

Abstract
Twenty-five years after its inception, the genetic engineering of T cells is now a therapeutic modality pursued at an increasing number of medical centers. This immunotherapeutic strategy is predicated on gene transfer technology to instruct T lymphocytes to recognize and reject tumor cells. Chimeric antigen receptors (CARs) are synthetic receptors that mediate antigen recognition, T cell activation, and — in the case of second-generation CARs — costimulation to augment T cell functionality and persistence. We demonstrated over a decade ago that human T cells engineered with a CD19-specific CAR eradicated B cell malignancies in mice. Several phase I clinical trials eventually yielded dramatic results in patients with leukemia or lymphoma, especially acute lymphoblastic leukemia (ALL). This review recounts the milestones of CD19 CAR therapy and summarizes lessons learned from the CD19 paradigm.
Introduction
The genetic engineering of T cells provides a means to rapidly generate antitumor T cells for any cancer patient. This approach is predicated on gene transfer technology that enables the expression of receptors and other gene products in primary T cells. This review focuses on how CD19 chimeric antigen receptors (CARs) came to be and what we’ve learned, to date, about CAR therapy owing to the CD19 paradigm.
Back in the late 1980s, the rationale for T cell engineering was to rapidly establish a potent immune attack on cancer cells. It had recently become apparent that adoptively transferred T cells could, in select circumstances, exert profound antitumor effects, as seen in graft-versus-host disease and graft-versus-leukemia in BM transplant recipients (1). The first attempts to isolate tumor- or virus-reactive T cells were underway (2, 3), hinting that the isolation of antigen-specific T cells would be feasible, although cumbersome. The discovery of the physiological receptor that mediates antigen recognition, known as the T cell receptor (TCR) (4–6), led to transgenic mouse studies that demonstrated that antigen specificity could be imparted to T cells through germline modification (7). The rationale for developing T cell engineering remains as compelling today as it was 25 years ago and is reinforced by the vast knowledge of T cell biology and tumor immunology that has since accumulated (Table 1).
Table 1. Rationale for T cell engineering in oncology.
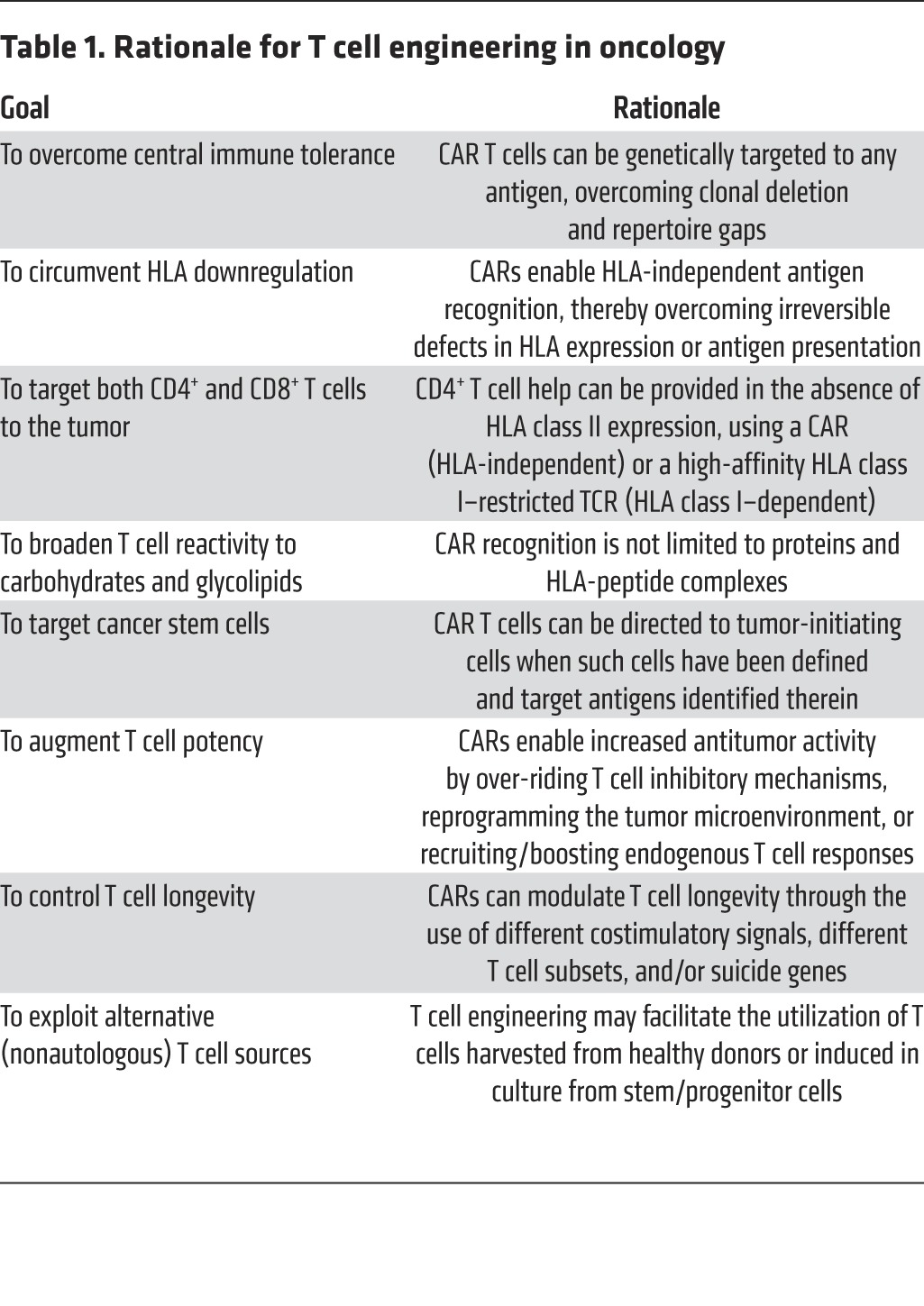
To contemplate T cell engineering, two primary requirements had to be fulfilled: it would be necessary to (i) establish gene transfer technology effective in primary T cells and (ii) identify receptor structures that enabled T cell reprogramming and were adapted to the available gene transfer technology. By the late 1980s, the use of replication-defective retroviruses to transduce mammalian cells was just starting to be applied to mouse hematopoietic cells (8). Retroviral-mediated gene transfer to mouse T lymphocytes proved to be challenging but was eventually feasible (9). By the mid-1990s, methods for the transduction of human T lymphocytes became available, based on the use of the gibbon ape leukemia virus envelope (GALV envelope) to mediate retroviral vector entry (10–12). This advance was pivotal for developing T cell engineering, which had been hitherto limited to transfection of surrogate leukemia cell lines or hybridomas that do not recapitulate several critical facets of normal T cell activation and function. Receptors and signaling molecules could now be studied in true human T cells harvested from peripheral blood. These methods remain the foundation for many of today’s clinical trials based on T cell engineering, which frequently make use of GALV envelope–pseudotyped packaging cell lines (13) and the SFG vector or variant γ-retroviral vectors (14–17). Improved packaging cell lines (18) and enhanced vector production processes (19) are available today, as are an array of T cell transduction methods, which utilize γ-retroviral, lentiviral, and nonviral DNA- or RNA-based vectors (reviewed in ref. 20).
