

Abstract
Major progress has been made toward our understanding of the programmed death-1/programmed death ligand-1 (PD-1/PD-L1) pathway (referred to as the PD pathway). mAbs are already being used to block the PD pathway to treat human cancers (anti-PD therapy), especially advanced solid tumors. This therapy is based on principles that were discovered through basic research more than a decade ago, but the great potential of this pathway to treat a broad spectrum of advanced human cancers is just now becoming apparent. In this Review, we will briefly review the history and development of anti-PD therapy, from the original benchwork to the most up-to-date clinical results. We will then focus the discussion on three basic principles that define this unique therapeutic approach and highlight how anti-PD therapy is distinct from other immunotherapeutic approaches, namely tumor site immune modulation, targeting tumor-induced immune defects, and repairing ongoing (rather than generating de novo) tumor immunity. We believe that these fundamental principles set the standard for future immunotherapies and will guide our efforts to develop more efficacious and less toxic immune therapeutics to treat human cancers.
The PD pathway
The cDNA of programmed cell death 1 (PD-1) was isolated in 1992 from a murine T cell hybridoma and a hematopoietic progenitor cell line undergoing apoptosis (1). Genetic ablation studies showed that deficiencies in PD-1 resulted in different autoimmune phenotypes in various mouse strains (2, 3). PD-1–deficient allogeneic T cells with transgenic T cell receptors exhibited augmented responses to alloantigens, indicating that the PD-1 on T cells plays a negative regulatory role in response to antigen (2).
Several studies contributed to the discovery of the molecules that interact with PD-1. In 1999, the B7 homolog 1 (B7-H1, also called programmed death ligand-1 [PD-L1]) was identified independently from PD-1 using molecular cloning and human expressed-sequence tag database searches based on its homology with B7 family molecules, and it was shown that PD-L1 acts as an inhibitor of human T cell responses in vitro (4). These two independent lines of study merged one year later when Freeman, Wood, and Honjo’s laboratories showed that PD-L1 is a binding and functional partner of PD-1 (5). Next, it was determined that PD-L1–deficient mice (Pdl1 KO mice) were prone to autoimmune diseases, although this strain of mice did not spontaneously develop such diseases (6). It became clear later that the PD-L1/PD-1 interaction plays a dominant role in the suppression of T cell responses in vivo, especially in the tumor microenvironment (7, 8).
In addition to PD-L1, another PD-1 ligand called B7-DC (also known as PD-L2) was also identified by the laboratories of Pardoll (9) and Freeman (10). This PD-1 ligand was found to be selectively expressed on DCs and delivered its suppressive signal by binding PD-1. Mutagenesis studies of PD-L1 and PD-L2 molecules guided by molecular modeling revealed that both PD-L1 and PD-L2 could interact with other molecules in addition to PD-1 and suggested that these interactions had distinct functions (11). The functional predictions from these mutagenesis studies were later confirmed when PD-L1 was found to interact with CD80 on activated T cells to mediate an inhibitory signal (12, 13). This finding came as a surprise because CD80 had been previously identified as a functional ligand for CD28 and cytotoxic T lymphocyte antigen-4 (CTLA-4) (14, 15). PD-L2 was also found to interact with repulsive guidance molecule family member b (RGMb), a molecule that is highly enriched in lung macrophages and may be required for induction of respiratory tolerance (16). With at least five interacting molecules in the PD-1/PD-L1 pathway (referred to as the PD pathway) (Figure 1), further studies will be required to understand the relative contributions of these molecules during activation or suppression of T cells.
Figure 1. The PD pathway.
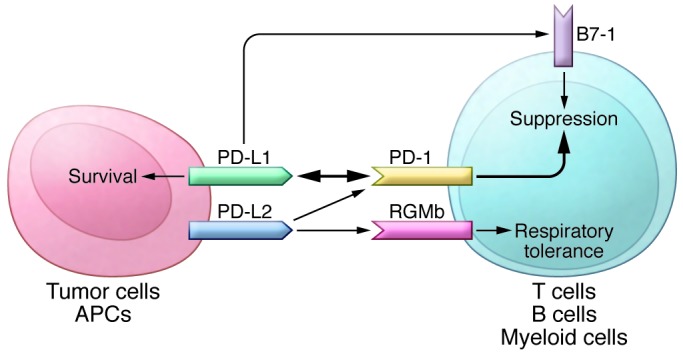
The PD pathway has at least 5 interacting molecules. PD-L1 and PD-L2, with different expression patterns, were identified as ligands of PD-1, and the interaction of PD-L1 or PD-L2 with PD-1 may induce T cell suppression. PD-L1 was found to interact with B7-1 (CD80) on activated T cells and inhibit T cell activity. PD-L2 has a second receptor, RGMb; initially, this interaction activates T cells, but it subsequently induces respiratory tolerance. PD-L1 on tumor cells can also act as a receptor, and the signal delivered from PD-1 on T cells can protect tumor cells from cytotoxic lysis.
