AL, Sullivan GJ, Carrasco M, Phatnani HP, Puddifoot CA, Story D, Fletcher J, Park IH, Friedman BA, Daley GQ, Wyllie DJA, Hardingham GE, Wilmut I, Finkbeiner S, Maniatis T, Shaw CE, Chandran S. Proc Natl Acad Sci. 2012;109:5803–5808. doi: 10.1073/pnas.1202922109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Serio A, Bilican B, Barmada SJ, Ando DM, Zhao C, Siller R, Burr K, Haghi G, Story D, Nishimura AL, Carrasco Ma, Phatnani HP, Shum C, Wilmut I, Maniatis T, Shaw CE, Finkbeiner S, Chandran S. Proc Natl Acad Sci U S A. 2013;110:4697–702. doi: 10.1073/pnas.1300398110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Byers B, Lee H, Reijo Pera R. Curr Neurol Neurosci Rep. 2012;12:237–242. doi: 10.1007/s11910-012-0270-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhang N, An MC, Montoro D, Ellerby LM. PLoS Curr. 2010:1–11. doi: 10.1371/currents.RRN1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.The HD iPSC Consortium. Cell Stem Cell. 2012;11:264–78. doi: 10.1016/j.stem.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Andorfer C, Kress Y, Espinoza M, De Silva R, Tucker KL, Barde YA, Duff K, Davies P. J Neurochem. 2003;86:582–590. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- 143.Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Biomol Eng. 2001;17:157–165. doi: 10.1016/s1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- 144.Garcia-Alloza M, Robbins EM, Zhang-Nunes SX, Purcell SM, Betensky RA, Raju S, Prada C, Greenberg SM, Bacskai BJ, Frosch MP. Neurobiol Dis. 2006;24:516–524. doi: 10.1016/j.nbd.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 145.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 146.Sasaki A, Shoji M, Harigaya Y, Kawarabayashi T, Ikeda M, Naito M, Matsubara E, Abe K, Nakazato Y. Virchows Arch. 2002;441:358–367. doi: 10.1007/s00428-002-0643-8. [DOI] [PubMed] [Google Scholar]
- 147.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 148.Tran HT, LaFerla FM, Holtzman DM, Brody DL. J Neurosci. 2011;31:9513–9525. doi: 10.1523/JNEUROSCI.0858-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Spires TL, Orne JD, SantaCruz K, Pitstick R, Carlson GA, Ashe KH, Hyman BT. Am J Pathol. 2006;168:1598–1607. doi: 10.2353/ajpath.2006.050840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I, Smits V, Ceuterick-de Groote C, Van Broeckhoven C, Kumar-Singh S. Proc Natl Acad Sci U S A. 2010;107:3858–3863. doi: 10.1073/pnas.0912417107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Mitchell JC, McGoldrick P, Vance C, Hortobagyi T, Sreedharan J, Rogelj B, Tudor EL, Smith BN, Klasen C, Miller CCJ, Cooper JD, Greensmith L, Shaw CE. Acta Neuropathol. 2013;125:273–288. doi: 10.1007/s00401-012-1043-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 154.Dobrowolny G, Giacinti C, Pelosi L, Nicoletti C, Winn N, Barberi L, Molinaro M, Rosenthal N, Musarò A. J Cell Biol. 2005;168:193–199. doi: 10.1083/jcb.200407021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 156.Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E. J Neurosci Res. 2002;68:568–578. doi: 10.1002/jnr.10231. [DOI] [PubMed] [Google Scholar]
