

Abstract
The relationship between cancer and the immune system is complex and provides unique therapeutic opportunities. Cytotoxic T lymphocyte antigen-4 (CTLA-4) is a regulatory molecule that suppresses T cell effector function following initial activation by costimulatory signals. Fully human monoclonal antibodies targeting CTLA-4 have been shown to increase T cell function and antitumor responses in patients with advanced metastatic melanoma. Responses observed with such immune checkpoint therapy can follow a different pattern from that seen with cytotoxic chemotherapy or targeted therapy and may continue after therapy is discontinued. In addition, the toxicities that are associated with anti–CTLA-4 therapy may differ from those of conventional therapies and consist of inflammatory events in parts of the body that do not contain cancerous cells. Early recognition of these inflammatory events and intervention is important, and the identification of predictive biomarkers continues to be an unfulfilled need in the field of immunotherapy. Combinatorial approaches with targeted therapies, radiation therapy, chemotherapy, or other immune checkpoint agonists/antagonists have the potential to increase the efficacy of CTLA-4 blockade.
Introduction
The immune system is an important defense against the development and progression of cancer (1). Historically, there has been a waxing and waning enthusiasm for immune therapies to treat cancer due to limited efficacy. Immunotherapies, such as IL-2 and adoptive transfer of autologous tumor-infiltrating lymphocytes, can induce durable tumor responses in a subset of patients, leading to long-term survival for patients with an otherwise poor prognosis (2, 3). With improved understanding of the importance for both the priming and effector phases of antitumor immunity, checkpoint blockade-based therapeutics have recently begun providing patients with durable benefits and appear to be applicable in a broad array of malignancies.
There are many mechanisms by which tumors evade destruction by the immune system. These mechanisms include recruitment of suppressor immune cells, such as Tregs, myeloid-derived suppressor cells that impair T cell proliferation, and tumor-associated macrophages, which appear to have both tumoricidal and tumorostatic functions. In addition, tumors upregulate programmed death ligand-1 (PD-L1, also known as B7-H1), which binds to the programmed death-1 receptor (PD-1) on the T cell surface to inhibit T cell function (4, 5). Tumors also produce factors associated with immunosuppression, such as TGF-β, IL-10, reactive oxygen species, and nitric oxide (6, 7).
Effective antitumor immunity is limited by insufficient costimulation of the immune system by tumor cells (8). These same costimulatory pathways can be involved in priming of the immune system as well as execution of tumor cell killing in the tumor microenvironment. As a result, the targeting of these costimulatory pathways has become a primary area of clinical investigation for cancer therapeutics.
Immune checkpoint blockade
The immune system is tightly regulated to respond to appropriate antigens without responding to self (9). T cells, one of the key mediators of the immune response, require costimulation for activation and can be inactivated by inhibitory signals (10). T cell activation is initiated when antigen is presented to the T cell receptor (TCR) complex by MHC class I or II on an antigen-presenting cell. Signals from the TCR are then amplified or counteracted by costimulatory molecules (11, 12). Binding of CD28 on the T cell to B7-1 (CD80) and B7-2 (CD86) molecules on the antigen-presenting cell creates an amplifying signal required for full T cell activation. This CD28/B7 binding leads to increased production of IL-2 and other stimulatory cytokines, enhances metabolism, facilitates cell cycle progression, upregulates cell survival genes, and results in T cell proliferation and differentiation (13).
Cytotoxic T lymphocyte antigen-4 (CTLA-4) is a key inhibitory receptor that affects T cell function and plays a critical role in the priming phase of the immune response (14). In resting T cells, CTLA-4 is found in the intracellular compartment. Following T cell activation through CD28 binding, CTLA-4 is transported to and expressed on the surface of T cells (15, 16). The stronger the stimulatory signal through the TCR, the more CTLA-4 is expressed and translocated to the T cell surface (17). Once at the cell surface, CTLA-4’s inhibitory signal is transmitted through the binding of B7-1 and B7-2 on activated B cells and monocytes. Compared with CD28, CTLA-4 binding is higher affinity and blocks further costimulation (Figure 1 and ref. 18). Additionally, CTLA-4–expressing cells capture and degrade B7-1 and B7-2 through a process called trans-endocytosis (19).
Figure 1. T cell activation requires costimulation through TCR and CD28.
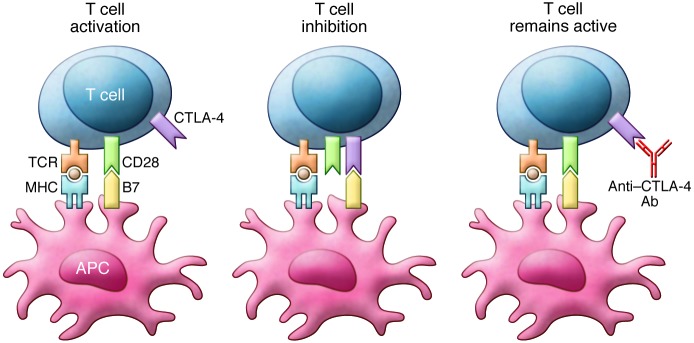
Binding of B7 to CTLA-4 inhibits T cell function. Anti–CTLA-4 antibodies block CTLA-4 binding and prevent inhibition of T cell function. This figure is adapted from Clinical Therapeutics.
The CTLA-4 inhibitory signaling goes beyond blocking costimulation, and the mechanisms underlying this inhibition are under active study. For example, CTLA-4 binding inhibits IL-2 transcription within activated T cells and prevents their progression through the cell cycle (20). Additionally, CTLA-4 interacts with Src homology domain–containing tyrosine phosphatases SHP-1, SHP-2, and PP2A, which dephosphorylate key TCR signaling kinases (FYN, LCK, ZAP-70) and members of the RAS pathway (21, 22). CTLA-4 is also constitutively expressed on Tregs, which control effector T cell functions and are involved in tolerance (23).