The second requirement for undertaking T cell engineering is the isolation or design of receptors for antigen that direct effective T cell responses. This goal has been pursued with two general approaches, one utilizing the physiological TCR as the tumor-targeting device (21) and the other using a variety of artificial receptors (22), starting from those described by Eshhar and Brocker (23, 24) and eventually encompassing a broadened range of structures that we regrouped under the general name of CAR (ref. 25). While the earliest artificial receptors attempted to reproduce a T cell activation signal similar to the TCR (see below), the main attraction of synthetic receptors was and remains to this date their potential to not only retarget T cells, but also to enhance T cell function and persistence (Table 1). This goal was eventually attained through the invention of receptors that provide three critical functions within a single molecule encoded by a single cDNA: targeting, activation, and costimulation. These receptors, known as second-generation CARs (25), comprise signaling domains derived from a T cell–activating molecule, such as the ζ-chain of the CD3 complex (CD3ζ), and a costimulatory receptor, such as CD28 or CD137. They entered the clinical arena in 2007, based on the CD19 paradigm, which is recounted below.
First drives
CARs are artificial antigen receptors that evolved in three critical steps. When CD3ζ was independently cloned by the Weiss, Seed, and Klausner groups, the newly discovered T cell–specific chain, which lacks a significant extracellular domain, was fused to either CD8, CD4, or CD25 (refs. 26–28 and Figure 1). These molecules bind to HLA class I, HLA class II, and IL-2, respectively. Cross-linking of these receptors by available antibodies targeting their extracellular domains resulted in calcium influx and other hallmarks of early T cell activation, establishing that cross-linked ζ-chain fusions could initiate T cell activation. The addition of a single-chain variable fragment (scFv) derived from an immunoglobulin specific for the 2,4,6-trinitrophenyl (TNP) hapten diversified the binding capacity of CD3ζ-chain fusion receptors (ref. 23 and Figure 1). Using leukemic T cells or cytotoxic cell lines, these fusion receptors were shown to activate T cells and redirect antigen specificity (29). In utilizing an scFv as their binding moiety, CARs target cell-surface antigens, unlike the physiological TCR, which binds to HLA-peptide complexes (21, 22). Thus, CARs engage molecules that do not depend on peptide processing or HLA expression in order to be recognized. Additionally, CARs do not need to be matched to the patient’s haplotype, as is the case for TCRs (21, 22). Furthermore, CARs can target tumor cells that have downregulated HLA expression or proteasomal antigen processing, two mechanisms that contribute to tumor escape from TCR-mediated immunity (30). Another feature of the broad applicability of CARs is their ability to bind not only to proteins, but also to carbohydrate and glycolipid structures, expanding the range of potential targets (31). Some CARs utilize receptor or ligand domains as their targeting moiety, such as heregulin (32), IL-13 (33), or NK cell lectin–like receptor NKG2D (34), which bind to their cognate ligands or receptor counterparts. Antibody-based CARs can also be utilized to target HLA-peptide complexes, allowing for recognition of antigens derived from intracellular proteins (35).
Figure 1. The first CARs and CCRs.

Top left: The first CD3ζ-chain fusions established the T cell–activating function of the CD3ζ-chain (26–28). The incorporation of an scFv diversified their antigen-binding capacity (23). Collectively, these receptors are now known as first-generation CARs. Bottom left: Derivation of the first CCR from the native CD28 costimulatory receptor (41). CCRs are not CARs, as they do not initiate T cell activation. The conflation of a first-generation CAR (top left) and a CCR (bottom left) resulted in the generation of a second-generation CAR capable of directing proliferation and sustained function of human peripheral blood T lymphocytes upon repeated exposure to antigen (right) (42).
While CD3ζ-chain aggregation is sufficient to enable lytic activity in cytotoxic T lymphocyte lines (CTL lines), it is important to bear in mind that the strength of signal required for cytotoxicity is lower than that needed for other T cell functions. This likely underlies the limited therapeutic responses reported with first-generation CARs, the antitumoral effects of which are often confined to local administration models (36, 37) or short-term systemic models (37). In transgenic mice, T cells expressing CARs that only comprise an activation domain within their cytoplasmic domain are prone to undergoing anergy (38). Transgenic mouse models later revealed that T cells expressing CD3ζ-chain–based CARs only modestly delayed tumor progression in vivo, as they produced low amounts of IFN-γ and rapidly became anergic (38, 39). Having established methods for the study of primary T cells (9, 12), we could test other essential features of CD3ζ-chain–based CARs. While these receptors effectively mediated cytotoxicity, we found that they were unable to direct T cell expansion upon repeated exposure to antigen in the absence of costimulatory support (40). Thus, CD3ζ-chain–based CARs mediate antigen-specific cytotoxicity but fail to support T cell expansion in the continued presence of antigen. A new strategy was therefore needed to prevent anergy or activation-induced cell death (AICD) and enable T cells to expand and persist in a functional state.
Second-generation CARs
In the 1990s, we designed chimeric costimulatory receptors (CCRs) to examine whether we could promote T cell expansion and offset AICD in an antigen-specific fashion. In these studies, human primary T cells were activated through their TCR under conditions that would limit survival and expansion by precipitating apoptosis. This AICD could be offset in the presence of a cell-surface antigen (in this case, the disialoganglioside GD2) when the T cells expressed a GD2-specific, CD28-based CCR (Figure 1). The CD28-based CCR was also able to direct IL-2 synthesis, similar to the natural CD28 costimulatory receptor (41). Having validated the CCR costimulatory signal in human primary T cells, we then fused a CCR with a ζ-chain–based CAR, thus creating a second-generation CAR (Figure 1) that supported human primary T cell expansion upon repeated exposure to antigen (42). These and other contemporary studies (43–45) paved the way for engineering persisting functional T cells.
Multiple costimulatory domains have since been introduced into CARs (46). The best known second-generation CARs incorporate CD28 or 4-1BB signaling elements (42, 45). Compared with first-generation CD3ζ-chain CARs, CD28/CD3ζ CARs induce more IL-2 secretion, increase T cell proliferation and persistence, and mediate greater tumor rejection (46–49). These results, which were obtained consistently in mouse models, were corroborated in a clinical study where both CAR T cell populations were coinfused, confirming the greater persistence of T cells expressing the CD28/CD3ζ CAR (17). 4-1BB costimulatory domains also extend T cell survival compared with first-generation CARs (45, 49, 50), albeit with different pharmacokinetics (51). 4-1BB/CD3ζ CARs impart greater longevity to T cells than do CD28/CD3ζ CARs, resulting in higher CAR T cell accumulation, which could potentially outlast tumor elimination (52). More studies are needed to better delineate the respective properties of these and other second-generation CARs that incorporate OX40, ICOS, DAP-10, NKG2D, or other costimulatory domains (51, 53).