- 157.Amschl D, Neddens J, Havas D, Flunkert S, Rabl R, Römer H, Rockenstein E, Masliah E, Windisch M, Hutter-Paier B. BMC Neurosci. 2013;14:6. doi: 10.1186/1471-2202-14-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chesselet MF, Richter F, Zhu C, Magen I, Watson MB, Subramaniam SR. Neurotherapeutics. 2012;9:297–314. doi: 10.1007/s13311-012-0104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jackson-Lewis V, Lester D, Kozina E, Przedborski S, Smeyne RJ. In: Movement Disorders. Second. LeDoux MS, editor. Academic Press; Boston: 2015. pp. 287–306. DOI: http://dx.doi.org/10.1016/B978-0-12-405195-9.00017-2. [Google Scholar]
- 160.Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Hum Mol Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- 161.Yamamoto A, Lucas JJ, Hen R. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- 162.Martín-Aparicio E, Yamamoto A, Hernández F, Hen R, Avila J, Lucas JJ. J Neurosci. 2001;21:8772–8781. doi: 10.1523/JNEUROSCI.21-22-08772.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Aldridge BB, Burke JM, Lauffenburger Da, Sorger PK. Nat Cell Biol. 2006;8:1195–1203. doi: 10.1038/ncb1497. [DOI] [PubMed] [Google Scholar]
- 164.Janes Ka, Yaffe MB. Nat Rev Mol Cell Biol. 2006;7:820–828. doi: 10.1038/nrm2041. [DOI] [PubMed] [Google Scholar]
- 165.Miller MB, Tang YW. Clin Microbiol Rev. 2009;22:611–633. doi: 10.1128/CMR.00019-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Dephoure N, Zhou C, Villén J, Beausoleil Sa, Bakalarski CE, Elledge SJ, Gygi SP. Proc Natl Acad Sci U S A. 2008;105:10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Autissier P, Soulas C, Burdo TH, Williams KC. Cytom Part A. 2010;77:410–419. doi: 10.1002/cyto.a.20859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lyons J, Herring CA, Banerjee A, Simmons AJ, Lau KS. Integr Biol. 2015 doi: 10.1039/C5IB00030K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.D'haeseleer P. Nat Biotechnol. 2005;23:1499–1501. doi: 10.1038/nbt1205-1499. [DOI] [PubMed] [Google Scholar]
- 170.Eriksson L. MKS Umetrics AB. Multi-and megavariate data analysis. 2006 [Google Scholar]
- 171.Janes KA, Kelly JR, Gaudet S, Albeck JG, Sorger PK, Lauffenburger DA. J Comput Biol. 2004;11:544–561. doi: 10.1089/cmb.2004.11.544. [DOI] [PubMed] [Google Scholar]
- 172.Jin L, Zuo X, Su W, Zhao X, Yuan M, Han L, Zhao X, Chen Y, Rao S. Genomics Proteomics Bioinformatics. 2014;12:210–220. doi: 10.1016/j.gpb.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Subramanian A, Subramanian A, Tamayo P, Tamayo P, Mootha VK, Mootha VK, Mukherjee S, Mukherjee S, Ebert BL, Ebert BL, Gillette Ma, Gillette Ma, Paulovich A, Paulovich A, Pomeroy SL, Pomeroy SL, Golub TR, Golub TR, Lander ES, Lander ES, Mesirov JP, Mesirov JP. Proc Natl Acad Sci U S A. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Strang G, Aarikka K. Introduction to applied mathematics. Vol. 16 Wellesley-Cambridge Press; Wellesley, MA: 1986. [Google Scholar]
- 175.Braak H, Braak E. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 176.Geladi P, Kowalski BR. Anal Chim Acta. 1986;185:19–32. [Google Scholar]
