

Abstract
Drug development has typically been a primary foundation of strategy for systematic, long-range management of pathogenic cells. However, drug development is limited in speed and flexibility when response is needed to changes in pathogenic cells, especially changes that produce drug-resistance. The high replication speed and high diversity of phages are potentially useful for increasing both response speed and response flexibility when changes occur in either drug resistance or other aspects of pathogenic cells. We present strategy, with some empirical details, for (1) using modern molecular biology and biophysics to access these advantages during the phage therapy of bacterial infections, and (2) initiating use of phage capsid-based drug delivery vehicles (DDVs) with procedures that potentially overcome both drug resistance and other present limitations in the use of DDVs for the therapy of neoplasms. The discussion of phage therapy includes (a) historical considerations, (b) changes that appear to be needed in clinical tests if use of phage therapy is to be expanded, (c) recent work on novel phages and its potential use for expanding the capabilities of phage therapy and (d) an outline for a strategy that encompasses both theory and practice for expanding the applications of phage therapy. The discussion of DDVs starts by reviewing current work on DDVs, including work on both liposomal and viral DDVs. The discussion concludes with some details of the potential use of permeability constrained phage capsids as DDVs.
Keywords: biofilms, cancer therapy, cryo-electron microscopy, drug-delivery vehicles, infectious diseases, novel bacteriophages
Introduction
Expansion of strategy is needed for systematic, long-range response to changes in pathogenic cells, especially changes that involve increase in drug-resistance. In pursuit of this expansion, our text is divided into 2 sections. The first section presents a proposal for expansion of strategy for phage therapy of bacterial disease. We will propose expansion that is based on data from recent studies of both isolation/propagation of novel phages and rapid characterization of all phages. The second section begins by describing recent work on drug delivery vehicles (DDVs). It continues by describing DDV-favorable characteristics of phages. It concludes by introducing the possibility of obtaining improved performance with a DNA-free DDV derived from a DNA packaging intermediate of the related phages, T3 and T7.
Phage Therapy
Historical considerations
Management of bacterial infections is compromised when bacteria become resistant to antibiotics either by entering biofilms or by mutating to antibiotic-resistance (or both).1-6 Bacteria resistant to all known antibiotics are typically called “superbugs” in the media.1,2 We will retain this terminology. Phage therapy can, in theory, respond to both superbug emergence and other barriers to management of bacterial infections, such as biofilm formation. Phage therapy is the use of bacterial viruses (phages) to clear a bacterial infection. Lytic phages are used. Phage therapy is normally done with a mixture of several phages (phage cocktail). Importantly, the phage cocktail can be rapidly changed in response to changes in bacteria. Changes in bacteria include development of drug resistance. No harm to humans from phage therapy has ever been reported, as far as we know (see references 7–13).
Sometimes phage therapy works; sometimes it does not work.7-13 Our response is that work should be done to increase success frequency by using modern molecular biology/biophysics to continuously re-optimize phage cocktail composition and use. We will discuss details, several based on characterization of phage DNA. To begin, we note that phage DNA can be obtained, free of significant host DNA, with rapid procedure that does not include phage purification.14 The DNA length can be rapidly determined by the ∼30 year-old procedure of pulsed field gel electrophoresis (PFGE). Finally, analysis of genome sequence can become a major factor in the design of phage cocktails. The reason is that, within the last 5 years, complete phage genome sequencing, with informatic analysis of the roles of genes, has become possible within weeks, if not days, after phage isolation.
Our proposed expansion of phage therapy has a foundation in current practice. The Eliava Institute in Georgia, former USSR,13 has implemented a strategy that includes re-optimizing their phage cocktails to respond to (1) evolution of any given bacterial target, including evolution to drug resistance, and (2) introduction of new bacterial targets. The deliberate changing of cocktails prevents the doing of the usual, clinical tests for effectiveness. These tests depend on a defined composition of therapeutic compound(s) and the use of negative controls, as also promoted in the popular literature.15 Tests of this latter type include those done with the Salk polio vaccine.16 Thus, before discussing details of constructing phage cocktails, we discuss how alternative clinical tests, i.e., tests without negative controls and with a phage cocktail of deliberately varied composition, might be performed.
Suggested expansion of the clinical tests performed
An alternative clinical test is suggested by the following example. A person decides to try phage therapy after being chronically infected for several years with, let's say, a biofilm-forming Staphylococcus.3-6 This person has a swab sent and tested and, then, goes for phage therapy. The phage therapy is time-correlated with a decrease in bacterial load. Within a couple of weeks after phage therapy, the person is no longer infected. That is to say, the original trajectory of the disease was changed at the point in time at which the phage therapy occurred. The possibility always exists that the trajectory change would have occurred without the phage therapy, although this idea strains credulity in the case of a person chronically infected for years.
To obtain statistics, this test is repeated. However, importantly, it is repeated with an optimized cocktail that will typically vary with details of infection. The optimization procedure (discussed below) is what (1) remains constant and (2) is being tested. As the optimization procedure becomes more extensively informed, its useful range will widen. Optimally, all bacterial infections would eventually be under a single procedure. Even at the beginning of optimization, the useful range should encompass emergence of antibiotic resistant versions of a single bacterial type, such as the sometimes lethal, carbapenem resistant (CRE) strains. Effective antibiotic therapy is not available for CRE strains.17,18
A second alternative test is to determine details of how the recovery occurs, rather than the percentage of recovered patients. Indeed, this latter test was used in the pre-antibiotic era. For example, in the case of typhoid fever, phage therapy altered the recovery trajectory so that fever spiked within 3–6 hours and then decreased to normal in another 9.5–24 hours, typically with elimination of Salmonella.19,20
For the skeptical reader, we note that, in the case of live polio vaccines, similar tests appear to have been major factors used to justify adoption in several countries, including the United States. What appears to have happened is the following (more work on the history is needed). (1) After attenuated, live virus vaccines were tested with animals, success with humans was initially monitored via establishment of an intestinal infection and, then, antibody response.22 (2) Subsequent field tests were based on the disease trajectory, without negative controls. These tests were conducted in the (then) Belgian Congo, Poland, Croatia and Switzerland.23 After a series of political intrigues, the (then) USSR conducted vaccination of a portion of its population with the live virus, Sabin vaccine. This vaccination was accompanied by improvement in the polio outcome, although the numerical details appear to have been sub-optimally documented.24,25 These results stimulated other countries to adopt the live virus, Sabin vaccine. That is to say, we are proposing use of an old concept re-cast in modern terms.
Indeed, as the superbug phenomenon becomes more frequent, the need for phage therapy and expanded testing procedures become more important. This conclusion is a product of common sense, especially given that new antibiotics take years to develop. Furthermore, new antibiotics are less effective, by a factor of 100–1,000, when the targeted bacteria form bacterial biofilms.26 Thus, for the Food and Drug Administration (FDA), advantages exist in the making of the needed additions to standards for testing of effectiveness.
The superbug phenomenon may already be approaching this level in the area of citrus trees, as illustrated by the following quotes from the popular press concerning citrus greening disease (a disease caused by Candidatus Liberibacter americanus):27 “If we don't find a cure soon, we won't be growing oranges in Florida much longer.” “The future of the citrus industry is at stake.”28 With the assistance of private foundation funding, we began a genomics- and phage therapy-motivated effort to isolate and characterize phages that are not detectable by conventional means (to be called novel phages) and, therefore, have the potential to provide the needed expansion of phage therapy.
Novel and unusual phage characteristics: expansion of phage therapy
The phages isolated in this project include a Bacillus thuringiensis phage (called 0305phi8–36) that we use here to illustrate how “novel” a newly isolated phage can be. Phage 0305phi8–36 is a myovirus with (1) a 218.948 Kb genome, (2) a long tail (486 nm) and (3) unusual (but not unique) curly tail fibers.29-32 Characteristics (2) and (3) were observed in images of negatively stained phage particles. Phage 0305phi8–36 has the following novel or unusual characteristics.
Response to changing the concentration of the supporting gel during propagation. Phage 0305phi8–36 is lytic and, like some Salmonella phages highlighted for high effectiveness in phage therapy (although without details),19 0305phi8–36 can form ultra-small (<1 mm in diameter)/clear plaques. But, plaques (24–48 hour, room temperature incubation) become larger as the supporting gel concentration is lowered. They can be 0.5–1.0 cm in diameter in 0.1% agarose gels.29,30 Among the well studied phages, even the relatively large coliphage, T4, is not large enough to exhibit such dramatic dependence of plaque size on supporting gel concentration.29
Phage particle aggregation. In situ (in-plaque) fluorescence microscopy reveals that extensive phage aggregation occurs during dilute gel (0.1% agarose) laboratory propagation, when 0305phi8–36 is first isolated.30 The larger phage aggregates consist of hundreds to thousands of phage particles and have not been observed for other well studied phages. But, repeated laboratory propagation eventually (> ∼ 5 × single-plaque propagation) causes loss of the aggregation phenotype. The apparent reason is the appearance of one or more mutations, site(s) not known, that were retained because aggregation does not provide selective advantage during laboratory propagation. Presumably aggregation does provide selective advantage in the wild.31 In general, minimizing laboratory propagation should reduce loss of characteristics specific to surviving in the wild. Thus, minimizing laboratory propagation should enhance phage therapy.
Genome permutation. Phage 0305phi8–36 is, to our knowledge, the only phage that has a non-permuted genome that is longer than 200 Kb.32 Non-permutation of a double-stranded DNA genome simplifies the isolation of deletion mutants. Deletion mutants are useful for making space for adding segments of DNA to the 0305phi8–36 genome,31 which might be useful for promoting phage therapy.
Look-alike phages in biofilms. Phage 0305phi8–36 can potentially provide clues for optimizing phage therapy of biofilms. The size and structure of 0305phi8–36 (reference 31 and Figure 1, below) are indistinguishable from those of an another aggregating phage observed in electron micrographs of thin sections of human dental plaque, a biofilm.33 The dental plaque-associated phage particles in reference 33 are not only aggregated, but appear to be part of the supporting structure of the biofilm. These observations suggest that biofilm-directed optimization of phage therapy might include either (1) increasing lytic character of endogenous phages of biofilms or (2) blocking the aggregation of the endogenous phages (or both). Neither of these methods has ever been attempted to our knowledge.