Immunology studies leading to anti-PD therapy for cancer
The discovery of the PD pathway did not automatically justify its application to cancer therapy, especially after the initial PD-1–deficient mouse studies, which suggested that PD-1 deficiency increases the incidence of autoimmune diseases (2, 3). In our initial work to characterize PD-L1 and its function, PDL1 mRNA was found to be broadly expressed in various tissues (17). However, normal human tissues seldom express PD-L1 protein on their cell surface, with the exception of tonsil (17), placenta (18), and a small fraction of macrophage-like cells in lung and liver (17), suggesting that, under normal physiological conditions, PDL1 mRNA is under tight posttranscriptional regulation. In sharp contrast, PD-L1 protein is abundantly expressed on the cell surface in various human cancers, as indicated by immunohistochemistry in frozen human tumor sections. Additionally, the pattern of PD-L1 expression was found to be focal rather than diffuse in most human cancers (17). In fact, the majority of in vitro–cultured tumor lines of both human and mouse origin are PD-L1–negative on the cell surface, despite overwhelming PD-L1 signal in specimens that are freshly isolated from patients with cancer (17, 19). This discrepancy was explained by the finding that IFN-γ upregulates PD-L1 on the cell surface of normal tissues and in various tumor lines (7, 17, 19). It was widely thought that IFN-γ typically promotes, rather than suppresses, T cell responses by stimulating antigen processing and presentation machinery (20, 21); therefore, the role of IFN-γ in downregulating immune responses in the tumor microenvironment via induction of PD-L1 was not well accepted until more recently. This finding is vital to our current understanding of the unique immunology that takes place in the tumor microenvironment and provided an important clue that led to the “adaptive resistance” hypothesis (see below) that explains this pathway’s mechanism of action to evade tumor immunity.
Due to the lack of cell surface expression of PD-L1 on most cultured tumor lines, it is necessary to reexpress PD-L1 on the surface using transfection to recapitulate the effects of cell surface PD-L1 in human cancers and to create models to study how tumor-associated PD-L1 interacts with immune cells. We now know that cancer cells and other cells in the tumor microenvironment can upregulate the expression of PD-L1 after encountering T cells, mostly via IFN-γ, which may make the transfection-mediated expression of PD-L1 unnecessary in some tumor models. Nevertheless, our results demonstrated that PD-L1+ human tumor cells could eliminate activated effector T cells (Teffs) via apoptosis in coculture systems, and this effect could be blocked by inclusion of an anti-human PD-L1 mAb (clone 2H1). Next, we generated a hamster mAb (clone 10B5) against mouse PD-L1 to block its interaction with T cells and test its role in tumor immunity in vitro and in vivo. We demonstrated that progressive growth of PD-L1+ murine P815 tumors in syngeneic mice could be suppressed using anti–PD-L1 mAb (17). Altogether, these studies represented the initial attempt at using mAb to block the PD pathway as an approach for cancer therapy. These proof-of-concept studies (17) were confirmed by several subsequent studies. A study from Nagahiro Minato’s laboratory showed that the J558L mouse myeloma line constitutively expressed high levels of cell surface PD-L1 and the growth of these cells in syngeneic BALB/c mice could be partially suppressed by administering anti–PD-L1 mAb (22). Our laboratory showed that regression of progressively growing squamous cell carcinomas in syngeneic mice could also be suppressed using a combination of adoptively transferred tumor-draining lymphocytes and anti–PD-L1 mAb (23). Furthermore, the Zou laboratory demonstrated that ovarian cancer–infiltrating human T cells could be activated in vitro using DCs, which showed enhanced activity in the presence of anti–PD-L1 mAb; upon transfer, these cells could eliminate established human ovarian cancers in immune-deficient mice (24). These early studies established the concept that the PD pathway could be used by tumors to escape immune attack in the tumor microenvironment. More importantly, these studies built a solid foundation for the development of anti-PD therapy for the treatment of human cancers.
Consequences of the PD-L1/PD-1 interaction
Our initial studies showed that tumor-associated PD-L1 facilitates apoptosis of activated T cells (17) and also stimulates IL-10 production in human peripheral blood T cells (4) to mediate immune suppression. We now know that the effects of PD-L1 on immune suppression are far more complicated. In addition to T cell apoptosis and IL-10 induction, PD-L1 can also induce T cell dysfunction through a variety of mechanisms. The PD pathway was also shown to promote T cell anergy in vitro and in vivo (25–27). In a mouse model of chronic lymphocytic choriomeningitis virus infection, persistent antigen exposure induced T cell exhaustion with either partial or complete loss of Teff function. Anti–PD-L1 mAb administration reversed exhaustion and restored Teff function (28). The PD pathway also regulates Treg functions (29, 30). In addition to T and B lymphocytes, PD-1 is upregulated on DCs by various inflammatory stimuli. PD-1–deficient DCs exhibit enhanced antibacterial function, indicating that PD-1 can also act as an inhibitory receptor on DCs (31). PD-L1 also serves as a receptor on cancer cells and can induce intrinsic resistance to T cell killing upon interaction with PD-1. These PD-L1 functions result in a “molecular shield” on cancer cells that prevents effector immune cells from killing cancer cells (32). The mechanisms of action of the PD pathway are summarized in Figure 2.
Figure 2. Mechanisms of PD pathway–induced immunosuppression in the tumor microenvironment.
