

Abstract
The generation of an inflammatory environment is favorable and often decisive for the growth of both primary tumors and metastases. Tumor cells either express membrane molecules or release tumor-derived soluble factors able to alter myelopoiesis. Tumor-reprogrammed myeloid cells not only create a tolerogenic environment by blocking T cell functions and proliferation, but also directly drive tumor growth by promoting cancer stemness, angiogenesis, stroma deposition, epithelial-to-mesenchymal transition, and metastasis formation. In this Review, we discuss the interplay between immunosuppressive and protumoral myeloid cells and detail their immune-regulatory mechanisms, the molecular pathways involved in their differentiation, as well as their potential role as prognostic and diagnostic biomarkers and prospective targets for innovative approaches to treat tumor-bearing hosts.
Tumor progression depends on the gradual accumulation of genetic and epigenetic aberrations in cancer cells that also modify the cellular composition of the tumor environment, establishing a state of chronic inflammation characterized by the stromal infiltration of immune cells. Myeloid cells play a critical role in sustaining cancer progression (1). Moreover, inflammatory myeloid cells help to create and fuel the mutagenic pressure underlying the genetic instability of neoplastic cells by both direct mechanisms, such as the production of free-radical compounds (2), and indirect processes, such as the disruption of host defense barriers (3).
Tumor growth is assisted by tumor-associated macrophages (TAMs), the major leukocyte population infiltrating cancers (4). Although macrophages have the potential to attack and eliminate tumor cells, TAMs exhibit many protumoral features that are partly shared by macrophages involved in tissue repair, and they interfere with the function and proliferation of immune effectors (5). Thus, a high frequency of TAMs is associated with poor prognosis in many but not all human tumors (6).
Myeloid-derived suppressor cells (MDSCs) have received increased attention, and their presence and frequency in the blood of patients with tumors is emerging as a potential and simple prognostic marker to monitor clinical outcome and response to therapy (7). MDSCs are characterized by their myeloid origin, heterogeneous cell composition, and ability to negatively regulate adaptive and innate immune responses to cancer. Although TAMs and MDSCs are regarded as separate entities (Figure 1), the boundaries between them are not clearly demarcated, and they share many characteristics (8). TAM accumulation in cancerous tissues is sustained by circulating inflammatory monocytes (CCR2+Ly6C+ cells in mice and CCR2+CD14+CD16– cells in humans; ref. 9), which are distinct from vessel-patrolling monocytes (Ly6CloCX3CR1hi in mice and CD14dimCD16–CX3CR1hi in humans). Interestingly, immunosuppressive MDSCs with monocytic features are able to traffic from BM to tumors, mainly through the same chemokine pathway (10). Therefore, the CCR2/CCL2 axis is required for MDSC and TAM accrual and functional specialization. Here, we review the distinctive and common characteristics of TAMs and MDSCs, their role in maintaining cancer growth, and the ongoing development of selective therapeutic approaches.
Figure 1. Common phenotypic markers of MDSCs and TAMs.

Several phenotypic markers of mouse and human MDSCs (A) and TAMs (B) have been identified (+ indicates expression, while – indicates lack of expression) and used to define specific cell subgroups, such as PMN-MDSCs, MO-MDSCs, and immature MDSCs (I-MDSCs), as well as M1-like and M2-like TAMs, by both cytofluorimetric and immunohistochemical analyses.
MDSCs and TAMs result from altered myelopoiesis
The most pervasive and efficient strategy of immune escape likely relies on cancer’s ability to create a widespread tolerogenic environment by altering normal hematopoiesis and promoting the expansion of myeloid cells through the constant and progressive release of tumor-derived factors (TDFs), which include metabolites, cytokines, and chemokines (ref. 11 and Figure 2). This “reactive myelopoiesis,” leading to MDSC and TAM accumulation, presents marked and distinct molecular features compared with emergency granulopoiesis (12), as emphasized below.
Figure 2. MDSC and TAM development in tumor-bearing mice.

Under steady-state conditions, resident macrophages may originate from either embryonic tissues or inflammatory monocytes. Resident macrophages are programmed by local factors, and molecular switches support their differentiation. Circulating monocytes can be divided into two subsets: patrolling monocytes (Ly6CloCX3CR1hi) and inflammatory monocytes (Ly6ChiCD11b+CD11c–MHCII–VCAM1–CCR2+), originating from macrophage and DC precursors (MDPs) in BM. Inflammatory monocytes migrate from blood to tissue under the guidance of CCL2/CCR2 chemokine signaling. Tumor cells secrete several factors that modify physiological myelopoiesis, promoting MDP differentiation into PMN-MDSCs (CD11b+Ly6G+) and MO-MDSCs (CD11b+Ly6ChiCCR2+CD115+F4/80lo). MO-MDSCs also originate from the spleen under conditions of emergency and reactive myelopoiesis. MO-MDSCs and inflammatory monocytes migrate to tumor tissues via CCL2/CCR2 and CSF1 signaling and differentiate into TAMs (Ly6C–CD11b+/loCD68+CD1d+MHCIIhi/loF4/80+VCAM1+) in the presence of specific signals released by tumor cells within the local environment. However, the TAM phenotypic profile depends on cancer histology and stage, which might influence marker distribution. TAMs also proliferate locally, with different rates in various tumors. Furthermore, TAMs are inherently plastic, with an activation state falling along a continuum between the two extremes of M1- and M2-like phenotypes. Rb, retinoblastoma.