Features of the DNA-enclosing shell of the capsid. The DNA-enclosing protein shell of phage 0305phi8–36 has protruding features in a position to regulate environmental interactions. This conclusion was drawn from cryo-electron microscopy of phage 0305phi8–38 after imaging thousands of particles, a few of which are in Figure 1A. After use of an advanced 3D reconstruction algorithm (Legend to Fig. 1), we obtained a 15Å 3D density map of the DNA-enclosing protein shell (Fig. 1B). The protruding features (a) extend from both hexamers (red) and pentamers (pink), arranged in a T = 13 icosahedral lattice, and (b) are most easily seen at the top right in Figure 1B. The lattice includes at least 3 proteins, the location of which is currently under investigation. One of these proteins is possibly gp81, a major capsid protein with a gene that is distant from the other genes with products involved in morphogenesis.32 Further work is needed to reveal whether the projections of Figure 1B do help to regulate environmental phage interactions. If they do, then cryo-electron microscopy adds to capacity for interpreting capsid gene sequences and optimizing phage cocktails.
Figure 1.
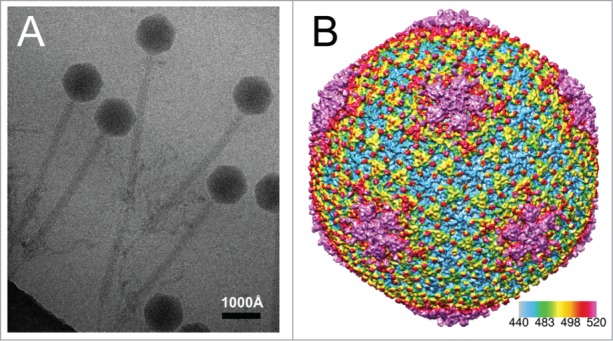
Cryo-EM reconstruction of the DNA-containing shell of phage 0305phi8–36. (A) A representative electron micrograph of frozen/hydrated (unstained) phage 0305phi8–36. (B) 3D view of the shell surface after reconstruction of the shell. Images were recorded in a 300 kV, JEM-3200FSC electron microscope with phage specimens at liquid nitrogen temperature. Thousands of particle images, as in (A), were subsequently processed by an image reconstruction method (MPSA software89), which includes icosahedral symmetry averaging, to generate the reconstruction in (B). The software, first, determines the alignment parameters of each image and then fills Fourier space from the 2D particle images. It subsequently generates a preliminary reconstruction by Fourier inversion. The software, then, iteratively improves the reconstruction until convergence. This type of structural approach has been previously used to determine the C-α backbone trace of the proteins forming capsid shells.89-91
In addition, as described in the next section, phage 0305phi8–36 has unusual propagation characteristics. Other phages have other novel and unusual characteristics of propagation, but have been less well characterized. These latter characteristics include propagation along a line (only) within the gel-containing medium of a Petri plate (unpublished observations). This characteristic suggests that propagation has a requirement for a gel structure that only exists along the line. These latter phages have all failed to re-propagate, thus far.
Improving the propagation of 0305phi8–36 and other large phages
In spite of their potential usefulness, in the past, both phage 0305phi8–36 and some other “large” phages are likely to have been invisible to the 2 most frequent phage detection procedures. Specifically, phage 0305phi8–36 could not have been detected by single plaque assay in the conventional 0.7% agar gels, given the non-formation of plaques in agarose (and agar) gels more concentrated than 0.4%. Pinpoint plaques are formed in 0.4% agarose gels.29 Thus, in the past, traditional single-plaque assays probably missed many, possibly most, phages of this type.
In addition, detection by liquid enrichment culture is also unlikely because 0305phi8–36 stops propagating before visible lysis in liquid cultures.29 Another “large” phage known to be in this category is Pseudomonas chlororaphis phage 201phi2-1, a lytic phage in the phiKZ family, genome length = 316.674 Kb.34-36 The largest known phage, G (GenBank: genome length = 497.513 Kb), is similar. Phage G propagates poorly in liquid culture and very well in medium with a dilute agarose gel.
A consequence is that, in liquid culture, 0305phi8–36 co-evolves with its host, as judged by presence of both phage and host in stationary cultures, after 12 consecutive 1/200 fresh medium dilutions, each followed by overnight propagation to stationary phase (unpublished observations). This phage/host co-evolution has been previously observed and thought to be a barrier for use in phage therapy of the phages involved.37,38
The factor limiting 0305phi8–36 propagation in liquid media (whatever it is) loses influence when dilute agarose is present. This is shown by the formation of clear, large plaques in overlays of dilute (0.05–2.0%) agarose.29,30 Empirically, ∼0.02% agarose (below the minimum gelling concentration) is the lower limit for the agarose concentration that is needed for phage-induced clearing of bacterial turbidity in the upper layer of Petri plates (unpublished data). Hydrated polymer-sensitivity of this type potentially provides the following advantage: increase in propagation when a phage particle receives a signal of the presence of susceptible host cells sufficiently concentrated to expand the phage population. The signal, in this case, would be the presence of hydrated polymer at relatively high concentration. This advantage would be especially strong for phages in the dry, hot (120–140°F on most summer days) soil from which 0305phi8–36 was isolated in Southern Texas, USA.
If improved phage therapy cocktails are to include 0305phi8-36-like (large) phages, then a phage propagation-stimulant, like dilute agarose, will be important, possibly necessary, for some of them. Other polymers and non-polymer compounds may also act as adjuvants and, indeed, phage stabilizers. For example, glucose (5%) was in the early cocktails used for phage therapy.20,21
Poultices and possibly related topics
Polymer-enhancement of phage propagation raises interest in the tradition of using polymer-containing poultices to manage surface infections. We note that, historically speaking, poultices, like phage therapy, sometimes worked and sometimes did not.39,40 Failures were caused, in part, by contaminating bacteria.39 This problem should be solvable by reducing the concentration of poultice-associated bacteria, while maintaining the active components of poultices, including phages (see reference 14). The underlying assumption is that a foundation for using poultices does exist. Poultices typically have soil and plant components and are probably rich in polymers that include at least starch and cellulose (example40). They are also possibly rich in phages. Nonetheless, use of polymer-enhancement of phage propagation has, to our knowledge, not been attempted during phage therapy.
Indeed, use of polymers might be more productive than one might think based on traditional phage therapy. The reason is that some polymers may, at the appropriate concentration, enhance propagation of phages endogenous to the biofilm(s) that is the target of therapy. Testing the therapeutic capacity of phage-free polymers is a rational strategy for improving the management of (intractable) biofilm-carried diseases such as citrus greening disease.
However, polymer-induced lysis of bacteria, including bacteria in biofilms, does not necessarily imply activation of phages. Such lysis can be achieved more directly with polymers that have both cationic and hydrophobic groups and, therefore, have cationic detergent character.41,42 This latter lysis is not analogous to the polymer-induced phage activation of the previous paragraphs. The reasons are that lysis by the cationic detergent-like polymers (1) occurs in biofilms generated by pure bacterial cultures, (2) often occurs “on contact” and (3) occurs via immediate damaging effects on bacterial membranes.41,42 However, cationic polymer-activation of endogenous phages is still a possible (apparently unexplored) factor in the wild.
Proposed strategy
Based on the above considerations, we propose that the following iteratively used strategy is reasonable for expansion of phage therapy.
Initially, rapidly isolate and characterize phages for the targeted hosts. For at least some of the initial isolations, use recently developed procedures29 for more comprehensive isolation of phages. These procedures are designed for (a) large and aggregating phages, (b) phages with long protruding fibers, (c) phages that adsorb to environmental particles and are released when in contact with a potential host bacterium, (d) phages that require hydrated polymer for aggressive propagation and (e) phages that typically exist in niches with other phages that outgrow them in conventional laboratory culture. These (i.e., a-e) and possibly other phages are more likely to be detected by the revised procedure than by conventional procedures of single-plaque and liquid enrichment culture.
After initial phage isolation, use previous data to make projections of effectiveness for various possible cocktails made from the phages in hand.
Test these projections by determining the minimal concentration of a cocktail that, in laboratory culture, (a) produces confluent lysis in the upper layer gel of a Petri plate and (b) prevents the emergence of bacterial mutants resistant to the cocktail.
By use of the data from (1)-(3), revise the projection procedure (step 2) for the next iteration.
Finally, test the iteratively derived, cocktail-making procedure in either infected animals or people, if appropriate. Isolate additional phages and repeat (1)-(4) if the final results are not up to standard.
This strategy has clinical goals and does not require knowing the underlying mechanisms. Speed is all-important, especially if used for superbugs. The basic science, including the construction of gene trees, comes later and presumably will inform step (2), above. Speed is increased by sequencing DNA that had been purified without purifying the phage.14 This latter aspect also bypasses the problem that many phages do not survive conventional purification in, for example, cesium chloride density gradients.
To select phages for a cocktail, the data to be integrated include at least the following. (1) Plaque turbidity provides a preliminary evaluation of whether a phage is lytic. Lytic phages are desired, possibly essential. Lytic character is further tested via sequencing/informatics. (2) Plaque size and border sharpness, coupled with response to changing the concentration of the plaque-supporting gel, provide a preliminary estimate of phage size.29 These results are tested and refined, within hours, by in situ (in-plaque) fluorescence microscopy30 and, within 2 days, by PFGE of expelled DNA obtained without isolating phage particles.14 (3) In situ fluorescence microscopy also provides a phage aggregation phenotype. A preliminary indication can also be obtained by visually observing plaques.30 (4) Informatic analysis of DNA sequences (again, obtained without phage purification) and cryo-EM analysis, together with 3D reconstruction at high resolution, will generate new criteria for selecting phages for phage cocktail construction. (5) Testing effects of both polymers and other non-phage cocktail components optimizes the type and amount of these components, given the phages of the cocktail and the information in (1)–(4). Polymers would presumably be effective only until dilution occurs. Dilution would occur relatively slowly during use with biofilms. (6) Finally, determination of the state of the bacterial target, including the degree of biofilm participation, will influence both phage and polymer selection.