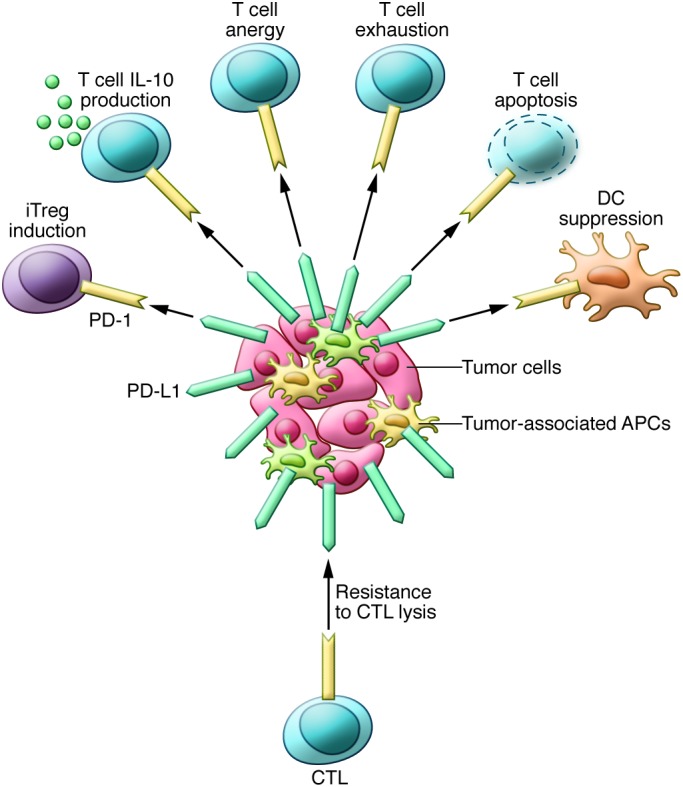
Tumor cells and other cells in tumor microenvironment can express high levels of PD-L1, which results in suppressed immunity upon interaction with PD-1. PD-L1–expressing cells use multiple mechanisms to suppress tumor immunity. PD-L1+ tumor cells and antigen-presenting cells (APCs) can induce T cell apoptosis, anergy, functional exhaustion, and IL-10 production. They can also mediate DC suppression and induce the differentiation of Tregs. PD-L1 can also act as a molecular shield on tumor cells and protect tumor cells from lysis by cytotoxic T lymphocytes (CTLs). iTreg, induced Treg.
The adaptive resistance hypothesis
Overall, PD pathway–mediated evasion of tumor immunity could be described as “adaptive resistance” (Figure 3), stemming from the original observations that PD-L1 is absent in most normal tissues, while its expression can be induced by IFN-γ (17) in virtually any nucleated cells (7, 8, 33). Because IFN-γ is mainly produced by inflammatory cells of hematopoietic origin, especially T cells, it is logical to propose that PD-L1 is upregulated in response to cancer-induced inflammation. Adaptive resistance may be initiated by the recognition of tumor antigens by tumor-infiltrating lymphocytes (TILs). In addition to tumor cells, tumor antigens could be presented by tumor stromal cells and hematopoietically derived infiltrating cells, including DCs, macrophages, neutrophils, and, even, lymphocytes. Upon specific recognition via T cell receptor, TILs release IFN-γ and may induce PD-L1 expression on these cells. This hypothesis is supported by immunohistochemistry-based observations that cell surface PD-L1 expression is detected only in cells that are adjacent to T cells (34). While IFN-γ can promote TIL effector functions by increasing TIL differentiation and stimulating antigen processing and presentation (20, 21), cell surface PD-L1 binds its receptors on Teffs, including PD-1 and B7-1, thereby paralyzing T cells. Therefore, while the physiological function of PD-L1’s upregulation is to prevent propagation of inflammation and to limit tissue damage, induced PD-L1 in the tumor microenvironment serves as a negative feedback mechanism to suppress tumor immunity. This hypothesis is further supported by studies showing a strong correlation between PD-L1 expression in human melanocytic lesions and the presence of TILs. PD-L1 expression on cancer cells is often clustered immediately adjacent to TILs. Using laser-capture microdissection and qPCR, IFN-γ was detected at the interface of TILs and PD-L1+ cells but remained undetectable within PD-L1– tumors (34). In a mouse tumor model, mAb-mediated neutralization of IFN-γ eliminated PD-L1 upregulation in the tumor microenvironment, indicating that IFN-γ is a major inducer of PD-L1 in vivo (35). Therefore, an adaptive resistance mechanism explains how cancer escapes immune destruction, despite endogenous antitumor immune responses. Furthermore, this model helps explain why various cancer immunotherapy approaches fail to control tumor growth (i.e., cancer vaccines) or may not reach maximal effect (i.e., adoptive cell therapy).
Figure 3. Tumor adaptive resistance model.

After activation in lymphoid organs, tumor-specific Teffs enter the tumor site to become TILs. Upon recognition of tumor antigens, TILs produce IFN-γ, which drives PD-L1 expression in the tumor microenvironment. Tumor antigens are presented by tumor cells, stromal cells, and infiltrating hematopoietic cells, including DCs, macrophages, neutrophils, and B lymphocytes; all of these cells can be induced to express PD-L1. Upon binding to PD-1, PD-L1 delivers a suppressive signal to T cells and an antiapoptotic signal to tumor cells, leading to T cell dysfunction and tumor survival.
Although induced expression by IFN-γ is a major driver of PD-L1 upregulation, cancer cells also express PD-L1 by an intrinsic, IFN-γ–independent mechanism. This is evidenced by the small fraction of human cancers that lack TILs in the tumor microenvironment but still express high levels of PD-L1 (34, 36). Several studies have demonstrated that the loss of phosphatase and tensin homolog (PTEN), constitutive anaplastic lymphoma kinase (ALK) signaling, and EGFR mutations can directly upregulate PD-L1 on cancer cells (37–39). Intrinsic induction of PD-L1 is present in approximately 1% of patients with melanoma (34) and occurs in up to 12% of patients with lung cancer (36).