The role of CTLA-4 in immune regulation has been demonstrated through the production of Ctla4 knockout mice. These animals exhibit diffuse tissue infiltration with polyclonal T cell blasts, which leads to death at 3 to 4 weeks (24). Moreover, Treg-specific CTLA-4 suppression resulted in spontaneous lymphoproliferation and fatal T cell–mediated autoimmune disease, suggesting that the development of these toxicities is related to CTLA-4 activity on Tregs (25). CTLA-4–targeted antibodies deplete intratumoral Tregs by an unknown mechanism, possibly through antibody-dependent cell-mediated cytotoxicity (26, 27).
There are likely several mechanisms by which CTLA-4 blockade increases antitumor immune responses. As noted above, CTLA-4–blocking antibodies increase CD8+ lymphocytes and deplete Tregs. However, depletion of the Tregs alone does not enhance antitumor responses (28). The combination of enhanced effector T cell function with inhibition of Treg cell activity seems to be necessary for maximal antitumor activity.
Role in cancer
Tumor cells can both inhibit and cause insufficient activation of immune costimulatory pathways to suppress antitumor immune function. Tumor cells that were transfected with B7-1 were rejected when placed into mice, and subsequent attempts at tumor growth in these mice were unsuccessful. These results suggest that activation of antitumor immunity through B7-1 signaling is durable, leading to the idea that targeting this signaling pathway through inhibition of CTLA-4 could be a tool in the treatment of malignancy (8).
Allison and colleagues developed CTLA-4–blocking antibodies and demonstrated that combining them with tumor lysate-loaded dendritic cells reduced Tregs, increased CD8+ lymphocytes, inhibited metastatic growth, and prolonged survival in murine colon carcinoma and fibrosarcoma models (29). In a syngeneic mouse model, CTLA-4 antibodies were found to enhance anti-prostate cancer immune responses against injected tumor cells, significantly delaying growth of tumors (30). Coadministration of anti–CTLA-4 antibodies and GM-CSF–producing vaccines induced rejection of a murine melanoma cell line, B16, in 80% of mice. In addition, those mice that survived an initial tumor cell injection rejected a subsequent challenge with B16 cells (31).
A human monoclonal IgG1κ antibody targeting CTLA-4 (MDX-010 also known as ipilimumab and Yervoy) and an IgG2 antibody targeting CTLA-4 (CP-675, also known as tremelimumab) were developed for use in patients (32). A trial of CTLA-4–blocking antibodies in prostate cancer demonstrated a ≥50% decline of prostate-specific antigen in 2 of 14 patients (33). A cohort of previously vaccinated patients with melanoma and ovarian carcinoma was treated with CTLA-4 antibody blockade. Reduced tumor markers were seen in two patients with ovarian carcinoma, and there was evidence of tumor necrosis in three patients with metastatic melanoma (34). This early indication that CTLA-4 blockade was safe and clinically active led to more intensive exploration of this therapy.
Success in melanoma
Melanoma is one of the diseases in which immunotherapy historically has shown promise. Interferon is used in the treatment of early-stage melanoma, and high-dose IL-2 can lead to durable responses in a subset of patients with metastatic melanoma (2, 35). Early trials found that CTLA-4 blockade was safe and had potential efficacy in patients with metastatic melanoma. In a multiple-dose phase I/II study of ipilimumab, a disease control rate (complete response plus partial response plus stable disease rates) of 39% was observed in the highest dose cohort (36, 37). Several dose-finding phase II studies showed an increase in inflammatory toxicity at higher doses, with efficacy at both the 3 mg/kg and 10 mg/kg dosage levels. As an example, in a double-blind study that tested doses of 0.3 mg/kg, 3.0 mg/kg, and 10 mg/kg, 11.1% of patients responded at the highest dosage (38–40). Based upon these results, both the 3 mg/kg and 10 mg/kg dosage have been used in subsequent clinical trials. Phase I/II testing of tremelimumab at 15 mg/kg every 90 days and 10 mg/kg monthly showed a 10% objective response rate with durable responses (41).
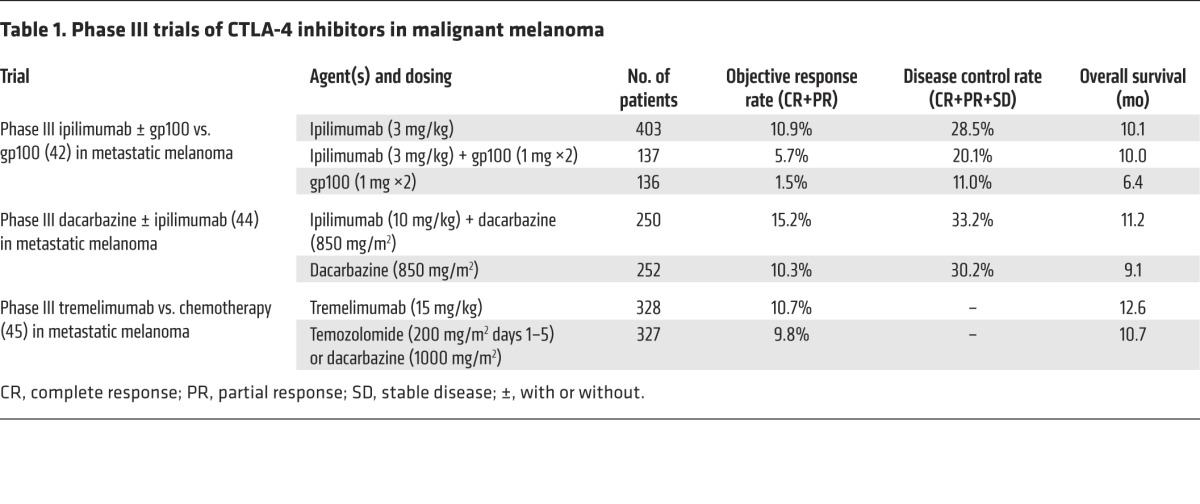
The pivotal phase III trial randomized untreated patients to ipilimumab at 3 mg/kg every three weeks for up to four doses in combination with glycoprotein 100 (gp100) peptide vaccine, ipilimumab alone, or gp100 alone. The gp100 vaccine consisted of a modified gp100 melanoma antigen with increased binding affinity for HLA-A2. An overall survival benefit was seen when the groups receiving ipilimumab plus gp100 and ipilimumab alone were compared with the group receiving gp100 alone. The overall survivals were 10.0 months, 10.1 months, and 6.4 months, respectively, with a hazard ratio for death between the two gp100 arms of 0.68. There was no difference in survival between the two groups that received ipilimumab (42). These results led to the FDA approval of ipilimumab in advanced melanoma as an intravenous infusion given at a dose of 3 mg/kg every three weeks for up to four doses.
