

Abstract
Parkinson’s disease (PD) is characterized by alpha-synucleinopathy that affects all levels of the brain-gut axis including the central, autonomic, and enteric nervous systems. Recently, it has been recognized that the brain-gut axis interactions are significantly modulated by the gut microbiota via immunological, neuroendocrine, and direct neural mechanisms. Dysregulation of the brain-gut-microbiota axis in PD may be associated with gastrointestinal manifestations frequently preceding motor symptoms, as well as with the pathogenesis of PD itself, supporting the hypothesis that the pathological process is spread from the gut to the brain. Excessive stimulation of the innate immune system resulting from gut dysbiosis and/or small intestinal bacterial overgrowth and increased intestinal permeability may induce systemic inflammation, while activation of enteric neurons and enteric glial cells may contribute to the initiation of alpha-synuclein misfolding. Additionally, the adaptive immune system may be disturbed by bacterial proteins cross-reacting with human antigens. A better understanding of the brain-gut-microbiota axis interactions should bring a new insight in the pathophysiology of PD and permit an earlier diagnosis with a focus on peripheral biomarkers within the enteric nervous system. Novel therapeutic options aimed at modifying the gut microbiota composition and enhancing the intestinal epithelial barrier integrity in PD patients could influence the initial step of the following cascade of neurodegeneration in PD.
Keywords: Brain-gut-microbiota axis, Enteric nervous system, Gastrointestinal dysfunction, Gut microbiota, Parkinson’s disease
Core tip: Parkinson’s disease (PD) is characterized by alpha-synucleinopathy affecting all levels of the brain-gut axis. Both clinical and neuropathological evidences indicate that neurodegenerative changes in PD are accompanied by gastrointestinal symptoms that may precede or follow the central nervous system impairment. Dysregulation of the brain-gut-microbiota axis may significantly contribute to the pathogenesis of PD. The gut seems to play a critical role in the pathophysiology of PD representing a rout of entry for a putative environmental factor to initiate the pathological process. The close relationship between gut dysbiosis, intestinal permeability and neurological dysfunction suggests that the gut microbiota modification may provide a promising therapeutic option in PD.
INTRODUCTION
Parkinson’s disease (PD) is a multicentric neurodegenerative disorder characterized by the accumulation and aggregation of alfa-synuclein (α-syn) in the substantia nigra in the central nervous system (CNS) and in other neural structures[1,2]. The classical motor symptoms like bradykinesia, resting tremor, rigidity and late postural instability result from the death of dopamine-generating cells in the substantia nigra. There is also a wide spectrum of non-motor manifestations involving for example olfactory (loss of smell), gastrointestinal (GI), cardiovascular, and urogenital systems[3]. It has become evident that the different levels of the brain-gut axis including the autonomic nervous system (ANS) and the enteric nervous system (ENS) may be affected in PD[4-8]. Recently, it has been also recognized that the brain-gut axis interactions may be essentially influenced by the gut microbiota[9-12]. On the one hand, dysregulation of the brain-gut-microbiota axis in PD may result in GI dysfunction, which is present in over 80% of PD subjects[13]. On the other hand, this dysregulation may also significantly contribute to the pathogenesis of PD itself, supporting the hypothesis that the pathological process is spread from the gut to the brain[4,7].
INTERACTIONS WITHIN THE BRAIN-GUT-MICROBIOTA AXIS
Bidirectional communication between the CNS and the GI tract - the brain-gut axis - occurs both in health and disease. The neural network for the control of GI functions involves the intrinsic and extrinsic nervous systems and forms a hierarchic four-level integrative organization[14,15].
The first level is the ENS represented by neurons of the myenteric (Auerbach’s) and submucosal (Meissner’s) plexi and enteric glial cells (EGCs)[16]. Local reflexes, such as the migrating motor complex and peristaltic reflex, are under the ENS control through intrinsic primary afferent neurons (IPANs). The IPANs, located in the myenteric and submucosal plexi, project dentritic processes that synapse with motor neurons and interneurons. The primary excitatory enteric motor neurons and interneurons are cholinergic. Neurons expressing VIP and/or NO elicit smooth muscle relaxation, and submucosal VIP neurons also stimulate intestinal secretion[17]. Enteric dopaminergic neurons, which may inhibit intestinal motility, are also present and distributed along an oral-aboral gradient within the GI tract[17]. Dopaminergic neurons account for 14%-20% of the enteric neurons of the upper GI tract, whereas their proportion decreases to 1%-6% in the lower small intestine and large bowel[17].
The second level is the prevertebral ganglia modulating many peripheral visceral reflex responses[18].
The third level is the ANS within the spinal cord [origin of the sympathetic (T5-L2) and sacral (S2-S4) parasympathetic nervous systems] and the brainstem with the nucleus tractus solitarius (NTS) and dorsal motor nucleus of the vagus nerve (DMVN), which receives and gives origin to the afferent and efferent fibers of the vagus nerve (VN), respectively. The DMVN influence is most prominent in the upper GI tract, where cholinergic myenteric neurons mediate vagal excitatory effect, and VIP/NO neurons mediate inhibitory reflexes[19].
The fourth level includes higher brain centers. Information from the cortical and subcortical centers, including the basal ganglia, funnels down to specific brainstem nuclei, from where many GI functions are controlled. Disturbances at every level of that neural control may affect modulation of the GI functions including mechanisms at the local enteric reflexes, and extrinsic neural control[15].
Recently, a role of the enteric microbiota, including both commensal and pathogenic organisms, in the brain-gut axis interactions has been essentially recognized[10]. This has been reflected by a revised nomenclature to the more inclusive brain-gut-enteric microbiota axis[9]. The impact of the gut microflora on the brain-gut axis regulation involves immunological, neuroendocrine, and direct neural mechanisms[12]. The gut microbiota is known to upregulate local and systemic inflammation due to lipopolysaccharides (LPS) from pathogenic bacteria and synthesis of pro-inflammatory cytokines. Excessive stimulation of the innate immune system resulting from gut dysbiosis and/or small intestinal bacterial overgrowth and increased intestinal permeability may produce systemic and/or CNS inflammation[20]. Additionally, the adaptive immune system may be disturbed by bacterial proteins cross-reacting with human antigens. The gut bacteria are able to synthesize numerous neurotransmitters and neuromodulators such as γ-aminobutyric acid, serotonin, dopamine or short-chain fatty acids[12,21]. The production of these neurochemicals enables also intracellular communication between the members of the microbiota. Therefore, the existence of so-called “microbial organ-specific nervous system” could be even speculated[21]. Bacterial enzymes may also produce neurotoxic metabolites such as D-lactic acid and ammonia[20]. The direct neural communication between the gut and the brain occurs via the VN, as bacteria can stimulate afferent neurons of the ENS[22]. Vagal signals from the gut can evoke an anti-inflammatory response protecting against microbial-induced sepsis in a nicotinic acetylcholine receptor α7 subunit-dependent manner. Many of the effects of the gut microbiota or potential probiotics on brain function have been shown to be dependent on vagal activation[10,11,22]. Furthermore, bacterial colonization of the gut plays a major role in the postnatal development and maturation of the immune, endocrine and even neural systems[23]. These processes are key factors underpinning CNS signaling. Dysfunction of the brain-gut-microbiota axis has been implicated in stress-related disorders such as depression, anxiety, irritable bowel syndrome, and inflammatory bowel disease, as well as neurodevelopmental disorders such as autism[12,23-26].
GASTROINTESTINAL DYSFUNCTION IN PARKINSON’S DISEASE
Among many causes of parkinsonism, including multiple system atrophy, progressive supranuclear palsy or corticobasal degeneration, GI symptoms have been best characterized in the classical PD[13]. In the study of Edwards et al[27] evaluating the frequency of various GI symptoms in 98 patients with PD, abnormal salivation, dysphagia, nausea, constipation and defecatory dysfunction were present in 70%, 52%, 24%, 29%, and 66% of subjects, respectively. Among the studied parameters, only PD activity and duration correlated with GI dysfunction. No correlation was found between the GI symptoms and patients’ age, gender, antiparkinsonian treatment, level of activity or dietary fiber intake[27].
Hypersalivation typical in PD results not from salivary hypersecretion (in fact saliva production is even diminished), but from decreased swallowing frequency. Swallowing dysfunction may be symptomatic in up to 50% of PD patients[28]. Apart from common oro-pharyngeal dysfunction described in videofluoroscopy in more than 85% of PD cases, significant dysfunction in either the esophageal body or lower esophageal sphincter has been also revealed in 61%-73% of patients during esophageal manometry[28-30]. A serious complication of dysphagia in PD is an aspiration pneumonia occuring with a frequency ranging from 15% to 56%[31].
Impaired gastric emptying is an important manifestation of PD and is characterized by symptoms such as postprandial bloating or abdominal discomfort, early satiety and nausea. In patients with both early and advanced PD electrogastrography has confirmed gastric motility abnormalities[32]. Reduced amplitude of stomach contractions in PD can be also detected using real-time magnetic resonance imaging[33]. However, symptoms of the upper GI dysfunction do not always correlate with objective measurements of gastroparesis[34]. Delayed gastric emptying may also have potentially relevant pharmacokinetic implication causing an impaired absorption of L-dopa and thus worsening motor fluctuations[35]. In addition, PD has been associated with a higher prevalence of ulcer disease and Helicobacter pylori infection[36].