Anti-PD therapy in the clinic
The FDA recently approved two PD-1 mAbs to treat human cancers, one from Bristol-Myers Squibb (Opdivo, also known as nivolumab, MDX-1106, BMS-936558, and ONO-4538) and another from Merck (Keytruda, also known as pembrolizumab, lambrolizumab, and MK-3475) (40). Additionally, multiple mAbs to either PD-1 or PD-L1 are under active development in hundreds of clinical trials involving thousands of patients. Thus far, anti-PD therapy has generated significant clinical benefits by inducing regression of advanced and metastatic tumors and improving survival. More importantly, anti-PD therapy can have durable effects, tolerable toxicity, and is applicable to a broad spectrum of cancer types, especially in solid tumors. These clinical findings further validate PD pathway blockade and put anti-PD therapy in a unique category, distinct from personalized or tumor type-specific therapy.
Nivolumab was the first mAb targeting PD-1 to show significant clinical activity in unresectable or metastatic melanomas, non–small-cell lung carcinoma (NSCLC), and metastatic renal cell carcinomas (41, 42). Nivolumab has induced a consistent objective response rate (ORR) of 30% to 40% in multiple clinical trials in patients with melanoma (NCT00730639, NCT01721772, NCT01844505) (43, 44). Nivolumab also extended overall survival in patients with metastatic squamous NSCLC compared with standard chemotherapy (NCT01642004). There was an ORR of 15% in patients who had progressed after receiving at least two systemic regimens (NCT01721759) (45). Nivolumab was approved by the FDA to treat both advanced melanoma and squamous NSCLC (40). A recent phase I study showed that nivolumab produced an ORR as high as 87% for the treatment of relapsed or refractory Hodgkin’s lymphoma (46). In an early combination trial using anti–CTLA-4 (ipilimumab) and nivolumab, the overall survival of patients with advanced melanoma on a concurrent regimen of nivolumab and ipilimumab was 94% after one year (NCT01024231) (47).
Pembrolizumab has shown similar efficacy and safety compared with nivolumab in a phase I clinical trial in advanced melanoma (NCT01295827) (48, 49) and is now an FDA-approved second-line drug for the treatment of melanoma (40). More recent results from a large phase III clinical trial on advanced melanoma indicate that pembrolizumab is far more effective than the anti–CTLA-4 mAb ipilimumab and has far better survival benefits and safety profiles as a first-line treatment agent for melanoma when compared with ipilimumab (NCT01866319) (50). Pembrolizumab is also effective in patients with advanced NSCLC (with an ORR of 19%; NCT01295827) (51) and has shown promising effects in other solid tumors, including advanced gastric cancer (with an ORR above 20%; NCT01848834) (52), advanced bladder cancer (with an ORR above 20%; NCT01848834) (53), head and neck cancer (with an ORR above 20%; NCT01848834) (54), classical Hodgkin’s lymphoma (NCT01953692) (55), and triple-negative breast cancer (56).
Other anti–PD-L1 mAbs have been shown to be efficacious in advanced human cancers. Bristol-Myers Squibb’s BMS-936559 (MDX-1105) demonstrated therapeutic efficacy in a phase I clinical trial (NCT00729664) (57). MPDL3280A (Genentech/Roche) is an engineered anti–PD-L1 IgG1 mAb that can inhibit PD-L1 interactions with both PD-1 and B7-1 (58). A study of metastatic urothelial bladder cancer (UBC) demonstrated that MPDL3280A has marked activity in controlling tumor growth; ORRs were 43% for those with PD-L1+ tumors and 11% for those with PD-L1– tumors. Moreover, owing to the mild side effects, including a lack of renal toxicity, patients with UBC, who are often older and have a higher incidence of renal impairment, are thought to tolerate MPDL3280A better than chemotherapy (NCT01375842) (58). In an expansion phase I trial across multiple cancer types, including NSCLC, melanoma, renal cell carcinoma, and other tumors, responses to MPDL3280A were observed in patients with tumors expressing high levels of PD-L1, especially when PD-L1 was expressed by TILs. For example, an ORR of 23% was shown in all patients with NSCLC, while ORRs of 85% were seen in patients with high PD-L1 expression (NCT01375842) (59). In June 2014, this drug received breakthrough designation status by the FDA for the treatment of metastatic UBCs.
Aside from the PD pathway inhibitors discussed above, there are several anti-PD agents currently being evaluated for cancer treatment. Pidilizumab (CT-011, anti–PD-1) by Medivation/CureTech (60–62), MEDI4736 (anti–PD-L1) by AstraZeneca (63, 64), and Avelumab (MSB0010718C, anti–PD-L1) by Merck-Sorono have all shown promise in the treatment of multiple human cancers (NCT01772004). There do not appear to be significant differences among these anti–PD-1/PD-L1 mAbs; however, there are currently no side-by-side comparison studies, making it difficult to determine how these new agents compare to approved PD pathway inhibitors.
Anti-PD therapy versus anti–CTLA-4 therapy
In the literature, the PD pathway has been broadly categorized together with CTLA-4 as an “immune checkpoint.” However, anti-PD therapy is based on completely different scientific principles compared with anti–CTLA-4 therapy. Early studies using conventional KO mice demonstrate that the major role of CTLA-4 is to regulate T cell responses to self-antigens because KO mice spontaneously develop massive infiltrating T cells to normal organs within weeks. These tissue-infiltrating T cells are highly active, cause damage to the normal tissue without specific antigen exposure, and eventually result in death (65, 66). In various mouse models, infusion of anti–CTLA-4 mAb induces various types of autoreactive T cell responses (67–70). Mice with Treg-selective ablation of CTLA-4 recapitulate the majority of autoimmune phenotypes observed in traditional KO mice, including tissue-infiltrating T cells and damage to normal tissues and organs (71). Therefore, the physiological function of CTLA-4 appears to be to suppress T cell responses to self-antigens by controlling Treg activity. These findings in mouse models are consistent with clinical observations of anti-human CTLA-4 mAbs in patients with cancer: rapid infiltration of T cells into normal tissues leads to broad and sometimes fatal damage to “off-target” normal organs, including skin, the gastrointestinal tract, adrenal glands, and liver (72, 73). Anti–CTLA-4–induced autoimmune toxicities are tightly associated with its antitumor effect (74). Currently, there is no evidence that Tregs are particularly active due to tumor growth, and it is unclear whether or not Tregs are responsible for immune evasion in melanoma (75). Therefore, anti–CTLA-4 mAbs work by promoting T cell responses that are largely nonspecific for tumor antigens (67–69, 73–75). Clinically broad toxicity in normal organs and tissue as well as limited efficacy are two major hurdles in the use of anti–CTLA-4 therapy in cancers beyond melanoma (76, 77).