Dacarbazine is an alkylating agent that destroys cancer cells by adding an alkyl group to DNA. Previous trials in melanoma demonstrated response rates to dacarbazine of between 8% and 20% with limited duration (43). A phase III trial compared ipilimumab plus dacarbazine with dacarbazine alone in previously untreated patients with metastatic melanoma. This trial used a higher dose of ipilimumab, 10 mg/kg, every three weeks for four doses, followed by maintenance therapy every twelve weeks. The overall survival was significantly longer in the ipilimumab group as compared with that in the dacarbazine group (44).
A phase III trial of tremelimumab versus chemotherapy showed an overall survival of 12.6 months in the tremelimumab arm and 10.7 months in the chemotherapy arm. These results failed to meet statistical significance (45). Phase III trial results are summarized in Table 1. Tremelimumab is being actively investigated in combination with angiopoietin 2 (ANG2) inhibition in melanoma and with PD-L1 inhibition in head and neck cancer, non–small-cell lung cancer, and other solid tumors (NCT02141542, NCT02369874, NCT02352948, NCT02261220).
Table 1. Phase III trials of CTLA-4 inhibitors in malignant melanoma.
Brain metastases frequently occur in patients with metastatic melanoma and are generally less responsive to systemic therapy than other distant metastasis due to the blood brain barrier. However, CTLA-4 blockade has proven to be effective in patients with brain metastasis from melanoma. An open-label phase II trial of ipilimumab in patients with brain metastasis showed a disease control rate of 18% in patients off steroids, with a 24% disease control rate in brain alone; the disease control rate was much lower in patients on systemic steroids. Thus, ipilimumab has some activity in patients with brain metastasis from melanoma, and its activity does not appear to be restricted by the blood brain barrier (46).
Data in non-melanoma cancers
Ipilimumab was evaluated in tumor types other than melanoma and showed some potential therapeutic benefit in a small fraction of patients (47). A randomized phase II trial of ipilimumab combined with carboplatin and paclitaxel in patients with non–small-cell lung cancer showed some improvement in progression-free survival when ipilimumab was given in a phased dosing regimen that started with chemotherapy alone; however, no overall survival benefit was observed (48).
Ipilimumab following radiation therapy was tested in patients with metastatic castration-resistant prostate cancer who progressed after treatment with docetaxel. The trial showed no difference in overall survival, and there were four deaths due to ipilimumab toxicity. However, there was a suggestion that some subgroups of patients with prostate cancer may benefit from ipilimumab (49).
Renal cell carcinoma (RCC) is another malignancy with a history of responses to immunotherapy. Ipilimumab was tested in patients with RCC in a phase II study in which 6 of 61 patients had a response to therapy, even if they had not previously responded to high-dose IL-2 treatment (50). Clinical trials are currently underway that examine the combination of ipilimumab with other immunotherapy in RCC and other malignancies (NCT02210117, NCT02231749, NCT02089685).
Patterns of response and patient selection
Patients with metastatic melanoma undergoing treatment with ipilimumab have patterns of response that are different from those typically seen with conventional chemotherapy or targeted therapies. Metastatic melanoma is monitored by obtaining CT scans, MRI scans, or PET scans at regular intervals. Responses to therapy are based upon the measurement of target lesions seen on these scans. Most patients treated with chemotherapy or targeted therapy have a response or lack of response at the first interval imaging that is reflective of the overall response to therapy. With CTLA-4 blockade, some responses were delayed, while other patients had initial progression of disease (characterized by an increase in target lesions or new sites of disease) and then went on to experience stabilization of their disease or decrease in overall tumor burden (51). Patterns of response to CTLA-4 blockade include (a) regression of baseline lesions with no new lesions; (b) stable disease followed by slow, steady decline in tumor burden; (c) delayed response after an initial increase in tumor burden; and (d) response after the appearance of new lesions (51).