The CD19 paradigm
CD19 is a cell-surface antigen found on most B-lineage lymphomas and leukemias (54). In the mid-1990s, we believed that the involvement of CD19 in B cell development and function (55–57) might extend to a role in tumor survival, which might in turn predict CD19 expression in most malignant cells, possibly including tumor-initiating cells. CD19 was chosen not only for its frequent and high-level expression in B cell malignancies, but also for its highly restricted expression in normal tissues, where it is confined to the B cell lineage. CD19 is also occasionally expressed in multiple myeloma (58). Thus, a successful therapy would be expected to induce B cell aplasia, as was indeed later observed in murine models (59, 60) and in patients given CD19 CAR therapy.
It has been almost 15 years since we reported that CD19 CAR therapy utilizing human peripheral blood T lymphocytes eradicated lymphoma and leukemia in immune-deficient mice, also providing the first demonstration that human CAR T cells of any specificity could eradicate systemic tumors established in mice (61). In these models, a single intravenous infusion of CD19 CAR T cells resulted in complete eradication of established, diffuse B cell malignancies (61). We later showed that the 19-28z CAR vastly outperformed a first-generation CD19 CAR in an aggressive acute lymphoblastic leukemia model (ALL model) (48). Successful B cell tumor eradication was eventually obtained with different CD19 CARs (36, 47–49, 59–61), paving the way for multiple, ongoing CD19 CAR clinical trials. The attractiveness of CD19 as a CAR target was confirmed by its subsequent selection by the groups at the National Cancer Institute (NCI), the University of Pennsylvania (UP), City of Hope National Medical Center, Fred Hutchinson Cancer Research Center (FHCRC), MD Anderson Cancer Center, and Baylor College of Medicine when they launched their clinical CAR programs. CD19 is by far the most investigated CAR target today.
Clinical trials for indolent B cell malignancies
Second-generation CARs have recently shown impressive clinical outcomes in trials that enrolled patients with relapsed or refractory B cell malignancies. Earlier studies utilizing first-generation CARs targeting the α-folate receptor in ovarian cancer (62), carbonic anhydrase IX in renal cancer (63), CD20 in lymphoma (64), and GD2 in neuroblastoma (65) yielded modest results, with the exception of one durable complete response (CR) in the neuroblastoma study.
The first clinical study to utilize second-generation CARs targeting CD19 enrolled patients with chronic lymphocytic leukemia (CLL; NCT 00466531), but the first publication of a CD19 CAR therapy came in the form of a case report from the NCI (66). In this report, a patient with advanced follicular lymphoma showed a durable partial remission lasting 32 weeks before progressing with CD19+ disease. Although the report did not document T cell presence or persistence, the response was attributed to the combined conditioning and CAR T cells. Later studies in lymphoma patients achieved several remissions, with a combined complete or partial remission rate reaching 60% in some reports (67, 68), as reviewed in ref. 69.
The initial results reported in CLL were dramatic and intriguing. Two of the three patients treated at UP showed profound complete remissions (70), which have lasted to this day. These two patients became tumor free and B cell aplastic within weeks/months following the T cell infusion. The CLL trial at MSKCC included a first arm, where T cells were given without prior conditioning as mandated by the FDA, which at that time considered conditioning and T cells to be a combination of two therapies. None of the patients who were given T cells without prior conditioning responded to the CAR therapy. The responses were modest in 4 evaluable patients who received cyclophosphamide conditioning (CY conditioning) (71). In comparison, the CLL studies at UP and the lymphoma study at the NCI utilized more potent conditioning regimens, consisting of either bendamustine or CY/fludarabine (FLU), respectively (68, 70). Although no comprehensive follow-up study in CLL has yet been published, recent meeting reports from UP and MSKCC suggest that approximately half the CLL patients have CR or partial response to the current form of CD19 CAR therapy (72, 73).
Clinical trials for adult and pediatric ALL
The most dramatic and consistent outcomes obtained with CD19 CAR therapy have occurred in ALL (74–78). The first published results were obtained in adult patients with relapsed, chemo-refractory disease who were infused with autologous peripheral-blood T cells collected by apheresis and transduced with the second-generation 19-28z CAR (16). These subjects were infused with 3 million autologous 19-28z+ T cells/kg following a single administration of CY as conditioning. Four of four patients with measurable disease went into complete, molecular remission within 4 weeks of the T cell infusion (74). MSKCC, the Children’s Hospital of Philadelphia (CHOP), and the NCI subsequently published follow-up studies in adult and pediatric ALL patients (75–78), which are summarized in Table 2. MSKCC next reported on 16 adult subjects, showing an 88% CR rate, which was obtained on average by day 24 and correlated with the detection of persisting T cells for 4–12 weeks following infusion (76). Five patients treated at UP showed similar results, resulting in 5 CRs and detectable T cells for 2–3 months after infusion (77). In pediatric ALL, the first report on two children showed two CRs, with one relapse due to a CD19-negative clone (75). A subsequent report from UP showed a CR rate of 90% in 25 children and notable T cell persistence, ranging from 1–10 months (76). The NCI also reported on 20 children or young adults with a CR rate of 70% in their intent-to-treat cohort (ref. 78 and Table 2).
Table 2. CD19 CAR therapy for ALL.