In contrast to CTLA-4 KO mice, PD-L1 KO mice do not spontaneously develop massive infiltration of inflammatory cells in normal organs, and these mice have an overall normal life span (6). However, PD-1 KO mice do spontaneously develop strain-specific autoimmune diseases over a period of several months (2, 3). Both PD-1 and PD-L1 KO mice are autoimmune prone because more severe autoimmune symptoms develop when they are challenged with autoantigens (6). These findings are consistent with clinical observations that anti-PD therapy has low autoimmune toxicity (50). Because PD-L1 has a limited distribution in normal tissues and is largely an inducible molecule under the control of IFN-γ, its major physiological function appears to mediate negative feedback control of tissue inflammation. In the context of tumor growth, which is often associated with or even promoted by chronic inflammation, anti-PD therapy selectively modulates inflammatory T cell responses at the tumor site, blocks the function of tumor- and chronic inflammation–induced PD-L1 interactions, and rescues naturally generated but often impaired tumor immunity (see below). Therefore, the main physiological function of the PD pathway is to control ongoing inflammatory responses and to prevent the spread of inflammation, rather than systemic regulation of autoreactive T cell responses. These features place anti-PD therapy in a different category scientifically and practically from anti–CTLA-4 therapy.
A recently completed clinical trial to treat advanced melanoma compared anti–PD-1 therapy to anti–CTLA-4 therapy (NCT01866319) (50). The results from this trial showed that anti–PD-1 therapy (pembrolizumab) outperforms anti–CTLA-4 therapy (ipilimumab) in the major clinical categories, including efficacy, survival, and adverse events. The trial was stopped ahead of schedule because patients reached the endpoints of progression-free survival and overall survival. With better therapeutic efficacy and safety profiles, anti-PD therapy will replace anti–CTLA-4 therapy as a first-line therapeutic agent for patients with melanoma in the near future.
The three principles of anti-PD therapy and perspectives
Anti-PD therapy has become the backbone of cancer immunotherapy and a major modality of cancer treatment. Establishment of basic principles and validation of the clinical results of anti-PD therapy have transformed our thinking of cancer immunotherapy in general. It is important to learn from this successful therapy to broaden our understanding of the immune system and its role in cancer and to improve future therapies. There are three essential principles that we have learned so far from anti-PD therapy, namely (a) tumor site immune modulation, (b) targeting of tumor-induced immune defects, and (c) repairing ongoing tumor immunity. We believe that these principles and the postulations extending from them will direct future efforts in developing better cancer immunotherapeutics.
Tumor site immune modulation.
The first and the most important principle of anti-PD therapy is its localized effect. The unique expression pattern of PD-L1 in the tumor microenvironment (8, 17) determines the location of PD-1+ Teff inhibition as well as the site at which anti-PD therapy predominately takes place. In the context of a broadly inducible pattern of PD-L1 expression by IFN-γ, it will not be a surprise to see the expression of PD-L1 on tumor cells, tumor stromal cells, or infiltrating hematopoietic cells in the tumor microenvironment as long as TILs can access the tumor site and be induced to release IFN-γ. PD-L1 expression in tumors is typically not diffuse but clustered or focal, a pattern that can be explained by direct contact with IFN-γ–producing TILs (17, 34). However, it is not impossible that other areas distal from TILs could also become positive for PD-L1 if a sufficiently high level of IFN-γ is released due to strong activation of T cells. Besides T cells, NK cells are also a main cellular source of IFN-γ (20), but we do not yet know whether this cell type contributes to PD-L1 expression in the tumor microenvironment. Analyses of immune responses during anti-PD therapy indicate that selective expansion and functional enhancement of T cells occur at the tumor site, while activity of immune cells in the bloodstream fails to correlate with tumor regression (78–80). This is in sharp contrast to other immunotherapy approaches that aim to boost systemic immunity. For example, tumor antigen-based cancer vaccines often enhance T cell responses in both quantity and quality; unfortunately, these responses have not been translated to tumor regression and clinical benefits (81). In this context, it is desirable for future cancer immunotherapies to specifically focus on the tumor site rather than systemic activation of the immune system. This selective activation can be achieved through the identification of immune modulatory targets that express or operate selectively at the tumor site, similar to PD-L1. Another approach would be to add tumor-targeting components for a systemic immune response activator in order to limit its off-target effects.
Targeting tumor-induced immune defects.