Macrophage composition in different tissues or inflammatory environments depends on a dynamic equilibrium between recruited and tissue-resident macrophages. Tissue-resident macrophages originate at the prenatal stage from the yolk sac and fetal liver (13–15) and acquire selective, tissue-dependent features through the activation of distinctive transcriptional profiles (16–20). During inflammation and under steady-state conditions in some tissues, macrophages are derived from circulating Ly6C+CCR2+ monocytes, as in the case of colonic mucosal macrophages (21).
In cancer, the evidence to date indicates that TAMs are dynamically replaced by circulating precursors. Both the tissue-resident macrophages present in normal mammary tissues and TAMs that develop during tumor progression in the MMTV-PyMT breast cancer model are derived from blood-circulating CCR2+ monocytes, but only TAMs display self-renewal capability (22). In fact, TAM differentiation relies on the NOTCH/recombination signal–binding protein for the Ig κ J region (RBPJ) signaling pathway and is cell restricted, as genetic ablation of RbpJ caused a reduction in both TAMs and tumor growth (22). In the MMTV-neu mouse model of autochthonous mammary carcinogenesis, in situ cell division of fully differentiated CD11bloF4/80hi macrophages was the main contributor to the rapid TAM expansion; however, circulating monocyte influx was required in the long term (23). TAM progenitors (Ly6C+ monocytes) can also arise from tumor-induced extramedullary hematopoiesis within the spleen (24), although the relative contribution of BM and spleen to the monocyte reservoir and tumor trafficking is not clear and might be tumor dependent (25).
MDSCs in tumor-bearing hosts: cellular heterogeneity.
Normal CD11b+Gr1+ cells in BM are multipotent cells that can differentiate, depending on the kind and/or extent of cytokine/chemokine stimulation, into cells able to either enhance (e.g., myeloid DCs) or restrain (MDSCs) the immune response (26, 27). However, even in tumor-bearing hosts, BM CD11b+Gr1+ cells are poorly suppressive, while the same cells isolated from liver, spleen, blood, and tumors are fully competent to inhibit T cell activation (28, 29). These findings suggest that the BM niche is not permissive for a complete, functional maturation of MDSCs.
As further detailed by Marvel and Gabrilovich (30), mouse MDSCs have been divided into two main subgroups with different phenotypic and biological properties: the monocytic (MO-MDSC) and polymorphonuclear/granulocytic (PMN-MDSC) subsets (31). In tumor-bearing mice, MO-MDSCs (Gr1lo/intCD11b+Ly6ChiLy6G−) are highly immunosuppressive and exert their effects largely in an antigen-nonspecific manner, whereas PMN-MDSCs (Gr1hiCD11b+Ly6CloLy6G+) are moderately immunosuppressive and promote T cell tolerance via antigen-specific mechanisms (32, 33). TDFs induce tumor-infiltrating MO-MDSC differentiation into immunosuppressive TAMs. This conversion is primarily mediated by CSF1 (34), but also by molecular pathways controlled by the hypoxia-inducible factor 1α (HIF-1α) (35). HIF-1α may also be stabilized by the lactic acid that is produced by aerobic glycolysis (Warburg effect) in cancer cells (36). Alternatively, lactic acid can be actively produced in immune-regulatory myeloid cells by cytokine-activated, anerobic glycolysis (28, 37).
TAMs in tumor-bearing hosts: cellular plasticity.
After arriving at the tumor site, Ly6C+CD11c–MHCII–CD11bhiVCAM– monocytes undergo sequential phenotypical changes characterized by the downregulation of Ly6C and CD11b and the upregulation of MHC class II (MHCII) molecules, VCAM, and CD11c (22). However, TAM differentiation and distribution is not a defined and preserved track but depends on both anatomical location and the tumor stage: cancers with different histology are infiltrated by TAMs with phenotypic and functionally distinct features (38). It is essential to avoid simplified conclusions regarding TAM ontogenetic analysis; for instance, the adoptive transfer of fully differentiated macrophages to alternate tissues demonstrated that the local environment is sufficient to reprogram both the macrophage chromatin landscape and gene expression, similar to what happens to less mature, BM-derived myeloid precursors (20).