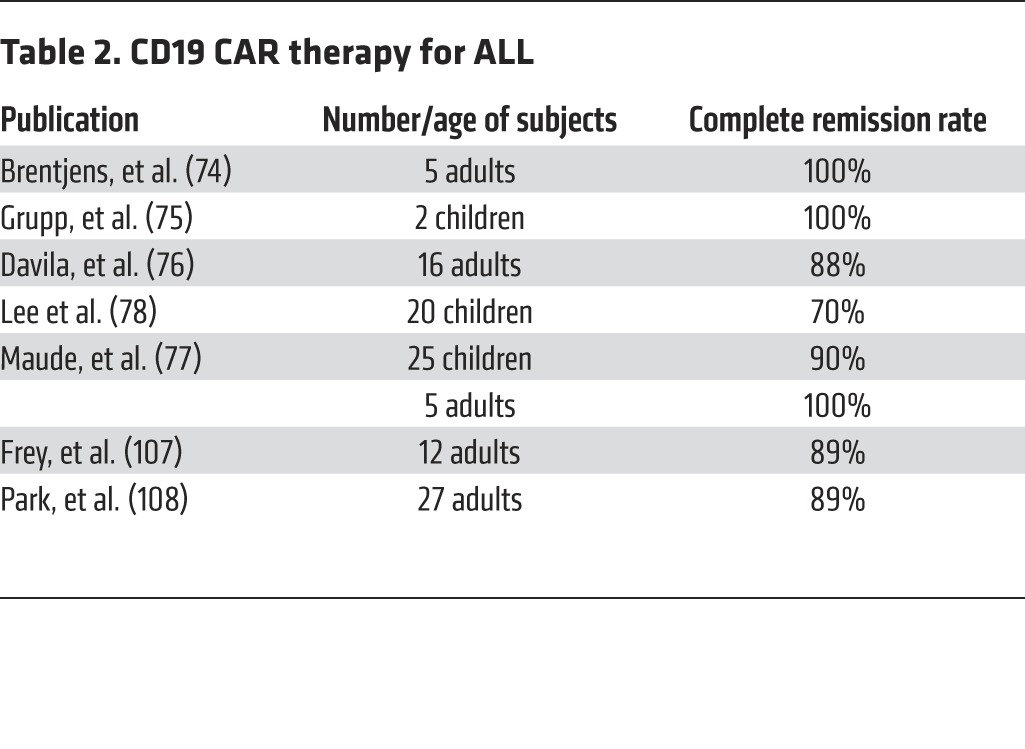
In summary, clinical results from three different centers all reported a remarkable CR rate — a rare occurrence for phase I studies in oncology, especially for patients with chemo-refractory leukemias. While these studies follow the same overall procedure (apheresis, CAR transduction, and conditioning therapy, followed by T cell infusion), they differ in several regards, including the CAR design (the CD28/CD3ζ dual-signaling domain utilized at the NCI and MSKCC, and the St. Jude 4-1BB/CD3ζ utilized at UP, Figure 2), T cell manufacturing, retroviral vector type, conditioning chemotherapy, patient age, tumor burden, tumor sensitivity to chemotherapy, and T cell dosage (79). Despite these differences, the comparable outcomes speak to the extraordinary robustness of CD19 CAR therapy in ALL.
Figure 2. Prototypic CD19 CARs and other CD19 CARs in clinical use.

Design of the first second-generation CARs to be used in the clinic: the MSKCC CD28-based CAR (42) and the St. Jude Children’s Research Hospital (SJCRH) 4-1BB CAR (45). CD28-based CARs have been utilized at MSKCC, the NCI, and Baylor College of Medicine (top). 4-1BB–based CARs have been utilized at CHOP/UP and the FHCRC/Seattle Children’s Hospital (FHCRC) (bottom).
The CD19 paradigm: five lessons learned over 20 years (1995–2015)
The enthusiasm of many groups for targeting CD19 with CARs is testimony to the attractive features of CD19 and its broad relevance as a CAR target. Its association with all B cell malignancies, its expression in most if not all tumor cells, its absence from vital tissues, and the demonstrated effectiveness of different second-generation CARs targeting CD19 have turned it into the most investigated CAR target and a paradigm for CAR therapy. Five major lessons have come out of the last two decades of CD19 CAR research.
Cells work where drugs have failed.
The most dramatic lesson learned from the CD19 paradigm is that engineered T cells induce complete remissions, including molecular remissions, in subjects for whom chemotherapies, often utilizing multiple drug combinations, have led to drug resistance and tumor progression. In essence, cells can succeed where chemicals have failed. This is a profound message that is still sending shock waves throughout academic medicine and the biopharmaceutical industry. CD19 CARs are the poster child for CAR therapy, for which ALL is, at present, the most resounding success. In the case of B cell lymphomas and CLL, the results are also very encouraging, even though CAR therapy will have to be evaluated against a broader field of competing or emerging treatments. CD19 CAR therapy can also be, and will be, further optimized for these B cell malignancies.
Academic T cell manufacturing is robust and dependable.
Another major lesson learned from the CD19 paradigm concerns feasibility. A handful of academic centers have successfully established manufacturing procedures that have proven to be reproducible and dependable (20). CAR T cell products have been successfully generated for the overwhelming majority of subjects enrolled at MSKCC and elsewhere. At MSKCC, the T cell engineering process starts from an apheresis product and utilizes a γ-retroviral vector for T cell transduction, yielding the prescribed cell doses in 8–12 days (16). Other variant cell-manufacturing processes have been developed and used successfully (20). Given the recent interest of biotech and large pharmaceutical companies in cell therapies (80), there is every reason to anticipate that increased funding for cell manufacturing will accelerate the pace of development for improved CAR T cell manufacturing platforms.
Second-generation CARs are potent.
The successes we have seen in CD19 CAR therapy were all obtained with second-generation CARs, supporting the critical importance of incorporating costimulatory signals in recombinant antigen receptors (Figure 1). Furthermore, two distinct designs utilizing either CD28 or 4-1BB signaling elements have yielded profound clinical results. In ALL, where both designs have been utilized (Figure 2), CARs utilizing either design elicit comparable CR rates (Table 2) and toxicities (see below); however, some nuances are emerging. The time until the onset of fever and the rapidity of CR induction seems to be faster with the CD28 CARs (74–78), while the persistence of T cells expressing 4-1BB CARs is greater, at least in children (77) compared with adults (76, 77). This is very encouraging, as it validates the general concept of incorporating costimulatory domains in engineered T cells and suggests that other costimulatory domains and domain combinations will yield T cells with tunable functional characteristics.
CAR T cells may induce toxicities.
The CD19 paradigm has revealed three potential toxicities of CAR therapy: B cell aplasia, severe cytokine release syndrome (CRS), and neurological toxicity. The first is an on-target/off-tumor effect that was predicted and observed in animal models (59, 60) and later in clinical trials (66–78). The anticipation of B cell aplasia was originally a reason for selecting CD19 as a target because a deficit in antibody production can be managed with intravenous immunoglobulin administration. Furthermore, B cell aplasia is reversible when the CAR T cells eventually vanish or when patients undergo hematopoietic stem cell transplantation. B cell aplasia is only a concern in those few patients who retain CD19 CAR T cells in the long-term and may be a greater concern when long-term T cell persistence occurs in children or in individuals in whom plasma cells are eventually eliminated.
The second major concern is that of severe CRS, which is associated with intense antitumor responses mediated by large numbers of activated T cells. Like B cell aplasia, it is an anticipated consequence of CAR T cell activity that exhibits variable magnitude. CD19 CAR T cells often induce fever accompanied by cytokine release, but a substantial cytokine response may induce hemodynamic and cardiac side effects requiring medical intervention. We (76) and others (81) have proposed definitions for CRS, which need to be reconciled in order to provide a standard definition and grading reference for clinicians and regulators. A conference addressing this issue was organized by the Recombinant DNA Advisory Committee in June 2015 (82). The management of severe CRS may require steroids, IL-6 receptor blockade (tocilizumab, as originally proposed by CHOP investigators), vasopressors, and/or supportive therapy delivered in the intensive care unit (74–78, 81–83). Importantly, in our first 5 ALL patients, we observed (74) that the likelihood of developing severe CRS is closely correlated with tumor burden, thereby providing a simple means to anticipate which patients may require medical intervention. This observation was later confirmed in a larger cohort (76) and has held up in pediatric patients (77, 78). Thus, the means to anticipate CRS and intervene therapeutically have made rapid progress. Approaches to treat or ultimately prevent severe CRS are reviewed elsewhere (76, 81–84).