Some aspects of criteria (1)–(3), previous paragraph, were accessible with 1920–1930s technology. They may have been used at that time, consciously or not, to optimize outcomes. In updated versions of this strategy, one might, for example, begin optimization at step (2) by selecting both (a) at least one large phage, like phages 0305phi8–36 and 201phi2-1, that is likely to have numerous genes for adapting to various bacterial niches, and (b) at least one small, rapidly propagating phage, like coliphages T3 and T7, that is likely to be relatively effective in rapidly killing bacterial cells in solution. One easily imagines synergism between phages of these 2 types.
A key reason for using the above strategy is that it provides a response to emerging superbugs, whatever the direction and the speed of superbug evolution. A major risk of focusing entirely on antibiotics is that the superbugs evolve more rapidly than we can produce new antibiotics.
Initiating Phage Capsid Therapy
We now turn the advantages of phage biology toward providing a “yes” answer to the following question. Can we use phages to significantly improve cancer therapies, at least for solid tumors?
The following is already on the “yes” side of the ledger for this question. (1) Chemotherapeutic drugs are effective, but their effectiveness has been limited by toxicity. (2) Nonetheless, lowering of drug toxicity can be achieved by drug packaging in a nano-sized drug delivery vehicle (DDV). An example is Doxil, a product already in use for ∼20 y. Doxil is based on loading the drug, doxorubicin, in liposomes.44-47 (3) The effectiveness of Doxil is increased by the hyper-loading of doxorubicin. The hyper-loading is performed by, first, generating an inside-liposome-to-outside-liposome decreasing ammonium sulfate concentration gradient and, then, allowing diffusion-driven “running down” of this gradient in the presence of doxorubicin. This “running down” drives the “running up” of a doxorubicin concentration gradient (remote loading).47 Other (in some cases simpler, as described below) types of chemical potential gradient-driven remote loading can be used, in theory. (4) Targeting to neoplastic cells has been achieved, for several DDVs, via either the relatively high permeability of tumor blood vessels (enhanced permeability and retention, or EPR, effect) or the covalent attachment of targeting ligands to the surface of the DDV. The targeting ligands include transferrin and folate.44-47 (5) Drug release from DDVs is, in theory, achievable via conditions in the lysosomes of targeted cells. These conditions include the lowered pH of lysosomes, typically lower than 4.48,49 In summary, investigators have already moved significantly toward success.
On the “no” side of the ledger, the use of a DDV both has encountered and will encounter 3 major limitations, no matter what DDV is used: (a) removal of the DDV to the spleen and liver, by what is usually called the reticulo-endothelial system (RES), (b) insufficient accuracy in targeting the DDV to neoplastic cells, which results in increased toxicity and (c) additional inaccuracy of both the time and place of drug release generated by, for example, DDV leaking. Leaking also causes increase in toxicity. Drug leaking occurs with those current DDVs loaded without covalent attachment to the DDV.44-47 However, covalent attachment is not necessary to prevent leaking (below).
Uniformity of DDVs
Although overall uniformity is not essential for an effective DDV, uniformity of some DDV characteristics assists achieving of both leak-tightness and targeting specificity. Some investigators have flagged both size heterogeneity and aggregation as major limiting factors, because of the influence on both toxicity control and the post-inoculation DDV fate. Storage stability is another, obvious factor.45,47 The National Institute of Standards (USA) allows use of the term, monodisperse, when size uniformity is equivalent to ±2% for a Gaussian distribution.45,50 Uniformity would presumably also be an advantage for obtaining FDA approval.
Also, particle heterogeneity limits quality control. One reason is the apparent absence of procedure for determining distributions of the following 2 characteristics of outer DDV edges, when heterogeneity exists: effective radius (RE) and average electrical surface charge density (σ; proportional to zeta potential) (see references 45 and 47).
The limitations caused by this absence are increased when the DDV is covalently coated with polyethylene glycol (PEG) at its surface. The reasons are the high water content and low density of the PEG layer.47 PEG has been coated on the surface of some DDVs primarily for the purpose of evading the RES.44-47 Nonetheless, for other particles, distribution of both RE and σ can be observed by use of a high-resolution, 2-dimensional, native gel electrophoresis.51 Investigators apparently have not yet tried this procedure for DDVs.
Leakiness of DDVs
Leakiness of DDVs has compromised all liposome-based preparations44-47 and presumably will do the same for other phospholipid vesicle-derived DDVs, such as “virosomes” made of phospholipids from influenza virus.52 Nonetheless, leakiness is essential for drug delivery with these DDVs because liposome-based DDVs do not have a gate that can be opened and closed accurately. In the case of Doxil, a delicate balance exists between leakiness (and toxicity) too high and leakiness (and delivery) too low.47
The best solution would be the development of a DDV that has a gate that is (1) opened to load the DDV and compatible with at least one form of remote loading, (2) closed after loading until (3) opened at the target site, possibly by the low pH of lysosomes (gated DDV). With a gated DDV, the drug(s) loaded would have diminished importance. Any cell-killing drug would work with a perfectly targeted and gated DDV.
Some advantages of phage-based DDVs
We project that use of phage-based DDVs will progressively whittle down limitations to the use of DDVs. To begin the presentation of details, phages make capsids with homogeneous, nucleic acid-containing shells. For icosahedral and related phages, the capsid size (without the tail) is in the 20–160 nm range. Typically, the size variability for these phages is less than 25% of what is defined above as monodisperse, based on cryo-electron microscopy (cryo-EM) reconstructions, as described below. Zeta potential (and σ) usually does not vary (±5%), as judged by dilute gel electrophoresis (review51).
Furthermore, phages are the most rapidly reproducing biological entities. Thus, phage-based DDV optimization can be performed via the use of directed laboratory evolution that is more rapid than, for example, the evolution of metastatic eukaryotic cancer cells. Phage T7 and the related phage, T3, only take 13–15 minutes at 37°C to reproduce by a factor of about 100; plaques form in 2.5 hr. Importantly, directed laboratory evolution bypasses the need for detailed knowledge in order to be successful in improving the (1) bypassing of the RES, (2) targeting to specific cells, and (3) opening and closing of a gate for the loading/sealing/delivery aspect of DDVs. By contrast, directed evolution is not an option with current liposome-based DDVs.
Finally, detailed structures are obtainable for phage capsids, as illustrated by Figure 1. These structures can help in the design of either a phage- or a phage capsid-based DDV. In the interest of genomic stability, DDVs from DNA phages are preferred to DDVs from RNA phages because of the higher fidelity of DNA replication and, therefore, lower genetic drift of DNA-based DDVs.53,54
In the most advanced previous, cancer therapy-directed use of directed viral evolution, the cell tropism of Sindbis virus (single-stranded RNA virus) was changed by surface peptide display, followed by directed evolution. Each half-round of a 4-round, bi-phasic selection required ∼2 days.54 So, minimally, 16 d were used to change tropism. This compares with less than 0.5 day projected for a T3/T7 phage counterpart. In addition, phage evolution can be accelerated via procedures (reference 55, for example) of continuous culture to select for specific tumor cell binding. Continuous cultures can run 24 hours per day. Phage-based DDV uptake would occur by endocytosis, which is not phage programmed and is different from Sindbis virus uptake.
RNA viral DDVs in practice
Currently, DDVs are neither uniform nor gated if made entirely in vitro via human design.45,47 By contrast, Mother Nature has provided uniformity in virus-based DDVs. Mother Nature has also provided gating for double-stranded DNA virus-based DDVs, as described below. Single-stranded RNA, icosahedral plant viruses have already been developed as potential DDVs. Drug loading was performed either (1) by non-covalently binding drugs to the packaged RNA of in vivo-assembled virus particles49 or (2) by covalently conjugating drugs to RNA and then packaging the conjugated RNA via the in vitro mimicking of in vivo co-assembly of shell protein and RNA. The in vitro product is called a virus-like particle (VLP).56
During this in vitro assembly, multiple RNAs are encapsulated in a single plant VLP, if each RNA has a stem-loop-containing nucleation sequence.56,57 Single-stranded RNA viruses, in general, co-assemble a protein shell and RNA (review57). The structure of the RNA-enclosing shell is uniform enough so that cryo-electron microscopy-based 3D reconstructions are performed at ∼3 Angstroms resolution without sorting of particles.58,59 Major disadvantages of plant viral DDVs are that (1) plant viral genetics is minimal and (2) directed evolution for plant viruses is slower than for phages.
In recent work, these limitations decrease when DDVs are based on single-stranded RNA, icosahedral phages, rather than plant viruses. The primary phage used is MS2. Phage MS2-based DDVs are similar to either previous plant viral or liposomal DDVs in the following ways. (1) The payload is typically covalently joined to either the viral RNA or a shortened version that has a stem-loop, assembly nucleation, RNA sequence. (2) The procedure for production is in vitro co-assembly of RNA and protein. (3) PEG, covalently linked to the shell, is used to evade the RES. (4) Targeting peptides, covalently linked to the shell, generate specificity. In one study, these peptides produce uptake by hepatocarcinoma cells, but not normal liver cells, in culture.60
A RNA phage-based DDV has the advantage of being genomically coded. Mature phage MS2 has been converted to a display vector.61 Thus, this RNA phage-based system appears to be the first for which one can obtain desired DDV characteristics via directed evolution of either displayed peptide or viral shell without displayed peptide.
However, all single-stranded RNA phage-based DDVs have the following limitations. (1) The single-stranded RNA genome is expected to have less genomic stability than a DNA genome.53,54 (2) Remote loading (for drug concentration increasing) is not simple and is not yet achieved. Typically, about 80 drug molecules are currently loaded in a shell 27.5 nm in diameter.60 The molar drug concentration is over 10× lower than for Doxil. (3) Drugs are covalently joined to RNA molecules, thereby generating complexity, a potential source of irreproducibility and problems in unloading.