Uncomfortable sensation of abdominal bloating experienced by some individuals with PD, especially as an “off” phenomenon, could be the consequence of small bowel dysmotility shown by manometry[37]. Moreover, observed dysmotility may predispose for small intestinal bacterial overgrowth (SIBO), the prevalence of which is increased in PD patients[38].
Constipation, the most prominent GI dysfunction of PD, seems to be an early manifestation of the disease process itself[6,13,37,39-41]. Increased colon transit time has been recorded in both treated and untreated subjects, and even in PD patients without symptomatic constipation[42]. In sever cases it may lead to megacolon[43]. Interestingly, a retrospective analysis exploring the association between the frequency of bowel movements and further risk for PD revealed an increased risk of PD in men with infrequent bowel movements[44].
Defecatory dysfunction characterized by excessive straining and incomplete evacuation are another common and distressing problems in PD[45,46]. Pelvic floor dyssynergia in PD has been confirmed using anorectal manometry, defecography, and anal sphincter electromyography in more than 65% of patients[27,42,47].
PATHOPHYSIOLOGY OF GASTROINTESTINAL DYSFUNCTION IN PARKINSON’S DISEASE
The pathophysiology of GI dysfunction in PD may reflect both central and peripheral derangements. While dopaminergic deficiency secondary to nigrostriatal damage may be responsible for some aspects of GI symptoms in PD, it is quite clear that the additional sites of involvement play an important role as well[13]. The lesions of the medullar, spinal and peripheral autonomic nervous system present in PD could be sufficient to induce GI disturbances[48,49]. The involvement of the basal ganglia, the central pattern generator of swallowing, the DMVN, and the median raphe nucleus of the pons have been described in PD. Neurodegeneration and dysfunction of the locus ceruleus (LC) in PD has been also observed[50]. The LC is the major brain noradrenergic candidate for modulating the migrating motor complex (MMC) pattern and it has been shown that lesions of the LC in rats increase the duration of MMC[51]. The LC has close connections with the Barrington nucleus that sends descending projections to the sacral parasympathetic command of the distal colon and bladder functions[52]. Disturbances in the neural network involving the LC and the frontal cortex, hippocampus, as well as the spinal cord may contribute to abdominal pain occuring in PD. Clinical presentation of autonomic dysfunction in PD includes not only GI dysfunction, but cardiovascular and urogenital system as well as sudomotor, thermoregulatory, pupillary, sleep and respiratory abnormalities[53].
The lesions of the ENS, affecting not only dopaminergic neurons, are also commonly considered as being responsible for the digestive symptoms. However, the available data on the structural and neurochemical alterations of enteric neurons are still incomplete[7,37]. Prolonged colonic transit time most likely reflects early involvement of the myenteric plexus neurons as supported by finding in a transgenic mouse model of PD[54]. In other studies conducted in mice which overexpressed α-syn driven by the Thy-1 promoter, alterations in propulsive colonic motor activity reminiscent of colonic dysmotility encountered by PD patients were displayed[55,56]. The respective contribution of intrinsic and extrinsic innervations in GI dysfunction has not been yet fully elucidated.
Noteworthy, GI dysfunction in PD, at least to some extent, may be associated with the side effects of the treatment. Anticholinergic agents, popular in the past, can delay GI transit, whereas dopaminergic compounds may induce nausea[57].
ALPHA-SYNUCLEINOPATHY SPREAD ALONG THE BRAIN-GUT AXIS
Under physiological conditions, α-syn is abundantly expressed in the CNS and involved in the regulation of neurotransmission. Insoluble fibrils of phosphorylated α-syn have been implicated in several neurodegenerative disorders, such as PD and Alzheimer’s disease[58]. The two pathological hallmarks of PD are a loss of dopaminergic neurons in the substantia nigra and the presence of cytoplasmatic eosinophilic α-syn-containing inclusions termed Lewy bodies (LBs) in perikarya and Lewy neurites (LNs) in neuronal process of the remaining neurons[7]. However, in PD these inclusions are not restricted to the CNS only, but present also in peripheral tissues and body fluids[59,60]. The association of α-syn and PD has been identified through the genetic finding reported in 1997 by Polymeropoulos et al[61], which concerns the A533T mutation of SNCA gene encoding α-syn in a family with autosomal dominant familial PD. Recent studies consistently show that the single nucleotide polymorphisms in SNCA gene are related to the risk for sporadic PD.
LBs can be seen in autonomic regulatory regions, including hypothalamus, sympathetic system (intermediolateral nucleus of thoracic cord and sympathetic ganglia), and parasympathetic system (vagal and sacral parasympathetic nuclei). LBs were also found in the adrenal medulla and in the neural plexi innervating the gut and the heart[53]. Thus GI dysfunction related to dysautonomia may result from central mechanisms mediated through the brainstem autonomic centers and/or peripheral mechanisms due to post-ganglionic lesions.
Recent reports have shown that lesions in the ENS occurred at a very early stage of the disease, even before the involvement of the CNS[7,62-64]. This led to Braak’s hypothesis according to which α-syn pathology starts in the submucosal plexus of the ENS and propagates retrogradely to the CNS via vagal preganglionic axons of the DMVN[4]. From the DMVN there would be predictable caudo-rostral spread of pathology to other areas of the brainstem including the substantia nigra and finally to the basal forebrain and neocortex[1,65-68]. It has been also suggested that the spread of pathology in PD may be from neuron to neuron in a prion-like fashion[69]. In fact, host-to-graft disease propagation has been reported based on the observation that LBs were detectable in grafted neurons in subjects with PD[70,71]. The dual-hit hypothesis presented by Hawkes et al[67] in 2007 supported their theory, indicating that α-syn pathology is transmitted to midbrain through two different pathways: (1) nasal, from the olfactory bulb to the temporal lobe; and (2) gastric, through the ENS, secondary to swallowing of nasal secretion in saliva.
The disease may start in the ENS and then spread retrogradely toward the CNS or vice versa. Retrograde transport (from gut to brain) of α-syn can be concomitant with anterograde (from brain to gut) diffusion[72]. The first direct experimental evidence of PD pathology spread from the GI tract to the brain in rats has been just proved[73]. However, there are also recent reports indicating that the GI dysfunction may result from CNS damage[74]. Zheng et al[74] using an animal model of PD showed that 6-hydroxydopamine-induced lesions of the substantia nigra resulted in impaired gastric motility and emptying that could be prevented by vagotomy. It is also possible, that in some cases the neuropathy first occurs in the ENS, while in others in the CNS, that α-syn can move between the ENS and CNS and that the pathology could arise in both places with different time courses (or reach thresholds for detection at different times in the ENS and CNS) in susceptible patients[72,75].
There is a rostrocaudal gradient of α-syn associated histopathology within the GI tract[76,77]. The submandibular gland and lower esophagus have the highest frequency of LBs, followed by the stomach, small intestine, colon and rectum. This rostrocaudal gradient coincides with the distribution of vagal innervation from the DMVN[78]. Moreover, the rostrocaudal gradient from ascending colon to the rectum in the distribution of Lewy pathology has been also demonstrated to have important practical implication, as rectal biopsies have substantially lower sensitivity than ascending colon biopsies to detect Lewy pathology in the gut[79]. Noteworthy, the presence of α-syn within the ENS may not be always regarded as a pathological correlate, as it is a regular finding in adults with increasing age[58].
Accumulating evidence shows that α-syn plays a crucial role in neuroinflammation by triggering and/or potentiating astroglial and microglial activation[80-82]. Recent studies have also shown that dysfunction of EGCs at the ENS level occurs in PD[83]. EGCs, which represent in the digestive tract counterpart for brain astrocytes, may be critically involved in gut inflammation and modulation of intestinal epithelial barrier integrity[84]. Devos et al[84] found that expression of pro-inflammatory cytokines and glial markers are increased in colonic biopsies from PD patients and that they are correlated with the disease duration.
Both clinical and neuropathological evidences indicate that neurodegenerative changes are accompanied by GI symptoms that may precede or follow the CNS impairment[72]. Based on these observations a mechanistic hypothesis presenting the gut as the gateway in neurodegenerative diseases has been proposed[85]. Accordingly, the ENS seems to play a critical role in the pathophysiology of PD representing a rout of entry for a putative environmental factor to initiate the pathological process. Furthermore, regarding the parallel manifestations of neuropathologies in the ENS and CNS, the ENS may provide a more accessible target for studies of neural function, histopathology, and biochemistry in PD[72]. Thus, the ENS can be considered not only as “the second brain”, but also as a window towards “the first brain”[63].
GUT MICROBIOTA AND PARKINSON’S DISEASE
Changes in the gut microbiota composition may cause alterations in the gut barrier function and intestinal permeability, affecting not only GI epithelial cells and immune system, but also the ENS including both neurons and glial cells[86]. The bidirectional brain-gut-microbiota axis interactions modulate pro- and anti-inflammatory responses[87]. It has been suggested that the gut microbiota changes associated with intestinal inflammation may contribute to the initiation of α-syn misfolding[84,88]. There is a growing number of evidence confirming that the gut microbiota alterations precede or occur during the course of PD[89]. However, the causal relationship between the microbiota changes and the pathogenesis of PD remains unclear.