The definition of TAM function that is based on a rigid dichotomy in which inducible NOS–positive (iNOS, also known as NOS2) macrophages (M1) are antitumoral and ARG1-positive macrophages (M2) are protumoral is no longer satisfactory and was recently revised (39). The M1 and M2 designations should only describe macrophages activated by either IFN-γ and LPS or IL-4 and IL-13, respectively, and M1 and M2 should be viewed as the extremes of a continuum that emphasize the extremes of macrophage plasticity. M1 and M2 extremes exhibit specific, characteristic expression of metabolic enzymes (iNOS vs. ARG1), cytokines (IL-12hiIL-10lo vs. IL-12loIL-10hi), chemokines (CXCL9 and CXCL10 vs. CCL17 and CCL22), and marker genes (Nos2, IL12b, and Ciita vs. Arg1, Retnla, and Chi3l3), as well as transcription factors (NF-κB, STAT1, and IRF5 vs. STAT6, MYC, IRF4, KLF4, and PPARγ) (39). M1 macrophages are functionally proinflammatory and cytotoxic, whereas M2 macrophages act preferentially in antiinflammatory responses and tissue repair; however, when applied to TAMs, this classification is excessively simplistic and can generate misunderstandings and serious errors in data interpretation. For instance, mammary carcinoma–derived TAMs exhibit M2-related gene expression that is IL-4 independent and primarily orchestrated by NOTCH signaling (22) or lactic acid–stabilized HIF-1α (36). M1-like TAMs are detectable in early-stage cancers as well as in regressing cancers and necrotic areas of growing tumors (40). Furthermore, monocytes isolated from the blood of patients with renal cell carcinoma (RCC) simultaneously express both tumor-suppressing genes, such as TNF and IL1A, and tumor-promoting genes, such as VEGFA, MMP9, and HIF1A, a mixed profile that was confirmed in macrophages of RCC specimens (41). Thus, TAM classification will require the integration of a multiparameter analysis of cell surface markers, exclusion of ambiguous identifications, and comparison of the TAM transcriptome with the gene profile of resident macrophages isolated from the same tissues (39).
Factors driving TAM and MDSC recruitment, expansion, and activation during tumor growth
In tumor-bearing hosts, MDSC and TAM generation requires the integration of at least two types of signals: factors that expand myeloid precursors, followed by factors that activate immune-regulatory programs. Myeloid cells are activated and localize to specific tumor areas with different kinetics during primary tumor formation. CSF1, granulocyte-CSF (G-CSF), and granulocyte-macrophage CSF (GM-CSF) are the three chief regulators of myeloid lineage proliferation and differentiation. G-CSF promotes the differentiation of myeloid precursors into PMN-MDSCs. Expansion of Ly6G+ PMN-MDSCs occurs very early during tumorigenesis in the MMTV-PyMT mouse model, and these cells are detectable in the blood, spleen, and lungs of mice at the onset of oncogene-driven malignant conversion (42). In this model, tumor-released G-CSF stimulated reactive granulopoiesis at the expense of erythropoiesis by expanding hematopoietic stem cells and granulocyte/macrophage progenitors, but not common myeloid progenitors. This peculiar precursor signature in the BM is reproduced by either G-CSF or GM-CSF inoculation (31, 42) as well as by transplantable, GM-CSF–secreting tumors (31), suggesting a shared action of both cytokines on myeloid progenitors. G-CSF also mediates the lung infiltration of PMN-MDSCs, a step required for the formation of the premetastatic niche (43).
GM-CSF and IL-6 activate the immune-suppressive program in BM-derived progenitors by regulating the C/EBPβ transcription factor (28) and affect myeloid function during very early stages of pancreatic ductal adenocarcinoma (PDAC) progression. After initiation of the transforming program controlled by the active KRAS oncogene in mouse PDAC models, there are progressive waves of myelomonocytic cell recruitment, with CD11b+Gr1+ cells and TAMs being among the first to be accrued (44). Along with transformed epithelial cells, CD11b+Gr1+ cells contribute to the local release of IL-6 and IL-11, which activate protumoral STAT3 in cancer cells (45, 46). Moreover, KRAS-dependent release of GM-CSF primed CD11b+Gr1+ cells to suppress tumor-specific CD8+ T cells and promoted progression to invasive PDAC; only the blockade of either GM-CSF production or CD11b+Gr1+ cell activity restored antitumor immunity (47). Other unknown factors might promote systemic CD11b+Gr1+ cell expansion in tumors driven by the viral SV40 oncogene, but GM-CSF was nonetheless required for the full in vivo maturation of CD11b+Gr1+ cell–suppressive activity (48). Further highlighting the role of GM-CSF, mesenchymal breast cancer cells activate TAMs by the combined activity of GM-CSF and lactate; in turn, TAMs release CCL18, which supports epithelial-to-mesenchymal transition (EMT) and metastasis formation (49).
The master factor for TAM recruitment and programming in the tumor microenvironment is CSF1. Genetic deletion of CSF1 either slowed tumor initiation or decreased disease progression and distal metastatic spread, both of which were associated with TAM loss or reduction (50, 51). Indeed, elevated CSF1 levels correlated with marked macrophage infiltration in human metastatic breast cancer (52). In addition to CSF1 and CCL2, several other TDFs attract circulating monocytes to the tumor site. For instance, chemokines, such as CCL5, CXCL12, and CX3CL1 (53) as well as growth factors and noncanonical chemotactic peptides, such as VEGF, TGF-β, bFGF, and the antimicrobial peptide β-defensin 3, are involved in monocyte recruitment and macrophage differentiation (54).