The potential of DVDs from double-stranded DNA phages
A more genomically stable, double-stranded DNA-encoded DDV can be developed. But, to do so, we must take account of the following observations. In contrast to what has been found for single-stranded RNA phages, double-stranded DNA phages (and double-stranded DNA viruses, in general) have been found to initially assemble a DNA-free protein shell that contains internal proteins (capsid). This pre-assembled capsid is called a procapsid (or capsid I in the case of the related phages, T3 and T7). The procapsid is typically not stable enough to be a DDV. But, the procapsid subsequently packages the DNA genome. During packaging, the genome-encapsidating protein shell changes in structure (reviews62-64). The changes in the shell include a dramatic increase in physical stability, as the shell becomes more like the shell of the mature phage.
We were not surprised to find that the mature shell of both phage T7 and its relative, T3, is stable to both proteases65 and ionic detergent (unpublished data). We expected these observations because intestinal content is a major native environment of the host, Escherichia coli. In any case, this stability is a major advantage for preventing off-target drug release by a DDV. But, neither mature phage T3 nor mature phage T7 would make a good DDV, because both are filled with DNA. Destabilization results from the pressure that the packaged DNA exerts on the protein shell. When a phage particle bursts, the DNA genome, and presumably other contents, exit.
By contrast, an empty (DNA-free) phage capsid can have the mature phage's stability to proteases and detergents. But, this capsid does not have the instability caused by packaged DNA. DDV prospects would be dramatically enhanced if a highly stable capsid retained some gating of the mature phage particle.
In a mature phage particle (T3/T7: Fig. 2A), a ring of 12 subunits (called portal ring or connector) forms the gate for infection-initiating exit of the double-stranded DNA genome. For T3/T7, the protein encoded by gene 8 (to be called gp8) forms the 12-subunit connector. Exterior to the connector is the tail, which participates in host-adsorption and triggers DNA injection. The tail of T3/T7 consists of gp11, 12 and 17 (recent reference66). Interior to the T3/T7 connector is a stack of layers, consisting of (in order) gp14, 15 and 16, as illustrated in Figure 2A without distinguishing the layers. Unfortunately, the connector and its gating potential are typically lost when DNA is expelled from a mature phage particle.
Figure 2.
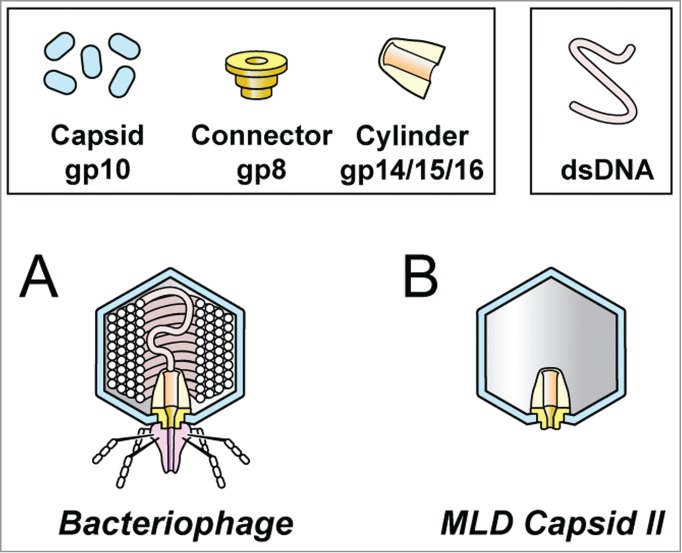
The structure of (A) bacteriophage T3/T7 and (B) T3/T7 MLD capsid II.64,67
But, fortunately, Mother Nature has provided an in vivo-produced (no in vitro assembly needed), stable, gated (connector intact), uniform-sized T3/T7 capsid that is an excellent DDV candidate, is produced in large amount and is not prone to aggregation. This candidate is a member of a sub-class of particles that are T3/T7 DNA packaging-associated, capsid I conversion products. They are all called capsid II, one version of which is illustrated in Figure 2B.
T3/T7 capsid II as a DDV
A potential capsid II-DDV is easily purified from a lysate of either phage T3- or phage T7-infected cells. Figure 3 shows purification of T3 capsid II through the following sequence: (1) a cesium chloride step gradient fractionation of an entire, pre-concentrated, T3-infected cell lysate (Fig. 3A), followed by (2) buoyant density centrifugation in a cesium chloride density gradient (Fig. 3B) of the capsid region of the step gradient (labeled CI + CII in Fig. 3A) and then (3) buoyant density centrifugation in a Metrizamide density gradient (Fig. 3C) of a fraction from the capsid II region of the second cesium chloride density gradient (labeled CII in Fig. 3B). The most visible capsid II band in Figure 3C was at a density of 1.086 g/cm, a density so low that the band-forming particle is called Metrizamide low density, or MLD, capsid II (MLD CII in Fig. 3C). MLD capsid II is the candidate DDV, as discussed below.
Figure 3.

Purification of MLD capsid II. Light scattering profiles were photographed for concentrated particles from (A) a phage T3-infected cell lysate after cesium chloride step gradient ultracentrifugation,69 (B) capsids I and II from the gradient in (A) after buoyant density centrifugation in a cesium chloride density gradient,69 (C) capsid II from the gradient in (B) after buoyant density centrifugation in a Metrizamide density gradient.67 M, meniscus; B, bottom of centrifuge tube; CI, capsid I; CII, capsid II, MLD, Metrizamide low density; MHD, Metrizamide high density.
The capsid II designation of the MLD particle is derived from native gel electrophoresis and electron microscopy for phage T767 and T3.68 Another capsid II band is at the position of MHD CII (Metrizamide high density CII) in Figure 2C, as seen by native gel electrophoresis (not shown). An illustration of the ∼15 Å-resolution structure of the gp10 shell of T7 MLD capsid II is in Figure 7a of reference 69. The resolution now extends to 3.5 Å, revealing an HK97-like gp10 conformation (F. Guo, Elena T. Wright, P. Serwer and W. Jiang, unpublished observations).
MLD capsid II is a potential DDV because the low density during Metrizamide-buoyant density centrifugation is caused by total impermeability to Metrizamide, as demonstrated in most detail for T7 MLD capsid II.67 The largest pore in the shell of T7 MLD capsid II can have a diameter no more than 1.2 nm, based on the molecular structure of Metrizamide.67 MLD capsid II retains its MLD character for at least 20 y during storage in Metrizamide solutions (data not shown). That is to say, Metrizamide is “sealed out” of MLD capsid II. This sealing out suggests that, if MLD capsid II can be drug-loaded via gate opening, procedures can be found to close the gate and store and use drug-loaded MLD capsid II as a DDV with the drug “sealed in.” In this case, the drug leakage problem is solved, in spite of the non-use of covalent drug-DDV joining. The barrier to leakage is steric, not solubility-based. Liposomal DDVs have a solubility-based barrier.
Metrizamide has a molecular weight = 789, but is no longer being sold. Nycodenz is, commercially speaking, replacing Metrizamide. Nycodenz is equivalent to Metrizamide for isolation of either T3 or T7 MLD capsid II, as far as we can tell. Nycodenz has a molecular weight of 821 and presumably is completely sealed out of MLD capsid II.
The potential for gate opening of an MLD capsid II-DDV
In contrast to the sealing out of Metrizamide and Nycodenz, slightly smaller compounds diffuse into MLD capsid II. For example, iothalamate anion (mass = 614) diffuses into T7 MLD capsid II in a period of about a day, as judged by buoyant density.67 Bis-ANS anion (5,5′-bis(8-(phenylamino)-1-naphthalenesulfonate); mass = 595) enters MLD capsid II in about 2 hours at 25°C, as judged by bis-ANS fluorescence enhancement generated when bis-ANS binds to hydrophobic regions of internal proteins. After covalent, ultraviolet light-induced cross-linking of the bound bis-ANS, we found these proteins to be the connector protein (gp8) and proteins stacked on the connector (gps 15 and 16).70
When bis-ANS is bound to the MLD capsid II interior, quenching of bis-ANS fluorescence is an assay for the entry of cationic dyes and related compounds. This assay was first demonstrated for ethidium cation (mass = 314). The entry time scale for ethidium was 0.5–2.0 hr. Binding of both bis-ANS and ethidium to external MLD capsid II sites occurred too rapidly to measure.70 This previous study also identified the axial hole of the connector/core stack as the most probable site at which both bis-ANS and ethidium diffuse into MLD capsid II.
Thus, a foundation exists for developing MLD capsid II into a connector-gated DDV. Our initial test for loading (gate open) was native agarose gel electrophoresis of MLD capsid II that had been stained with bis-ANS before electrophoresis. The bis-ANS remained with MLD capsid II during electrophoresis and was detected by UV light induced visible fluorescence (not shown).
We then (1) incubated the bis-ANS-stained MLD capsid II with 0.64 M Nycodenz at the temperatures (°C) indicated above lanes in Figure 4 and (2) subjected the Nycodenz-incubated, bis-ANS-stained MLD capsid II to native agarose gel electrophoresis. An image of the fluorescence signal from the bis-ANS-stained MLD capsid II is in Figure 4A; an image of protein-specific Coomassie staining of the same gel is in Figure 4B. In relation to capsid protein, the fluorescence signal in Figure 4A underwent decrease when the temperature of incubation was either 40°C or above. By contrast, this decrease began at 51–55°C in the absence Nycodenz (bis-ANS fluorescence: Figure 4C; protein staining: Fig. 4D). The difference in minimum temperature of fluorescence loss is likely to be the result of the entry of Nycodenz (gate opening) and, therefore, the Nycodenz-quenching of bis-ANS fluorescence. The 51–55°C-loss of fluorescence in the absence of Nycodenz is an assay imperfection.
Figure 4.