The interesting concept of molecular mimicry involving the microbiota in neurodegeneration has been also proposed. Indeed, Friedland[90] suggested that bacterial proteins may elicit cross-seeded misfolding, inflammation and oxidative stress, and cellular toxicity in neurodegeneration, initiating or otherwise influencing the development of PD, Alzheimer’s disease and other related disorders. Pathways of molecular mimicry processes induced by bacterial amyloid may involve TLR2/1, C14, and NFκB among others. Priming of the innate immune system by the microbiota (residing in the gut and oral/nasal cavities) may enhance the inflammatory response to cerebral amyloids such as α-syn. Trudler et al[91] postulated that cerebral amyloid may mimic viral or bacterial infection resulting in glial cell activation through TLRs. Specifically, it has been documented that neuroinflammation in PD is associated with upregulation of TLR2 signaling and activation of microglia[92]. TLR2, playing an important role in the regulation of intestinal barrier integrity, has been also found to activate microglial cells in the CNS[92]. It has been suggested that the peripheral immune response characterized by the presence of pro-inflammatory cytokines such as TNF-α, IL-1β and IL-8 in the serum induces a disruption of the blood-brain barrier and promotes microglia-mediated inflammation and neurotoxicity[36,93]. In a germ-free animal model, it has been found that the gut microbiota influences the blood-brain barrier permeability associated with reduced expression of the tight junction proteins in a homological way as it affects the intestinal epithelial barrier[94]. Very recently, Sui et al[95] proved that a bidirectional transport of α-syn into and out of the brain by the blood-brain barrier is possible and suggested that LPS-induced inflammation could increase α-syn uptake by the brain by disrupting the blood-brain barrier.
In an animal model of PD, peripherally-induced inflammation was shown to induce the microglial complement pathway to damage dopaminergic neurons[96]. Several studies have demonstrated that pro-inflammatory factors associated with chronic GI diseases induce brain inflammation and the death of dopaminergic neurons and could eventually be responsible for parkinsonism[97,98].
Importantly, some genetic risk factors may play a crucial role in the interactions between the brain-gut-microbiota axis with respect to gut inflammation. It has been also shown that the methylation status in the SNCA promoter region may affect α-syn expression and the risk for PD[99]. Therefore, a potential role of the gut microbiota as an epigenetic factor influencing DNA methylation may be speculated. Moreover, genetic variant of the component of innate immune system - TREM2 (Triggering Receptor Expressed on Myeloid cells) has been reported to be associated with a higher risk for PD[100]. The ε4 allele of apolipoprotein E (ApoE) has been shown to increase the risk for dementia in synucleinopathies such as PD[101]. Potentially, ApoE genotype, by influencing bile acid secretion, could affect the composition of the gut microbiota to favor the development of organisms triggering misfolding[90]. Moreover, three single nucleotide polymorphisms in CARD15 gene, known to be associated with Crohn’s disease, have been also shown to be over-expressed in PD patients, supporting the observation that GI inflammation contributes to the pathogenesis of PD[102].
Quantitative and qualitative changes in the gut microbiota in Parkinson’s disease
Just recently, the first report on alterations in the gut microbiota composition in PD and its association with clinical phenotype of the disease has been published by Scheperjans and colleagues[103]. They showed a reduced abundance of the Prevotellaceae bacteria family in PD patients compared with healthy controls, and greater abundance of Enterobacteriaceae among those patients with the postural instability and gait difficulty phenotype compared to those with tremor-dominant PD[103]. Prevotellaceae bacteria as commensals are involved in mucin synthesis in the gut mucosal layer and production of neuroactive short-chain fatty acids (SCFA) through fiber fermentation[104]. Thus the reduced abundance of Prevotellaceae could result in decreased mucin synthesis and increased intestinal permeability leading to the greater local and systemic exposure to bacterial antigens and endotoxins, which in turn would trigger or maintain excessive α-syn expression in the colon or even promote its misfolding[105,106]. The presence of inflammation and evidence of EGCs dysregulation in colonic biopsies from patients with PD have been shown[83,84,107]. The inflammatory changes in PD are associated with increased colonic permeability, which was observed both in a mouse model of the disease as well as in PD patients[105,108]. Another suggested possibility, by which the gut microbiota could affect α-syn pathology, is that α-syn intra- and extraneuronal clearance mechanisms are impaired by SCFA-dependent modulation of gene expression[86].
Recent studies reported high prevalence of SIBO in PD, ranging from 54% to 67%[38,109,110]. PD being associated with gastroparesis and impaired GI motility may predispose to SIBO. In the study by Fasano et al[38], the presence of SIBO reported in 54% of PD subjects, was associated not only with the GI symptoms but also with the motor symptoms. Interestingly, the improvement in the motor fluctuations following treatment with rifaximin was observed[38]. In another study SIBO was detected in 25% of PD patients and occurred early in the disease course[109]. According to that report, SIBO was not associated with worse GI function, but independently predisposed to worse motor function[109]. It is possible that SIBO contributes to motor dysfunction by disrupting small intestinal integrity leading to immune stimulation and/or alteration in L-dopa absorption. SIBO may cause changes in the gut permeability which promote translocation of bacteria and endotoxins across the intestinal epithelium, inducing the pro-inflammatory response[111]. A recent study involving newly-diagnosed PD patients confirmed that intestinal permeability in these subjects was markedly increased compared with healthy controls, and this was associated with more intense staining of Escherichia coli in the intestinal mucosa and with systemic exposure to LPS[105]. Noteworthy, these alterations were correlated with abnormal accumulation of α-syn in enteric neurons[105]. LPS is a gut-derived, pro-inflammatory bacterial endotoxin that may cause delayed and progressive nigral pathology when administered systematically and serves even as a progressive model of PD[108,112,113]. These data reinforce the link between gut dysbiosis, intestinal permeability and neurological dysfunction (Figure 1). The potential role of antibiotic therapy in improving motor symptoms has generated a considerable interest in the interplay between the gut microbiota and PD, but well designed treatment trials are needed to fully elucidate a causal link between SIBO and motor function in PD. It should also encourage further studies on a new therapeutic approach based on the manipulation of the gut microbiota with probiotics, prebiotics, or even fecal microbiota transplantation. Some preliminary reports on favorable outcome of fecal microbiota transplantation in PD already exists[114].
Figure 1.
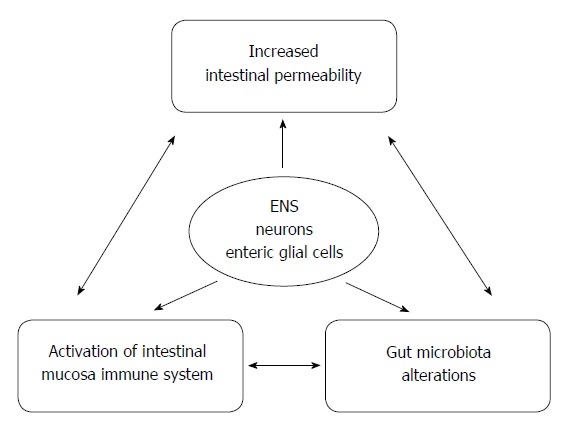
The link between gut dysbiosis, intestinal permeability and neurological dysfunction. There is a close mutual relationship between gut dysbiosis, intestinal permeability and gut inflammation. The enteric nervous system (ENS) is a key player participating in these interactions. Changes in the intestinal permeability may promote translocation of bacteria and endotoxins across the epithelial barrier inducing immunological response associated with the production of pro-inflammatory cytokines. The activation of both enteric neurons and glial cells may result in neurological dysfunction spreading along the brain-gut axis.
Specific pathogens as potential triggering factors
Helicobacter pylori: The potential role of Helicobacter pylori (HP) in PD, both with regard to the pathogenesis of PD itself and the development of motor symptoms fluctuation, remains controversial[36,115-119]. Gastric ulcers, strongly related to HP infection, have been associated with PD since 1960s[117]. Based on the observation of an age-associated increase in the levels of antibodies against HP in PD patients, Dobbs et al[120] proposed that HP infection predisposes to autoimmunity that results in neuronal damage leading to eventual parkinsonism. In fact, HP infection may increase the risk of PD[117]. Nielsen et al[117] showed that prescription of HP eradication drugs and proton pump inhibitors five or more years prior to the diagnosis of PD were associated with 45% and 23% increase in the PD risk, respectively. At the same time eradication of HP infection has been shown to ameliorate symptoms of PD[121]. Currently, HP eradication in PD patients treated with L-dopa is recommended as it may improve the bioavailability of the drug and reduce motor fluctuation[38,119,122-124].
Moreover, there is an increased mortality from PD amongst livestock farmers, which has been associated with Helicobacter suis being the most common zoonotic Helicobacter in man[125]. That observation is supported by the recent finding that a significantly higher frequency of Helicobacter suis is observed in patients with idiopathic parkinsonism then in control patients[125].
Mycobacterium paratuberculosis: An association between Mycobacterium avium ss. paratuberculosis (MAP) and several inflammatory diseases including Crohn’s disease has been suggested. As mentioned above, genetic studies concerning the polymorphism of CARD15 gene revealed a link between PD and Crohn’s disease[102]. Moreover, the polymorphisms of LRRK2 and PARK genes are associated both with PD and susceptibility to mycobacterial infection, as they are related to xenophagy, an autophagic pathway involved in the removal of intracellular pathogens[126-128]. The same genetic defects are associated with disruption of cellular homeostasis resulting in protein (e.g., α-syn) folding abnormalities. Dow[126] proposed that genetic defects associated with PD allow persistent infection with MAP, and that MAP is the triggering factor of α-syn aggregation. He postulated that beginning as an enteric infection, MAP - via the VN - initiates a pathological process that results in a targeted neuroinvasion of the CNS. The iron concentration in the substantia nigra in PD subjects could be caused by sequestered iron in MAP spheroplasts. According to Dow[126], protein aggregation associated with LBs formation is due to iron toxicity and/or to “consumptive exhaustion” of the processes that both maintain cellular protein homeostasis and influence removal of intracellular pathogens. Importantly, as MAP is competent as a bioaerosol, the additional/alternative route of neuroinvasion via the olfactory nerve does not eliminate MAP from consideration as the environmental triggering agent. To support his hypothesis Dow points at the recent findings: anti-mycobacterial rifampicin has a protective function in PD[129], mycobacterial heat shock proteins (HPS 65 and HPS 70) have been found in the cerebral spinal fluid of PD patients[130], and BCG vaccination partially preserves the substantia nigra integrity in an animal model of PD[131].
Influence of age, diet, coffee and smoking on the gut microbiota composition
The age-related changes in the gut microbiota, in particular a decreased diversity of species, together with other factors including impaired gut motility, impaired gut-blood barrier, and suppressed immune function may be linked to the age-related neurodegeneration[23,90]. Recent studies reported a great inter-individual variation among elderly regarding the gut microbiota composition, and a significant relationship between microbiota, diet and institution or community living[132]. The gut microbiota-related mechanisms could also explain the differences in PD prevalence between rural and urban environment, between countries or even sexes[86]. Fiber-rich diet enhances the growth of colonic bacteria that produce SCFA, which have systemic anti-inflammatory effect[133]. Therefore, intervention studies with probiotics and prebiotics offer promising way to bring benefits in elderly’s health.
Although unequivocal epidemiologic evidence indicates that the risk of PD is lower in smokers and coffee drinkers, explanations for these findings remain controversial[134,135]. In the large meta-analysis of 61 case-control and cohort studies, the risk of PD was found to be 60% lower among current cigarette smokers than among never smokers, and 30% lower among coffee drinkers than among non-drinkers[136]. It has been also shown that consumption of caffeine-containing beverages other than coffee, such as black tea and Japanese and Chinese teas, is also inversely related to the PD risk[137]. Derkinderen et al[134] proposed that the beneficial effects of smoking and coffee consumption on PD may be mediated through the modulation of the brain-gut-microbiota axis. Both cigarette and coffee consumption can alter the composition of the gut microbiota in a way that mitigates intestinal inflammation. This, in turn, could lead to less misfolding of α-syn in the ENS, reducing the risk of PD by minimizing propagation of the protein to the CNS[134]. In fact, a marked shift in the composition of the intestinal microbiota was observed in humans after smoking cessation[138]. The potential immunomodulatory effect of smoking, possibly via alteration of the gut microbiota, is also observed in patients with ulcerative colitis in whom smoking cessation may be linked to the disease onset[139]. It has been also shown that consumption of coffee in both mice and humans induces a significant increase in the number of Bifidobacteria, which exert anti-inflammatory properties[140,141]. Additionally, coffee and tobacco might promote bacteria that counteract certain forms of chronic GI infection, such as that caused by HP, the presence of which has been associated with the increased risk for PD[117,134].
Putative role of viral infection or neurotoxins
Dysfunction of the intestinal epithelial barrier resulting from dysregulation of the brain-gut-microbiota axis could promote invasion of neuroactive substances, including neurotropic viruses, unconventional pathogens with prion-like properties, or slow neurotoxins, as suggested by Braak[67]. Some environmental toxins, as the pesticide rotenone used in animal models of PD, could also induce the release of α-syn by the enteric neurons[142]. Numerous studies showed that environmental factors such as herbicides, pesticides, and metal pollution may be related to the occurrence of PD[68,143-145].
The possible role of viral invasion (e.g., with influenza virus of herpes simplex virus) has been suggested[67]. As it has been demonstrated in a mouse model, the H5N1 influenza virus may travel from the periphery to the CNS and induce neuroinflammation, accumulation of phosphorylated α-syn and dopaminergic cell loss[146]. Hepatitis C virus (HCV) has been also shown to invade the CNS and release the inflammatory cytokines, which may play a role in the pathogenesis of PD. Recently, Wu et al[147] have corroborated the dopaminergic toxicity of HCV (but not HBV) in the midbrain neuron-glia coculture system in rats and demonstrated a significantly positive epidemiological association between HCV infection and PD in human.
CONCLUSION
A better understanding of the brain-gut-microbiota axis interactions should bring a new insight in the pathophysiology of PD, permit an earlier diagnosis with a focus on peripheral biomarkers within the ENS, as well as lead to novel therapeutic options in PD. Dietary or pharmacological interventions should be aimed at modifying the gut microbiota composition and enhancing the intestinal epithelial barrier integrity in PD patients or subjects at higher risk for the disease. This could influence the initial step of the following cascade of neurodegeneration in PD. The elucidation of the temporal and casual relationship between the gut microbiota alterations and the pathogenesis of PD will be of great clinical relevance. Further studies on a new therapeutic approach in PD based on the modification of the gut microbiota with probiotics, prebiotics, or even fecal microbiota transplantation are awaited.
Footnotes
Conflict-of-interest statement: There is no conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: March 3, 2015
First decision: May 18, 2015
Article in press: August 31, 2015
P- Reviewer: Cao HL, de Castro CG S- Editor: Yu J L- Editor: A E- Editor: Wang CH
References
- 1.Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 2.Dickson DW, Fujishiro H, Orr C, DelleDonne A, Josephs KA, Frigerio R, Burnett M, Parisi JE, Klos KJ, Ahlskog JE. Neuropathology of non-motor features of Parkinson disease. Parkinsonism Relat Disord. 2009;15 Suppl 3:S1–S5. doi: 10.1016/S1353-8020(09)70769-2. [DOI] [PubMed] [Google Scholar]
- 3.Cersosimo MG, Raina GB, Pecci C, Pellene A, Calandra CR, Gutiérrez C, Micheli FE, Benarroch EE. Gastrointestinal manifestations in Parkinson’s disease: prevalence and occurrence before motor symptoms. J Neurol. 2013;260:1332–1338. doi: 10.1007/s00415-012-6801-2. [DOI] [PubMed] [Google Scholar]
- 4.Braak H, de Vos RA, Bohl J, Del Tredici K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett. 2006;396:67–72. doi: 10.1016/j.neulet.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 5.Bloch A, Probst A, Bissig H, Adams H, Tolnay M. Alpha-synuclein pathology of the spinal and peripheral autonomic nervous system in neurologically unimpaired elderly subjects. Neuropathol Appl Neurobiol. 2006;32:284–295. doi: 10.1111/j.1365-2990.2006.00727.x. [DOI] [PubMed] [Google Scholar]
- 6.Cersosimo MG, Benarroch EE. Autonomic involvement in Parkinson’s disease: pathology, pathophysiology, clinical features and possible peripheral biomarkers. J Neurol Sci. 2012;313:57–63. doi: 10.1016/j.jns.2011.09.030. [DOI] [PubMed] [Google Scholar]
- 7.Lebouvier T, Chaumette T, Paillusson S, Duyckaerts C, Bruley des Varannes S, Neunlist M, Derkinderen P. The second brain and Parkinson’s disease. Eur J Neurosci. 2009;30:735–741. doi: 10.1111/j.1460-9568.2009.06873.x. [DOI] [PubMed] [Google Scholar]
- 8.Cersosimo MG, Benarroch EE. Neural control of the gastrointestinal tract: implications for Parkinson disease. Mov Disord. 2008;23:1065–1075. doi: 10.1002/mds.22051. [DOI] [PubMed] [Google Scholar]
- 9.Rhee SH, Pothoulakis C, Mayer EA. Principles and clinical implications of the brain-gut-enteric microbiota axis. Nat Rev Gastroenterol Hepatol. 2009;6:306–314. doi: 10.1038/nrgastro.2009.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grenham S, Clarke G, Cryan JF, Dinan TG. Brain-gut-microbe communication in health and disease. Front Physiol. 2011;2:94. doi: 10.3389/fphys.2011.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borre YE, Moloney RD, Clarke G, Dinan TG, Cryan JF. The impact of microbiota on brain and behavior: mechanisms & amp; therapeutic potential. Adv Exp Med Biol. 2014;817:373–403. doi: 10.1007/978-1-4939-0897-4_17. [DOI] [PubMed] [Google Scholar]
- 12.Mayer EA, Tillisch K, Gupta A. Gut/brain axis and the microbiota. J Clin Invest. 2015;125:926–938. doi: 10.1172/JCI76304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pfeiffer RF. Gastrointestinal dysfunction in Parkinson’s disease. Parkinsonism Relat Disord. 2011;17:10–15. doi: 10.1016/j.parkreldis.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 14.Aziz Q, Thompson DG. Brain-gut axis in health and disease. Gastroenterology. 1998;114:559–578. doi: 10.1016/s0016-5085(98)70540-2. [DOI] [PubMed] [Google Scholar]
- 15.Mulak A, Bonaz B. Irritable bowel syndrome: a model of the brain-gut interactions. Med Sci Monit. 2004;10:RA55–RA62. [PubMed] [Google Scholar]
- 16.Schemann M, Neunlist M. The human enteric nervous system. Neurogastroenterol Motil. 2004;16 Suppl 1:55–59. doi: 10.1111/j.1743-3150.2004.00476.x. [DOI] [PubMed] [Google Scholar]
- 17.Anlauf M, Schäfer MK, Eiden L, Weihe E. Chemical coding of the human gastrointestinal nervous system: cholinergic, VIPergic, and catecholaminergic phenotypes. J Comp Neurol. 2003;459:90–111. doi: 10.1002/cne.10599. [DOI] [PubMed] [Google Scholar]
- 18.Szurszewski JH. Physiology of mammalian prevertebral ganglia. Annu Rev Physiol. 1981;43:53–68. doi: 10.1146/annurev.ph.43.030181.000413. [DOI] [PubMed] [Google Scholar]
- 19.Chang HY, Mashimo H, Goyal RK. Musings on the wanderer: what’s new in our understanding of vago-vagal reflex? IV. Current concepts of vagal efferent projections to the gut. Am J Physiol Gastrointest Liver Physiol. 2003;284:G357–G366. doi: 10.1152/ajpgi.00478.2002. [DOI] [PubMed] [Google Scholar]
- 20.Galland L. The gut microbiome and the brain. J Med Food. 2014;17:1261–1272. doi: 10.1089/jmf.2014.7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lyte M. Microbial endocrinology: Host-microbiota neuroendocrine interactions influencing brain and behavior. Gut Microbes. 2014;5:381–389. doi: 10.4161/gmic.28682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Forsythe P, Bienenstock J, Kunze WA. Vagal pathways for microbiome-brain-gut axis communication. Adv Exp Med Biol. 2014;817:115–133. doi: 10.1007/978-1-4939-0897-4_5. [DOI] [PubMed] [Google Scholar]
- 23.Borre YE, O’Keeffe GW, Clarke G, Stanton C, Dinan TG, Cryan JF. Microbiota and neurodevelopmental windows: implications for brain disorders. Trends Mol Med. 2014;20:509–518. doi: 10.1016/j.molmed.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 24.Dinan TG, Cryan JF. Melancholic microbes: a link between gut microbiota and depression? Neurogastroenterol Motil. 2013;25:713–719. doi: 10.1111/nmo.12198. [DOI] [PubMed] [Google Scholar]
- 25.Bonaz BL, Bernstein CN. Brain-gut interactions in inflammatory bowel disease. Gastroenterology. 2013;144:36–49. doi: 10.1053/j.gastro.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 26.Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, Codelli JA, Chow J, Reisman SE, Petrosino JF, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155:1451–1463. doi: 10.1016/j.cell.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edwards LL, Quigley EM, Pfeiffer RF. Gastrointestinal dysfunction in Parkinson’s disease: frequency and pathophysiology. Neurology. 1992;42:726–732. doi: 10.1212/wnl.42.4.726. [DOI] [PubMed] [Google Scholar]
- 28.Bushmann M, Dobmeyer SM, Leeker L, Perlmutter JS. Swallowing abnormalities and their response to treatment in Parkinson’s disease. Neurology. 1989;39:1309–1314. doi: 10.1212/wnl.39.10.1309. [DOI] [PubMed] [Google Scholar]
- 29.Castell JA, Johnston BT, Colcher A, Li Q, Gideon RM, Castell DO. Manometric abnormalities of the oesophagus in patients with Parkinson’s disease. Neurogastroenterol Motil. 2001;13:361–364. doi: 10.1046/j.1365-2982.2001.00275.x. [DOI] [PubMed] [Google Scholar]
- 30.Monte FS, da Silva-Júnior FP, Braga-Neto P, Nobre e Souza MA, de Bruin VM. Swallowing abnormalities and dyskinesia in Parkinson’s disease. Mov Disord. 2005;20:457–462. doi: 10.1002/mds.20342. [DOI] [PubMed] [Google Scholar]
- 31.Ali GN, Wallace KL, Schwartz R, DeCarle DJ, Zagami AS, Cook IJ. Mechanisms of oral-pharyngeal dysphagia in patients with Parkinson’s disease. Gastroenterology. 1996;110:383–392. doi: 10.1053/gast.1996.v110.pm8566584. [DOI] [PubMed] [Google Scholar]
- 32.Soykan I, Lin Z, Bennett JP, McCallum RW. Gastric myoelectrical activity in patients with Parkinson’s disease: evidence of a primary gastric abnormality. Dig Dis Sci. 1999;44:927–931. doi: 10.1023/a:1026648311646. [DOI] [PubMed] [Google Scholar]
- 33.Unger MM, Hattemer K, Möller JC, Schmittinger K, Mankel K, Eggert K, Strauch K, Tebbe JJ, Keil B, Oertel WH, et al. Real-time visualization of altered gastric motility by magnetic resonance imaging in patients with Parkinson’s disease. Mov Disord. 2010;25:623–628. doi: 10.1002/mds.22841. [DOI] [PubMed] [Google Scholar]
- 34.Goetze O, Nikodem AB, Wiezcorek J, Banasch M, Przuntek H, Mueller T, Schmidt WE, Woitalla D. Predictors of gastric emptying in Parkinson’s disease. Neurogastroenterol Motil. 2006;18:369–375. doi: 10.1111/j.1365-2982.2006.00780.x. [DOI] [PubMed] [Google Scholar]
- 35.Djaldetti R, Baron J, Ziv I, Melamed E. Gastric emptying in Parkinson’s disease: patients with and without response fluctuations. Neurology. 1996;46:1051–1054. doi: 10.1212/wnl.46.4.1051. [DOI] [PubMed] [Google Scholar]
- 36.Alvarez-Arellano L, Maldonado-Bernal C. Helicobacter pylori and neurological diseases: Married by the laws of inflammation. World J Gastrointest Pathophysiol. 2014;5:400–404. doi: 10.4291/wjgp.v5.i4.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pfeiffer RF. Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol. 2003;2:107–116. doi: 10.1016/s1474-4422(03)00307-7. [DOI] [PubMed] [Google Scholar]
- 38.Fasano A, Bove F, Gabrielli M, Petracca M, Zocco MA, Ragazzoni E, Barbaro F, Piano C, Fortuna S, Tortora A, et al. The role of small intestinal bacterial overgrowth in Parkinson’s disease. Mov Disord. 2013;28:1241–1249. doi: 10.1002/mds.25522. [DOI] [PubMed] [Google Scholar]
- 39.Siddiqui MF, Rast S, Lynn MJ, Auchus AP, Pfeiffer RF. Autonomic dysfunction in Parkinson’s disease: a comprehensive symptom survey. Parkinsonism Relat Disord. 2002;8:277–284. doi: 10.1016/s1353-8020(01)00052-9. [DOI] [PubMed] [Google Scholar]
- 40.Jost WH, Eckardt VF. Constipation in idiopathic Parkinson’s disease. Scand J Gastroenterol. 2003;38:681–686. doi: 10.1080/00365520310003200. [DOI] [PubMed] [Google Scholar]
- 41.Sakakibara R, Kishi M, Ogawa E, Tateno F, Uchiyama T, Yamamoto T, Yamanishi T. Bladder, bowel, and sexual dysfunction in Parkinson’s disease. Parkinsons Dis. 2011;2011:924605. doi: 10.4061/2011/924605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sakakibara R, Odaka T, Uchiyama T, Asahina M, Yamaguchi K, Yamaguchi T, Yamanishi T, Hattori T. Colonic transit time and rectoanal videomanometry in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2003;74:268–272. doi: 10.1136/jnnp.74.2.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kupsky WJ, Grimes MM, Sweeting J, Bertsch R, Cote LJ. Parkinson’s disease and megacolon: concentric hyaline inclusions (Lewy bodies) in enteric ganglion cells. Neurology. 1987;37:1253–1255. doi: 10.1212/wnl.37.7.1253. [DOI] [PubMed] [Google Scholar]
- 44.Abbott RD, Petrovitch H, White LR, Masaki KH, Tanner CM, Curb JD, Grandinetti A, Blanchette PL, Popper JS, Ross GW. Frequency of bowel movements and the future risk of Parkinson’s disease. Neurology. 2001;57:456–462. doi: 10.1212/wnl.57.3.456. [DOI] [PubMed] [Google Scholar]
- 45.Kim JS, Sung HY, Lee KS, Kim YI, Kim HT. Anorectal dysfunctions in Parkinson’s disease. J Neurol Sci. 2011;310:144–151. doi: 10.1016/j.jns.2011.05.048. [DOI] [PubMed] [Google Scholar]
- 46.Sung HY, Choi MG, Kim YI, Lee KS, Kim JS. Anorectal manometric dysfunctions in newly diagnosed, early-stage Parkinson’s disease. J Clin Neurol. 2012;8:184–189. doi: 10.3988/jcn.2012.8.3.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang CP, Sung WH, Wang CC, Tsai PY. Early recognition of pelvic floor dyssynergia and colorectal assessment in Parkinson‘s disease associated with bowel dysfunction. Colorectal Dis. 2013;15:e130–e137. doi: 10.1111/codi.12105. [DOI] [PubMed] [Google Scholar]
- 48.Wakabayashi K, Takahashi H. Neuropathology of autonomic nervous system in Parkinson’s disease. Eur Neurol. 1997;38 Suppl 2:2–7. doi: 10.1159/000113469. [DOI] [PubMed] [Google Scholar]
- 49.Benarroch EE, Schmeichel AM, Low PA, Boeve BF, Sandroni P, Parisi JE. Involvement of medullary regions controlling sympathetic output in Lewy body disease. Brain. 2005;128:338–344. doi: 10.1093/brain/awh376. [DOI] [PubMed] [Google Scholar]
- 50.McNaught KS, Perl DP, Brownell AL, Olanow CW. Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson’s disease. Ann Neurol. 2004;56:149–162. doi: 10.1002/ana.20186. [DOI] [PubMed] [Google Scholar]
- 51.Bonaz B, Martin L, Beurriand E, Manier M, Hostein J, Feuerstein C. Locus ceruleus modulates migrating myoelectric complex in rats. Am J Physiol. 1992;262:G1121–G1126. doi: 10.1152/ajpgi.1992.262.6.G1121. [DOI] [PubMed] [Google Scholar]
- 52.Valentino RJ, Chen S, Zhu Y, Aston-Jones G. Evidence for divergent projections to the brain noradrenergic system and the spinal parasympathetic system from Barrington’s nucleus. Brain Res. 1996;732:1–15. doi: 10.1016/0006-8993(96)00482-9. [DOI] [PubMed] [Google Scholar]
- 53.Micieli G, Tosi P, Marcheselli S, Cavallini A. Autonomic dysfunction in Parkinson’s disease. Neurol Sci. 2003;24 Suppl 1:S32–S34. doi: 10.1007/s100720300035. [DOI] [PubMed] [Google Scholar]
- 54.Kuo YM, Li Z, Jiao Y, Gaborit N, Pani AK, Orrison BM, Bruneau BG, Giasson BI, Smeyne RJ, Gershon MD, et al. Extensive enteric nervous system abnormalities in mice transgenic for artificial chromosomes containing Parkinson disease-associated alpha-synuclein gene mutations precede central nervous system changes. Hum Mol Genet. 2010;19:1633–1650. doi: 10.1093/hmg/ddq038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang L, Fleming SM, Chesselet MF, Taché Y. Abnormal colonic motility in mice overexpressing human wild-type alpha-synuclein. Neuroreport. 2008;19:873–876. doi: 10.1097/WNR.0b013e3282ffda5e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang L, Magen I, Yuan PQ, Subramaniam SR, Richter F, Chesselet MF, Taché Y. Mice overexpressing wild-type human alpha-synuclein display alterations in colonic myenteric ganglia and defecation. Neurogastroenterol Motil. 2012;24:e425–e436. doi: 10.1111/j.1365-2982.2012.01974.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Woitalla D, Goetze O. Treatment approaches of gastrointestinal dysfunction in Parkinson’s disease, therapeutical options and future perspectives. J Neurol Sci. 2011;310:152–158. doi: 10.1016/j.jns.2011.06.050. [DOI] [PubMed] [Google Scholar]
- 58.Böttner M, Zorenkov D, Hellwig I, Barrenschee M, Harde J, Fricke T, Deuschl G, Egberts JH, Becker T, Fritscher-Ravens A, et al. Expression pattern and localization of alpha-synuclein in the human enteric nervous system. Neurobiol Dis. 2012;48:474–480. doi: 10.1016/j.nbd.2012.07.018. [DOI] [PubMed] [Google Scholar]
- 59.Malek N, Swallow D, Grosset KA, Anichtchik O, Spillantini M, Grosset DG. Alpha-synuclein in peripheral tissues and body fluids as a biomarker for Parkinson’s disease - a systematic review. Acta Neurol Scand. 2014;130:59–72. doi: 10.1111/ane.12247. [DOI] [PubMed] [Google Scholar]
- 60.Kim HJ. Alpha-Synuclein Expression in Patients with Parkinson’s Disease: A Clinician’s Perspective. Exp Neurobiol. 2013;22:77–83. doi: 10.5607/en.2013.22.2.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 62.Paillusson S, Clairembault T, Biraud M, Neunlist M, Derkinderen P. Activity-dependent secretion of alpha-synuclein by enteric neurons. J Neurochem. 2013;125:512–517. doi: 10.1111/jnc.12131. [DOI] [PubMed] [Google Scholar]
- 63.Lebouvier T, Neunlist M, Bruley des Varannes S, Coron E, Drouard A, N’Guyen JM, Chaumette T, Tasselli M, Paillusson S, Flamand M, et al. Colonic biopsies to assess the neuropathology of Parkinson’s disease and its relationship with symptoms. PLoS One. 2010;5:e12728. doi: 10.1371/journal.pone.0012728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grathwohl SA, Steiner JA, Britschgi M, Brundin P. Mind the gut: secretion of α-synuclein by enteric neurons. J Neurochem. 2013;125:487–490. doi: 10.1111/jnc.12191. [DOI] [PubMed] [Google Scholar]
- 65.Del Tredici K, Rüb U, De Vos RA, Bohl JR, Braak H. Where does Parkinson disease pathology begin in the brain? J Neuropathol Exp Neurol. 2002;61:413–426. doi: 10.1093/jnen/61.5.413. [DOI] [PubMed] [Google Scholar]
- 66.Braak H, Rüb U, Gai WP, Del Tredici K. Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm. 2003;110:517–536. doi: 10.1007/s00702-002-0808-2. [DOI] [PubMed] [Google Scholar]
- 67.Hawkes CH, Del Tredici K, Braak H. Parkinson’s disease: a dual-hit hypothesis. Neuropathol Appl Neurobiol. 2007;33:599–614. doi: 10.1111/j.1365-2990.2007.00874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reichmann H. View point: etiology in Parkinson’s disease. Dual hit or spreading intoxication. J Neurol Sci. 2011;310:9–11. doi: 10.1016/j.jns.2011.04.016. [DOI] [PubMed] [Google Scholar]
- 69.Olanow CW, Brundin P. Parkinson’s disease and alpha synuclein: is Parkinson’s disease a prion-like disorder? Mov Disord. 2013;28:31–40. doi: 10.1002/mds.25373. [DOI] [PubMed] [Google Scholar]
- 70.Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Björklund A, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–503. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- 71.Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 2008;14:504–506. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- 72.Natale G, Pasquali L, Paparelli A, Fornai F. Parallel manifestations of neuropathologies in the enteric and central nervous systems. Neurogastroenterol Motil. 2011;23:1056–1065. doi: 10.1111/j.1365-2982.2011.01794.x. [DOI] [PubMed] [Google Scholar]
- 73.Holmqvist S, Chutna O, Bousset L, Aldrin-Kirk P, Li W, Björklund T, Wang ZY, Roybon L, Melki R, Li JY. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014;128:805–820. doi: 10.1007/s00401-014-1343-6. [DOI] [PubMed] [Google Scholar]
- 74.Zheng LF, Song J, Fan RF, Chen CL, Ren QZ, Zhang XL, Feng XY, Zhang Y, Li LS, Zhu JX. The role of the vagal pathway and gastric dopamine in the gastroparesis of rats after a 6-hydroxydopamine microinjection in the substantia nigra. Acta Physiol (Oxf) 2014;211:434–446. doi: 10.1111/apha.12229. [DOI] [PubMed] [Google Scholar]
- 75.Visanji NP, Marras C, Hazrati LN, Liu LW, Lang AE. Alimentary, my dear Watson? The challenges of enteric α-synuclein as a Parkinson’s disease biomarker. Mov Disord. 2014;29:444–450. doi: 10.1002/mds.25789. [DOI] [PubMed] [Google Scholar]
- 76.Beach TG, Adler CH, Sue LI, Vedders L, Lue L, White Iii CL, Akiyama H, Caviness JN, Shill HA, Sabbagh MN, et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010;119:689–702. doi: 10.1007/s00401-010-0664-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wakabayashi K, Takahashi H, Takeda S, Ohama E, Ikuta F. Parkinson’s disease: the presence of Lewy bodies in Auerbach’s and Meissner’s plexuses. Acta Neuropathol. 1988;76:217–221. doi: 10.1007/BF00687767. [DOI] [PubMed] [Google Scholar]
- 78.Cersosimo MG, Benarroch EE. Pathological correlates of gastrointestinal dysfunction in Parkinson’s disease. Neurobiol Dis. 2012;46:559–564. doi: 10.1016/j.nbd.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 79.Pouclet H, Lebouvier T, Coron E, Des Varannes SB, Neunlist M, Derkinderen P. A comparison between colonic submucosa and mucosa to detect Lewy pathology in Parkinson’s disease. Neurogastroenterol Motil. 2012;24:e202–e205. doi: 10.1111/j.1365-2982.2012.01887.x. [DOI] [PubMed] [Google Scholar]
- 80.Hansen C, Li JY. Beyond α-synuclein transfer: pathology propagation in Parkinson’s disease. Trends Mol Med. 2012;18:248–255. doi: 10.1016/j.molmed.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 81.Gao HM, Zhang F, Zhou H, Kam W, Wilson B, Hong JS. Neuroinflammation and α-synuclein dysfunction potentiate each other, driving chronic progression of neurodegeneration in a mouse model of Parkinson’s disease. Environ Health Perspect. 2011;119:807–814. doi: 10.1289/ehp.1003013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sanchez-Guajardo V, Tentillier N, Romero-Ramos M. The relation between α-synuclein and microglia in Parkinson’s disease: Recent developments. Neuroscience. 2015;302:47–58. doi: 10.1016/j.neuroscience.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 83.Clairembault T, Leclair-Visonneau L, Neunlist M, Derkinderen P. Enteric glial cells: new players in Parkinson’s disease? Mov Disord. 2015;30:494–498. doi: 10.1002/mds.25979. [DOI] [PubMed] [Google Scholar]
- 84.Devos D, Lebouvier T, Lardeux B, Biraud M, Rouaud T, Pouclet H, Coron E, Bruley des Varannes S, Naveilhan P, Nguyen JM, et al. Colonic inflammation in Parkinson’s disease. Neurobiol Dis. 2013;50:42–48. doi: 10.1016/j.nbd.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 85.Natale G, Pasquali L, Ruggieri S, Paparelli A, Fornai F. Parkinson’s disease and the gut: a well known clinical association in need of an effective cure and explanation. Neurogastroenterol Motil. 2008;20:741–749. doi: 10.1111/j.1365-2982.2008.01162.x. [DOI] [PubMed] [Google Scholar]
- 86.Vizcarra JA, Wilson-Perez HE, Espay AJ. The power in numbers: gut microbiota in Parkinson’s disease. Mov Disord. 2015;30:296–298. doi: 10.1002/mds.26116. [DOI] [PubMed] [Google Scholar]
- 87.Hollister EB, Gao C, Versalovic J. Compositional and functional features of the gastrointestinal microbiome and their effects on human health. Gastroenterology. 2014;146:1449–1458. doi: 10.1053/j.gastro.2014.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Olanow CW, Wakeman DR, Kordower JH. Peripheral alpha-synuclein and Parkinson’s disease. Mov Disord. 2014;29:963–966. doi: 10.1002/mds.25966. [DOI] [PubMed] [Google Scholar]
- 89.Pfeiffer R. Beyond here be dragons: SIBO in Parkinson’s disease. Mov Disord. 2013;28:1764–1765. doi: 10.1002/mds.25705. [DOI] [PubMed] [Google Scholar]
- 90.Friedland RP. Mechanisms of molecular mimicry involving the microbiota in neurodegeneration. J Alzheimers Dis. 2015;45:349–362. doi: 10.3233/JAD-142841. [DOI] [PubMed] [Google Scholar]
- 91.Trudler D, Farfara D, Frenkel D. Toll-like receptors expression and signaling in glia cells in neuro-amyloidogenic diseases: towards future therapeutic application. Mediators Inflamm. 2010;2010 doi: 10.1155/2010/497987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Béraud D, Maguire-Zeiss KA. Misfolded α-synuclein and Toll-like receptors: therapeutic targets for Parkinson’s disease. Parkinsonism Relat Disord. 2012;18 Suppl 1:S17–S20. doi: 10.1016/S1353-8020(11)70008-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 94.Braniste V, Al-Asmakh M, Kowal C, Anuar F, Abbaspour A, Tóth M, Korecka A, Bakocevic N, Ng LG, Kundu P, et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci Transl Med. 2014;6:263ra158. doi: 10.1126/scitranslmed.3009759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sui YT, Bullock KM, Erickson MA, Zhang J, Banks WA. Alpha synuclein is transported into and out of the brain by the blood-brain barrier. Peptides. 2014;62:197–202. doi: 10.1016/j.peptides.2014.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bodea LG, Wang Y, Linnartz-Gerlach B, Kopatz J, Sinkkonen L, Musgrove R, Kaoma T, Muller A, Vallar L, Di Monte DA, et al. Neurodegeneration by activation of the microglial complement-phagosome pathway. J Neurosci. 2014;34:8546–8556. doi: 10.1523/JNEUROSCI.5002-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dobbs RJ, Charlett A, Purkiss AG, Dobbs SM, Weller C, Peterson DW. Association of circulating TNF-alpha and IL-6 with ageing and parkinsonism. Acta Neurol Scand. 1999;100:34–41. doi: 10.1111/j.1600-0404.1999.tb00721.x. [DOI] [PubMed] [Google Scholar]
- 98.Villarán RF, Espinosa-Oliva AM, Sarmiento M, De Pablos RM, Argüelles S, Delgado-Cortés MJ, Sobrino V, Van Rooijen N, Venero JL, Herrera AJ, et al. Ulcerative colitis exacerbates lipopolysaccharide-induced damage to the nigral dopaminergic system: potential risk factor in Parkinson`s disease. J Neurochem. 2010;114:1687–1700. doi: 10.1111/j.1471-4159.2010.06879.x. [DOI] [PubMed] [Google Scholar]
- 99.Matsumoto L, Takuma H, Tamaoka A, Kurisaki H, Date H, Tsuji S, Iwata A. CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS One. 2010;5:e15522. doi: 10.1371/journal.pone.0015522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, Hatanpaa KJ, Lomen-Hoerth C, Kertesz A, Bigio EH, et al. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol Neurodegener. 2013;8:19. doi: 10.1186/1750-1326-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tsuang D, Leverenz JB, Lopez OL, Hamilton RL, Bennett DA, Schneider JA, Buchman AS, Larson EB, Crane PK, Kaye JA, et al. APOE ε4 increases risk for dementia in pure synucleinopathies. JAMA Neurol. 2013;70:223–228. doi: 10.1001/jamaneurol.2013.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bialecka M, Kurzawski M, Klodowska-Duda G, Opala G, Juzwiak S, Kurzawski G, Tan EK, Drozdzik M. CARD15 variants in patients with sporadic Parkinson’s disease. Neurosci Res. 2007;57:473–476. doi: 10.1016/j.neures.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 103.Scheperjans F, Aho V, Pereira PA, Koskinen K, Paulin L, Pekkonen E, Haapaniemi E, Kaakkola S, Eerola-Rautio J, Pohja M, et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov Disord. 2015;30:350–358. doi: 10.1002/mds.26069. [DOI] [PubMed] [Google Scholar]
- 104.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Forsyth CB, Shannon KM, Kordower JH, Voigt RM, Shaikh M, Jaglin JA, Estes JD, Dodiya HB, Keshavarzian A. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLoS One. 2011;6:e28032. doi: 10.1371/journal.pone.0028032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Niehaus I, Lange JH. Endotoxin: is it an environmental factor in the cause of Parkinson’s disease? Occup Environ Med. 2003;60:378. doi: 10.1136/oem.60.5.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Clairembault T, Kamphuis W, Leclair-Visonneau L, Rolli-Derkinderen M, Coron E, Neunlist M, Hol EM, Derkinderen P. Enteric GFAP expression and phosphorylation in Parkinson’s disease. J Neurochem. 2014;130:805–815. doi: 10.1111/jnc.12742. [DOI] [PubMed] [Google Scholar]
- 108.Kelly LP, Carvey PM, Keshavarzian A, Shannon KM, Shaikh M, Bakay RA, Kordower JH. Progression of intestinal permeability changes and alpha-synuclein expression in a mouse model of Parkinson’s disease. Mov Disord. 2014;29:999–1009. doi: 10.1002/mds.25736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tan AH, Mahadeva S, Thalha AM, Gibson PR, Kiew CK, Yeat CM, Ng SW, Ang SP, Chow SK, Tan CT, et al. Small intestinal bacterial overgrowth in Parkinson’s disease. Parkinsonism Relat Disord. 2014;20:535–540. doi: 10.1016/j.parkreldis.2014.02.019. [DOI] [PubMed] [Google Scholar]
- 110.Gabrielli M, Bonazzi P, Scarpellini E, Bendia E, Lauritano EC, Fasano A, Ceravolo MG, Capecci M, Rita Bentivoglio A, Provinciali L, et al. Prevalence of small intestinal bacterial overgrowth in Parkinson’s disease. Mov Disord. 2011;26:889–892. doi: 10.1002/mds.23566. [DOI] [PubMed] [Google Scholar]
- 111.Chen WC, Quigley EM. Probiotics, prebiotics & amp; synbiotics in small intestinal bacterial overgrowth: opening up a new therapeutic horizon! Indian J Med Res. 2014;140:582–584. [PMC free article] [PubMed] [Google Scholar]
- 112.Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liu Y, Qin L, Wilson B, Wu X, Qian L, Granholm AC, Crews FT, Hong JS. Endotoxin induces a delayed loss of TH-IR neurons in substantia nigra and motor behavioral deficits. Neurotoxicology. 2008;29:864–870. doi: 10.1016/j.neuro.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xu MQ, Cao HL, Wang WQ, Wang S, Cao XC, Yan F, Wang BM. Fecal microbiota transplantation broadening its application beyond intestinal disorders. World J Gastroenterol. 2015;21:102–111. doi: 10.3748/wjg.v21.i1.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dobbs RJ, Charlett A, Dobbs SM, Weller C, Iguodala O, Smee C, Bowthorpe J, Taylor D, Bjarnason IT. Towards defining a rigidity-associated pathogenic pathway in idiopathic parkinsonism. Neurodegener Dis. 2012;10:183–186. doi: 10.1159/000332807. [DOI] [PubMed] [Google Scholar]
- 116.Rees K, Stowe R, Patel S, Ives N, Breen K, Clarke CE, Ben-Shlomo Y. Helicobacter pylori eradication for Parkinson’s disease. Cochrane Database Syst Rev. 2011;(11):CD008453. doi: 10.1002/14651858.CD008453.pub2. [DOI] [PubMed] [Google Scholar]
- 117.Nielsen HH, Qiu J, Friis S, Wermuth L, Ritz B. Treatment for Helicobacter pylori infection and risk of Parkinson’s disease in Denmark. Eur J Neurol. 2012;19:864–869. doi: 10.1111/j.1468-1331.2011.03643.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tan HJ, Goh KL. Extragastrointestinal manifestations of Helicobacter pylori infection: facts or myth? A critical review. J Dig Dis. 2012;13:342–349. doi: 10.1111/j.1751-2980.2012.00599.x. [DOI] [PubMed] [Google Scholar]
- 119.Rahne KE, Tagesson C, Nyholm D. Motor fluctuations and Helicobacter pylori in Parkinson’s disease. J Neurol. 2013;260:2974–2980. doi: 10.1007/s00415-013-7089-6. [DOI] [PubMed] [Google Scholar]
- 120.Dobbs SM, Dobbs RJ, Weller C, Charlett A. Link between Helicobacter pylori infection and idiopathic parkinsonism. Med Hypotheses. 2000;55:93–98. doi: 10.1054/mehy.2000.1110. [DOI] [PubMed] [Google Scholar]
- 121.Bjarnason IT, Charlett A, Dobbs RJ, Dobbs SM, Ibrahim MA, Kerwin RW, Mahler RF, Oxlade NL, Peterson DW, Plant JM, et al. Role of chronic infection and inflammation in the gastrointestinal tract in the etiology and pathogenesis of idiopathic parkinsonism. Part 2: response of facets of clinical idiopathic parkinsonism to Helicobacter pylori eradication. A randomized, double-blind, placebo-controlled efficacy study. Helicobacter. 2005;10:276–287. doi: 10.1111/j.1523-5378.2005.00330.x. [DOI] [PubMed] [Google Scholar]
- 122.Lee WY, Yoon WT, Shin HY, Jeon SH, Rhee PL. Helicobacter pylori infection and motor fluctuations in patients with Parkinson’s disease. Mov Disord. 2008;23:1696–1700. doi: 10.1002/mds.22190. [DOI] [PubMed] [Google Scholar]
- 123.Lyte M. Microbial endocrinology as a basis for improved L-DOPA bioavailability in Parkinson’s patients treated for Helicobacter pylori. Med Hypotheses. 2010;74:895–897. doi: 10.1016/j.mehy.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 124.Hashim H, Azmin S, Razlan H, Yahya NW, Tan HJ, Manaf MR, Ibrahim NM. Eradication of Helicobacter pylori infection improves levodopa action, clinical symptoms and quality of life in patients with Parkinson’s disease. PLoS One. 2014;9:e112330. doi: 10.1371/journal.pone.0112330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Blaecher C, Smet A, Flahou B, Pasmans F, Ducatelle R, Taylor D, Weller C, Bjarnason I, Charlett A, Lawson AJ, et al. Significantly higher frequency of Helicobacter suis in patients with idiopathic parkinsonism than in control patients. Aliment Pharmacol Ther. 2013;38:1347–1353. doi: 10.1111/apt.12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Dow CT. M. paratuberculosis and Parkinson’s disease-is this a trigger. Med Hypotheses. 2014;83:709–712. doi: 10.1016/j.mehy.2014.09.025. [DOI] [PubMed] [Google Scholar]
- 127.Li JQ, Tan L, Yu JT. The role of the LRRK2 gene in Parkinsonism. Mol Neurodegener. 2014;9:47. doi: 10.1186/1750-1326-9-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Winklhofer KF. Parkin and mitochondrial quality control: toward assembling the puzzle. Trends Cell Biol. 2014;24:332–341. doi: 10.1016/j.tcb.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 129.Bi W, Zhu L, Jing X, Liang Y, Tao E. Rifampicin and Parkinson’s disease. Neurol Sci. 2013;34:137–141. doi: 10.1007/s10072-012-1156-0. [DOI] [PubMed] [Google Scholar]
- 130.Fiszer U, Fredrikson S, Członkowska A. Humoral response to hsp 65 and hsp 70 in cerebrospinal fluid in Parkinson’s disease. J Neurol Sci. 1996;139:66–70. [PubMed] [Google Scholar]
- 131.Yong J, Lacan G, Dang H, Hsieh T, Middleton B, Wasserfall C, Tian J, Melega WP, Kaufman DL. BCG vaccine-induced neuroprotection in a mouse model of Parkinson’s disease. PLoS One. 2011;6:e16610. doi: 10.1371/journal.pone.0016610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Voreades N, Kozil A, Weir TL. Diet and the development of the human intestinal microbiome. Front Microbiol. 2014;5:494. doi: 10.3389/fmicb.2014.00494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, Thangaraju M, Prasad PD, Manicassamy S, Munn DH, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40:128–139. doi: 10.1016/j.immuni.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Derkinderen P, Shannon KM, Brundin P. Gut feelings about smoking and coffee in Parkinson’s disease. Mov Disord. 2014;29:976–979. doi: 10.1002/mds.25882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wirdefeldt K, Adami HO, Cole P, Trichopoulos D, Mandel J. Epidemiology and etiology of Parkinson’s disease: a review of the evidence. Eur J Epidemiol. 2011;26 Suppl 1:S1–58. doi: 10.1007/s10654-011-9581-6. [DOI] [PubMed] [Google Scholar]
- 136.Hernán MA, Takkouche B, Caamaño-Isorna F, Gestal-Otero JJ. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann Neurol. 2002;52:276–284. doi: 10.1002/ana.10277. [DOI] [PubMed] [Google Scholar]
- 137.Tanaka K, Miyake Y, Fukushima W, Sasaki S, Kiyohara C, Tsuboi Y, Yamada T, Oeda T, Miki T, Kawamura N, et al. Intake of Japanese and Chinese teas reduces risk of Parkinson’s disease. Parkinsonism Relat Disord. 2011;17:446–450. doi: 10.1016/j.parkreldis.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 138.Biedermann L, Brülisauer K, Zeitz J, Frei P, Scharl M, Vavricka SR, Fried M, Loessner MJ, Rogler G, Schuppler M. Smoking cessation alters intestinal microbiota: insights from quantitative investigations on human fecal samples using FISH. Inflamm Bowel Dis. 2014;20:1496–1501. doi: 10.1097/MIB.0000000000000129. [DOI] [PubMed] [Google Scholar]
- 139.Takahashi H, Matsui T, Hisabe T, Hirai F, Takatsu N, Tsurumi K, Kanemitsu T, Sato Y, Kinjyo K, Yano Y, et al. Second peak in the distribution of age at onset of ulcerative colitis in relation to smoking cessation. J Gastroenterol Hepatol. 2014;29:1603–1608. doi: 10.1111/jgh.12616. [DOI] [PubMed] [Google Scholar]
- 140.Nakayama T, Oishi K. Influence of coffee (Coffea arabica) and galacto-oligosaccharide consumption on intestinal microbiota and the host responses. FEMS Microbiol Lett. 2013;343:161–168. doi: 10.1111/1574-6968.12142. [DOI] [PubMed] [Google Scholar]
- 141.Jaquet M, Rochat I, Moulin J, Cavin C, Bibiloni R. Impact of coffee consumption on the gut microbiota: a human volunteer study. Int J Food Microbiol. 2009;130:117–121. doi: 10.1016/j.ijfoodmicro.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 142.Drolet RE, Cannon JR, Montero L, Greenamyre JT. Chronic rotenone exposure reproduces Parkinson’s disease gastrointestinal neuropathology. Neurobiol Dis. 2009;36:96–102. doi: 10.1016/j.nbd.2009.06.017. [DOI] [PubMed] [Google Scholar]
- 143.Sanyal J, Chakraborty DP, Sarkar B, Banerjee TK, Mukherjee SC, Ray BC, Rao VR. Environmental and familial risk factors of Parkinsons disease: case-control study. Can J Neurol Sci. 2010;37:637–642. doi: 10.1017/s0317167100010829. [DOI] [PubMed] [Google Scholar]
- 144.Vlajinac HD, Sipetic SB, Maksimovic JM, Marinkovic JM, Dzoljic ED, Ratkov IS, Kostic VS. Environmental factors and Parkinson’s disease: a case-control study in Belgrade, Serbia. Int J Neurosci. 2010;120:361–367 . doi: 10.3109/00207451003668374. [DOI] [PubMed] [Google Scholar]
- 145.Willis AW, Evanoff BA, Lian M, Galarza A, Wegrzyn A, Schootman M, Racette BA. Metal emissions and urban incident Parkinson disease: a community health study of Medicare beneficiaries by using geographic information systems. Am J Epidemiol. 2010;172:1357–1363. doi: 10.1093/aje/kwq303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jang H, Boltz D, Sturm-Ramirez K, Shepherd KR, Jiao Y, Webster R, Smeyne RJ. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci USA. 2009;106:14063–14068. doi: 10.1073/pnas.0900096106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wu WY, Kang KH, Chen SL, Chiu SY, Yen AM, Fann JC, Su CW, Liu HC, Lee CZ, Fu WM, et al. Hepatitis C virus infection: a risk factor for Parkinson’s disease. J Viral Hepat. 2015;22:784–791. doi: 10.1111/jvh.12392. [DOI] [PubMed] [Google Scholar]