The last toxicity is neurological, consisting of confusion, obtundation, seizures, or aphasia. The mechanism underlying this response is unclear. It does not appear to be related to CNS leukemia. Neurological toxicity is also seen in therapies utilizing CD19/CD3ζ bispecific protein conjugates (85). More studies are needed to devise preventive or therapeutic interventions for neurological toxicity (83, 84).
Antigen escape may occur.
CAR T cells most often target a single antigen. One must therefore seek to identify target antigens that, ideally, are present in all tumor cells, including cancer stem cells, if possible. A broad effort is now underway to identify suitable targets for a variety of cancers. It is expected that the selective pressure imparted by CAR T cells, as for any targeted therapy, will sometimes yield antigen escape variants. This was first observed in pediatric ALL, where one of the first two subjects treated with CD19 CAR therapy at CHOP eventually relapsed with CD19-negative disease (75). Antigen escape of CD19-negative ALL has been observed at all centers. Targeting a second ALL antigen, such as CD22 (86), is one strategy that is poised to offset this risk. The mechanism for epitope loss remains to be fully elucidated. Interestingly, this occurrence has not been reported to date in CLL or non-Hodgkin lymphoma.
Conclusions and perspectives
CAR therapy lies at the intersection of cell therapy, gene therapy, and immunotherapy. It is rooted in the principles of T cell biology, gene transfer biology, and tumor immunology. The CD19 paradigm is the culmination of 25 years of research on T cell engineering. Collectively, the preclinical and clinical studies on CD19 CARs have validated the invention of second-generation CARs, established the feasibility of implementing T cell engineering in the clinic, and demonstrated the effective potency of CAR therapy in at least one cancer, ALL. Current CD19 CAR therapy is, at the very least, a bridge to allogeneic hematopoietic stem cell transplantation in ALL patients for whom this potentially curative intervention is no longer an option due to the advanced stage of their disease. Further follow-up of those subjects who received CD19 CAR therapy without a subsequent transplantation will eventually establish whether CAR therapy alone may be curative. In due course, CAR therapy also has the potential to become a front-line therapy for ALL and other B cell malignancies.
CD19 CAR therapy in its present form or an enhanced form will likely deliver similar outcomes for B cell lymphomas and CLL. CAR therapy, based on second-generation CARs, can be further enhanced in several ways. CARs may be combined with costimulatory ligands (87), CCRs (88–91), or cytokines such as IL-15 (92, 93) or IL-12 (59, 94, 95) to further enhance T cell potency, specificity, and/or safety. CARs may also be combined with other immune enhancers, such as anti–PD-1/PD-L1 antibodies (96), lenalinomide (97), or indoleamine 2,3-dioxygenase (IDO) inhibitors (98).
The optimal conditioning for CD19 CAR therapy is not yet fully understood. Without any conditioning, CD19 CAR therapy is not effective (17, 71), as is the case in the mouse (59, 60). Whereas in ALL, conditioning consisting of either CY alone or CY and FLU has yielded similar outcomes (74–78), this has not been the case in CLL, where bendamustine conditioning has been associated with better outcomes than CY alone (70, 71) and in non-Hodgkin lymphoma, where more intense conditioning (CY [120 mg/kg] in combination with FLU [25 mg/m2 × 5], compared with CY [300 mg/m2 × 3] in combination with FLU [30 mg/m2 × 3]) has resulted in better outcomes but also higher toxicities (78, 99). An ideal conditioning regimen remains to be defined. Interestingly, Brentjens and colleagues have shown in an immunocompetent murine model that CAR T cells expressing IL-12 could mediate tumor rejection in the absence of prior conditioning (59).
Another major question about the optimal delivery of CAR therapy awaiting resolution is the identity of the optimal T cell substrates. Strikingly, the consistent clinical outcomes obtained in ALL patients have been obtained with bulk peripheral blood T cells comprising variable CD4+/CD8+ T cell ratios starting from variable mixtures of naive and antigen-experienced T cells (74–78). The quasi uniformity of successful outcomes has thus not revealed an optimal CD4+/CD8+ T cell ratio or a superior T cell subset, yet we know from murine studies that, although CD8+ or CD4+ CAR T cells alone can exert significant therapeutic effects (61, 100), a mixture of both subsets displays superior efficacy (100, 101). Much remains to be learned about the potential of virus-specific T cells (102) and defined T cell subsets, in particular the central memory (TCM) and the stem cell–like memory (TSCM) subsets (103, 104). It is likely that the selection of optimal T cell subsets for CAR therapy will impact the efficacy, consistency, and safety of CAR therapy (104, 105). The principles to be uncovered in clinical studies utilizing defined T cell products will also inform the choice of alternative (i.e., nonautologous) T cell sources that are likely to emerge in the future (106).
CD19 CARs have taught us a great deal about the enormous potential and current limitations of CAR technology, providing insights into how to tackle solid tumors, which is one of the major next steps for the CAR field. CAR targets will have to be chosen thoughtfully to limit damage to normal tissue, which will not always be as tolerable as B cell aplasia. T cells will also have to be selected and engineered to overcome inhospitable tumor microenvironments and persist long enough to induce deep remissions without causing severe CRS. All of these goals are attainable in principle, making CAR T cell biology and CAR therapy exciting scientific and clinical pursuits for the next decade and beyond.
Acknowledgments
I thank all the lab members and MSKCC collaborators who contributed to developing CAR therapy over the past 20 years. First, Renier Brentjens — originally a fellow in the lab, then the PI of our first CD19 CAR trial, and now the director of our newly established Cell Therapy Center at MSKCC — and Isabelle Rivière, who made numerous key contributions to developing T cell engineering, and established and directs vector and CAR T cell manufacturing in the Cell Therapy and Cell Engineering Facility at MSKCC. I also thank all other past and present lab members who worked with me on CARs (Prasad Adusumilli, Maud Condomines, Marco Davila, Justin Eyquem, Victor Federov, Theodoros Giavridis, Michael Gong, Gertrude Gunset, Mohamad Hamieh [also thanked for help with figures], Sun Jie, Chris Kloss, Anja Krause, Jean-Baptiste Latouche, John Maher, Jorge Mansilla-Soto, John Markley, Maiko Matsushita, Marcela Maus, Chad May, Aurore Morello, Eirini Papapetrou, Fabiana Perna, Jason Plotkin, Steve Poon, Benjamin Reuben, Gerson Rothschild, Elliot Servais, Sjoukje van der Stegen, Matthias Stephan, Cuiwen Tan, Maria Themeli, Laure Ysebrant, Xiuyan Wang, Zeguo Zhao, and Xiao-Song Zhong), as well as the principal investigators of our CD19 CAR clinical trials at MSKCC: Marco Davila, Kevin Curran, Jae Park, and Craig Sauter.