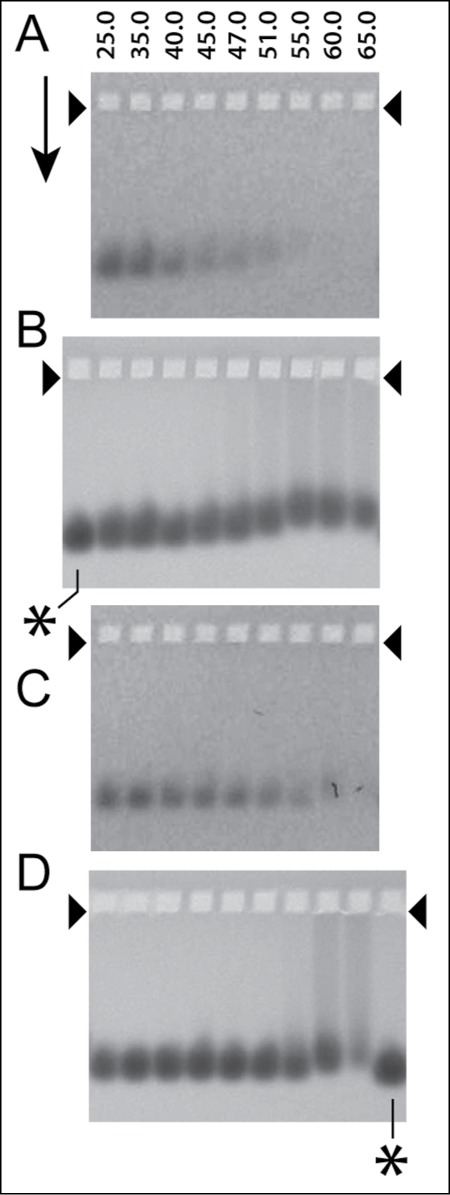
Native gel electrophoresis for the monitoring of the loading of Nycodenz in the cavity of T3 MLD capsid II. The MLD capsid II had been stained by incubation with 100 μg/ml bis-ANS for 2 hr. at 30°C and then exposure to ultraviolet light for 1 min at a distance of 9 cm from the surface of a Foto/UV 300 (Fotodyne, Hartland, WI, USA) uv transilluminator. The samples were incubated with either (A and B) 0.64 M Nycodenz for 2 hr., in 0.05 M sodium phosphate, pH 7.4, 0.001 M MgCl2 or (C and D) the same buffer without Nycodenz at the temperature (°C) indicated at the top of lanes. Native agarose gel electrophoresis was then performed at 2.0 V/cm and 25°C for 14.0 hr. through a submerged, 1.0% Seakem LE agarose gel (Lot#602696) in the following electrophoresis buffer: 0.09 M Tris-Acetate, pH 8.4, 0.001 M MgCl2. Particles in the gel were initially visualized via the fluorescence of the capsid-bound bis-ANS, without further staining (A and C), and subsequently (in the same gel) by the protein-specific dye, Coomassie blue (B and D). The arrow indicates the direction of electrophoresis; the arrowheads indicate the forward edges of the sample wells. The asterisks indicate lanes for which the sample was MLD capsid II that had not been incubated.
To further test for loading, we performed electron microscopy after the following treatment: (1) first, a 2 hr. incubation of MLD capsid II with 0.64 M Nycodenz (2) and, then, dialysis of the Nycodenz and storage in the absence of Nycodenz for about 24 hr. When the temperature of Nycodenz-incubation was 25°C, the MLD capsid II appeared empty (Fig. 5A). By contrast, when the temperature of incubation was 51°C, the MLD capsid II appeared filled with Nycodenz. This was the case even after the following 3 Nycodenz-removing steps: (1) post-Nycodenz dialysis, (2) subsequent purification by rate zonal centrifugation in a sucrose gradient, followed by (3) a second dialysis to remove sucrose (Fig. 5B). That is to say, a gate (presumably the connector) had been opened by the elevated temperature and then closed, so that the Nycodenz remained in the MLD capsid II after 2x-dialysis and purification.
Figure 5.
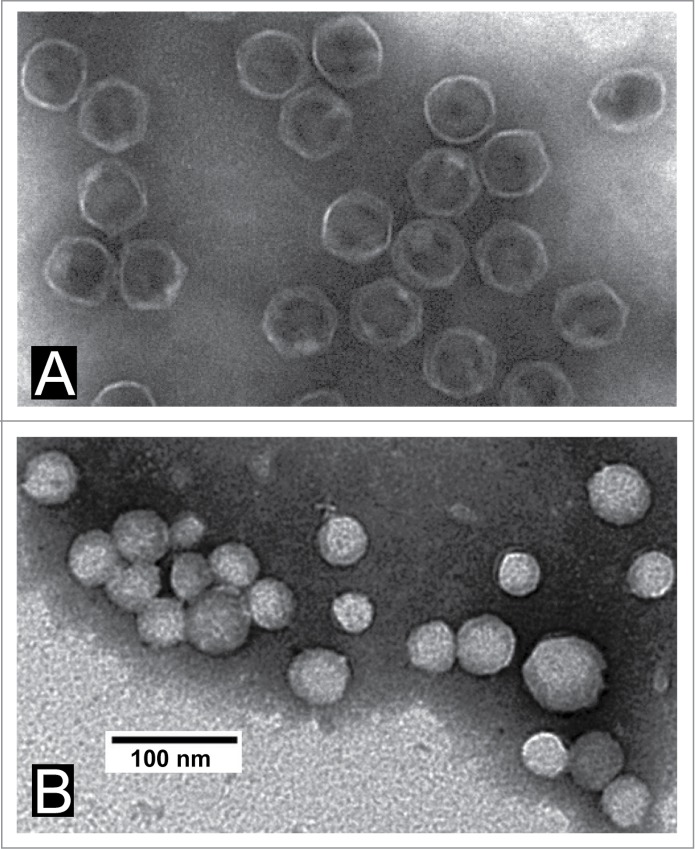
Electron microscopy of negatively stained MLD capsid II incubated with 0.64 M Nycodenz. After staining as in Figure 4 with Bis-ANS at 250 μg/ml, MLD capsid II was incubated with 0.64 M Nycodenz at (A) 25°C and (B) 51°C. The Nycodenz was removed by dialysis and the capsids were left at 4°C for a total of 24 hr. The 51°C-incubated sample was further purified by rate zonal centrifugation in a sucrose gradient and then sucrose was removed by a second dialysis. The dialyzed MLD capsid II was, then, negatively stained with 1% sodium phosphotungstate, pH 8.4. The specimen was observed in a JEOL 100CX electron microscope.
But, surprisingly, most Nycodenz-loaded MLD capsid II particles appeared to have become smaller and more heterogeneous in size. This latter aspect is unfavorable for a DDV because the targeting properties are likely to change. A change in capsid structure is also suggested by an elevated temperature-associated lowering of mobility in Figure 4A, B. Loading-associated change in structure is a potential limitation that must be monitored after drug loading. Nonetheless, molar drug concentrations are likely to be at least 10× lower than the Nycodenz concentration used for Figures 4 and 5.
Generating specificity for MLD capsid II
In theory, directed evolution can be used to generate the 2 most difficult DDV capacities to obtain: RES bypass and tumor cell-specific uptake. A specific, swinging-for-the-fences example is that one might iteratively (1) inject phage T3 or T7 (infective) into a tumor-bearing organism and then (2) isolate and single-plaque purify phages that migrate to the tumor until one isolates a mutant (presumably multi-site) that preferentially accumulates in the tumor!
If directed evolution were to yield a T3 or T7 phage mutant of this type, then the DDV is MLD capsid II from the mutant. A primary job of the researcher is to develop increasingly proficient procedures of directed evolution. The one proposed here should be considered only a beginning.
In previous studies, directed evolution has produced (1) phage lambda multi-site, shell protein mutants with increased survival time in live mice, apparently by reducing uptake by the RES71, (2) phage T3 multi-site mutants with increased ability to propagate in the presence of elevated [NaCl]72 and (3) phage T7 host range mutants selected for propagation on mixtures of hosts of different quality,73 among other directed phage evolution produced characteristics (review74). We note that sequencing technology has advanced to the point that a complete phage genotype takes, at most, a couple of weeks to obtain, independent of the number and types of mutations present.
The procedure of the previous 3 paragraphs has limitations the most obvious of which is that some of the directed evolution-derived mutations will be in the tail. The tail is not present in MLD capsid II. To bypass this limitation, one could perform random PCR mutagenesis of part of the shell protein gene only (gene 10 for T3/T7; right of Fig. 6A) before performing the directed evolution. To transfer mutagenized DNA to infective phage particles one might, first, PCR-fuse non-mutagenized and mutagenized DNA segments (Fig. 6B) so that homologous recombination can be used for mutagenized DNA integration and, then, recombine the fusion product into a mature genome. To increase the speed of doing this, one might further develop in vitro recombination (see reference 75 for results with T3/T7). Alternatively, one might use the more traditional technique of transfer to a plasmid and, then, in vivo recombination-transfer to infective phages.
Figure 6.
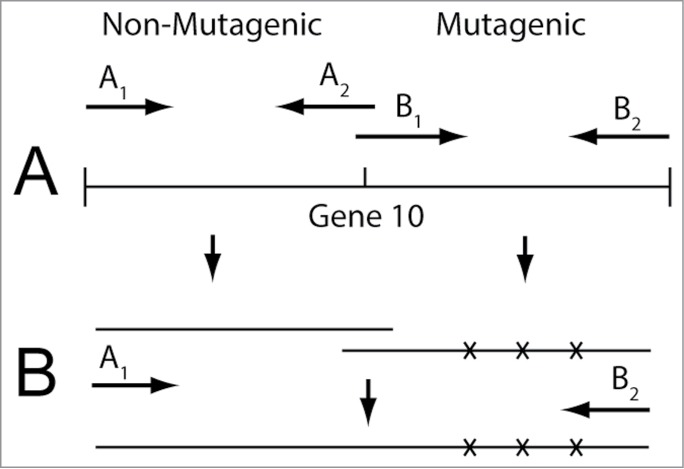
(A) PCR mutagenesis and (B) subsequent PCR fusion of the mutagenized DNA product. The fusion is to non-mutagenized DNA segments that provide homology for incorporation of the mutagenized DNA into a mature T3 or T7 genome.
Ultimately, the connector-gate must open for drug release when MLD capsid II reaches its target. The low pH of lysosomes might be exploited to do this job. One might also make MLD capsid II DDVs that are unloaded by an external signal, possibly radiation (microwaves or visible light, for example), depending on the type of tumor. An advantage of a MLD capsid II-based DDV is that one can modify the gate protein(s) (connector and possibly core stack) without modifying the specificity-determining protein, gp10. Thus, we minimize the chance that, when we modify specificity, we accidentally modify gating and vice versa.