The second critical point we have learned from anti-PD therapy is the importance of targeting tumor-induced immune defects. As described above, PD-L1 is largely induced at the tumor site by TIL-derived IFN-γ. While PD-L1 expression represents an effort by the host to protect its tissues from excessive damage caused by ongoing T cell–mediated inflammation, PD-L1 expression at the tumor site could be viewed as a “misunderstanding” of the host immune response toward tumor growth. This is a problem created initially by tumor growth and subsequently by host inflammatory responses. Therefore, anti-PD therapy targets a specific defect of the immune response during the fight against cancer, while such events occur minimally in cancer-free tissues. Targeting tumor-induced alternations insures the specificity of anti-PD therapy, prevents damage to normal tissues, and allows a focused, efficient, and more precise immune response. In addition to abnormal PD-L1 expression, various tumor-associated immune defects have also been identified, including elevated immune inhibitory cytokines (IL-10, TGF-β, etc.), expansion of Tregs, accumulation and proliferation of myeloid-derived suppressor cells (MDSCs), and increased tumor metabolites (82). While these findings help us understand possible immune-suppressive mechanisms in general, it is important to critically evaluate these targets to make sure that they are present or operate at the tumor site but are absent or limited in normal tissues.
Repairing ongoing tumor immunity.
The third principle of anti-PD therapy is its capacity to repair ongoing tumor immunity. During tumor progression, host immune responses are constantly fighting cancer. Progression of cancer to a late stage often suggests a failure or insufficiency of the ongoing immune response. Current anti-PD therapy studies indicate that the impaired immune response could be repaired so that it was capable of eradicating a tumor (83). Future studies will be needed to determine how the quantity and quality of TILs and other infiltrating immune cells contribute to the antitumor immune response. Immune infiltration is a reliable indicator of tumor immunogenicity, and it has been shown that the presence of gene signatures for CD8+ T cells and Th1 cells are associated with a better prognosis for several types of cancers, including colorectal and lung cancers, whereas Tregs, MDSCs, Th2 cells, and Th17 cells are not (84). Additionally, the presence of Th17 cells was shown to correlate with a good prognosis for ovarian cancer (85, 86). The effects of selectively eliminating “bad cells” or promoting “good cells” in the tumor microenvironment on the efficacy of anti-PD therapy has not been tested yet. A large fraction of patients with cancer do not have significant infiltration of inflammatory cells (34), suggesting a fundamental defect in the recruitment of inflammatory cells. Thus, it is important to understand and identify specific defects that prevent recruitment of inflammatory cells. Molecular targets could be identified based on these findings. Although patients with low levels of immune infiltration in their tumors are unlikely to benefit from anti-PD therapy, anti-PD therapy may be enhanced by combining it with therapies that promote recruitment of inflammatory cells into the tumor (83). For example, it would be particularly interesting to see whether adoptive transfer of in vitro–expanded TILs could be used in combination with anti-PD therapy. Localized radiation therapy is an efficient method to induce inflammation, and this approach could potentially benefit patients without ongoing immune responses. However, radiation therapy should not be used for the patients who have significant infiltration in the tumor site because this may impair ongoing immune responses. Similar concerns should also be taken into account when considering other combination therapies, such as chemotherapy and targeted therapy, to make sure that these therapies will not impair ongoing immune response.
Conclusion
Anti-PD therapy has taken center stage in immunotherapies for human cancer, especially for solid tumors. This therapy is distinct from the prior immune therapeutic agents, which primarily boost systemic immune responses or generate de novo immunity against cancer; instead, anti-PD therapy modulates immune responses at the tumor site, targets tumor-induced immune defects, and repairs ongoing immune responses. While the clinical success of anti-PD therapy for the treatment of a variety of human cancers has validated this approach, we are still learning from this pathway and the associated immune responses, which will aid in the discovery and design of new clinically applicable approaches in cancer immunotherapy.
Acknowledgments
We thank Beth Cadugan for editing the manuscript. This work was partially supported by NIH grants CA142779 and CA016359, Yale SPORE in Skin Cancer (CA121974), Yale SPORE in Lung Cancer (CA177444), and funding from a United Technologies Corporation endowed chair.
Footnotes
Conflict of interest: Lieping Chen is a scientific consultant for Pfizer and MedImmune; receives licensing fees from Bristol-Myers Squibb, Amplimmune, Ventana, and ImmuNext; and receives research funding from Boehringer Ingelheim.
Reference information:J Clin Invest. 2015;125(9):3384–3391. doi:10.1172/JCI80011.