IL-4 and IL-13 participate in both TAM and MDSC survival and the acquisition of an immune-suppressive phenotype. They bind different receptors sharing the IL-4Rα chain that is responsible for recruiting and phosphorylating STAT6, which induces the transcription of genes involved in the immune-suppressive program, including Arg1 (55). GM-CSF released by mouse and human gliomas upregulate IL-4Rα in MDSCs (56), which further fuel a positive loop for MDSC-mediated immune-suppressive activity by releasing IL-13 and IFN-γ, with the last cytokine maintaining IL-4Rα surface expression (57). Accordingly, IL-4R genetic depletion impaired MDSC-dependent immune suppression in vivo (57), and administration of aptamers targeting IL-4Rα triggered MDSC and TAM apoptosis and delayed tumor progression (58). Additionally, IL-4 in the tumor microenvironment (secreted by tumor cells or Th2-polarized infiltration T cells) (59, 60) induces local macrophages to produce WNT7β, thereby promoting tumor invasion (61).
Metabolic environmental signals can also modulate the intratumoral distribution of myeloid cells. Macrophages can survive in a hypoxic environment, but the high lactate levels produced via the Warburg effect can influence their spatial dissemination within specific areas of tumors as well as their dismissal (62). Hypoxia induces semaphorin 3A (SEMA3A), which interacts with a holoreceptor composed of neuropilin 1 (NRP1) and plexin A1/A4 to trigger VEGFR1 phosphorylation and macrophage recruitment (63). A TAM retention signal within hypoxic areas is delivered by SEMA3A through plexin A1/A4; conversely, NRP1 is downregulated in cancer, and its genetic inactivation in macrophages enhances TAM trapping within normoxic areas, resulting in the ablation of their immunosuppressive and proangiogenic activity (63). Partial correction of tumor hypoxia did not affect the relative distribution of TAM subsets or overall M2 marker expression, but rather downregulated the hypoxia-sensitive genes and proangiogenic activity of TAMs residing in the hypoxic areas (64).
Myeloid cells and cancer promotion
MDSC and TAM activity is not simply a buildup of an immune-suppressive environment that keeps T cells at bay and protects tumors from the effector arm of the immune system, but includes mechanisms that sustain and promote tumor growth and metastasis (Figure 3), as detailed below.
Figure 3. TAM- and MDSC-dependent mechanisms driving tumor progression.

TAMs and MDSCs sustain tumor growth, progression, and dissemination by promoting immune dysfunction (green slices) but also by nonimmune-related mechanisms (yellow slices). (A) TAMs alter immune responses in tumor-bearing hosts by four main mechanisms: 1) inhibition of T cell activation; 2) inhibition of T cell viability; 3) promotion of Treg induction and recruitment; and 4) consumption of metabolites essential for T cell fitness. TAMs promote tumor angiogenesis and vasculogenesis by the release of VEGF and WNT7β, which favor the generation of new blood vessels and sustain metastasis. Finally, TAMs maintain the cancer cell reservoir by secreting IL-6 and TNF-α and produce MFG-E8 to protect CSCs from chemotherapy. (B) MDSCs inhibit the immune response in tumor-bearing mice by four processes: 1) MDSCs drive the differentiation of immune cells toward regulatory cells; 2) MDSCs interfere with T cell migration and viability; 3) MDSCs alter T cell fitness by turning on intracellular ARG1, NOS2, and NOX2 expression to produce NO, ROS, and RNS (ONOO–, O2–, H2O2); and 4) MDSCs deplete essential metabolites for T lymphocyte fitness. MDSCs can also promote tumor angiogenesis and vasculogenesis via VEGF and MMP9 secretion. MDSCs produce elevated levels of TGF-β and HGF in primary tumors, inducing EMT, and secrete versican in the metastatic niche, promoting MET. Finally, MDSCs maintain tumor cell stemness by both IL-1RA production and by inducing the upregulation of miR-101 in cancer stem cells. cGMP, cyclic GMP; βcat, β-catenin; N, nitrosylated/nitrated; Tcf, HNF1 homeobox A.
MDSC- and TAM-induced immune dysfunction.
TAMs and MDSCs exert their immunosuppressive effects in an antigen-specific and -nonspecific manner, deploying strategies that can be either direct or indirect, with the latter involving the generation or expansion of other regulatory cell populations, such as CD4+CD25+ Tregs (65).
Indirect strategies of immune suppression.