I thank the NCI, the Center for Experimental Therapeutics of MSKCC (ETC), the Alliance for Cancer Gene Therapy (ACGT), the V Foundation, Stand Up To Cancer (a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research, AACR), the Cancer Research Institute (CRI), Golfers against Cancer, The Lake Road Foundation, Mr. and Mrs. Mallah, William H. and Alice Goodwin and the Commonwealth Foundation for Cancer Research, and Lewis Sanders for their support of our research or clinical studies conducted in the the Center for Cell Engineering at MSKCC.
Footnotes
Conflict of interest: Michel Sadelain is a scientific cofounder of Juno Therapeutics.
Reference information:J Clin Invest. 2015;125(9):3392–3400. doi:10.1172/JCI80010.
References
- 1.Horowitz MM, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood. 1990;75(3):555–562. [PubMed] [Google Scholar]
- 2.Greenberg PD, Cheever MA, Fefer A. Specific adoptive immunotherapy: experimental basis and future potential. Surv Immunol Res. 1982;1(1):85–90. doi: 10.1007/BF02918244. [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. 1986;233(4770):1318–1321. doi: 10.1126/science.3489291. [DOI] [PubMed] [Google Scholar]
- 4.Allison JP, Ridge L, Lund J, Gross-Pelose J, Lanier L, McIntyre BW. The murine T cell antigen receptor and associated structures. Immunol Rev. 1984;81:145–160. doi: 10.1111/j.1600-065X.1984.tb01108.x. [DOI] [PubMed] [Google Scholar]
- 5.Yanagi Y, Yoshikai Y, Leggett K, Clark SP, Aleksander I, Mak TW. A human T cell-specific cDNA clone encodes a protein having extensive homology to immunoglobulin chains. Nature. 1984;308(5955):145–149. doi: 10.1038/308145a0. [DOI] [PubMed] [Google Scholar]
- 6.Davis MM, Bjorkman PJ. T-cell antigen receptor genes and T-cell recognition. Nature. 1988;334(6181):395–402. doi: 10.1038/334395a0. [DOI] [PubMed] [Google Scholar]
- 7.Dembic Z, et al. Transfer of specificity by murine alpha and β T-cell receptor genes. Nature. 1986;320(6059):232–238. doi: 10.1038/320232a0. [DOI] [PubMed] [Google Scholar]
- 8.Keller G, Paige C, Gilboa E, Wagner EF. Expression of a foreign gene in myeloid and lymphoid cells derived from multipotent haematopoietic precursors. Nature. 1985;318(6042):149–154. doi: 10.1038/318149a0. [DOI] [PubMed] [Google Scholar]
- 9. Sadelain M, Mulligan RC. Efficient transduction of murine primary T lymphocytes. Presented at: International Congress of Immunology, Magyar Immunológiai Társaság, International Union of Immunological Societies (IUIS) eds. August 23–28, 1992; Budapest, Hungary. Abstracts. [Google Scholar]
- 10.Mavilio F, et al. Peripheral blood lymphocytes as target cells of retroviral vector-mediated gene transfer. Blood. 1994;83(7):1988–1997. [PubMed] [Google Scholar]
- 11.Bunnell BA, Muul LM, Donahue RE, Blaese RM, Morgan RA. High- efficiency retroviral-mediated gene transfer into human and nonhuman primate peripheral blood lymphocytes. Proc Natl Acad Scie U S A. 1995;92(17):7739–7743. doi: 10.1073/pnas.92.17.7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gallardo HF, Tan C, Ory D, Sadelain M. Recombinant retroviruses pseudotyped with the vesicular stomatitis virus G glycoprotein mediate both stable gene transfer and pseudotransduction in human peripheral blood lymphocytes. Blood. 1997;90(3):952–957. [PubMed] [Google Scholar]
- 13.Miller AD, Garcia JV, von Suhr N, Lynch CM, Wilson C, Eiden MV. Construction and properties of retrovirus packaging cells based on gibbon ape leukemia virus. J Virol. 1991;65(5):2220–2224. doi: 10.1128/jvi.65.5.2220-2224.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Riviere I, Brose K, Mulligan RC. Effects of retroviral vector design on expression of human adenosine deaminase in murine bone marrow transplant recipients engrafted with genetically modified cells. Proc Natl Acad Sci U S A. 1995;92(15):6733–6737. doi: 10.1073/pnas.92.15.6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morgan RA, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hollyman D, et al. Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. J Immunother. 2009;32(2):169–180. doi: 10.1097/CJI.0b013e318194a6e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Savoldo B, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121(5):1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghani K, et al. Efficient human hematopoietic cell transduction using RD114- and GALV-pseudotyped retroviral vectors produced in suspension and serum-free media. Hum Gene Ther. 2009;20(9):966–974. doi: 10.1089/hum.2009.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, et al. Large-scale clinical-grade retroviral vector production in a fixed-bed bioreactor. J Immunother. 2015;38(3):127–135. doi: 10.1097/CJI.0000000000000072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, Riviere I. Manufacture of tumor- and virus-specific T lymphocytes for adoptive cell therapies. Cancer Gene Ther. 2015;22(2):85–94. doi: 10.1038/cgt.2014.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ho WY, Blattman JN, Dossett ML, Yee C, Greenberg PD. Adoptive immunotherapy: engineering T cell responses as biologic weapons for tumor mass destruction. Cancer Cell. 2003;3(5):431–437. doi: 10.1016/S1535-6108(03)00113-2. [DOI] [PubMed] [Google Scholar]
- 22.Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3(1):35–45. doi: 10.1038/nrc971. [DOI] [PubMed] [Google Scholar]
- 23.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T- cell receptors. Proc Natl Acad Sci U S A. 1993;90(2):720–724. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brocker T, Peter A, Traunecker A, Karjalainen K. New simplified molecular design for functional T cell receptor. Eur J Immunol. 1993;23(7):1435–1439. doi: 10.1002/eji.1830230705. [DOI] [PubMed] [Google Scholar]
- 25.Sadelain M, Brentjens R, Riviere I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009;21(2):215–223. doi: 10.1016/j.coi.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Irving BA, Weiss A. The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell. 1991;64(5):891–901. doi: 10.1016/0092-8674(91)90314-O. [DOI] [PubMed] [Google Scholar]
- 27.Romeo C, Seed B. Cellular immunity to HIV activated by CD4 fused to T cell or Fc receptor polypeptides. Cell. 1991;64(5):1037–1046. doi: 10.1016/0092-8674(91)90327-U. [DOI] [PubMed] [Google Scholar]
- 28.Letourneur F, Klausner RD. T-cell and basophil activation through the cytoplasmic tail of T-cell-receptor zeta family proteins. Proc Natl Acad Sci U S A. 1991;88(20):8905–8909. doi: 10.1073/pnas.88.20.8905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. doi: 10.1007/BF00820666. Eshhar Z, et al. The T-body approach: potential for cancer immunotherapy. Springer Semin Immunopathol. 1996:18(2):199-209. [DOI] [PubMed] [Google Scholar]
- 30.Zhou G, Levitsky H. Towards curative cancer immunotherapy: overcoming posttherapy tumor escape. Clin Dev Immunol. 2012;2012:124187. doi: 10.1155/2012/124187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Riviere I, Sadelain M, Brentjens RJ. Novel strategies for cancer therapy: the potential of genetically modified T lymphocytes. Curr Hematol Rep. 2004;3(4):290–297. [PubMed] [Google Scholar]