- 177.Khatri P, Sirota M, Butte AJ. PLoS Comput Biol. 2012;8 doi: 10.1371/journal.pcbi.1002375. Article no: e1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kanehisa M, Goto S. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, Richter J, Rubin GM, Blake JA, Bult C, Dolan M, Drabkin H, Eppig JT, Hill DP, Ni L, Ringwald M, Balakrishnan R, Cherry JM, Christie KR, Costanzo MC, Dwight SS, Engel S, Fisk DG, Hirschman JE, Hong EL, Nash RS, Sethuraman A, Theesfeld CL, Botstein D, Dolinski K, Feierbach B, Berardini T, Mundodi S, Rhee SY, Apweiler R, Barrell D, Camon E, Dimmer E, Lee V, Chisholm R, Gaudet P, Kibbe W, Kishore R, Schwarz EM, Sternberg P, Gwinn M, Hannick L, Wortman J, Berriman M, Wood V, de la Cruz N, Tonellato P, Jaiswal P, Seigfried T, White R. Nucleic Acids Res. 2004;32:D258–D261. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Holmans P, Green EK, Pahwa JS, Ferreira MAR, Purcell SM, Sklar P, Owen MJ, O'Donovan MC, Craddock N. Am J Hum Genet. 2009;85:13–24. doi: 10.1016/j.ajhg.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Friedman N, Linial M, Nachman I, Pe'er D. J Comput Biol. 2000;7:601–20. doi: 10.1089/106652700750050961. [DOI] [PubMed] [Google Scholar]
- 182.Charniak E. AI Mag. 1991;12:50. [Google Scholar]
- 183.Larjo A, Shmulevich I, Lähdesmäki H. Methods Mol Biol. 2013;939:35–45. doi: 10.1007/978-1-62703-107-3_4. [DOI] [PubMed] [Google Scholar]
- 184.Sachs K, Perez O, Pe'er D, Lauffenburger DA, Nolan GP. Science. 2005;308:523–529. doi: 10.1126/science.1105809. [DOI] [PubMed] [Google Scholar]
- 185.Mitra R, Müller P, Qiu P, Ji Y. Cancer Inform. 2014;13:79–89. doi: 10.4137/CIN.S13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Larjo A, Shmulevich I, Lähdesmäki H. Methods Mol Biol. 2013;939:35–45. doi: 10.1007/978-1-62703-107-3_4. [DOI] [PubMed] [Google Scholar]
- 187.Chickering D. In: Learning from Data SE - 12. Fisher D, Lenz HJ, editors. Vol. 112. Springer; New York: 1996. pp. 121–130. [Google Scholar]
- 188.Pe'er D. Sci STKE. 2005;2005:l4. [Google Scholar]
- 189.Friedman N, Nachman I, Peér D. Proceedings of the Fifteenth conference on Uncertainty in artificial intelligence. 1999;0:206–215. [Google Scholar]
- 190.Holmans P, Moskvina V, Jones L, Sharma M, Vedernikov A, Buchel F, Sadd M, Bras JM, Bettella F, Nicolaou N, Simón-sánchez J, Mittag F, Gibbs JR, Schulte C, Durr A, Guerreiro R, Hernandez D, Brice A, Stefánsson H, Majamaa K, Gasser T, Heutink P, Wood NW, Martinez M, Singleton AB, Nalls MA, Hardy J, Morris HR, Williams NM. Hum Mol Genet. 2013;22:1039–1049. doi: 10.1093/hmg/dds492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Neueder A, Bates GP. BMC Med Genomics. 2014;7:60. doi: 10.1186/s12920-014-0060-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Wexler EM, Rosen E, Lu D, Osborn GE, Martin E, Raybould H, Geschwind DH. Sci Signal. 2011;4:ra65–ra65. doi: 10.1126/scisignal.2002282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.IGAP. Alzheimer's Dement. 2014:1–14. [Google Scholar]
- 194.Gjoneska E, Pfenning AR, Mathys H, Quon G, Kundaje A, Tsai LH, Kellis M. Nature. 2015;518:365–369. doi: 10.1038/nature14252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Neuron. 2003;40:471–483. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- 196.Miller JA, Oldham MC, Geschwind DH. J Neurosci. 2008;28:1410–1420. doi: 10.1523/JNEUROSCI.4098-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zhang B, Horvath S. Stat Appl Genet Mol Biol. 2005;4 doi: 10.2202/1544-6115.1128. Article 17. [DOI] [PubMed] [Google Scholar]