We conclude by stating that the risk of attempting a MLD capsid II-based strategy is relatively small, given the relatively low expense and time needed for work with phages. If successfully implemented, this strategy can be pursued no matter how the pathology evolves, an advantage shared with phage therapy.
Discussion
Phage therapy
We have outlined a strategy, with details, for obtaining more effective phage therapy cocktails, especially for drug-resistant bacterial infections. Presumably, the best time for implementation is before a (more) serious superbug plague arrives. Cocktail optimization procedures will provide intellectual property, the lack of which appears to have been inhibitory to commercialization of phage therapy in the past.
The optimal area for initial implementation appears to be the phage therapy of agricultural plants. Reasons are the following: (1) The need exists and has a major business and societal component. (2) The bacterial diseases involved are typically biofilm-carried. Managing biofilms is also the current area of apparent greatest need for infections of humans. Success with a plant-model system is likely to improve the odds of success with humans. (3) Problems with investigator safety are minimal in that diseases of plants do not transfer to humans. Thus, the speed of laboratory work will not be slowed by safety precautions beyond those taken for current work with phages in the area of basic science. The use of cell envelope and other biofilm-degrading enzymes76-78 has potential to increase effectiveness at the point of application of a phage cocktail used for biofilm-associated bacteria.
On the negative side of the ledger is the fact that biofilm-forming bacteria of plants often propagate very slowly in the laboratory. Xylella fastidiosa (the xylem-inhabiting cause of both Pierce's disease of grape vines and almond leaf scorch disease), for example, takes 5–30 d to form colonies, depending on the detail being investigated.79 Hopefully, we can find more rapidly propagating bacterial strains for the detection and propagation of phages that are also active on either slowly propagating pathogenic strains or strains not yet cultivatable. Whether or not this cross-plating strategy is feasible, the activation of endogenous phages, as discussed above, is a strategy that has the potential to be effective.
Finally, we note that we have not found evidence of any effort to determine whether herbicides have negative effects on phage propagation. One possibility is that the use of herbicides is, indeed, aggravating the problem of biofilm-carried bacterial infections of plants, by inhibiting the propagation of endogenous phages.
Managing of Neoplasms
The use of phage capsids as DDVs has been tried with filamentous DNA phages, which have single-stranded DNA. But, the strategy used for filamentous phages is more complex than the one described above. Specifically, the drugs either have to be individually covalently linked to the phage80 or diffused into non-filamentous, capsid protein vesicles that were assembled in vitro, are non-uniform and are leaky.81 Even at the drug-loading/sealing stage, this strategy is basically unrelated to and has success-limiting aspects more potent than those of the strategy that we have outlined. Nonetheless, implementation of this strategy has already had some success with cultured tumor cells.82 We note that, independent of DDV-activity, phages have been found to have additional, probably immune stimulatory, therapeutic effects on tumors.83
The loading studies performed here are the first to indicate the possibility of an in vivo-generated, highly uniform, stable, non-leaky, gated DDV. In addition, remote loading via an osmotic pressure gradient should be possible with MLD capsid II. The gradient would be generated by a MLD capsid II-impermeable compound and would increase the loading of a MLD capsid II-permeable drug. We obtain 10–20 mg of MLD capsid II from a 6-liter lysate. We have not tested phages other than T3 and T7 for the production of a MLD capsid II-like particle.
Parenthetically, we note, first, that the observed (Fig. 5) change in shell structure potentially reflects shell flexibility that facilitates DNA packaging (review64). The contraction of T3 shells to sub-phage RE has previously been observed both during analysis of T3 DNA packaging intermediates84 and after the expulsion of packaged DNA from shells that initially had a complete genome.85 Thus, shell contraction, in general, is not surprising. But, loss of shell subunits is an alternative explanation for size decrease and this alternative explanation has not been empirically excluded for Figure 5. Second, we note that MLD capsid II-targeting of bacteria, rather than cancer cells, might be used to manage intracellular bacteria, when bacteria are not manageable with either antibiotics or conventional phage therapy.
Finally, we note that, although all MLD capsid II-based DDVs will have a structural component in common, they will also, by necessity, have variability in their structure. This variability could extend to a single preparation, designed to manage a tumor in which tumor cells differ from each other, which often they do.86,87 The variability raises questions of cost. We suggest that the achieving of viable economics is dependent on automation. In the case of both phage therapy and phage-based DDVs, automated storage and retrieval of all strains previously generated will, with help of a database, reduce costs by producing progressively more effective starting material. In the case of DDVs, automating of the directed evolution will be critical to increasing the speed and lowering the cost. In this area, art88 appears to have gone before science.
Acknowledgments
The authors thank Drs. Mzia Kutateladze and Revaz Adamia for updates on the strategy of the courageous effort in phage therapy at the Eliava Institute in Tblisi, Georgia. We thank Julie A. Thomas and Saurav Pathria for preparing phage 0305phi8–36. We thank the following investigators for comments on drafts of this manuscript: Drs. Hans Ackermann, Wen Jiang, Mzia Kutateladze, Betty Kutter and William C. Summers. As this manuscript was being completed, we were saddened by the news that Dr. Adamia died from cancer. Cryo-EM was performed in National Center for Macromolecular Imaging, Baylor College of Medicine. Electron micrographs of negatively stained specimens were taken in the Electron Microscopy Facility in the Department of Pathology at the University of Texas Health Science Center at San Antonio.
Funding
This work was supported by grants from the Welch Foundation (AQ-764 to PS and Q1242 to Wah Chiu), The Robert J. Kleberg, Jr. and Helen C. Kleberg Foundation (to PS), and the National Institutes of Health (P41GM103832 to Wah Chiu).
References
- 1. Spellberg B, Gilbert D. Rising Plague. Amherst, NY: Prometheus Books; 2009. [Google Scholar]
- 2. Bartlett JG, Gilbert DN, Spellberg B. Seven ways to preserve the miracle of antibiotics. Clin Infectious Dis 2013; 56:1445-50; PMID:23403172; http://dx.doi.org/ 10.1093/cid/cit070 [DOI] [PubMed] [Google Scholar]
- 3. Cogan NG, Gunn JS, Wozniak DJ. Biofilms and infectious diseases: biology to mathematics and back again. FEMS Microbiol Lett 2011; 322:1-7; PMID:21595745; http://dx.doi.org/ 10.1111/j.1574-6968.2011.02314.x [DOI] [PubMed] [Google Scholar]
- 4. Lewis K. Persister cells. Ann Rev Microbiol 2010; 64:357-72; PMID:20528688; http://dx.doi.org/ 10.1146/annurev.micro.112408.134306 [DOI] [PubMed] [Google Scholar]
- 5. Wu C, Labrie J, Tremblay YD, Haine D, Mourez M, Jacques M. Zinc as an agent for the prevention of biofilm formation by pathogenic bacteria. J Appl Microbiol 2013; 115:30-40; PMID:23509865; http://dx.doi.org/ 10.1111/jam.12197 [DOI] [PubMed] [Google Scholar]
- 6. Romling U, Balsalobre C. Biofilm interactions, their resilience to therapy and innovative treatment strategies. J Intern Med 2012; 272:541-61; PMID:23025745; http://dx.doi.org/ 10.1111/joim.12004 [DOI] [PubMed] [Google Scholar]
- 7. Sulakvelidze A, Alavidze Z, Morris J. Bacteriophage therapy. Antimicrob Agents Chemother 2001; 45:649-59; PMID:11181338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Summers WC. Bacteriophage therapy. Annu Rev Microbiol 2001; 55:437-51; PMID:11544363 [DOI] [PubMed] [Google Scholar]
- 9. Rhoads D, Wolcott R, Kuskowski M, Wolcott B, Ward L, Sulakvelidze A. Bacteriophage therapy of various leg ulcers in humans: results of a phase I safety trial. J Wound Care 2009; 18:237-43; PMID:19661847 [DOI] [PubMed] [Google Scholar]
- 10. Summers WC. The strange history of phage therapy. Bacteriophage 2012; 2:130-3; http://dx.doi.org/ 10.4161/bact.20757; PMID:23050223; PMCID: PMC3442826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schoolnik GK, Summers WC, Watson JD. Phage offer a real alternative. Nat Biotechnol 2004; 22:505-6; http://dx.doi.org/ 10.1038/nbt0504-505 [DOI] [PubMed] [Google Scholar]
- 12. Örmälä A-M, Jalasvuori M. Phage therapy: should bacterial resistance to phages be a concern, even in the long run? Bacteriophage 2013; 3:e24219; PMID:23819105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pirnay JP, De Vos D, Verbeken G, Merabishvili M, Chanishvili N, Vaneechoutte M, Zizi M, Laire G, Lavigne R, Huys I, et al. . The Phage therapy paradigm: pret-a-porter or sur-mesure? Pharmaceut Res 2011; 4:934-7; PMID:21063753; http://dx.doi.org/ 10.1007/s11095-010-0313-5 [DOI] [PubMed] [Google Scholar]