- 32.Altenschmidt U, et al. Cytolysis of tumor cells expressing the Neu/erbB-2, erbB-3, erbB-4 receptors by genetically targeted naive T lymphocytes. Clin Cancer Res. 1996;2(6):1001–1008. [PubMed] [Google Scholar]
- 33.Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13- zetakine redirected cytolytic T cells. Cancer Res. 2004;64(24):9160–9166. doi: 10.1158/0008-5472.CAN-04-0454. [DOI] [PubMed] [Google Scholar]
- 34.Barber A, Rynda A, Sentman CL. Chimeric NKG2D expressing T cells eliminate immunosuppression activate immunity within the ovarian tumor microenvironment. J Immunol. 2009;183(11):6939–6947. doi: 10.4049/jimmunol.0902000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stewart-Jones G, et al. Rational development of high- affinity T-cell receptor-like antibodies. Proc Natl Acad Scie U S A. 2009;106(14):5784–5788. doi: 10.1073/pnas.0901425106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cooper LJ, et al. T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood. 2003;101(4):1637–1644. doi: 10.1182/blood-2002-07-1989. [DOI] [PubMed] [Google Scholar]
- 37.Hwu P, et al. In vivo antitumor activity of T cells redirected with chimeric antibody/T-cell receptor genes. Cancer Res. 1995;55(15):3369–3373. [PubMed] [Google Scholar]
- 38.Brocker T, Karjalainen K. Signals through T cell receptor-ζ chain alone are insufficient to prime resting T lymphocytes. J Exp Med. 1995;181(5):1653–1659. doi: 10.1084/jem.181.5.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brocker T. Chimeric Fv-ζ or Fv-ε receptors are not sufficient to induce activation or cytokine production in peripheral T cells. Blood. 2000;96(5):1999–2001. [PubMed] [Google Scholar]
- 40.Gong MC, Latouche JB, Krause A, Heston WD, Bander NH, Sadelain M. Cancer patient T cells genetically targeted to prostate-specific membrane antigen specifically lyse prostate cancer cells and release cytokines in response to prostate-specific membrane antigen. Neoplasia. 1999;1(2):123–127. doi: 10.1038/sj.neo.7900018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krause A, Guo HF, Latouche JB, Tan C, Cheung NK, Sadelain M. Antigen-dependent CD28 signaling selectively enhances survival proliferation in genetically modified activated human primary T lymphocytes. J Exp Med. 1998;188(4):619–626. doi: 10.1084/jem.188.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T- lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ/CD28 receptor. Nat Biotechnol. 2002;20(1):70–75. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 43.Finney HM, Lawson AD, Bebbington CR, Weir AN. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol. 1998;161(6):2791–2797. [PubMed] [Google Scholar]
- 44.Hombach A, et al. Tumor-specific T cell activation by recombinant immunoreceptors: CD3ζ signaling CD28 costimulation are simultaneously required for efficient IL-2 secretion can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J Immunol. 2001;167(11):6123–6131. doi: 10.4049/jimmunol.167.11.6123. [DOI] [PubMed] [Google Scholar]
- 45.Imai C, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18(4):676–684. doi: 10.1038/sj.leu.2403302. [DOI] [PubMed] [Google Scholar]
- 46.Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3(4):388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brentjens RJ, et al. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2007;13(18 pt 1):5426–5435. doi: 10.1158/1078-0432.CCR-07-0674. [DOI] [PubMed] [Google Scholar]
- 48.Kowolik CM, et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66(22):10995–11004. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- 49.Milone MC, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells increased antileukemic efficacy in vivo. Mol Ther. 2009;17(8):1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation CD8+ T cell-mediated tumor eradication. Mol Ther. 2010;18(2):413–420. doi: 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov. 2015;14(7):499–509. doi: 10.1038/nrd4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carpenito C, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009;106(9):3360–3365. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jensen MC, Riddell SR. Designing chimeric antigen receptors to effectively and safely target tumors. Curr Opin Immunol. 2015;33:9–15. doi: 10.1016/j.coi.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. 2008;112(5):1570–1580. doi: 10.1182/blood-2008-02-078071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Engel P, Zhou LJ, Ord DC, Sato S, Koller B, Tedder TF. Abnormal B lymphocyte development, activation, and differentiation in mice that lack or overexpress the CD19 signal transduction molecule. Immunity. 1995;3(1):39–50. doi: 10.1016/1074-7613(95)90157-4. [DOI] [PubMed] [Google Scholar]
- 56.Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature. 1995;376(6538):352–355. doi: 10.1038/376352a0. [DOI] [PubMed] [Google Scholar]
- 57.Carter RH, Fearon DT. CD19: lowering the threshold for antigen receptor stimulation of B lymphocytes. Science. 1992;256(5053):105–107. doi: 10.1126/science.1373518. [DOI] [PubMed] [Google Scholar]
- 58.Ghosh N, Matsui W. Cancer stem cells in multiple myeloma. Cancer Lett. 2009;277(1):1–7. doi: 10.1016/j.canlet.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pegram HJ, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119(18):4133–4141. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Davila ML, Kloss CC, Gunset G, Sadelain M. CD19 CAR-targeted T cells induce long-term remission B cell aplasia in an immunocompetent mouse model of B cell acute lymphoblastic leukemia. PLoS One. 2013;8(4):e61338. doi: 10.1371/journal.pone.0061338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brentjens RJ, et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med. 2003;9(3):279–286. doi: 10.1038/nm827. [DOI] [PubMed] [Google Scholar]
- 62.Kershaw MH, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12(20 pt 1):6106–6115. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lamers CH, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24(13):e20–e22. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 64.Till BG, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;112(6):2261–2271. doi: 10.1182/blood-2007-12-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pule MA, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14(11):1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kochenderfer JN, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116(20):4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kochenderfer JN, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kochenderfer JN, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33(6):540–549. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kochenderfer JN1, Rosenberg SA. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat Rev Clin Oncol. 2012;10(5):267–276. doi: 10.1038/nrclinonc.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kalos M, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brentjens RJ, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118(18):4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Porter DL, et al. Randomized, Phase II Dose Optimization Study of Chimeric Antigen Receptor Modified T Cells Directed Against CD19 (CTL019) in Patients with Relapsed, Refractory CLL. Paper presented at: 56th ASH Annual Meeting and Exposition; December 6–9, 2014; San, Francisco, California, USA. Paper 68877. [Google Scholar]