The mechanisms for Treg expansion and conversion are not completely understood but involve cell-to-cell contact (including CD40 and CD40L interactions) and the production of soluble factors such as TGF-β, IFN-γ, and IL-10 (66–68). To sustain the immune-suppressive environment, TAMs and MDSCs secrete an array of chemokines acting on CCR5 and CCR6, which are involved in Treg recruitment (67–69). MDSCs also skew macrophages toward an M2 phenotype, characterized by impaired production of functional IL-12, through a cell contact–dependent mechanism (70). The downregulation of IL-12 is further exacerbated by the macrophages themselves, because TAMs stimulate an additional IL-10 release by MDSCs, thereby creating a self-perpetuating negative loop. Therefore, both MDSCs and TAMs can regulate the intratumoral IL-10/IL-12 balance, which is critical for priming T lymphocyte responses, as reviewed elsewhere (54, 71–73). Interestingly, IL-10 receptor blockade enhanced tumor responses to paclitaxel and carboplatin, enabling CD103+ DCs to produce IL-12 and support antitumor CD8+ T cells (74).
Direct immune suppression strategies.
Direct immune-suppressive mechanisms rely on the activity of enzymes, chemokines, and receptors in myeloid cells. L-arginine and L-tryptophan consumption — which is dependent on the activity of ARG1 (73) and iNOS (75) or indoleamine 2,3-dioxygenase 1 (IDO1) and IDO2 (76), respectively — or L-cysteine deprivation (77) promotes T cell proliferation arrest and functional inhibition by downregulation of the CD3ζ chain in the T cell receptor (TCR) complex. The production of NO can inhibit T cell signaling downstream of IL-2R and induce T cell apoptosis by different mechanisms in an antigen-independent manner (78, 79). Another TAM/MDSC-related immune-suppressive mechanism is based on the production of ROS and reactive nitrogen species (RNS). ROS comprise superoxide anion (O2–) and hydrogen peroxide (H2O2) and are generated in high amounts by the activity of NADPH oxidase (NOX) family members, in which NOX2 is the key player (80). ROS affect T cell fitness by downregulating CD3ζ chain expression and reducing cytokine secretion, as observed in pancreatic cancer (81). RNS, such as peroxynitrite (ONOO–), are byproducts of the combined activity of iNOS, ARG1, and NOX2 and can alter the formation of a correct peptide-MHC complex in MHCI molecules or induce modification of the immunodominant tumor-antigen peptides, thereby affecting TCR recognition and T cell activation (82). RNS can act on α and β chains of the TCR, promoting dissociation of the CD3ζ chain from the TCR complex and preventing TCR signaling (83). Last, RNS also modify trafficking of leukocytes that promote homing of immune-suppressive subsets (but not T cells) through aromatic amino acid nitration and nitrosylation of chemokines (CCL2, CCL5, CCL21, CXCL12) or chemokine receptors (CXCR4) (84, 85). Myeloid cells also promote immune dysfunction by expressing membrane surface ligands of T cell–inhibitory receptors, such as programmed death ligand 1/2 (PD-L1/2), which bind programmed death 1 (PD-1) (86–88) and B7-1/2, which bind to cytotoxic T lymphocyte antigen 4 (CTLA4) (89) and CD28 as well as FASL (90). Moreover, TAMs express nonclassical HLA-G and HLA-E molecules that can inhibit T cell activation upon their ligation to the inhibitory leukocyte Ig–like receptor LIT-2 (91).
MDSC- and TAM-dependent protumoral aid
Cancer stemness.
MDSCs finely tune tumor senescence by promoting cellular stemness. At tumor onset in different autochthonous tumor models, neoplastic cells showed a senescent phenotype, a condition limiting tumor progression that was reversed by MDSCs (92). MDSC-secreted IL-1RA was the main molecular mediator of this reprogramming activity, and interference with MDSC trafficking to the tumor (i.e., by CXCL1/2 and CXCR2 targeting) enhanced chemotherapy-induced cellular senescence (92). In human ovarian carcinoma, MDSCs regulated senescence by inducing tumor cell expression of miR-101, which downregulated the stemness repressor C-terminal–binding protein 2 (CTBP2), ultimately triggering cancer stem cell (CSC) sphere formation and enhancing metastatic potential (93). Finally, in a mouse model of pancreatic cancer, MO-MDSCs directly induced expansion of aldehyde dehydrogenase 1+ (ALDH1A1+) pancreatic CSCs; a similar effect was observed with human CD14+HLA-DR− MDSCs from patients with PDAC (94).
In pancreatic tumors, TAM depletion arrests the proliferation of tumor-initiating cells (95). Indeed, TAMs can sustain CSC proliferation by releasing proinflammatory cytokines such as TNF-α and IL-6, which reinforce tumor cell proliferation through NF-κB and STAT3 signaling pathways (96, 97). These same molecular pathways may be activated through a direct TAM-to-CSC contact via CD90 and ephrin A4 receptors (98). Finally, the crosstalk between CSCs and TAMs induced TAM secretion of milk fat globule EGF factor 8 (MFGE8) and IL-6, which favored CSC reservoir survival during chemotherapeutic treatment (99).
Angiogenesis.