- 14. Serwer P, Hayes SJ, Thomas JA, Griess GA, Hardies SC. Rapid determination of genomic DNA length for new bacteriophages. Electrophoresis 2007; 28:1896-902; PMID:17480041; http://dx.doi.org/ 10.1002/elps.200600672 [DOI] [PubMed] [Google Scholar]
- 15. Lewis S. Arrowsmith. New York, N.Y: Harcourt Brace & Co., 1925. [Google Scholar]
- 16. Monto AS. Francis field trial of inactivated poliomyelitis vaccine: background and lessons for today. Epidemiol Rev 1999; 21:7-23; PMID:10520470 [DOI] [PubMed] [Google Scholar]
- 17. Perez F, Van Duin D. Carbapenem-resistant Enterobacteriaceae: a menace to our most vulnerable patients. Cleveland Clin J Med 2013; 80:225-33; PMID:23547093; http://dx.doi.org/ 10.3949/ccjm.80a.12182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Logan LK. Carbapenem-resistant enterobacteriaceae: an emerging problem in children. Clin Infect Dis 2012; 55:852-9; PMID:22700827; http://dx.doi.org/ 10.1093/cid/cis543 [DOI] [PubMed] [Google Scholar]
- 19. Desranleau J-M. Progress in the treatment of typhoid fever with Vi bacteriophages. Can J Public Health 1949; 40:473-8; PMID:15408422 [PubMed] [Google Scholar]
- 20. Knouf EG, Ward WE, Reichle PA, Bower AG, Hamilton PM. Treatment of typhoid fever with type specific bacteriophage. J Am Med Assoc 1946; 132:134-8; PMID:20997193 [DOI] [PubMed] [Google Scholar]
- 21. Slopek S, Weber-Dabrowska B, Dabrowski M, Kucharewicz-Krukowska A. 1987. Results of bacteriophage treatment of suppurative bacterial infections in the years 1981–1986. Arch Immunol Ther Exp (Warsz) 1987; 35:569-83; PMID:3455647 [PubMed] [Google Scholar]
- 22. Koprowski H. Historical aspects of the development of live virus vaccine in poliomyelitis. British Med J 1960; 2:85-91; PMID:13856034; PMCID: PMC2096806 [PubMed] [Google Scholar]
- 23. Koprowski H. First decade (1950–1960) of studies and trials with the polio vaccine. Biologicals 2006; 34:81-6; PMID:16682219 [DOI] [PubMed] [Google Scholar]
- 24. Swanson W. Birth of a cold war vaccine. Sci Am 2012; 306:66-9; PMID:22486119 [DOI] [PubMed] [Google Scholar]
- 25. Sabin AB, Ramos-Alvarez M, Alvarez-Amezquita J, Pelon W, Michaels RH, Spigland I, Koch MA, Barnes JM, Rhim JS. Live, orally given poliovirus vaccine. Effects of rapid mass immunization on population under conditions of massive enteric infection with other viruses 1960. Bull World Health Organ 1999; 77:196-201; PMCID: PMC2557586 [PMC free article] [PubMed] [Google Scholar]
- 26. Høiby N, Ciofu O, Johansen HK, Song ZJ, Moser C, Jensen PØ, Molin S, Givskov M, Tolker-Nielsen T, Bjarnsholt T. The clinical impact of bacterial biofilms. Int J Oral Sci 2011; 3:55-65; PMID:21485309; http://dx.doi.org/ 10.4248/IJOS11026; PMCID: PMC3469878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mafra V, Martins PK, Francisco CS, Ribeiro-Alves M, Freitas-Astúa J, Machado MA. Candidatus liberibacter americanus induces significant reprogramming of the transcriptome of the susceptible citrus genotype. BMC Genomics 2013; 14:247; http://dx.doi.org/ 10.1186/1471-2164-14-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lush T. Fed joins fight on citrus greening. Miami Herald Business December 12 2012; http://www.miamiherald.com/2013/12/12/3814810/feds-joins-battle-on-citrus-greening.html [Google Scholar]
- 29. Serwer P, Hayes SJ, Thomas JA, Hardies SC. Propagating the missing bacteriophages: a large bacteriophage in a new class. Virol J 2007; 4:21; PMID:17324288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Serwer P, Hayes SJ, Lieman K, Griess GA. In situ fluorescence microscopy of bacteriophage aggregates. J Microsc 2007; 228:309-21; PMID:18045325; http://dx.doi.org/ 10.1111/j.1365-2818.2007.01855.x [DOI] [PubMed] [Google Scholar]
- 31. Pathria S, Rolando M, Lieman K, Hayes S, Hardies S, Serwer P. Islands of non-essential genes, including a DNA translocation operon, in the genome of bacteriophage 0305φ8–36. Bacteriophage 2012; 2:25-35; PMID:22666654; PMCID: PMC3357382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thomas JA, Hardies SC, Rolando M, Hayes SJ, Lieman K, Carroll CA, Weintraub ST, Serwer P. Complete genomic sequence and mass spectrometric analysis of highly diverse, atypical Bacillus thuringiensis phage 0305phi8–36. Virology 2007; 368:405-21; PMID:17673272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brady JM, Gray WA, Caldwell MA. The electron microscopy of bacteriophage-like particles in dental plaque. J Dent Res 1977; 56:991-3; http://dx.doi.org/ 10.1177/00220345770560082901 [DOI] [PubMed] [Google Scholar]
- 34. Serwer P, Hayes SJ, Zaman S, Lieman K, Rolando M, Hardies SC. Improved isolation of undersampled bacteriophages: finding of distant terminase genes. Virology 2004; 329:412-24; PMID:15518819 [DOI] [PubMed] [Google Scholar]
- 35. Walmagh M, Briers Y, Branco dos Santos S, Azeredo J, Lavigne R. Characterization of modular bacteriophage endolysins from Myoviridae phages OBP, 201φ2-1 and PVP-SE1. PLoS One 2012; 7:e36991; PMID:22615864; http://dx.doi.org/ 10.1371/journal.ponqe.0036991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kraemer JA, Erb ML, Waddling CA, Montabana EA, Zehr EA, Wang H, Nguyen K, Pham DSL, Agard DA, Pogliano J. A phage tubulin assembles dynamic filaments by a novel mechanism to center viral DNA within the host cell. Cell 2012; 149:1488-99; PMID:22726436; http://dx.doi.org/ 10.1016/j.cell.2012.04.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Quigley BJZ, López DG, Buckling A, McKane AJ, Brown SP. The mode of host-parasite interaction shapes coevolutionary dynamics and the fate of host cooperation. Proc Royal Soc B 2012; 279:3742-8; http://dx.doi.org/ 10.1098/rspb.2012.0769; PMCID: PMC3415897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hall AR, Scanlan PD, Morgan AD, Buckling A. Host–parasite coevolutionary arms races give way to fluctuating selection. Ecol Lett 2011; 14:635-42; PMID:21521436; http://dx.doi.org/ 10.1111/j.1461-0248.2011.01624.x [DOI] [PubMed] [Google Scholar]
- 39. Millard C. Destiny of the Republic: A Tale of Madness, Medicine and the Murder of a President. New York, NY: Anchor Books; 2011. [Google Scholar]
- 40. Njoroge GN, Bussmann RW. Ethnotherapeautic management of skin diseases among the Kikuyus of Central Kenya. J Ethnopharmacol 2007; 111:303-7; PMID:17207950 [DOI] [PubMed] [Google Scholar]
- 41. Carmona-Ribeiro AM, de Melo Carrasco LD. Cationic antimicrobial polymers and their assemblies. Int J Mol Sci 2013; 14:9906-46; http://dx.doi.org/ 10.3390/ijms14059906; PMCID: PMC3676821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Horvath R, Kobzi B, Keul H, Moeller M, Kiss E. Molecular interaction of a new antibacterial polymer with a supported lipid bilayer measured by an in situ label-free optical technique. Int J Mol Sci 2013; 14:9722-36; http://dx.doi.org/ 10.3390/ijms14059722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thomas JA, Hardies SC, Rolando M, Hayes SJ, Lieman K, Carroll CA, Weintraub ST, Serwer P. Complete genomic sequence and mass spectrometric analysis of highly diverse, atypical Bacillus thuringiensis phage 0305phi8–36. Virology 2007; 368:405-21; PMID:17673272; http://dx.doi.org/ 10.1016/j.virol.2007.06.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Torchilin VP. Micellar nanocarriers: Pharmaceutical perspectives. Pharm Res 2007; 24:1-16; PMID:17109211 [DOI] [PubMed] [Google Scholar]
- 45. Wei A, Mehtala JG, Patri AK. Challenges and opportunities in the advancement of nanomedicines. J Controlled Release 2012; 164:236-48; PMID:23064314; http://dx.doi.org/ 10.1016/j.jconrel.2012.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Uchegbu IF, Siew A. Nanomedicines and nanodiagnostics come of age. J Pharm Sci 2013; 102:305-10; PMID:23175462; http://dx.doi.org/ 10.1002/jps.23377 [DOI] [PubMed] [Google Scholar]
- 47. Barenholz Y. Doxil® – the first FDA-approved nano-drug: Lessons learned. J Control Release 2012; 160:117-34; PMID:22484195; http://dx.doi.org/ 10.1016/j.jconrel.2012.03.020 [DOI] [PubMed] [Google Scholar]
- 48. Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov 2005; 4:145-60; PMID:15688077 [DOI] [PubMed] [Google Scholar]
- 49. Yildiz I, Lee KL, Chen K, Shukla S, Steinmetz MF. Infusion of imaging and therapeutic molecules into the plant virus-based carrier cowpea mosaic virus: cargo-loading and delivery. J Control Release 2013; 172:568-78; PMID:23665254; http://dx.doi.org/ 10.1016/j.jconrel.2013.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu S, Nie Z, Seo M, Lewis P, Kumacheva E, Stone HA, Garstecki P, Weibel DB, Gitlin I, Whitesides GM. Generation of monodisperse particles by using microfluidics: Control over size, shape, and composition. Angew Chem Int Ed 2005; 44:3799; http://dx.doi.org/ 10.1002/anie.200462226 [DOI] [PubMed] [Google Scholar]
- 51. Serwer P, Easom RA, Hayes SJ, Olson MS. Rapid detection and characterization of multimolecular cellular constituents by two-dimensional agarose gel electrophoresis. Trends Biochem Sci 1989; 14:4-7; http://dx.doi.org/http://dx.doi.org/ 10.1016/0968-0004(89)90076-5 [DOI] [Google Scholar]
- 52. Battacharya S, Mazumder B. Virosomes: A novel strategy for drug delivery and targeting. Biopharm Int 2011; 24:s9-14 [Google Scholar]