- 198.Fu H, Subramanian RR, Masters SC. Annu Rev Pharmacol Toxicol. 2000;40:617–647. doi: 10.1146/annurev.pharmtox.40.1.617. [DOI] [PubMed] [Google Scholar]
- 199.Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov Aa, Zhang C, Xie T, Tran L, Dobrin R, Fluder E, Clurman B, Melquist S, Narayanan M, Suver C, Shah H, Mahajan M, Gillis T, Mysore J, MacDonald ME, Lamb JR, Bennett Da, Molony C, Stone DJ, Gudnason V, Myers AJ, Schadt EE, Neumann H, Zhu J, Emilsson V. Cell. 2013;153:707–20. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Xu XL, Olson JM, Zhao LP. Hum Mol Genet. 2002;11:1977–1985. doi: 10.1093/hmg/11.17.1977. [DOI] [PubMed] [Google Scholar]
- 201.Chen JY, Shen C, Sivachenko AY. Pac Symp Biocomput. 2006:367–378. [PubMed] [Google Scholar]
- 202.Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Nucleic Acids Res. 2005;33:D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Brown KR, Jurisica I. Bioinformatics. 2005;21:2076–2082. doi: 10.1093/bioinformatics/bti273. [DOI] [PubMed] [Google Scholar]
- 204.Krauthammer M, Kaufmann CA, Gilliam TC, Rzhetsky A. Proc Natl Acad Sci U S A. 2004;101:15148–15153. doi: 10.1073/pnas.0404315101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Caberlotto L, Lauria M, Nguyen TP, Scotti M. PLoS One. 2013;8 doi: 10.1371/journal.pone.0078919. Article no: e78919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kohalmi S, Reader LV, Samach A, Nowak J, Haughn G, Crosby W. In: Plant Molecular Biology Manual SE - 6. Gelvin S, Schilperoort R, editors. Springer; Netherlands: 1998. pp. 95–124. [Google Scholar]
- 207.Liu B, Jiang T, Ma S, Zhao H, Li J, Jiang X, Zhang J. Biochem Biophys Res Commun. 2006;349:1308–1314. doi: 10.1016/j.bbrc.2006.08.168. [DOI] [PubMed] [Google Scholar]
- 208.Soler-López M, Zanzoni A, Lluís R, Stelzl U, Aloy P. Genome Res. 2011;21:364–376. doi: 10.1101/gr.114280.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Ramanan VK, Saykin AJ. Am J Neurodegener Dis. 2013;2:145–75. [PMC free article] [PubMed] [Google Scholar]
- 210.Limviphuvadh V, Tanaka S, Goto S, Ueda K, Kanehisa M. Bioinformatics. 2007;23:2129–2138. doi: 10.1093/bioinformatics/btm307. [DOI] [PubMed] [Google Scholar]
- 211.Nguyen TP, Caberlotto L, Morine MJ, Priami C. Biomed Res Int. 2014;2014:686505. doi: 10.1155/2014/686505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Caberlotto L, Nguyen TP. BMC Syst Biol. 2014;8:65. doi: 10.1186/1752-0509-8-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Nixon Ra. Nat Med. 2013;19:983–97. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 214.Harris H, Rubinsztein DC. Nat Rev Neurol. 2011;8:108–117. doi: 10.1038/nrneurol.2011.200. [DOI] [PubMed] [Google Scholar]
- 215.Valdor R, Macian F. Pharmacol Res. 2012;66:475–83. doi: 10.1016/j.phrs.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Vasaikar SV, Padhi AK, Jayaram B, Gomes J. BMC Neurosci. 2013;14:3. doi: 10.1186/1471-2202-14-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Janes KA, Albeck JG, Gaudet S, Sorger PK, Lauffenburger DA, Yaffe MB. Science. 2005;310:1646–1653. doi: 10.1126/science.1116598. [DOI] [PubMed] [Google Scholar]
- 218.Lau KS, Cortez-Retamozo V, Philips SR, Pittet MJ, Lauffenburger DA, Haigis KM. PLoS Biol. 2012;10 doi: 10.1371/journal.pbio.1001393. Article no: e1001393. [DOI] [PMC free article] [PubMed] [Google Scholar]