References
- 1.Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11(11):3887–3895. doi: 10.1002/j.1460-2075.1992.tb05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11(2):141–151. doi: 10.1016/S1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 3.Nishimura H, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291(5502):319–322. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 4.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5(12):1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 5.Freeman GJ, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dong H, Zhu G, Tamada K, Flies DB, van Deursen JM, Chen L. B7-H1 determines accumulation and deletion of intrahepatic CD8(+) T lymphocytes. Immunity. 2004;20(3):327–336. doi: 10.1016/S1074-7613(04)00050-0. [DOI] [PubMed] [Google Scholar]
- 7.Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol. 2004;4(5):336–347. doi: 10.1038/nri1349. [DOI] [PubMed] [Google Scholar]
- 8.Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. 2008;8(6):467–477. doi: 10.1038/nri2326. [DOI] [PubMed] [Google Scholar]
- 9.Tseng SY, et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med. 2001;193(7):839–846. doi: 10.1084/jem.193.7.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Latchman Y, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2(3):261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 11.Wang S, Bajorath J, Flies DB, Dong H, Honjo T, Chen L. Molecular modeling and functional mapping of B7-H1 and B7-DC uncouple costimulatory function from PD-1 interaction. J Exp Med. 2003;197(9):1083–1091. doi: 10.1084/jem.20021752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27(1):111–122. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park JJ, et al. B7-H1/CD80 interaction is required for the induction and maintenance of peripheral T-cell tolerance. Blood. 2010;116(8):1291–1298. doi: 10.1182/blood-2010-01-265975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Linsley PS, Clark EA, Ledbetter JA. T-cell antigen CD28 mediates adhesion with B cells by interacting with activation antigen B7/BB-1. Proc Natl Acad Sci U S A. 1990;87(13):5031–5035. doi: 10.1073/pnas.87.13.5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. 1991;174(3):561–569. doi: 10.1084/jem.174.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiao Y, et al. RGMb is a novel binding partner for PD-L2 and its engagement with PD-L2 promotes respiratory tolerance. J Exp Med. 2014;211(5):943–959. doi: 10.1084/jem.20130790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dong H, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 18.Petroff MG, Chen L, Phillips TA, Hunt JS. B7 family molecules: novel immunomodulators at the maternal-fetal interface. Placenta. 2002;23(suppl A):S95–S101. doi: 10.1053/plac.2002.0813. [DOI] [PubMed] [Google Scholar]
- 19.Hirano F, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65(3):1089–1096. [PubMed] [Google Scholar]
- 20.Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- 21.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75(2):163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 22.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99(19):12293–12297. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strome SE, et al. B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Res. 2003;63(19):6501–6505. [PubMed] [Google Scholar]
- 24.Curiel TJ, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9(5):562–567. doi: 10.1038/nm863. [DOI] [PubMed] [Google Scholar]
- 25.Selenko-Gebauer N, et al. B7-H1 (programmed death-1 ligand) on dendritic cells is involved in the induction and maintenance of T cell anergy. J Immunol. 2003;170(7):3637–3644. doi: 10.4049/jimmunol.170.7.3637. [DOI] [PubMed] [Google Scholar]
- 26.Tsushima F, et al. Interaction between B7-H1 and PD-1 determines initiation and reversal of T-cell anergy. Blood. 2007;110(1):180–185. doi: 10.1182/blood-2006-11-060087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldberg MV, et al. Role of PD-1 and its ligand, B7-H1, in early fate decisions of CD8 T cells. Blood. 2007;110(1):186–192. doi: 10.1182/blood-2006-12-062422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barber DL, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439(7077):682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 29.Francisco LM, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206(13):3015–3029. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amarnath S, et al. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci Transl Med. 2011;3(111): doi: 10.1126/scitranslmed.3003130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yao S, et al. PD-1 on dendritic cells impedes innate immunity against bacterial infection. Blood. 2009;113(23):5811–5818. doi: 10.1182/blood-2009-02-203141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Azuma T, Yao S, Zhu G, Flies AS, Flies SJ, Chen L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood. 2008;111(7):3635–3643. doi: 10.1182/blood-2007-11-123141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flies DB, Sandler BJ, Sznol M, Chen L. Blockade of the B7-H1/PD-1 pathway for cancer immunotherapy. Yale J Biol Med. 2011;84(4):409–421. [PMC free article] [PubMed] [Google Scholar]
- 34.Taube JM, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4(127): doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spranger S, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. 2013;5(200): doi: 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Velcheti V, et al. Programmed death ligand-1 expression in non-small cell lung cancer. Lab Invest. 2014;94(1):107–116. doi: 10.1038/labinvest.2013.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parsa AT, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13(1):84–88. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 38.Akbay EA, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3(12):1355–1363. doi: 10.1158/2159-8290.CD-13-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marzec M, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1) Proc Natl Acad Sci U S A. 2008;105(52):20852–20857. doi: 10.1073/pnas.0810958105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hematology/Oncology (Cancer) Approvals & Safety Notifications. [April 24, 2015]; [June 23, 2015];FDA Web site. http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm279174.htm
- 41.Brahmer JR, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28(19):3167–3175. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Topalian SL, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Topalian SL, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32(10):1020–1030. doi: 10.1200/JCO.2013.53.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robert C, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–330. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 45.Rizvi NA, et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol. 2015;16(3):257–265. doi: 10.1016/S1470-2045(15)70054-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ansell SM, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2014;372(4):311–319. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sznol M, et al. Survival, response duration, and activity by BRAF mutation (MT) status of nivolumab (NIVO, anti-PD-1, BMS-936558, ONO-4538) and ipilimumab (IPI) concurrent therapy in advanced melanoma (MEL) J Clin Oncol. 2014;32(5s suppl): [Google Scholar]