The observations described above showed that the conventional Response Evaluation Criteria in Solid Tumors (RECIST) would not be an effective way to monitor for responses in all patients, and alternative criteria, termed immune-related response criteria (irRC), were proposed (51). The irRC consider total tumor burden regardless of the growth of new disease, with higher maximum tumor growth allowed within the definition of stable disease. These criteria permit increases in tumor burden, with a requirement for confirmation of progression with subsequent CT, MRI, or PET imaging. Based on this, it has been recommended that no imaging be obtained until the end of the four-dose treatment period with anti–CTLA-4 antibodies in melanoma (12–16 weeks) and that disease status be confirmed with the same type of imaging at least 4 weeks later (51, 52). Anti–CTLA-4 antibody treatment may be discontinued due to toxicity during the twelve-week treatment period but would not be discontinued for disease progression unless there was marked symptomatic progression. Another set of ipilimumab treatments may be considered for select patients who experience no significant systemic toxicity during prior ipilimumab therapy and who relapse after initial clinical response or progress after stable disease for over three months (53, 54).
Five-year survival rates of patients treated with ipilimumab have been reported to be between 16.5% and 25%. Some of these patients did not display an objective response to ipilimumab but did not exhibit disease progression in the long term (55). Survival curves show a plateau at three years that is ongoing, and initial reports suggest that similar findings will be seen with analysis of the larger phase III trials (56). It appears that a minority of patients receives the majority of the long-term benefit, with a tail-of-the-curve phenomenon seen in the Kaplan-Meier analyses characterized by very prolonged survival for a small proportion of patients.
One area of active investigation includes the identification of biomarkers to track responses to anti–CTLA-4 therapy as well as the development of prospective markers that can better predict which patients might benefit from anti–CTLA-4 therapy. There has been an association between response and baseline expression of immune-related tumor markers and an increase in tumor-infiltrating lymphocytes after treatment (57). Some markers identified retrospectively that reflect immune activation by ipilimumab in responders include absolute lymphocyte count, upregulation of the T cell activation marker inducible costimulator (ICOS), and the development of a polyfunctional T cell response to the tumor antigen NY-ESO-1 (58).
Some promising mechanisms that are currently being explored in the realm of predictive markers of response include looking at genetic factors within the tumor or the host. High baseline expression levels of immune-related genes predicted response to ipilimumab treatment in a cohort of 45 patients (59). Additionally, the tumor mutation volume may help predict activity of checkpoint blockade (60, 61). In a recent study, whole-exome sequencing was performed in tumors and matched blood samples from 64 patients treated with CTLA-4 blockade. Mutational load was associated with the degree of clinical benefit but was not able to predict benefit on its own. Candidate tumor neoantigens were identified for each patient, and those that were correlated with a strong response to CTLA-4 blockade were validated with a second patient set (62). Similar approaches may lead to better patient selection in the future once prospective clinical testing validates their use.
Toxicity
Immune therapy with CTLA-4 blockade induces drug-related adverse events that are different from those observed with other types of treatment. These toxicities are related to immune effects and are classified as immune-related adverse events (irAEs; also now called adverse events of special interest), with an emphasis on end-organ inflammatory events.
The most prevalent toxicity with ipilimumab is skin rash, with 47% to 66% of patients experiencing a pruritic maculopapular rash (63). The most concerning toxicity observed is ipilimumab-induced inflammatory colitis, which initially manifests as diarrhea and can lead to death from bowel perforation. Overall, diarrhea was observed in 44% of patients receiving ipilimumab at 10 mg/kg, and severe diarrhea was reported in 18% of patients. Hepatotoxicity was observed in 3% to 9% of patients receiving anti–CTLA-4 antibodies, and the presentation was often an asymptomatic elevation of liver enzymes. Hypophysitis occurs in 1% to 9% of patients treated with 3 or 10 mg/kg ipilimumab, respectively, and develops an average of 6 weeks after the initiation of therapy. Pancreatitis, iridocyclitis, lymphadenopathy, neuropathies, and nephritis have also been reported with ipilimumab (64). An analysis of patients in a phase III trial of ipilimumab showed that the majority of these irAEs developed within 12 weeks of initial dosing and resolved within 12 weeks of onset (64).
Early diagnosis, vigilant follow up, and early treatment are essential to minimizing morbidity from these inflammatory adverse events (51). Systemic corticosteroids and other immunosuppressive drugs can be used in the treatment of inflammatory toxicities. Although rash, when it appears, is generally mild, there have been rare cases of toxic epidermal necrolysis and Stevens-Johnson syndrome, which require systemic steroids and discontinuation of ipilimumab (63). Given the potential severity of ipilimumab-induced colitis, an algorithm to treat diarrhea has been developed and published to help guide clinicians on dosing of corticosteroids and the use of the tumor necrosis inhibitor infliximab in patients with steroid refractory diarrhea (64). Budesonide, a nonabsorbable oral steroid, was initially used to treat the colitis; however, a randomized phase II study comparing the efficacy of ipilimumab with or without prophylactic budesonide showed no change in the rate of grade 2 or higher diarrhea between the two arms (65). Hypophysitis can present with somewhat nonspecific symptoms, including headache, double vision, nausea, weakness, and fatigue. In patients with these symptoms, this toxicity should be confirmed with lab tests and a brain MRI, which may show pituitary swelling and/or enlargement of the sella turcica. Patients with evidence of hypophysitis often require long-term hormone replacement, including systemic corticosteroids (63, 66). Severe hepatotoxicity can develop and should be treated with systemic corticosteroids. If patients do not respond to systemic corticosteroids, then oral mycophenolate mofetil should be initiated. Infliximab should not be used in hepatic toxicity, as it can elevate liver function tests (63).
The toxicities observed with CTLA-4 blockade are typically transient, reversible inflammatory events. However, many of the patients require high doses of systemic corticosteroids that cause significant immunosuppression. In addition, patients with hypophysitis can remain on lifelong corticosteroid supplementation. The effect of this immunosuppression on the tumor immune response is unclear, as these patients still respond to therapy, and further analysis will be needed to determine the long-term effects on tumor growth. However, in the short term, there are cases of opportunistic infections associated with the use of high-dose corticosteroids for irAEs, and these must be considered when patients present with new or worsening symptoms (67).
