

Abstract
Recessive mutations in the ion channel-encoding KCNQ1 gene may cause Jervell and Lange-Nielsen syndrome (JLNS), a fatal cardiac disease leading to arrhythmia and sudden cardiac death in young patients. Mutations in KCNQ1 may also cause a milder and dominantly inherited form of the disease, long QT syndrome 1 (LQT1). However, why some mutations cause LQT1 and others cause JLNS can often not be understood a priori. In a recent study,1 we have generated human induced pluripotent stem cell (hiPSC) models of JLNS. Our work mechanistically revealed how distinct classes of JLNS-causing genetic lesions, namely, missense and splice-site mutations, may promote the typical severe features of the disease at the cellular level. Interestingly, the JLNS models also displayed highly sensitive responses to pro-arrhythmic stresses. We hence propose JLNS hiPSCs as a powerful system for evaluating both phenotype-correcting as well as cardiotoxicity-causing drug effects.
Keywords: cardiac differentiation, disease modeling, drug testing, human iPS cells; Jervell and Lange-Nielsen syndrome
Potassium ion influx plays a key role in repolarizing the cardiomyocyte outer membrane during action potential generation. Mutations in genes encoding the major potassium ion channels mediating this event, KCNQ1 and HERG, therefore account for ∼90% of cases of inherited long QT syndrome.2 The latter term is based on the defining diagnostic feature of the disease, a prolongation of the QT interval in a patient's electrocardiogram. As a consequence of defective ventricular repolarization, the T wave signal is shifted toward a later time-point (frequency-corrected QT interval ≥ 440 ms). Hence, LQT patients frequently display a high incidence of cardiac events including ventricular fibrillation, syncope, or life-threatening torsade de pointes (TdP) tachycardia. The two potassium channels encoded by the above-mentioned genes, KvLQT1 and hERG, serve similar functions and conduct the slow and fast delayed rectifier potassium currents IKs and IKr, respectively. Nonetheless, their physiological roles appear to be somewhat different. While hERG is strictly required under resting conditions, KvLQT1 (encoded by KCNQ1) becomes rapidly activated under physical exercise and other adrenergic stress conditions–when there is a need for accelerated repolarization at concomitantly increased heart beat rates. Consequently, LQT1 patients with dominant mutations in KCNQ1 are at high risk for cardiac events under catecholaminergic stress conditions and hence, these individuals are frequently treated with beta-blockers.3 JLNS patients with recessive KCNQ1 mutations, however, display particularly severe cardiac symptoms as well as inner ear deafness, and cannot be sufficiently protected by beta-blocker therapy.4 Alternative therapeutic strategies to treat JLNS are therefore required.
Predicting the severity of the consequences of a given KCNQ1 mutation and its mechanism of action a priori, however, is not a trivial task. This is because KvLQT1 is a multimeric channel and mutations in KCNQ1 may compromise function at different levels, i.e. affect channel trafficking through the ER/Golgi apparatus, interfere with tetrameric channel assembly, impact channel functionality, or affect a combination of these mechanisms. Insights in this context may be obtained from studying the disease at the cellular level, in a human cardiac context. Patient-specific hiPSC models, from which cardiomyocytes harbouring a given mutation can be derived, have proven to be a powerful system for studying various variants of cardiac channelopathies.5 In this regard, Moretti et al.6 as well as Egashira et al.7 previously investigated disease mechanisms underlying the dominant LQT1 syndrome, using patient-derived hiPSCs with heterozygous point mutations in KCNQ1. Both groups found that the point mutations were causing defects in channel biogenesis, i.e., retention of KvLQT1 in the ER/Golgi apparatus. In heterozygous hiPSC-derived cardiomyocytes, however, wild-type subunits of the channel would also be affected by this trafficking defect if the mutant proteins maintain the capability of co-assembling. In this way, a point mutation on just one allele may compromise overall channel function–i.e. the amount of KvLQT1 reaching the outer membrane in these cases–by more than 50% and consequently exert a dominant negative effect.6,7 How, then, does this compare to JLNS-causing mutations that tend to evoke recessive symptoms?