MDSCs and TAMs play a crucial role in promoting the angiogenic switch. During hypoxia adaptation, tumor cells, which sense O2 levels through HIF prolyl hydroxylase 1–3 (PHD1–3) to control HIF-1α stability, release VEGF and thereby stimulate angiogenesis (100). Similarly, TAMs, in response to hypoxia, release mediators such as VEGF, bFGF, CXCL8/IL-8, and glycolytic enzymes (101, 102). Secreted VEGF also orchestrates peripheral expansion, trafficking, and functional commitment of MDSCs (103). In the tumor microenvironment, TAMs and MDSCs release proteases (cathepsin and MMP9), which support angiogenesis by freeing heparin-bound growth factors, such as VEGF-A, and by inducing extracellular matrix remodeling, which promotes invasion (51). Recruitment of MDSCs mediates resistance to anti-VEGF Ab–mediated therapy, as MDSCs can support new vessel growth, even in the presence of anti-VEGF Ab (104), by releasing the proangiogenic bombina variegata peptide 8 (105).
EMT–mesenchymal-to-epithelial transition and metastatic spreading.
Myeloid cells play an active role in promoting the spread of distal tumor cells. In mammary tumors, TAMs promote metastatic diffusion via a paracrine loop involving CSF1 and EGF, which induces macrophages and tumor cells to cluster around blood vessels, where macrophages create a gate for tumor cell intravasation into the circulation, thus producing a tumor microenvironment for metastasis (TMEM) (106–108). The proinflammatory proteins S100A8 and S100A9, potent MDSC chemoattractants, have been implicated in tumor progression (109); S100A8/A9-induced serum amyloid A3 directly recruited MDSCs to premetastatic lungs, stimulated NF-κB signaling in a TLR4-dependent manner, and facilitated metastatic spreading (110). Moreover, MO-MDSCs and inflammatory monocytes are recruited through the CCL2/CCR2 axis to a metastatic environment in which they can differentiate into metastasis-associated macrophages (MAMs) (52, 111). Hypoxia in primary tumors can trigger MDSC-induced dysfunction in NK cells within the lung premetastatic niche, a defined site to which hematopoietic cells migrate before the tumor cells can seed the niche (112). PMN-MDSCs can also be armed by IL-17 released from γδ T cells infiltrating the primary breast cancers and assist lymph node and lung metastasis, in part through the inhibition of CD8+ T cell function (113). MDSCs and TAMs also assist the metastatic process by inducing tumor cell EMT. MDSCs attracted by CXCL5 induced EMT in melanoma cells by releasing HGF and TGF-β at the primary tumor site; targeting of PMN-MDSCs in this model resulted in marked impairment of primary tumor growth (114). TAM recruitment induces EMT by both TGF-β release in a variety of solid tumors (115) and IL-8 in hepatocellular carcinoma (116). Additionally, a positive correlation was found between intratumoral macrophage densities, EMT markers, intraepithelial TGF-β levels, and tumor grade of non–small-cell lung cancer (NSCLC) patient samples (115). Because metastatic cells reacquire morphological and phenotypic traits of epithelial cells at the metastatic site, it is conceivable that premetastatic myeloid cells also control a mesenchymal-to-epithelial transition (MET) that promotes cancer cell colonization of and survival in the new organ, likely by releasing the proteoglycan versican (117).
Prognostic significance of myeloid cells in cancer patients
Three main myeloid classes with distinct lineage commitments have been identified in the blood of cancer patients: monocytic, granulocytic, and immature MDSCs. Each class contains more than one subset (118). Although the role of MDSCs has been acknowledged in primary tumor formation (119), extensive data connect MDSC expansion to more advanced cancer stages (120). MDSC numbers are associated with clinical stage in bladder carcinoma (121), pancreatic adenocarcinoma (122), hepatocellular carcinoma (123, 124), gastric cancer (125), NSCLC (126), and head and neck squamous cell carcinoma (127), as well as in hematological malignancies such as non-Hodgkin lymphoma (128). Collectively, these results indicate that expansion of MDSCs in cancer patients is a general phenomenon accompanying tumor progression. MDSC levels also correlated with response to therapy (126, 129, 130) or surgery (121); however, a deep analysis of clinical outcome in patients showed that MDSC frequency in blood is associated with prognosis, independent of tumor burden (131, 132). In patients with either stage IV breast cancer or stage IV colorectal cancer (CRC), a significant correlation was observed between high numbers of circulating MDSCs and poor prognosis. In fact, survival estimates for patients with high numbers of immature MDSCs (lineage–HLA-DRlo/–CD11b+CD33+) in the blood prior to starting standard chemotherapy were associated with shorter overall survival (OS) (133). Finally, high levels of MDSCs, cytokines, and chemokines (PDGF, IL-4, IL-8, IL-17, FGF-2, CCL5, and VEGF) in patients with PDAC are associated with progressive disease (134).