Combination therapies and future directions
The ultimate goal with checkpoint blockade therapy is long-term disease control in patients with advanced malignancy. While this is unlikely to be attained through the use of CTLA-4 therapy alone, it may be achieved through appropriate combinations of different therapeutics. Selecting combinations with some scientific rationale involves identifying combinations that may enhance the environment for immunomodulatory therapy by releasing antigens from tumors or by changing the tumor microenvironment. Potential combinations for CTLA-4 blockade include conventional cytotoxic chemotherapy, radiation therapy, targeted therapy, and traditional immune therapy as well as use of other checkpoint blockade agents.
Evidence suggests that radiation therapy may activate the immune system and thus prime it for greater activation by immunotherapy. The abscopal effect, by which local radiation therapy causes tumor regression at a distant site, may be mediated by an immune response. In patients treated with radiation therapy with or without ipilimumab, changes in peripheral blood immune cells and increased antibodies to known tumor antigens were observed (68, 69). Further studies are ongoing to determine how radiation therapy may be used to enhance responses to anti–CTLA-4 antibodies (NCT01689974, NCT02239900, NCT02107755, NCT01970527, NCT01996202).
BRAF-targeted therapy has been proven to prolong survival in patients with melanoma with BRAF mutations (70), and the combination of BRAF inhibitors and MEK inhibitors has proven even more effective (71, 72). BRAF inhibition enhances melanoma antigen expression and facilitates T cell cytotoxicity, suggesting that combinations of BRAF inhibitors with immunotherapy may be synergistic (73, 74); however, attenuation of the MAPK pathway using MEK inhibitors may impair T cell effector function, although this appears to be more related to MEK inhibition than BRAF inhibition alone (75). Unfortunately, the initial combination of BRAF inhibitor therapy and ipilimumab led to hepatoxicity in 8 of 12 patients enrolled in a phase I trial test of this combination. The trial was closed to further accrual due to this toxicity (76). Additional trials combining MAPK-targeted therapies with immunotherapy are underway. A trial of sequential vemurafenib followed by ipilimumab has completed accrual, and results are pending (NCT01673854). Trials of dabrafenib plus ipilimumab or dabrafenib/trametinib/ipilimumab and sequencing trials are ongoing (NCT01940809, NCT02224781, NCT01767454).
VEGF plays an immunologic role by suppressing dendritic cell activation. Additionally, studies in preclinical animal models have shown that VEGF blockade may play a role in lymphocyte trafficking across endothelium (77). Bevacizumab is an anti–VEGF-A antibody that is widely used in the treatment of malignancy. A phase I study of ipilimumab plus bevacizumab treated 46 patients and showed that the combination was safe for further testing, with a disease control rate of 67.4% (78). Based upon these results, patients are currently being enrolled in a randomized phase II trial of this combination (NCT01950390).
The ability to induce a higher rate of durable responses with ipilimumab is dependent upon immune activation, suggesting that combinations with other immune modulators may increase efficacy. The combination of IL-2 and ipilimumab has shown an overall response rate of 22%, with three complete responses (79). Trials to further explore this combination are ongoing (NCT01856023, NCT02203604). GM-CSF is known to enhance dendritic cell activation and potentiate antitumor T and B cell responses and is hypothesized to synergize with CTLA-4 blockade. In a randomized phase II trial, 245 patients with melanoma were treated with ipilimumab with and without GM-CSF. While no difference in progression-free survival was observed, the one-year survival rate for GM-CSF plus ipilimumab was 68.9% versus 52.9% for ipilimumab alone. Additionally, there was a reduction in grade 3 to 5 adverse events in patients on ipilimumab plus GM-CSF compared with patients on ipilimumab monotherapy (80). Further trials to evaluate the addition of GM-CSF to ipilimumab will be needed to confirm these findings.
Recently, fully human monoclonal antibodies that target PD-1 have been developed in a variety of tumor types. Two PD-1–targeting drugs, nivolumab and pembrolizumab, have demonstrated single-agent activity in melanoma and are currently approved by the FDA for patients with ipilimumab-refractory disease (81, 82). A phase I trial combining nivolumab with ipilimumab in 53 patients with advanced melanoma showed evidence of clinical activity (conventional, unconfirmed, or immune-related response or stable disease for ≥24 weeks) in 65% of patients. Many of the responding patients had a reduction in tumor volume of 80% or greater (83). Survival updates for these patients showed 1- and 2-year overall survival rates of 82% and 75%, respectively. The objective response rate was 40%, and the median duration of response was not reached, indicating ongoing responses for many patients (84).
It is still unclear which sequence of checkpoint blockade therapies is the most effective. Trials are underway to determine whether the benefit is greater if ipilimumab and nivolumab are given concurrently or whether they could be given sequentially with similar clinical activity and long-term benefits (NCT01844505, NCT01927419, NCT01783938). In addition, understanding the sequence of immune checkpoint blockade may improve our understanding of how targeting one checkpoint may affect the activity of another and what roles immune infiltrates, lymphocytes, and other stromal elements, such as dendritic cells and myeloid-derived suppressor cells, play.