We sought to address this question by selecting 2 representative types of JLNS-causing mutations–a missense mutation8 and a newly identified putative splice-site mutation. Using a combination of approaches, hiPSC derivation from patient biopsies and genetic engineering, we thus generated 2 sets of cell lines. These collections comprised heterozygous and homozygous hiPSCs with the missense mutation plus wild-type control, as well as heterozygous and homozygous hiPSCs with the putative splice mutation plus wild-type control. All hiPSC lines showed typical features of pluripotency and could readily be differentiated into cardiomyocytes. Because the serverity of cardiac phenotypes could potentially be influenced by the expression levels of ion channels in the differentiated progeny, we carefully assessed the kinetics of ion channel induction. Both the HERG and KCNQ1 genes became induced to rather stable expression levels over a time-period of ∼3–4 weeks after the induction of differentiation (Fig. 1A). These induction kinetics highlighted the need for longer term culture-induced maturation9 of the hiPSC-derived cardiomyocytes (CMs) although the spontaneous contractions actually tended to commence much earlier, already after ∼1 week of differentiation. Such early-stage hiPSC-CMs, however, may be unsuitable for phenotype analysis since KvLQT1 is undetectable by pharmacological assays at this stage.10 In contrast, later stage hiPSC-CMs express both HERG and KCNQ1 at physiological levels.1 This was also demonstrated using hiPSC-CM analysis on multielectrode arrays (MEAs) as the T wave-like signals could be shifted after blocking the 2 potassium channels (Fig. 1B).
Figure 1.
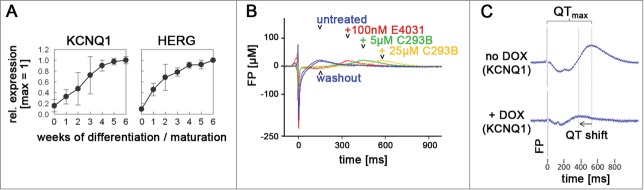
Functional expression of the KCNQ1 and HERG ion channel genes in hiPSC-CMs and rescue of JLNS hiPSC-CMs. (A) Induction kinetics of the KCNQ1 and HERG genes upon long-term culture of hiPSC-CMs. Spontaneous beating is first observed at ∼1 wk. 0 wk cells are undifferentiated hiPSCs. Data denote mean values ± SEM from independent experiments (qRT-PCR data). (B) Drug testing of ∼4 wk-old wild-type hiPSC-CMs on MEA chips using specific hERG (E4031) and KvLQT1 (C293B, chromanol 293B) inhibitors. Note that the T wave-like peak is reversibly shifted toward increased field potential (FP) durations upon addition of these molecules (representative MEA recordings). (C) Transgenic rescue of JLNS hiPSC-CMs using inducible KCNQ1 expression. Doxycycline (DOX) addition induces KCNQ1 leading to FPD shortening (representative MEA spectra).
Moreover, there is a peculiar phenomenon concerning the allele-specific expression of KCNQ1: This is an imprinted gene and, accordingly, it is expressed from only one allele in the early embryo. Later in development, however, in the mouse heart, KCNQ1 expression becomes biallelic.11 Hence, for comparing cardiac phenotypes between heterozygous and homozygous disease hiPSC-CMs, it was important for us to check whether this also holds true for our human in-vitro system. Strikingly, matured hiPSC-CMs indeed showed an epigenetic activation of the second KCNQ1 allele as compared to undifferentiated hiPSCs, similar to the in-vivo scenario.1 We have recently made similar observations comparing immature and longer-term cultured CMs derived from human ES cells.10 These results suggest that KCNQ1 imprinting erasure is a universal feature of human pluripotent stem cell-derived CMs cultured long-term, again highlighting the requirement for in-vitro maturation for functional downstream analyses.