In recent years, immunotherapy has emerged as a therapeutic option for the treatment of cancer. IMA901 is a therapeutic vaccine for RCC that consists of HLA-A*02–restricted, tumor-derived peptides. In patients with advanced RCC, the levels of five of six MDSC subsets were expanded at baseline, and two of these subsets were prognostic for OS following IMA901 administration. These results indicate that MDSCs are potential biomarkers of response to the vaccine (135).
Immune checkpoint inhibitors represent a new drug category that is dramatically changing the treatment options for cancer (136). Lower MDSC frequencies correlated with prolonged OS in ipilimumab-treated patients (132, 137), whereas a decrease in MDSCs after treatment correlated with improved progression-free survival (PFS) in advanced melanoma patients receiving neoadjuvant ipilimumab (138). To date, it is not clear whether ipilimumab targets MDSCs or, conversely, whether the lower MDSC levels observed following ipilimumab treatment simply reflect tumor shrinkage in response to immune-mediated rejection.
While some studies demonstrated a correlation of extensive TAM infiltration with poor prognosis in breast, cervix, and bladder carcinomas, conflicting results were obtained in other solid tumors like prostate, NSCLC, and brain cancers (139). Along the same line, a recent meta-analysis of the literature showed inconsistent results (6), since elevated TAM numbers were associated with worse OS in patients with gastric, urogenital, or head and neck cancers, but with better prognosis in patients with CRC.
It appears that, while the expansion of MDSCs is often associated with poor prognosis, expansion of TAMs is not always a negative prognostic factor. When TAM evaluation is carried out at the molecular level, another layer of complexity appears. As discussed above, monocytes from patients with RCC have a distinct transcriptional profile, with upregulation of protumor and antitumor genes. The tumor-promoting function of RCC monocytes and TAMs required IL-1/IL-1R signaling, which also supported progression of RCC xenografts (41). These results are the first indication in human cancers that TAM induction is not mediated by the tumor microenvironment and suggest that patients’ monocytes are already primed in the blood. Finally, CSF1R inhibition in a mouse model of proneural glioblastoma (GBM) increased survival by inducing regression of established tumors. Interestingly, a gene signature induced by CSF1R inhibition in murine TAMs was associated with increased survival in patients with proneural GBM (140).
Conclusions and future perspective
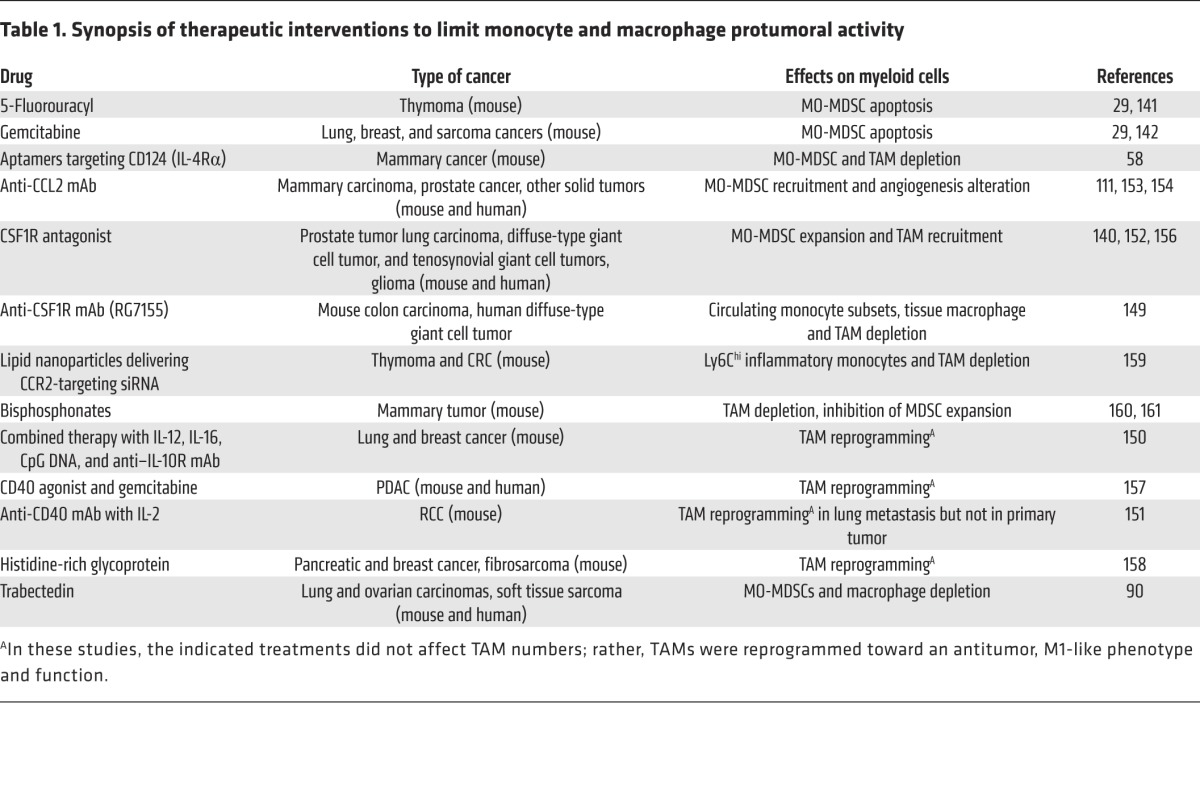
Targeting MO-MDSCs and TAMs can open new therapeutic opportunities to control tumor progression and block metastatic diffusion. The main strategies used thus far involve the inhibition of recruitment, depletion, or reprogramming of target cell populations. Some first-generation chemotherapeutic agents, such as 5-fluorouracil (141) and gemcitabine (29, 142), are able to control MO-MDSC accumulation, probably because these cells are more sensitive than tumor cells to low-dose chemotherapy (29). Low-dose irradiation also increases CD8+ T cell trafficking and normalizes tumor vasculature in many cancer models by reprogramming TAMs toward a more inflammatory M1 type that releases NO (143). However, TAMs can either positively or negatively influence the antitumor activity of cytotoxic chemotherapy and radiotherapy (144), and targeting of immunosuppressive myeloid cells can have different effects on cancer progression (145, 146). Additionally, the microbiome can condition different myeloid cells, including TAMs, within murine tumors to contribute to the antitumor efficacy of both chemotherapy and immunotherapy (147, 148). Novel biologic drugs recognizing MDSC and TAM antigens or disrupting their function have been developed for selective targeting of these cell populations. As shown in Table 1, these compounds include Abs and/or aptamers (58, 111, 