- 73.Sauter CS, et al. Phase I trial of 19-28z chimeric antigen receptor modified T cells (19-28z CAR) post-high dose therapy and autologous stem cell transplant (HDT-ASCT) for relapsed and refractory (rel/ref) aggressive B-cell non-Hodgkin lymphoma (B-NHL) J Clin Oncol. 2015;33(suppl):abstr 8515. [Google Scholar]
- 74.Brentjens RJ, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5(177):177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grupp SA, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Davila ML, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Maude SL, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee DW, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Davila ML, Brentjens R, Wang X, Rivière I, Sadelain M. How do CARs work?: Early insights from recent clinical studies targeting CD19. Oncoimmunology. 2012;1(9):1577–1583. doi: 10.4161/onci.22524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.June CH, Riddell SR, Schumacher TN. Adoptive cellular therapy: a race to the finish line. Sci Transl Med. 2015;7(280):280ps7. doi: 10.1126/scitranslmed.aaa3643. [DOI] [PubMed] [Google Scholar]
- 81.Lee DW, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–195. doi: 10.1182/blood-2014-05-552729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Recombinant DNA Advisory Committee at the National Institutes of Health Cytokine Release Syndrome after T Cell Immunotherapy Meeting. [June 10, 2015]; [July 2, 2015];NIH Web site. http://videocast.nih.gov/Summary.asp?File=19063&bhcp=1
- 83.Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014;20(2):119–122. doi: 10.1097/PPO.0000000000000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fedorov VD, Sadelain M, Kloss CC. Novel approaches to enhance the specificity and safety of engineered T cells. Cancer J. 2014;20(2):160–165. doi: 10.1097/PPO.0000000000000040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Topp MS, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16(1):57–66. doi: 10.1016/S1470-2045(14)71170-2. [DOI] [PubMed] [Google Scholar]
- 86.Haso W, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121(7):1165–1174. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stephan MT, et al. T cell-encoded CD80 and 4-1BBL induce auto-transcostimulation, resulting in potent tumor rejection. Nat Med. 2007;13(12):1440–1449. doi: 10.1038/nm1676. [DOI] [PubMed] [Google Scholar]
- 88.Duong CP, Westwood JA, Berry LJ, Darcy PK, Kershaw MH. Enhancing the specificity of T-cell cultures for adoptive immunotherapy of cancer. Immunotherapy. 2011;3(1):33–48. doi: 10.2217/imt.10.81. [DOI] [PubMed] [Google Scholar]
- 89.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31(1):71–75. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Prosser ME, Brown CE, Shami AF, Forman SJ, Jensen MC. Tumor PD-L1 co-stimulates primary human CD8(+) cytotoxic T cells modified to express a PD1:CD28 chimeric receptor. Mol Immunol. 2012;51(3–4):263–272. doi: 10.1016/j.molimm.2012.03.023. [DOI] [PubMed] [Google Scholar]
- 91.Wilkie S, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. 2012;32(5):1059–1070. doi: 10.1007/s10875-012-9689-9. [DOI] [PubMed] [Google Scholar]
- 92.Hoyos V, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects safety. Leukemia. 2010;24(6):1160–1170. doi: 10.1038/leu.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Markley JC, Sadelain M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood. 2010;115(17):3508–3519. doi: 10.1182/blood-2009-09-241398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chmielewski M, Abken H. CAR T cells transform to trucks: chimeric antigen receptor-redirected T cells engineered to deliver inducible IL-12 modulate the tumour stroma to combat cancer. Cancer Immunol Immunother. 2012;61(8):1269–1277. doi: 10.1007/s00262-012-1202-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chinnasamy D, et al. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res. 2012;18(6):1672–1683. doi: 10.1158/1078-0432.CCR-11-3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ramsay AG, Clear AJ, Fatah R, Gribben JG. Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: establishing a reversible immune evasion mechanism in human cancer. Blood. 2012;120(7):1412–1421. doi: 10.1182/blood-2012-02-411678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Munn DH. Blocking IDO activity to enhance anti-tumor immunity. Front Biosci (Elite Ed) 2012;4:734–745. doi: 10.2741/e414. [DOI] [PubMed] [Google Scholar]
- 99. Schuster SJ, et al. Phase IIa Trial of Chimeric Antigen Receptor Modified T Cells Directed Against CD19 (CTL019) in Patients with Relapsed or Refractory CD19+ Lymphomas. Paper presented at: 56th ASH Annual Meeting and Exposition; December 6–9, 2014; San, Francisco, California, USA. Paper 3087. [Google Scholar]
- 100.Adusumilli PS, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. 2014;6(261):261ra151. doi: 10.1126/scitranslmed.3010162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Moeller M, et al. Sustained antigen-specific antitumor recall response mediated by gene-modified CD4+ T helper-1 and CD8+ T cells. Cancer Res. 2007;67(23):11428–37. doi: 10.1158/0008-5472.CAN-07-1141. [DOI] [PubMed] [Google Scholar]
- 102.Cruz CR, et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood. 2013;122(17):2965–2973. doi: 10.1182/blood-2013-06-506741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gattinoni L, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17(10):1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Riddell SR, et al. Adoptive therapy with chimeric antigen receptor-modified T cells of defined subset composition. Cancer J. 2014;20(2):141–144. doi: 10.1097/PPO.0000000000000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Klebanoff CA, Gattinoni L, Restifo NP. Sorting through subsets: which T-cell populations mediate highly effective adoptive immunotherapy? J Immunother. 2012;35(9):651–699. doi: 10.1097/CJI.0b013e31827806e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Themeli M, Riviere I, Sadelain M. New cell sources for T cell engineering and adoptive immunotherapy. Cell Stem Cell. 2015;16(4):357–366. doi: 10.1016/j.stem.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Frey NV, et al. Refractory Cytokine Release Syndrome in Recipients of Chimeric Antigen Receptor (CAR) T Cells. Paper presented at: 56th ASH Annual Meeting and Exposition; December 6–9, 2014; San, Francisco, California, USA. Paper 76315. [Google Scholar]
- 108. Pak JH, et al. CD19-Targeted 19-28z CAR-Modified Autologous T Cells Induce High Rates of Refractory B-Cell ALL. Paper presented at: 56th ASH Annual Meeting and Exposition; December 6–9, 2014; San, Francisco, California, USA. Paper 76573. [Google Scholar]