- 48.Hamid O, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Robert C, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384(9948):1109–1117. doi: 10.1016/S0140-6736(14)60958-2. [DOI] [PubMed] [Google Scholar]
- 50.Robert C, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372(26):2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 51.Garon EB, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 52.Bang Y-J, et al. Relationship between PD-L1 expression and clinical outcomes in patients with advanced gastric cancer treated with the anti-PD-1 monoclonal antibody pembrolizumab (MK-3475) in KEYNOTE-012. J Clin Oncol. 2015;33(suppl 3) abstr 3. [Google Scholar]
- 53.O’Donnell PH, et al. Pembrolizumab (Pembro; MK-3475) for advanced urothelial cancer: Results of a phase IB study. J Clin Oncol. 2015;33(suppl 7) abstr 296. [Google Scholar]
- 54.Seiwert TY, et al. Inflamed-phenotype gene expression signatures to predict benefit from the anti-PD-1 antibody pembrolizumab in PD-L1+ head and neck cancer patients. J Clin Oncol. 2015;33(suppl) abstr 6017. [Google Scholar]
- 55. Moskowitz CH, et al. PD-1 Blockade with the Monoclonal Antibody Pembrolizumab (MK-3475) in Patients with Classical Hodgkin Lymphoma after Brentuximab Vedotin Failure: Preliminary Results from a Phase 1b Study (KEYNOTE-013). Presented at: 56th ASH Annual Meeting; December 8, 2014; San Francisco, California, USA. Abstract 290. [Google Scholar]
- 56. Nanda R, et al. A phase Ib study of pembrolizumab (MK-3475) in patients with advanced triple-negative breast cancer. Presented at: 2014 San Antonio Breast Cancer Symposium; December 10, 2014; San Antonio, Texas, USA. Abstract S1-09. [Google Scholar]
- 57.Brahmer JR, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Powles T, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515(7528):558–562. doi: 10.1038/nature13904. [DOI] [PubMed] [Google Scholar]
- 59.Herbst RS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Armand P, et al. Disabling immune tolerance by programmed death-1 blockade with pidilizumab after autologous hematopoietic stem-cell transplantation for diffuse large B-cell lymphoma: results of an international phase II trial. J Clin Oncol. 2013;31(33):4199–4206. doi: 10.1200/JCO.2012.48.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Westin JR, et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: a single group, open-label, phase 2 trial. Lancet Oncol. 2014;15(1):69–77. doi: 10.1016/S1470-2045(13)70551-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bryan LJ, Gordon LI. Pidilizumab in the treatment of diffuse large B-cell lymphoma. Expert Opin Biol Ther. 2014;14(9):1361–1368. doi: 10.1517/14712598.2014.942637. [DOI] [PubMed] [Google Scholar]
- 63.Lee SM, Chow LQ. A new addition to the PD-1 checkpoint inhibitors for non-small cell lung cancer-the anti-PDL1 antibody-MEDI4736. Transl Lung Cancer Res. 2014;3(6):408–410. doi: 10.3978/j.issn.2218-6751.2014.11.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zielinski CC. A phase I study of MEDI4736, NNT-PD-L1 antibody in patients with advanced solid tumors. Transl Lung Cancer Res. 2014;3(6):406–407. doi: 10.3978/j.issn.2218-6751.2014.08.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3(5):541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 66.Waterhouse P, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270(5238):985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 67.Karandikar NJ, Vanderlugt CL, Walunas TL, Miller SD, Bluestone JA. CTLA-4: a negative regulator of autoimmune disease. J Exp Med. 1996;184(2):783–788. doi: 10.1084/jem.184.2.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perrin PJ, Maldonado JH, Davis TA, June CH, Racke MK. CTLA-4 blockade enhances clinical disease and cytokine production during experimental allergic encephalomyelitis. J Immunol. 1996;157(4):1333–1336. [PubMed] [Google Scholar]
- 69.Luhder F, Hoglund P, Allison JP, Benoist C, Mathis D. Cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) regulates the unfolding of autoimmune diabetes. J Exp Med. 1998;187(3):427–432. doi: 10.1084/jem.187.3.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang HB, Shi FD, Li H, Chambers BJ, Link H, Ljunggren HG. Anti-CTLA-4 antibody treatment triggers determinant spreading and enhances murine myasthenia gravis. J Immunol. 2001;166(10):6430–6436. doi: 10.4049/jimmunol.166.10.6430. [DOI] [PubMed] [Google Scholar]
- 71.Wing K, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322(5899):271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 72.Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691–2697. doi: 10.1200/JCO.2012.41.6750. [DOI] [PubMed] [Google Scholar]
- 73.Tarhini A. Immune-mediated adverse events associated with ipilimumab ctla-4 blockade therapy: the underlying mechanisms and clinical management. Scientifica (Cairo) 2013;2013:857519. doi: 10.1155/2013/857519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maker AV, Attia P, Rosenberg SA. Analysis of the cellular mechanism of antitumor responses and autoimmunity in patients treated with CTLA-4 blockade. J Immunol. 2005;175(11):7746–7754. doi: 10.4049/jimmunol.175.11.7746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jacobs JF, Nierkens S, Figdor CG, de Vries IJ, Adema GJ. Regulatory T cells in melanoma: the final hurdle towards effective immunotherapy? Lancet Oncol. 2012;13(1):e32–e42. doi: 10.1016/S1470-2045(11)70155-3. [DOI] [PubMed] [Google Scholar]
- 76.Lynch TJ, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol. 2012;30(17):2046–2054. doi: 10.1200/JCO.2011.38.4032. [DOI] [PubMed] [Google Scholar]
- 77.Reese Z, Straubhar A, Pal SK, Agarwal N. Ipilimumab in the treatment of prostate cancer. Future Oncol. 2015;11(1):27–37. doi: 10.2217/fon.14.196. [DOI] [PubMed] [Google Scholar]
- 78.Rosenberg SA, et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol. 2005;175(9):6169–6176. doi: 10.4049/jimmunol.175.9.6169. [DOI] [PubMed] [Google Scholar]
- 79.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10(9):909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schwartzentruber DJ, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364(22):2119–2127. doi: 10.1056/NEJMoa1012863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Butterfield LH. Cancer vaccines. BMJ. 2015;350:h988. doi: 10.1136/bmj.h988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5(4):263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 83.Sznol M, Chen L. Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer. Clin Cancer Res. 2013;19(5):1021–1034. doi: 10.1158/1078-0432.CCR-12-2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Galon J, Angell HK, Bedognetti D, Marincola FM. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity. 2013;39(1):11–26. doi: 10.1016/j.immuni.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 85.Kryczek I, et al. Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood. 2009;114(6):1141–1149. doi: 10.1182/blood-2009-03-208249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kryczek I, et al. Human TH17 cells are long-lived effector memory cells. Sci Transl Med. 2011;3(104): doi: 10.1126/scitranslmed.3002949. [DOI] [PMC free article] [PubMed] [Google Scholar]