As with many therapies that demonstrate activity in the metastatic setting, investigation in the high-risk adjuvant setting is underway. A European Organisation for Research and Treatment of Cancer (EORTC) study randomized 951 patients with high-risk resected melanoma to placebo or ipilimumab at 10 mg/kg every three weeks for four doses, followed by treatment every three months for three years. This trial demonstrated a recurrence-free survival of 26.1 months on the ipilimumab arm as compared with a recurrence-free survival of 17.1 months in the placebo arm (85). Unfortunately, there were five deaths due to ipilimumab-related immune toxicity during the trial; three were due to colitis, one was due to myocarditis, and one was due to Guillain-Barré syndrome. In the US, high-dose interferon-α2b is approved by the FDA for use in this setting and has demonstrated benefit previously (35, 86). The Eastern Cooperative Oncology Group (ECOG) recently completed enrollment for a study (NCT01274338) comparing high-dose interferon-α2b against ipilimumab at 3 mg/kg and 10 mg/kg, respectively.
Conclusion
Immunotherapy is rapidly changing the landscape of cancer therapy. Patients with advanced malignancies are looking for effective therapies with durable disease control and manageable toxicity. Anti–CTLA-4 therapy with ipilimumab is the first example of a new class of therapeutics providing such opportunities. The future of immunotherapy is in the development of combination therapies and improving the numbers of patients experiencing durable benefit. In addition, investigation continues in biomarker development to identify the patients that are most likely to respond to therapy and those at highest risk of toxicity. Further roles for CTLA-4 blockade in the adjuvant setting as well as in other cancers and combinations are still to be determined.
Footnotes
Conflict of interest: The authors received research support from Bristol-Myers Squibb, Merck, Pfizer, and Genentech.
Reference information:J Clin Invest. 2015;125(9):3377–3383. doi:10.1172/JCI80012.
References
- 1.Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–271. doi: 10.1146/annurev-immunol-031210-101324. [DOI] [PubMed] [Google Scholar]
- 2.Atkins MB, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17(7):2105–2116. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg SA, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17(13):4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fong L, Small EJ. Anti-cytotoxic T-lymphocyte antigen-4 antibody: the first in an emerging class of immunomodulatory antibodies for cancer treatment. J Clin Oncol. 2008;26(32):5275–5283. doi: 10.1200/JCO.2008.17.8954. [DOI] [PubMed] [Google Scholar]
- 5.Mocellin S, Nitti D. CTLA-4 blockade and the renaissance of cancer immunotherapy. Biochim Biophys Acta. 2013;1836(2):187–196. doi: 10.1016/j.bbcan.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Poschke I, Mougiakakos D, Kiessling R. Camouflage and sabotage: tumor escape from the immune system. Cancer Immunol Immunother. 2011;60(8):1161–1171. doi: 10.1007/s00262-011-1012-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bronte V, Mocellin S. Suppressive influences in the immune response to cancer. J Immunother. 2009;32(1):1–11. doi: 10.1097/CJI.0b013e3181837276. [DOI] [PubMed] [Google Scholar]
- 8.Allison JP, Hurwitz AA, Leach DR. Manipulation of costimulatory signals to enhance antitumor T-cell responses. Curr Opin Immunol. 1995;7(5):682–686. doi: 10.1016/0952-7915(95)80077-8. [DOI] [PubMed] [Google Scholar]
- 9.Bretscher P, Cohn M. A theory of self-nonself discrimination. Science. 1970;169(3950):1042–1049. doi: 10.1126/science.169.3950.1042. [DOI] [PubMed] [Google Scholar]
- 10.Sharpe AH, Abbas AK. T-cell costimulation — biology, therapeutic potential, and challenges. N Engl J Med. 2006;355(10):973–975. doi: 10.1056/NEJMp068087. [DOI] [PubMed] [Google Scholar]
- 11.Kroczek RA, Mages HW, Hutloff A. Emerging paradigms of T-cell co-stimulation. Curr Opin Immunol. 2004;16(3):321–327. doi: 10.1016/j.coi.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 12.Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol. 2002;2(2):116–126. doi: 10.1038/nri727. [DOI] [PubMed] [Google Scholar]
- 13.Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev. 2008;224:166–182. doi: 10.1111/j.1600-065X.2008.00662.x. [DOI] [PubMed] [Google Scholar]
- 14.Scalapino KJ1, Daikh DI. CTLA-4: a key regulatory point in the control of autoimmune disease. Immunol Rev. 2008;223:143–155. doi: 10.1111/j.1600-065X.2008.00639.x. [DOI] [PubMed] [Google Scholar]
- 15.Walunas TL, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1(5):405–413. doi: 10.1016/1074-7613(94)90071-X. [DOI] [PubMed] [Google Scholar]
- 16.Linsley PS, Bradshaw J, Greene J, Peach R, Bennett KL, Mittler RS. Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity. 1996;4(6):535–543. doi: 10.1016/S1074-7613(00)80480-X. [DOI] [PubMed] [Google Scholar]
- 17.Egen JG, Allison JP. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity. 2002;16(1):23–35. doi: 10.1016/S1074-7613(01)00259-X. [DOI] [PubMed] [Google Scholar]
- 18.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182(2):459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qureshi OS, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332(6029):600–603. doi: 10.1126/science.1202947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brunner MC, Chambers CA, Chan FK, Hanke J, Winoto A, Allison JP. CTLA-4-mediated inhibition of early events of T cell proliferation. J Immunol. 1999;162(10):5813–5820. [PubMed] [Google Scholar]
- 21.Marengère LE, Waterhouse P, Duncan GS, Mittrücker HW, Feng GS, Mak TW. Regulation of T cell receptor signaling by tyrosine phosphatase SYP association with CTLA-4. Science. 1996;272(5265):1170–1173. doi: 10.1126/science.272.5265.1170. [DOI] [PubMed] [Google Scholar]
- 22.Parry RV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25(21):9543–9553. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takahashi T, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192(2):303–310. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Waterhouse P, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270(5238):985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 25.Wing K, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322(5899):271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 26.Selby MJ, et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013;1(1):32–42. doi: 10.1158/2326-6066.CIR-13-0013. [DOI] [PubMed] [Google Scholar]
- 27.Simpson TR, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210(9):1695–1710. doi: 10.1084/jem.20130579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med. 2009;206(8):1717–1725. doi: 10.1084/jem.20082492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 30.Kwon ED, et al. Manipulation of T cell costimulatory and inhibitory signals for immunotherapy of prostate cancer. Proc Natl Acad Sci U S A. 1997;94(15):8099–8103. doi: 10.1073/pnas.94.15.8099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190(3):355–366. doi: 10.1084/jem.190.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keler T, et al. Activity and safety of CTLA-4 blockade combined with vaccines in cynomolgus macaques. J Immunol. 2003;171(11):6251–6259. doi: 10.4049/jimmunol.171.11.6251. [DOI] [PubMed] [Google Scholar]
- 33.Small EJ, Tchekmedyian NS, Rini BI, Fong L, Lowy I, Allison JP. A pilot trial of CTLA-4 blockade with human anti-CTLA-4 in patients with hormone-refractory prostate cancer. Clin Cancer Res. 2007;13(6):1810–1815. doi: 10.1158/1078-0432.CCR-06-2318. [DOI] [PubMed] [Google Scholar]
- 34.Hodi FS, et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc Natl Acad Sci U S A. 2003;100(8):4712–4717. doi: 10.1073/pnas.0830997100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kirkwood JM, Strawderman MH, Ernstoff MS, Smith TJ, Borden EC, Blum RH. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol. 1996;14(1):7–17. doi: 10.1200/JCO.1996.14.1.7. [DOI] [PubMed] [Google Scholar]
- 36.Tchekmedyian S, Glaspy J, Korman A, Keler T, Deo Y, Davis T. MDX-010 (human anti-CTLA4): a phase I trial in malignant melanoma. Proc Am Soc Clin Oncol. 2002 (Abstract 56) [Google Scholar]
- 37.Weber JS, et al. Phase I/II study of ipilimumab for patients with metastatic melanoma. J Clin Oncol. 2008;26(36):5950–5956. doi: 10.1200/JCO.2008.16.1927. [DOI] [PubMed] [Google Scholar]
- 38.Maker AV, et al. Intrapatient dose escalation of anti-CTLA-4 antibody in patients with metastatic melanoma. J Immunother. 2006;29(4):455–463. doi: 10.1097/01.cji.0000208259.73167.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolchok JD, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010;11(2):155–164. doi: 10.1016/S1470-2045(09)70334-1. [DOI] [PubMed] [Google Scholar]
- 40.O’Day SJ, et al. Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: a multicenter single-arm phase II study. Ann Oncol. 2010;21(8):1712–1717. doi: 10.1093/annonc/mdq013. [DOI] [PubMed] [Google Scholar]
- 41.Camacho LH, et al. Phase I/II trial of tremelimumab in patients with metastatic melanoma. J Clin Oncol. 2009;27(7):1075–1081. doi: 10.1200/JCO.2008.19.2435. [DOI] [PubMed] [Google Scholar]
- 42.Hodi FS, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patel PM, et al. Extended schedule, escalated dose temozolomide versus dacarbazine in stage IV melanoma: final results of a randomised phase III study (EORTC 18032) Eur J Cancer. 2011;47(10):1476–1483. doi: 10.1016/j.ejca.2011.04.030. [DOI] [PubMed] [Google Scholar]
- 44.Robert C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517–2526. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 45.Ribas A, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol. 2013;31(5):616–622. doi: 10.1200/JCO.2012.44.6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Margolin K, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol. 2012;13(5):459–465. doi: 10.1016/S1470-2045(12)70090-6. [DOI] [PubMed] [Google Scholar]
- 47.Calabrò L, Danielli R, Sigalotti L, Maio M. Clinical studies with anti-CTLA-4 antibodies in non-melanoma indications. Semin Oncol. 2010;37(5):460–467. doi: 10.1053/j.seminoncol.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 48.Lynch TJ, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol. 2012;30(17):2046–2054. doi: 10.1200/JCO.2011.38.4032. [DOI] [PubMed] [Google Scholar]
- 49.Kwon ED, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15(7):700–712. doi: 10.1016/S1470-2045(14)70189-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang JC, et al. Ipilimumab (anti-CTLA4 antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J Immunother. 2007;30(8):825–830. doi: 10.1097/CJI.0b013e318156e47e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wolchok JD, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15(23):7412–7420. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 52.Wolchok JD, et al. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci. 2013;1291:1–13. doi: 10.1111/nyas.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coit DG, et al. Melanoma, version 4.2014. J Natl Compr Canc Netw. 2014;12(5):621–629. doi: 10.6004/jnccn.2014.0066. [DOI] [PubMed] [Google Scholar]
- 54.Robert C, Schadendorf D, Messina M, Hodi FS, O’Day S, MDX010-20 investigators Efficacy and safety of retreatment with ipilimumab in patients with pretreated advanced melanoma who progressed after initially achieving disease control. Clin Cancer Res. 2013;19(8):2232–2239. doi: 10.1158/1078-0432.CCR-12-3080. [DOI] [PubMed] [Google Scholar]
- 55.Prieto PA, et al. CTLA-4 blockade with ipilimumab: long-term follow-up of 177 patients with metastatic melanoma. Clin Cancer Res. 2012;18(7):2039–2047. doi: 10.1158/1078-0432.CCR-11-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McDermott D, Haanen J, Chen TT, Lorigan P, O’Day S, MDX010-20 Investigators Efficacy and safety of ipilimumab in metastatic melanoma patients surviving more than 2 years following treatment in a phase III trial (MDX010-20) Ann Oncol. 2013;24(10):2694–2698. doi: 10.1093/annonc/mdt291. [DOI] [PubMed] [Google Scholar]
- 57.Hamid O, et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J Transl Med. 2011;9:204. doi: 10.1186/1479-5876-9-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Callahan MK, Postow MA, Wolchok JD. Immunomodulatory therapy for melanoma: ipilimumab and beyond. Clin Dermatol. 2013;31(2):191–199. doi: 10.1016/j.clindermatol.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ji RR, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother. 2012;61(7):1019–1031. doi: 10.1007/s00262-011-1172-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Charen AS, et al. The neoantigen landscape underlying clinical response to ipilimumab. J Clin Oncol. 2014;32 :5s(suppl; abstr 3003) [Google Scholar]
- 61.Adaniel C, et al. Germline genetic determinants of immunotherapy response in metastatic melanoma. J Clin Oncol. 2014;32 :5s(suppl; abstr 3004) [Google Scholar]
- 62.Snyder A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weber JS, Kähler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691–2697. doi: 10.1200/JCO.2012.41.6750. [DOI] [PubMed] [Google Scholar]
- 64.Weber JS, Dummer R, de Pril V, Lebbé C, Hodi FS, MDX010-20 Investigators Patterns of onset and resolution of immune-related adverse events of special interest with ipilimumab: detailed safety analysis from a phase 3 trial in patients with advanced melanoma. Cancer. 2013;119(9):1675–1682. doi: 10.1002/cncr.27969. [DOI] [PubMed] [Google Scholar]
- 65.Weber J, et al. A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res. 2009;15(17):5591–5598. doi: 10.1158/1078-0432.CCR-09-1024. [DOI] [PubMed] [Google Scholar]
- 66.Blansfield JA, et al. Cytotoxic T-lymphocyte-associated antigen-4 blockage can induce autoimmune hypophysitis in patients with metastatic melanoma and renal cancer. J Immunother. 2005;28(6):593–598. doi: 10.1097/01.cji.0000178913.41256.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kyi C, Hellmann MD, Wolchok JD, Chapman PB, Postow MA. Opportunistic infections in patients treated with immunotherapy for cancer. J Immunother Cancer. 2014;2:19. doi: 10.1186/2051-1426-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Postow MA, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. 2012;366(10):925–931. doi: 10.1056/NEJMoa1112824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stamell EF, Wolchok JD, Gnjatic S, Lee NY, Brownell I. The abscopal effect associated with a systemic anti-melanoma immune response. Int J Radiat Oncol Biol Phys. 2013;85(2):293–295. doi: 10.1016/j.ijrobp.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chapman PB, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Robert C, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372(1):30–39. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 72.Larkin J, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867–1876. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- 73.Boni A, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010;70(13):5213–5219. doi: 10.1158/0008-5472.CAN-10-0118. [DOI] [PubMed] [Google Scholar]
- 74.Frederick DT, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19(5):1225–1231. doi: 10.1158/1078-0432.CCR-12-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gargett T, Fraser CK, Dotti G, Yvon ES, Brown MP. BRAF and MEK inhibition variably affect GD2-specific chimeric antigen receptor (CAR) T-cell function in vitro. J Immunother. 2015;38(1):12–23. doi: 10.1097/CJI.0000000000000061. [DOI] [PubMed] [Google Scholar]
- 76.Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368(14):1365–1366. doi: 10.1056/NEJMc1302338. [DOI] [PubMed] [Google Scholar]
- 77.Motz GT, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. 2014;20(6):607–615. doi: 10.1038/nm.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hodi FS, et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res. 2014;2(7):632–642. doi: 10.1158/2326-6066.CIR-14-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Maker AV, et al. Tumor regression and autoimmunity in patients treated with cytotoxic T lymphocyte-associated antigen 4 blockade and interleukin 2: a phase I/II study. Ann Surg Oncol. 2005;12(12):1005–1016. doi: 10.1245/ASO.2005.03.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hodi FS, et al. Ipilimumab plus sargramostim vs ipilimumab alone for treatment of metastatic melanoma: a randomized clinical trial. JAMA. 2014;312(17):1744–1753. doi: 10.1001/jama.2014.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Topalian SL, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32(10):1020–1030. doi: 10.1200/JCO.2013.53.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Robert C, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384(9948):1109–1117. doi: 10.1016/S0140-6736(14)60958-2. [DOI] [PubMed] [Google Scholar]
- 83.Wolchok JD, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sznol M, et al. Survival, response duration, and activity by BRAF mutation (MT) status of nivolumab (NIVO, anti-PD-1, BMS-936558, ONO-4538) and ipilimumab (IPI) concurrent therapy in advanced melanoma (MEL) J Clin Oncol. 2014;32 :5s(suppl; abstr LBA9003) [Google Scholar]
- 85.Eggermont AM, et al. Ipilimumab versus placebo after complete resection of stage II melanoma: Initial efficacy and safety results from EORTC 18071 phase III trial. J Clin Oncol. 2014;32 :5s(suppl; abstr LBA9008) [Google Scholar]
- 86.Jonasch E, et al. Adjuvant high-dose interferon alfa-2b in patients with high-risk melanoma. Cancer J. 2000;6(3):139–145. [PubMed] [Google Scholar]
- 87.Buchbinder EI, McDermott DF. Cytotoxic T-lymphocyte antigen-4 blockade in melanoma. Clin Ther. 2015;37(4):755–763. doi: 10.1016/j.clinthera.2015.02.003. [DOI] [PubMed] [Google Scholar]