- 53. Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. New York, N.Y: Garland Science, 2002; ISBN 0-8153-3218-1 [Google Scholar]
- 54. Dai H-S, Liu Z, Jiang W, Kuhn RJ. Directed evolution of a virus exclusively utilizing human epidermal growth factor receptor as the entry receptor. J Virol 2013; 87:11231-43; PMID:23926357; http://dx.doi.org/ 10.1128/JVI.01054-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Esvelt KM, Carlson JC, Liu DR. A system for the continuous directed evolution of biomolecules. Nature 2011; 472:499-505; PMID:21478873; http://dx.doi.org/ 10.1038/nature09929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. North EH. Command to robot, Gort, In: The day the earth stood still, a science fiction movie. 1951, 20th Century Fox. [Google Scholar]
- 57. Stockley PG, Twarock R, Bakker SE, Barker AM, Borodavka A, Dykeman E, Ford RJ, Pearson AR, Phillips SEV, Ranson NA, et al. Packaging signals in single-stranded RNA viruses: nature's alternative to a purely electrostatic assembly mechanism. J Biol Phys 2013; 39:277-87; http://dx.doi.org/ 10.1007/s10867-013-9313-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jiang W, Chiu W. Cryoelectron microscopy of icosahedral virus particles. Meth Mol Biol 2007; 369:345-63; PMID:17656759 [DOI] [PubMed] [Google Scholar]
- 59. Johnson JE. Confessions of an icosahedral virus crystallographer. Microscopy (Oxf) 2013; 62:69-79; PMID:23291268; http://dx.doi.org/ 10.1093/jmicro/dfs097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ashley CE, Carnes EC, Phillips GK, Durfee PN, Buley MD, Lino CA, Padilla DP, Phillips B, Carter MB, Willman CL, et al. . Cell-specific delivery of diverse cargos by bacteriophage MS2 virus-like particles. ACS Nano 2011; 5:5729-45; PMID:21615170; http://dx.doi.org/ 10.1021/nn201397z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Peabody DS, Manifold-Wheeler B, Medford A, Jordan SK, do Carmo Caldeira J, Chackerian B. Immunogenic display of diverse peptides on virus-like particles of RNA phage MS2. J Mol Biol 2008; 380:252-63; PMID:18508079; http://dx.doi.org/ 10.1016/j.jmb.2008.04.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Smith DE. Single-molecule studies of viral DNA packaging. Curr Opin Virol 2011; 1:134-41; PMID:22440623; http://dx.doi.org/ 10.1016/j.coviro.2011.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Casjens SR. The DNA-packaging nanomotor of tailed bacteriophages. Nat Rev Microbiol 2011; 9:647-57; PMID:21836625; http://dx.doi.org/ 10.1038/nrmicro2632 [DOI] [PubMed] [Google Scholar]
- 64. Serwer P, Jiang W. Dualities in the analysis of phage DNA packaging motors. Bacteriophage 2012; 2:239-55; PMID:23532204; http://dx.doi.org/ 10.4161/bact.23829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Serwer P, Hayes SJ, Watson RH. The structure of a bacteriophage T7 procapsid and its in vivo conversion product probed by digestion with trypsin. Virology 1982; 122:392-401; PMID:7147708 [DOI] [PubMed] [Google Scholar]
- 66. Cuervo A, Pulido-Cid M, Chagoyen M, Arranz R, González-García VA, Garcia-Doval C, Castón JR, Valpuesta JM, van Raaij MJ, Martín-Benito J, et al. . Structural characterization of the bacteriophage T7 tail machinery. J Biol Chem 2013; 288:26290-9; PMID:23884409; http://dx.doi.org/ 10.1074/jbc.M113.491209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Serwer P. A metrizamide-impermeable capsid in the DNA packaging pathway of bacteriophage T7. J Mol Biol 1980; 138:65-91; PMID:7411607; http://dx.doi.org/ 10.1016/S0022-2836(80)80005-2 [DOI] [PubMed] [Google Scholar]
- 68. Serwer P, Watson RH, Hayes SJ, Allen JL. Comparison of the physical properties and assembly pathways of the related bacteriophages T7, T3 and phiII. J Mol Biol 1983; 170:447-69; PMID:6631966; http://dx.doi.org/ 10.1016/S0022-2836(83)80157-0 [DOI] [PubMed] [Google Scholar]
- 69. Fang PA, Wright ET, Weintraub ST, Hakala K, Wu W, Serwer P, et al. . Visualization of bacteriophage T3 capsids with DNA incompletely packaged in vivo. J Mol Biol 2008; 384:1384-99; PMID:18952096; http://dx.doi.org/ 10.1016/j.jmb.2008.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Khan SA, Griess GA, Serwer P. Assembly-associated structural changes of bacteriophage T7 capsids. Detection by use of a protein-specific probe. Biophys J 1992; 63:1286-92; PMID:1477280; http://dx.doi.org/ 10.1016/S0006-3495(92)81724-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Merril CR, Biswas B, Carlton R, Jensen NC, Creed GJ, Zullo S, Adhya S. Long-circulating bacteriophage as antibacterial agents. Proc Natl Acad Sci U S A 1996; 93:3188-92; PMID:8622911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Serwer P, Wright ET, Liu Z, Jiang W. Length quantization of DNA partially expelled from heads of a bacteriophage T3 mutant. Virology 2014; 456-457:157-70; PMID:24889235; http://dx.doi.org/ 10.1016/j.virol.2014.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Heineman RH, Springman R, Bull JJ. Optimal foraging by bacteriophages through host avoidance. Am Nat 2008; 171:E149-157; PMID:18254683; http://dx.doi.org/ 10.1086/528962 [DOI] [PubMed] [Google Scholar]
- 74. Keen EC. Tradeoffs in bacteriophage life histories. Bacteriophage 2014; 4: e28365; PMID:24616839; http://dx.doi.org/ 10.4161/bact.28365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Khan SA, Watson RH, Hayes SJ, Serwer P. PCR-directed formation of viral hybrids in vitro. Virology 1997; 227:409-19; PMID:9018140 [DOI] [PubMed] [Google Scholar]
- 76. Fischetti V. Phage factor. Interview by Brendan Borrell. Sci Am 2012; 307:80-3; PMID:22844857 [PubMed] [Google Scholar]
- 77. Oliveira H, Azeredo J, Lavigne R, Kluskens LD. Bacteriophage endolysins as a response to emerging foodborne pathogens. Trends Food Sci Technol 2012; 28:103-115 [Google Scholar]
- 78. Kaplan JB. Therapeutic potential of biofilm-dispersing enzymes. Int J Artif Organs 2009; 32:545-54; PMID:19851978 [DOI] [PubMed] [Google Scholar]
- 79. Chen J, Groves R, Zheng Y, Civerlo EL, Viveros M, Freeman M. Colony morphology of Xylella fastidiosa almond leaf scorch strains. Phytopathol 2005; 95:708-14; PMID:18943788; http://dx.doi.org/ 10.1094/PHYTO-95-0708 [DOI] [PubMed] [Google Scholar]
- 80. Bar H, Yacoby I, Benhar I. Killing cancer cells by targeted drug-carrying phagenanomedicines. BMC Biotechnol 2008; 8: 37; PMID:18387177; http://dx.doi.org/ 10.1186/1472-6750-8-37; PMCID: PMC2323368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Suthiwangcharoen N, Li T, Li K, Thompson P, You S, Wang Q. M13 nanoassemblies as drug delivery vehicles. NANO Res 2011; 4:483-493; http://dx.doi.org/ 10.1007/s12274-011-0104-2 [DOI] [Google Scholar]
- 82. Vaks L, Benhar I. In vivo characteristics of targeted drug-carrying filamentous bacteriophage nanomedicines. J Nanobiotechnol 2011; 9:58; http://dx.doi.org/ 10.1186/1477-3155-9-58; PMCID: PMC3281789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Budynek P, Dabrowska K, Skaradzinski G, Gorski A. Bacteriophages and cancer. Arch Microbiol 2010; 192:315-20; PMID:20232198; http://dx.doi.org/ 10.1007/s00203-010-0559-7 [DOI] [PubMed] [Google Scholar]
- 84. Serwer P, Wright ET. Agarose gel electrophoresis reveals structural fluidity of a phage T3 DNA packaging intermediate. Electrophoresis 2012; 33:352-65; PMID:22222979; http://dx.doi.org/ 10.1002/elps.201100326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Casjens S, Wyckoff E, Hayden M, Sampson L, Eppler K, Randall S, Moreno ET, Serwer P. Bacteriophage P22 portal protein is part of the gauge that regulates packing density of intravirion DNA. J Mol Biol 1992; 224:1055-74; PMID:1569567 [DOI] [PubMed] [Google Scholar]
- 86. Bernstein C, Nfonsam V, Prasad AR, Bernstein H. Epigenetic field defects in progression to cancer. World J Gastrointest Oncol 2013; 5:43-9; PMID 23671730; http://dx.doi.org/ 10.4251/wjgo.v5.i3.43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lahtz C, Pfeifer GP. Epigenetic changes of DNA repair genes in cancer. J Mol Cell Biol 2011; 3:51-88; PMID:21278452; http://dx.doi.org/ 10.1093/jmcb/mjq053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Command to robot , Gort, in the science fiction movie, The Day the Earth Stood Still, 20th Century Fox 1951 [Google Scholar]
- 89. Liu X, Jiang W, Jakana J, Chiu W. Averaging tens to hundreds of icosahedral particle images to resolve protein secondary structure elements using a multi-path simulated annealing optimization algorithm. J Struct Biol 2007; 160:11-27; PMID:17698370; http://dx.doi.org/ 10.1016/j.jsb.2007.06.009; PMCID: PMC2039893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Liu X, Zhang Q, Murata K, Baker ML, Sullivan MB, Fu C, Dougherty MT, Schmid MF, Osburne MS, Chisholm SW, et al. . Structural changes in a marine podovirus associated with release of its genome into Prochlorococcus. Nat Struct Mol Biol 2010; 17:830-6; PMID:20543830; http://dx.doi.org/ 10.1038/nsmb.1823; PMCID: PMC2924429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Baker ML, Hryc CF, Zhang Q, Wu W, Jakana J, Haase-Pettingell C, Afonine PV, Adams PD, King JA, Jiang W, et al. . Validated near-atomic resolution structure of bacteriophage epsilon15 derived from cryo-EM and modeling. Proc Natl Acad Sci U S A 2013; 110:12301-6; PMID:23840063; http://dx.doi.org/ 10.1073/pnas.1309947110; PMCID: PMC3725109 [DOI] [PMC free article] [PubMed] [Google Scholar]