Subsequently, electrophysiological analysis of CMs derived from the different hiPSC lines indeed revealed severe repolarization defects in the JLNS hiPSC-CMs. Action potential durations at the single-cell level as well as field potential durations monitoring bulk cultures were strongly prolonged as compared to wild-type hiPSC-CMs.1 Interestingly, in case of the putative splice mutation, these parameters were entirely unchanged in the heterozygous state, in agreement with the recessive symptoms in JLNS. In comparison, the heterozygous missense hiPSC-CMs showed somewhat intermediate phenotypes, clustering in-between the JLNS-CMs and the wild-type controls. Nonetheless, these observations appeared to be in agreement with the patient data from which these hiPSCs were derived: This individual was originally diagnosed as an LQT1 patient on the basis of a prolonged QT interval. She was, however, asymptomatic, implying that the heterozygous phenotype was mild, like in several other reported cases.12
How do different classes of JLNS-causing KCNQ1 mutations lead to similarly severe phenotypes at the cellular level? We subsequently addressed this question by investigating the molecular consequences of our 2 mutations. The putative splice-site mutation, disrupting the splice acceptor site of KCNQ1 intron 2, was indeed confirmed to cause the skipping of the downstream exon at the RNA level. This defect also induced a shift of the open reading frame and, consequently, mRNA degradation by the nonsense-mediated decay machinery.13 As a result, we were unable to detect any functional channel at the protein level, and the disease phenotype could readily be rescued using KCNQ1 transgene delivery (Fig. 1C). Hence, considering the lack of phenotypes in the corresponding heterozygous hiPSC-CMs, the remaining wild-type allele in these cells could apparently fully compensate for the knock-out mutation on the second gene copy. Such allelic compensation is very common for most genes and may seem rather expected at first sight. However, in comparison to the above-mentioned paradigms of dominant-negative trafficking mutations in KCNQ1, this finding provided a straightforward–albeit trivial–explanation for our phenotypic observations: The JLNS mutant allele is unable to interfere with the function of the wild-type one in heterozygous hiPSC-CMs simply because it gives rise to an unstable message and a nonsense protein. Despite only being demonstrated for a single case, we think these findings may have universal validity for most–if not all–JLNS-causing frameshift mutations in KCNQ1.2
In contrast, our missense mutation causes an arginine-to-glutamine substitution in the C-terminal intracellular domain of KvLQT1 (R594Q). Cellular analysis revealed a moderate trafficking defect leading to reduced amounts of channel reaching the outer membrane even in heterozygous hiPSC-CMs. This observation was also confirmed using isolated trafficking assays in a heterologous system.1 The severe cardiac phenotype seen in homozygous hiPSC-CMs may therefore be a result of an additive, gene dosage-dependent mechanism. Considering the moderateness of the phenotypes in our heterozygous point mutant, we feel inclined to agree with a view by Schwarz et al.:4 JLNS-causing mutations may in general cause milder phenotypes in heterozygosity as compared to LQT1-causing ones. The fact that they also occur as homozygous mutations–and then cause severe symptoms–may hence reflect that milder or asymptomatic, heterozygous defects in KCNQ1 are more likely to breed to homozygosity in the human population. As cautionary note, the fact that in our cellular system the homozygous-mutated genotype showed some residual IKs current (∼20%) would also support the possibility that homozygous patients harbouring the R594Q amino acid exchange could also present with autosomic recessive (AR) LQTS (i.e., without deafness).
The different mechanisms revealed by our investigation may also have consequences for putative therapeutic strategies. For instance, while in case of a missense mutation in KCNQ1, the KvLQT1 channel itself could be considered as a drug target, in case of frameshift mutations it cannot.14 We therefore challenged our system and asked whether our JLNS hiPSC-CMs could be used to evoke arrhythmia in response to drug-induced or adrenergic stresses, similar to the patient scenario. MEAs, monitoring spontaneous beating behavior of bulk hiPSC-CM cultures, proved to be an ideal system for recording these integrated responses. Indeed, administration of the cardiotoxic compound cisapride caused arrhythmic responses preferably in JLNS-CMs, i.e. at significantly lower dosages than in wild-type or heterozygous hiPSC-CMs (Fig. 2, top panel). Recently, we were also able to reproducibly and specifically cause arrhythmia in JLNS-CMs by using physiological adrenergic stress induced using isoprenaline administration. Interestingly, in this case, the cellular responses showed similarity with life-threatening torsade de pointes-like arryhthmia that is commonly associated with LQT syndrome (Fig. 2, bottom panels).15 In an effort to correct these induced-arrhythmia phenotypes in our cellular models, we furthermore considered pharmacological activation of the cooperating potassium channel, hERG.16-18 Indeed, the drug-induced arrhythmic responses could be fully blocked using this strategy, although these beneficial effects required high concentrations of the molecule.1 Therefore, more potent hERG activators will need to be developed to potentially consider applications in JLNS patients. Furthermore, undesired drug-induced cardiotoxicity is a major hurdle in drug development and hiPSC-CMs are emerging as an attractive test system–as an alternative to animal trials.19-21 Our preliminary data suggests that JLNS hiPSC-CMs may be a particularly well-suited model in this regard because of their extraordinary sensitivity and propensity to display arrhythmia as an integrated physiological response.1
Figure 2.
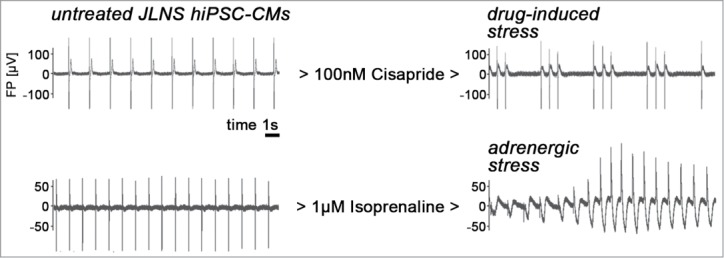
Stress-induced arrhythmia in JLNS hiPSC-CMs (representative MEA recordings). Left: Untreated cells beating spontaneously at a frequency of ∼0.5–1 Hz. Right: Administration of cisapride or isoprenaline induces arrhythmia in JLNS hiPSC-CMs. Note the torsade de point-like shape of the MEA spectrum under adrenergic stress conditions.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We acknowledge the contributions of all the co-authors of our recent PNAS publication.
Funding
Grant support: EU Marie Curie FP7-people-2011-IEF program (MB); Eva Luise and Horst Köhler Foundation (BG); Bundesinstitut für Risikobewertung, FK-3-1329-471 (BG); Chemical Genomics Center of the Max Planck Society (BG).
References
- 1. Zhang M, D'Aniello C, Verkerk AO, Wrobel E, Frank S, Ward-van Oostwaard D, Piccini I, Freund C, Rao J, Seebohm G, et al. . Recessive cardiac phenotypes in induced pluripotent stem cell models of Jervell and Lange-Nielsen syndrome: disease mechanisms and pharmacological rescue. Proc Natl Acad Sci U S A 2014; 111:E5383-92; PMID:25453094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, et al. . Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 2000; 102:1178-85; PMID:10973849; http://dx.doi.org/ 10.1161/01.CIR.102.10.1178 [DOI] [PubMed] [Google Scholar]
- 3. Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, Denjoy I, Guicheney P, Breithardt G, Keating MT, et al. . Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 2001; 103:89-95; PMID:11136691; http://dx.doi.org/ 10.1161/01.CIR.103.1.89 [DOI] [PubMed] [Google Scholar]
- 4. Schwartz PJ, Spazzolini C, Crotti L, Bathen J, Amlie JP, Timothy K, Shkolnikova M, Berul CI, Bitner-Glindzicz M, Toivonen L, et al. . The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation 2006; 113:783-90; PMID:16461811; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.105.592899 [DOI] [PubMed] [Google Scholar]
- 5. Bellin M, Marchetto MC, Gage FH, Mummery CL. Induced pluripotent stem cells: the new patient? Nat Rev Mol Cell Biol 2012; 13:713-26; PMID:23034453; http://dx.doi.org/ 10.1038/nrm3448 [DOI] [PubMed] [Google Scholar]
- 6. Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flugel L, Dorn T, Goedel A, Hohnke C, Hofmann F, et al. . Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med 2010; 363:1397-409; PMID:20660394; http://dx.doi.org/ 10.1056/NEJMoa0908679 [DOI] [PubMed] [Google Scholar]
- 7. Egashira T, Yuasa S, Suzuki T, Aizawa Y, Yamakawa H, Matsuhashi T, Ohno Y, Tohyama S, Okata S, Seki T, et al. . Disease characterization using LQTS-specific induced pluripotent stem cells. Cardiovasc Res 2012; 95:419-29; PMID:22739119; http://dx.doi.org/ 10.1093/cvr/cvs206 [DOI] [PubMed] [Google Scholar]
- 8. Huang L, Bitner-Glindzicz M, Tranebjaerg L, Tinker A. A spectrum of functional effects for disease causing mutations in the Jervell and Lange-Nielsen syndrome. Cardiovasc Res 2001; 51:670-80; PMID:11530100; http://dx.doi.org/ 10.1016/S0008-6363(01)00350-9 [DOI] [PubMed] [Google Scholar]
- 9. Yang X, Pabon L, Murry CE. Engineering adolescence: maturation of human pluripotent stem cell-derived cardiomyocytes. Circ Res 2014; 114:511-23; PMID:24481842; http://dx.doi.org/ 10.1161/CIRCRESAHA.114.300558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang M, Schulte JS, Heinick A, Piccini I, Rao J, Quaranta R, Zeuschner D, Malan D, Kim KP, Ropke A, et al. Universal cardiac induction of human pluripotent stem cells in 2D and 3D formats - Implications for in-vitro maturation. Stem cells 2015; doi: 10.1002/stem.1964. [DOI] [PubMed] [Google Scholar]
- 11. Lee MP, Hu RJ, Johnson LA, Feinberg AP. Human KVLQT1 gene shows tissue-specific imprinting and encompasses Beckwith-Wiedemann syndrome chromosomal rearrangements. Nat Genet 1997; 15:181-5; PMID:9020845; http://dx.doi.org/ 10.1038/ng0297-181 [DOI] [PubMed] [Google Scholar]
- 12. Splawski I, Timothy KW, Vincent GM, Atkinson DL, Keating MT. Molecular basis of the long-QT syndrome associated with deafness. N Engl J Med 1997; 336:1562-7; PMID:9164812; http://dx.doi.org/ 10.1056/NEJM199705293362204 [DOI] [PubMed] [Google Scholar]
- 13. Maquat LE. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol 2004; 5:89-99; PMID:15040442; http://dx.doi.org/ 10.1038/nrm1310 [DOI] [PubMed] [Google Scholar]
- 14. Nattel S, Carlsson L. Innovative approaches to anti-arrhythmic drug therapy. Nat Rev Drug Discov 2006; 5:1034-49; PMID:17139288; http://dx.doi.org/ 10.1038/nrd2112 [DOI] [PubMed] [Google Scholar]
- 15. Morita H, Wu J, Zipes DP. The QT syndromes: long and short. Lancet 2008; 372:750-63; PMID:18761222; http://dx.doi.org/ 10.1016/S0140-6736(08)61307-0 [DOI] [PubMed] [Google Scholar]
- 16. Hansen RS, Diness TG, Christ T, Demnitz J, Ravens U, Olesen SP, Grunnet M. Activation of human ether-a-go-go-related gene potassium channels by the diphenylurea 1,3-bis-(2-hydroxy-5-trifluoromethyl-phenyl)-urea (NS1643). Mol Pharmacol 2006; 69:266-77; PMID:16219910 [DOI] [PubMed] [Google Scholar]
- 17. Casis O, Olesen SP, Sanguinetti MC. Mechanism of action of a novel human ether-a-go-go-related gene channel activator. Mol Pharmacol 2006; 69:658-65; PMID:16284303; http://dx.doi.org/ 10.1124/mol.105.019943 [DOI] [PubMed] [Google Scholar]
- 18. Xu X, Salata JJ, Wang J, Wu Y, Yan GX, Liu T, Marinchak RA, Kowey PR. Increasing I(Ks) corrects abnormal repolarization in rabbit models of acquired LQT2 and ventricular hypertrophy. Am J Physiol Heart Circ Physiol 2002; 283:H664-70; PMID:12124214 [DOI] [PubMed] [Google Scholar]
- 19. Navarrete EG, Liang P, Lan F, Sanchez-Freire V, Simmons C, Gong T, Sharma A, Burridge PW, Patlolla B, Lee AS, et al. . Screening drug-induced arrhythmia [corrected] using human induced pluripotent stem cell-derived cardiomyocytes and low-impedance microelectrode arrays. Circulation 2013; 128:S3-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chi KR. Revolution dawning in cardiotoxicity testing. Nature Rev Drug Discov 2013; 12:565-7; PMID:23903208; http://dx.doi.org/ 10.1038/nrd4083 [DOI] [PubMed] [Google Scholar]
- 21. Braam SR, Tertoolen L, van de Stolpe A, Meyer T, Passier R, Mummery CL. Prediction of drug-induced cardiotoxicity using human embryonic stem cell-derived cardiomyocytes. Stem Cell Res 2010; 4:107-16; PMID:20034863; http://dx.doi.org/ 10.1016/j.scr.2009.11.004 [DOI] [PubMed] [Google Scholar]