149–151) as well as molecular antagonists of essential receptors and/or molecular pathways (152). Among chemokines, targeting of CCL2 with a mAb (carlumab, CNTO 888) has proven to be beneficial in patients (153, 154); however, abrupt discontinuation of the therapy may result in a rebound effect causing increased metastatic disease (155). The inhibition of the CSF1/CSF1R axis with Abs (RG7155) or RTK inhibitors (imatinib mesylate) affects macrophage recruitment and differentiation and has shown encouraging results in clinical trials (149, 156). Considering the role of macrophages in regulating the tissue architecture and in mediating innate immune defense, there are concerns about side effects from the extended depletion of these cells. In this context, Abs activating immune stimulators (CD40), combinations of cytokines and Abs, or administration of histidine-rich glycoprotein appeared to modify macrophage polarization toward an antitumor phenotype, without affecting overall macrophage levels (150, 151, 157, 158).
Table 1. Synopsis of therapeutic interventions to limit monocyte and macrophage protumoral activity.
Future investigations will need to focus on the mechanisms driving macrophage polarization toward either proimmune or protumoral phenotypes. Gene expression, proteomic, and metabolomic profiles are increasing our understanding of TAM and MDSC biology and offer potential therapeutic strategies for impeding tumor-induced immune dysfunctions. The identification of functional markers could guide the development of a new class of drugs targeting specific subsets of macrophages and MDSCs, thereby reducing the side effects of ablative therapy. In conclusion, while MDSC/TAM targeting will likely be insufficient to eradicate tumors, interference with patients’ immune dysfunctions is a prerequisite and fundamental step for improving the efficacy of passive and active immunotherapeutic protocols.
Acknowledgments
This work was supported by grants from the Italian Ministry of Health; the Italian Ministry of Education, Universities and Research (FIRB cup: B31J11000420001); the Italian Association for Cancer Research (AIRC) (6599, 12182, 14103, and 12886); and the Fondazione Cassa di Risparmio di Verona, Vicenza, Belluno e Ancona.
Footnotes
Conflict of interest: The authors have declared the no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(9):3365–3376. doi:10.1172/JCI80006.
References
- 1.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 2.Mangerich A, Dedon PC, Fox JG, Tannenbaum SR, Wogan GN. Chemistry meets biology in colitis-associated carcinogenesis. Free Radic Res. 2013;47(11):958–986. doi: 10.3109/10715762.2013.832239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer. 2013;13(11):800–812. doi: 10.1038/nrc3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balkwill FR, Mantovani A. Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol. 2012;22(1):33–40. doi: 10.1016/j.semcancer.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 5.Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41(1):49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang QW, et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS One. 2012;7(12):e50946. doi: 10.1371/journal.pone.0050946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Montero AJ, Diaz-Montero CM, Kyriakopoulos CE, Bronte V, Mandruzzato S. Myeloid-derived suppressor cells in cancer patients: a clinical perspective. J Immunother. 2012;35(2):107–115. doi: 10.1097/CJI.0b013e318242169f. [DOI] [PubMed] [Google Scholar]
- 8.Yu J, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. 2013;190(7):3783–3797. doi: 10.4049/jimmunol.1201449. [DOI] [PubMed] [Google Scholar]
- 9.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19(1):71–82. doi: 10.1016/S1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 10.Lesokhin AM, et al. Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 2012;72(4):876–886. doi: 10.1158/0008-5472.CAN-11-1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 12.Manz MG, Boettcher S. