

Abstract
Antibodies can swiftly provide therapeutics to target disease-related molecules discovered in genomic research. Antibody engineering techniques have been actively developed and these technological innovations have intensified the development of therapeutic antibodies. From the mid-1990’s, a series of therapeutic antibodies were launched that are now being used in clinic. The disease areas that therapeutic antibodies can target have subsequently expanded, and antibodies are currently utilized as pharmaceuticals for cancer, inflammatory disease, organ transplantation, cardiovascular disease, infection, respiratory disease, ophthalmologic disease, and so on. This paper briefly describes the modes of action of therapeutic antibodies. Several non-clinical study results of the pathological changes induced by therapeutic antibodies are also presented to aid the future assessment of the toxic potential of an antibody developed as a therapeutic.
Keywords: therapeutic antibody, mode of action, pathological findings, toxicity study
Antibodies can swiftly provide therapeutics to target the disease-related molecules that have been discovered in genomic research because 1) the high level of specificity and affinity to the target molecule or antigen achieves a high level of efficacy and fewer adverse events, 2) their ability to target diverse molecules and the modes of action of the antibodies allow them to be applied to a wide range of therapeutic targets, and 3) modification and refinement by genetic engineering technology and the establishment of recombinant manufacturing technology has made industrial manufacturing possible.
Development of therapeutic antibodies boomed in the 1980’s, and the first therapeutic antibody, a mouse antibody, was launched in 1986 as an immunosuppressive agent used during organ transplantation1,2,3. Although problems, such as mouse antibodies expressing antigenicity in humans, prevented any therapeutic antibodies being launched in the next 10 years, antibody engineering techniques continued to be actively developed and resulted in techniques to produce chimeric antibodies and humanized antibodies from mouse antibodies 4,5,6,7,8. In chimeric antibodies, 33% of the structure originates from mouse, with variable regions from mouse and constant regions from human, and in humanized antibodies, up to 90% of the structure originates from human, with only the antigen binding site in the variable region (complementarity-determining region) originating from mouse. Furthermore, new techniques made it possible to obtain human antibodies from human antibody phage libraries and human antibody–producing mice9,10,11,12,13,14,15. These technological innovations intensified the development of therapeutic antibodies, and from the mid-1990’s, a series of therapeutic antibodies were launched that are now being used in clinic. The disease areas that therapeutic antibodies can target have subsequently expanded, and antibodies are currently utilized as pharmaceuticals for cancer, inflammatory disease, organ transplantation, cardiovascular disease, infection, respiratory disease, ophthalmologic disease, and so on (Table 1).
Table 1. Antibody Type, Target Molecule, Mechanism of Action, and Major Indication of Antibody Pharmaceuticals.
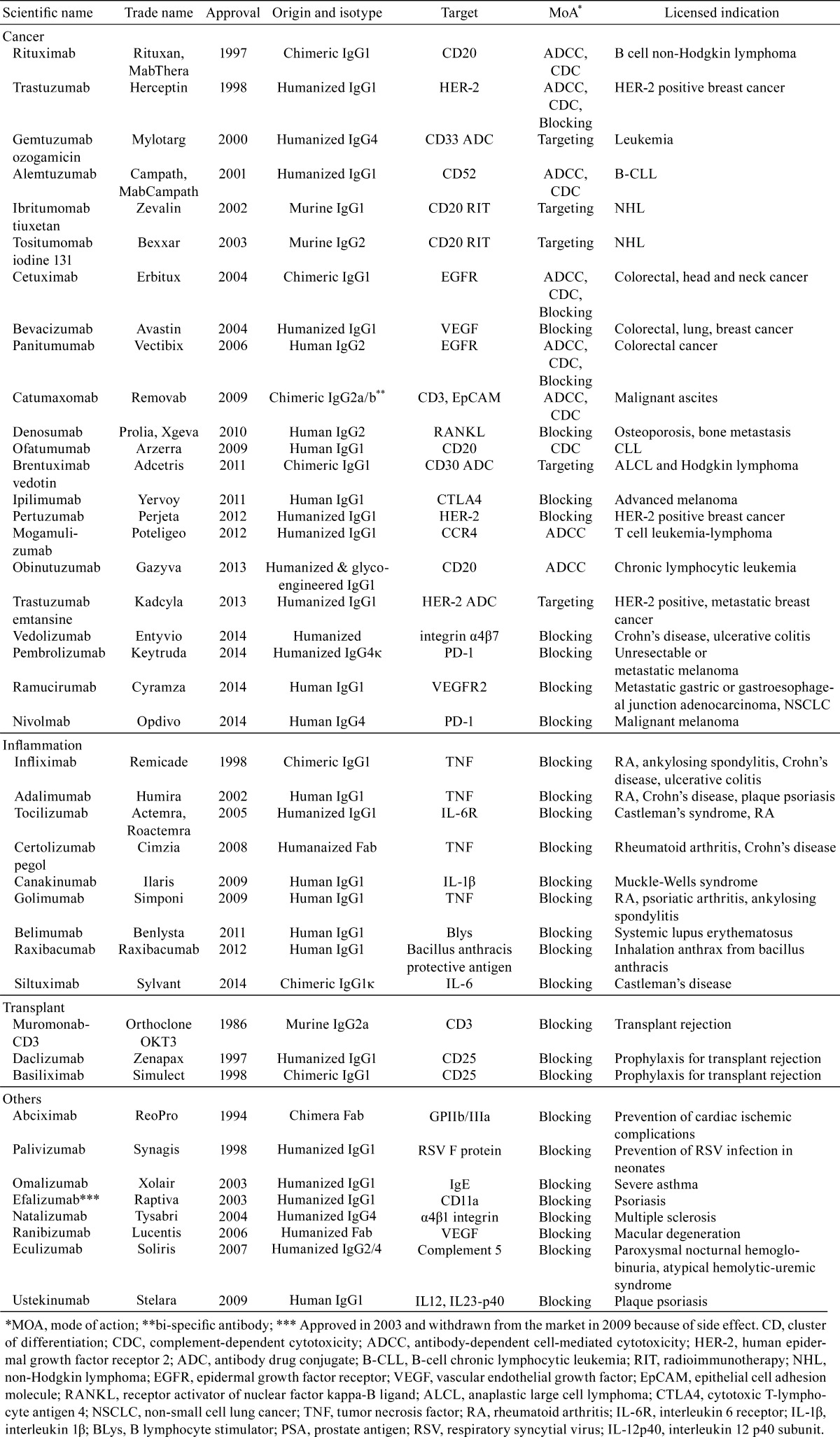
This paper briefly describes the modes of action of therapeutic antibodies. Several non-clinical study results of the pathological changes induced by therapeutic antibodies are also presented to aid the future assessment of the toxic potential of an antibody that is being developed as a therapeutic.
Mechanisms of Action of Therapeutic Antibodies
The efficacy of therapeutic antibodies stems from various natural functions of antibodies — neutralization, antibody-dependent cell-mediated cytotoxic (ADCC) activity, or complement-dependent cytotoxic (CDC) activity —, or the antibody can be utilized as a drug delivery carrier (missile therapy)1 (Fig. 1).
Fig. 1.

Mechanisms of action of therapeutic antibodies.
Neutralization: Many therapeutic antibodies utilize neutralization to block the pathophysiological function of their target molecules1. In this case, antibodies bind to the ligand or receptor that is expressed on the cell surface and block the target signaling pathway. When the signaling in the tumor through these ligands or receptors is diminished, it can result in cellular activity being lost, proliferation being inhibited, pro-apoptotic programs being activated, or cells being resensitized to cytotoxic agents16.
ADCC: To trigger ADCC, the Fv binding domain of an antibody binds to a specific antigen expressed on the surface of a target cell. The antibody is then able to recruit immune-effector cells (such as macrophages and NK cells) that express various receptors able to bind to the Fc and thus activate the immune-effector cells to lyze the target cell17.
CDC: CDC is triggered when the C1 complex binds the antibody–antigen complex, activates a cascade of complement proteins, and causes a complex to form that attacks the membrane. This results in lysis of the target cell17. Both ADCC and CDC are interactions that involve components of the host immune system and, among the therapeutic antibodies being developed for cancer, there are presumably products that utilize more than one mechanism (ADCC, CDC, and neutralizing functions) in their pharmacological actions.
Drug delivery carrier: Antibodies can be applied as drug delivery carriers when conjugated to radioisotopes, toxins, drugs or cytokines17. The advantage of these conjugates over conventional drugs is that cytotoxic agents can be delivered directly and at higher local concentrations to tumor tissues, without causing damage to normal cells.
Antibodies that bind and/or cross-link to target molecules and thus stimulate several signal pathways are also under research. However, these agonistic antibodies have not been placed on the market at this point.
Pathological Findings Induced by Therapeutic Antibodies in Toxicity Studies
Below are examples of the histopathological changes induced by therapeutic antibodies in non-clinical studies. As examples of therapeutic antibodies that use neutralization to block the pathophysiological function of their target antigens, we will show the changes caused by an anti-vascular endothelial growth factor (VEGF) antibody and by an epidermal growth factor receptor (EGFR) antibody. For those that use ADCC and CDC, we will give examples of biological reactions to an anti-CD20 antibody.
Anti-VEGF antibody
Bevacizumab (Avastin®) is an anti-VEGF humanized monoclonal antibody. It binds to VEGF and blocks VEGF from uniting with its receptors (VEGFR-1 and -2), which then blocks the signal transduction of VEGF18. VEGF is the main factor that controls angiogenesis, and its expression is increased in most human tumors and is related to tumor proliferation/metastasis. Hence, bevacizumab was approved for colorectal cancer, non-small cell lung cancer except squamous cell carcinoma, breast cancer, and so on18. Because the therapeutic blocks all the signaling transduced by VEGF, angiogenesis is inhibited in normal organs as well as in tumors.
Cynomolgus monkeys treated repeatedly with bevacizumab via intravenous injection exhibited several pathological adverse effects on the epiphyseal growth plate, ovary, and uterus19. Lesions on the epiphyseal growth plate were characterized by a linear cessation of growth line and chondrocyte hyperplasia20. In the ovary, arrested follicular development and absent corpora lutea were shown, and in the uterus, a decrease in endometrial proliferation and in the number of menstrual cycles were also seen 19, 21.
It is well known that vascularization of the epiphyseal growth plate region represents a key mechanism for chondrogenesis (cartilage production) and osteogenesis (bone formation)22, 23. A small-molecule VEGF inhibitor that inhibited angiogenesis in rats showed epiphyseal growth plate lesions that were characterized by thickening due to the retention of hypertrophic chondrocytes 24, 25. It is reported that vascularization is essential for corpus luteum and endometrial formation26,27,28; therefore, biological reactions caused by an anti-VEGF antibody are considered to be specific reactions by the target molecule in the organs and tissues in which vascularization was constantly maintained.
Anti-EGFR antibody
Cetuximab (Erbitux®) is a recombinant human/mouse chimeric anti-EGFR monoclonal antibody29. Cetuximab binds to EGFR selectively, blocks EGFR from uniting with its ligand, EGF, and then blocks the signal transduction of EGF. EGFR is a transmembrane glycoprotein that is expressed in epithelial tissues and acts as a receptor. Binding of EGFR to EGF induces receptor dimerization and tyrosine autophosphorylation and leads to cell proliferation and differentiation30, 31. EGFR is expressed in normal tissues and also in many solid tumors, including colorectal cancer. Hence, cetuximab is approved for colorectal cancer and squamous cell carcinoma of the head and neck30, 31.
In cynomolgus monkeys, cetuximab was given by repeated intravenous injection and it resulted in dermatologic lesions characterized by hyperkeratosis, parakeratosis, abscess, and acantholysis with bullosa at the external integument. Similar changes were observed in the epithelial mucosa of the nasal passage, esophagus, and tongue at the highest dose level32, 33. In addition, deaths due to sepsis associated with ulcerative dermatitis were observed in the animals at the highest dose level 32, 33.
Anti-CD20 antibody
Rituximab (Rituxan®) is a chimeric murine/human monoclonal antibody targeted against the pan-B-cell marker CD20. Rituximab binds to B cells that express CD20 and induces cell death through CDC or ADCC34. CD20 is expressed in non-neoplastic B cells (pre, immature, mature, and activated) and neoplastic cells derived from B cells. Rituximab is indicated for the treatment of patients with non-Hodgkin’s lymphoma (NHL), chronic lymphocytic leukemia (CLL) and rheumatoid arthritis35,36,37.
In a non-clinical study, rituximab was administered to cynomolgus monkeys repeatedly via intravenous injection (1/ week), and changes were found in immune-hematopoietic tissues. The total number of lymphocytes decreased in peripheral blood owing to a decrease of B cells, and atrophy of lymphoid follicles and a decrease of CD20-positive B cells were seen in the spleen and systemic lymph node38. All of the cells affected by cytotoxicity were B cells that express CD20, and the reaction is considered to be specific to the target molecule.
The changes induced by a therapeutic antibody in non-clinical study are thought of as biological reactions that are dependent on the target molecule39, 40. For example, with a blocking antibody the changes occur in the tissues and organs in which the targeted pathway functions. With antibodies that target specific ligands, changes are found in organs and tissues that express the receptor of the targeted ligand, and with antibodies that target specific receptors, changes are found in organs and tissues that express the targeted receptor. With a cytotoxic antibody the changes are found in the tissues and organs that express the target molecule.
Although the biological reactions induced by a therapeutic antibody are dependent on the target molecule and the target molecules selected in this paper, VEGFR and EGFR, were expressed broadly in normal tissues, the biological changes were not observed in all the organs and tissues that express the target molecule19, 20. With a blocking antibody, differences in the biological reactions may depend not only on expression of the target molecule but also on how the target pathway contributes to maintenance of homeostasis21,22,23. The existence of alternative systems that compensate for the blocked pathway is thought to be an important factor of toxicologic changes.
Cytotoxicity antibodies are reported to have biological reactions that are not induced in all the cells in which antigen is expressed41, 42. We analyzed CDC induction in a non-clinical in vivo model and demonstrated that the biological response to an antibody with a CDC mechanism is regulated not only by the distribution of the target molecule but also by various other factors, ranging from antibody distribution to the nature of the host immune system and the presence of membrane complement regulatory proteins43, 44. Hence, when a therapeutic antibody induces cytotoxic change via the host immune system, CDC, or ADCC, the immune regulatory system is an important factor on the occurrence of toxic effects43.
Future Trends of Therapeutic Antibodies and Pathological Evaluation
Recently, antibody engineering techniques have progressed and it is now possible to create antibodies with a diverse selection of functions, such as antibodies with more efficient and long-lasting neutralizing effects, agents that cause cytotoxicity at lower molecule expression levels, or bispecific antibodies that can recognize two different molecules simultaneously to induce new biological responses45,46,47,48. These recent advances along with the discovery of novel target molecules shed light on the possibility of new therapies. As the functions and target molecules of antibodies become more and more diverse, it becomes increasingly necessary to understand how the target molecule functions biologically and what will be the biological response to the modified functions induced by the antibody. The toxicological pathology associated with these issues will also need to be evaluated and researched most carefully.
Footnotes
Disclosure of Potential Conflicts of Interest: The authors are employees of Chugai Pharmaceutical Co., Ltd., and declare no other potential conflict of interest.
References
- 1.Buss NA, Henderson SJ, McFarlane M, Shenton JM, and de Haan L. Monoclonal antibody therapeutics: history and future. Curr Opin Pharmacol. 12: 615–622. 2012. [DOI] [PubMed] [Google Scholar]
- 2.Emmons C, and Hunsicker LG. Muromonab-CD3 (Orthoclone OKT3): the first monoclonal antibody approved for therapeutic use. Iowa Med. 77: 78–82. 1987. [PubMed] [Google Scholar]
- 3.Goldstein G. Overview of the development of Orthoclone OKT3: monoclonal antibody for therapeutic use in transplantation. Transplant Proc. 19(Suppl 1): 1–6. 1987. [PubMed] [Google Scholar]
- 4.Morrison SL, Johnson MJ, Herzenberg LA, and Oi VT. Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Proc Natl Acad Sci USA. 81: 6851–6855. 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Presta LG. Engineering of therapeutic antibodies to minimize immunogenicity and optimize function. Adv Drug Deliv Rev. 58: 640–656. 2006. [DOI] [PubMed] [Google Scholar]
- 6.Jones PT, Dear PH, Foote J, Neuberger MS, and Winter G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature. 321: 522–525. 1986. [DOI] [PubMed] [Google Scholar]
- 7.Boulianne GL, Hozumi N, and Shulman MJ. Production of functional chimaeric mouse/human antibody. Nature. 312: 643–646. 1984. [DOI] [PubMed] [Google Scholar]
- 8.Green LL, Hardy MC, Maynard-Currie CE, Tsuda H, Louie DM, Mendez MJ, Abderrahim H, Noguchi M, Smith DH, Zeng Y, David NE, Sasai H, Garza D, Brenner DG, Hales JF, McGuinness RP, Capon DJ, Klapholz S, and Jakobovits A. Antigen-specific human monoclonal antibodies from mice engineered with human Ig heavy and light chain YACs. Nat Genet. 7: 13–21. 1994. [DOI] [PubMed] [Google Scholar]
- 9.Hoet RM, Cohen EH, Kent RB, Rookey K, Schoonbroodt S, Hogan S, Rem L, Frans N, Daukandt M, Pieters H, van Hegelsom R, Neer NC, Nastri HG, Rondon IJ, Leeds JA, Hufton SE, Huang L, Kashin I, Devlin M, Kuang G, Steukers M, Viswanathan M, Nixon AE, Sexton DJ, Hoogenboom HR, and Ladner RC. Generation of high-affinity human antibodies by combining donor-derived and synthetic complementarity-determining-region diversity. Nat Biotechnol. 23: 344–348. 2005. [DOI] [PubMed] [Google Scholar]
- 10.Jostock T, Vanhove M, Brepoels E, Van Gool R, Daukandt M, Wehnert A, Van Hegelsom R, Dransfield D, Sexton D, Devlin M, Ley A, Hoogenboom H, and Müllberg J. Rapid generation of functional human IgG antibodies derived from Fab-on-phage display libraries. J Immunol Methods. 289: 65–80. 2004. [DOI] [PubMed] [Google Scholar]
- 11.Lonberg N, Taylor LD, Harding FA, Trounstine M, Higgins KM, Schramm SR, Kuo CC, Mashayekh R, Wymore K, McCabe JG, Munoz-O’regan D, O’Donnell SL, Lapachet ESG, Bengoechea T, Fishwild DM, Carmack CE, Kay RM, and Huszar D. Antigen-specific human antibodies from mice comprising four distinct genetic modifications. Nature. 368: 856–859. 1994. [DOI] [PubMed] [Google Scholar]
- 12.McCafferty J, Griffiths AD, Winter G, and Chiswell DJ. Phage antibodies: filamentous phage displaying antibody variable domains. Nature. 348: 552–554. 1990. [DOI] [PubMed] [Google Scholar]
- 13.Vaughan TJ, Williams AJ, Pritchard K, Osbourn JK, Pope AR, Earnshaw JC, McCafferty J, Hodits RA, Wilton J, and Johnson KS. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat Biotechnol. 14: 309–314. 1996. [DOI] [PubMed] [Google Scholar]
- 14.Vaughan TJ, Osbourn JK, and Tempest PR. Human antibodies by design. Nat Biotechnol. 16: 535–539. 1998. [DOI] [PubMed] [Google Scholar]
- 15.Winter G, Griffiths AD, Hawkins RE, and Hoogenboom HR. Making antibodies by phage display technology. Annu Rev Immunol. 12: 433–455. 1994. [DOI] [PubMed] [Google Scholar]
- 16.Cavallo F, Calogero RA, and Forni G. Are oncoantigens suitable targets for anti-tumour therapy? Nat Rev Cancer. 7: 707–713. 2007. [DOI] [PubMed] [Google Scholar]
- 17.Zafir-Lavie I, Michaeli Y, and Reiter Y. Novel antibodies as anticancer agents. Oncogene. 26: 3714–3733. 2007. [DOI] [PubMed] [Google Scholar]
- 18.Lyseng-Williamson KA, and Robinson DM. Spotlight on bevacizumab in advanced colorectal cancer, breast cancer, and non-small cell lung cancer. BioDrugs. 20: 193–195. 2006. [DOI] [PubMed] [Google Scholar]
- 19.Ryan AM, Eppler DB, Hagler KE, Bruner RH, Thomford PJ, Hall RL, Shopp GM, and O’Neill CA. Preclinical safety evaluation of rhuMAbVEGF, an antiangiogenic humanized monoclonal antibody. Toxicol Pathol. 27: 78–86. 1999. [DOI] [PubMed] [Google Scholar]
- 20.Hall AP, Westwood FR, and Wadsworth PF. Review of the effects of anti-angiogenic compounds on the epiphyseal growth plate. Toxicol Pathol. 34: 131–147. 2006. [DOI] [PubMed] [Google Scholar]
- 21.Ferrara N, Chen H, Davis-Smyth T, Gerber HP, Nguyen TN, Peers D, Chisholm V, Hillan KJ, and Schwall RH. Vascular endothelial growth factor is essential for corpus luteum angiogenesis. Nat Med. 4: 336–340. 1998. [DOI] [PubMed] [Google Scholar]
- 22.Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, and Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med. 5: 623–628. 1999. [DOI] [PubMed] [Google Scholar]
- 23.Gerber HP, and Ferrara N. Angiogenesis and bone growth. Trends Cardiovasc Med. 10: 223–228. 2000. [DOI] [PubMed] [Google Scholar]
- 24.Wedge SR, Ogilvie DJ, Dukes M, Kendrew J, Curwen JO, Hennequin LF, Thomas AP, Stokes ES, Curry B, Richmond GH, and Wadsworth PF. ZD4190: an orally active inhibitor of vascular endothelial growth factor signaling with broad-spectrum antitumor efficacy. Cancer Res. 60: 970–975. 2000. [PubMed] [Google Scholar]
- 25.Beebe JS, Jani JP, Knauth E, Goodwin P, Higdon C, Rossi AM, Emerson E, Finkelstein M, Floyd E, Harriman S, Atherton J, Hillerman S, Soderstrom C, Kou K, Gant T, Noe MC, Foster B, Rastinejad F, Marx MA, Schaeffer T, Whalen PM, and Roberts WG. Pharmacological characterization of CP-547,632, a novel vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for cancer therapy. Cancer Res. 63: 7301–7309. 2003. [PubMed] [Google Scholar]
- 26.Maas JW, Groothuis PG, Dunselman GA, de Goeij AF, Struyker Boudier HA, and Evers JL. Endometrial angiogenesis throughout the human menstrual cycle. Hum Reprod. 16: 1557–1561. 2001. [DOI] [PubMed] [Google Scholar]
- 27.Reynolds LP, Grazul-Bilska AT, and Redmer DA. Angiogenesis in the female reproductive organs: pathological implications. Int J Exp Pathol. 83: 151–163. 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson RS, Woad KJ, Hammond AJ, Laird M, Hunter MG, and Mann GE. Angiogenesis and vascular function in the ovary. Reproduction. 138: 869–881. 2009. [DOI] [PubMed] [Google Scholar]
- 29.Blick SK, and Scott LJ. Cetuximab: a review of its use in squamous cell carcinoma of the head and neck and metastatic colorectal cancer. Drugs. 67: 2585–2607. 2007. [DOI] [PubMed] [Google Scholar]
- 30.Harding J, and Burtness B. Cetuximab: an epidermal growth factor receptor chemeric human-murine monoclonal antibody. Drugs Today (Barc). 41: 107–127. 2005. [DOI] [PubMed] [Google Scholar]
- 31.Cohen MH, Chen H, Shord S, Fuchs C, He K, Zhao H, Sickafuse S, Keegan P, and Pazdur R. Approval summary: Cetuximab in combination with cisplatin or carboplatin and 5-fluorouracil for the first-line treatment of patients with recurrent locoregional or metastatic squamous cell head and neck cancer. Oncologist. 18: 460–466. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pharmaceuticals and medical devices agency. Interview form: Erbitux. 2015, from Pharmaceuticals and medical devices agency website: http://www.info.pmda.go.jp/go/interview/1/380079_4291415A1021_2_1F.
- 33.U.S. food and drug administration. Erbitux (cetuximab) prescribing information, 2015, from U.S. food and drug administration website: http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/125084s262lbl.pdf.
- 34.Cerny T, Borisch B, Introna M, Johnson P, and Rose AL. Mechanism of action of rituximab. Anticancer Drugs. 13(Suppl 2): S3–S10. 2002. [DOI] [PubMed] [Google Scholar]
- 35.Leget GA, and Czuczman MS. Use of rituximab, the new FDA-approved antibody. Curr Opin Oncol. 10: 548–551. 1998. [DOI] [PubMed] [Google Scholar]
- 36.Plosker GL, and Figgitt DP. Rituximab: a review of its use in non-Hodgkin’s lymphoma and chronic lymphocytic leukaemia. Drugs. 63: 803–843. 2003. [DOI] [PubMed] [Google Scholar]
- 37.Buch MH, Smolen JS, Betteridge N, Breedveld FC, Burmester G, Dörner T, Ferraccioli G, Gottenberg JE, Isaacs J, Kvien TK, Mariette X, Martin-Mola E, Pavelka K, Tak PP, van der Heijde D, van Vollenhoven RF, and Emery P. Rituximab Consensus Expert Committee. Updated consensus statement on the use of rituximab in patients with rheumatoid arthritis. Ann Rheum Dis. 70: 909–920. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mao CP, Brovarney MR, Dabbagh K, Birnböck HF, Richter WF, and Del Nagro CJ. Subcutaneous versus intravenous administration of rituximab: pharmacokinetics, CD20 target coverage and B-cell depletion in cynomolgus monkeys. PLoS ONE. 8: e80533 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Toma MB, and Medina PJ. Update on targeted therapy - Focus on monoclonal antibodies. J Pharm Pract. 21: 4–16. 2008. [Google Scholar]
- 40.Hansel TT, Kropshofer H, Singer T, Mitchell JA, and George AJ. The safety and side effects of monoclonal antibodies. Nat Rev Drug Discov. 9: 325–338. 2010. [DOI] [PubMed] [Google Scholar]
- 41.Horvat M, Kloboves Prevodnik V, Lavrencak J, and Jezersek Novakovic B. Predictive significance of the cut-off value of CD20 expression in patients with B-cell lymphoma. Oncol Rep. 24: 1101–1107. 2010. [PubMed] [Google Scholar]
- 42.Perz J, Topaly J, Fruehauf S, Hensel M, and Ho AD. Level of CD 20-expression and efficacy of rituximab treatment in patients with resistant or relapsing B-cell prolymphocytic leukemia and B-cell chronic lymphocytic leukemia. Leuk Lymphoma. 43: 149–151. 2002. [DOI] [PubMed] [Google Scholar]
- 43.Kato C, Kato A, Adachi K, Fujii E, Isobe K, Matsushita T, Watanabe T, and Suzuki M. Anti-Thy-1 Antibody-mediated Complement-dependent Cytotoxicity is Regulated by the Distribution of Antigen, Antibody and Membrane Complement Regulatory Proteins in Rats. J Toxicol Pathol. 26: 41–49. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kato C, Kato A, Adachi K, Fujii E, Isobe K, Watanabe T, Ito T, and Suzuki M. Expression of Membrane Complement Regulatory Proteins Crry and CD55 in Normal Rats. J Toxicol Pathol. 26: 223–226. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Igawa T, Mimoto F, and Hattori K. pH-dependent antigen-binding antibodies as a novel therapeutic modality. Biochim Biophys Acta. 1844: 1943–1950. 2014. [DOI] [PubMed] [Google Scholar]
- 46.Igawa T, Tsunoda H, Kuramochi T, Sampei Z, Ishii S, and Hattori K. Engineering the variable region of therapeutic IgG antibodies. MAbs. 3: 243–252. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.May C, Sapra P, and Gerber HP. Advances in bispecific biotherapeutics for the treatment of cancer. Biochem Pharmacol. 84: 1105–1112. 2012. [DOI] [PubMed] [Google Scholar]
- 48.Liu JK. The history of monoclonal antibody development - Progress, remaining challenges and future innovations. Ann Med Surg (Lond). 3: 113–116. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]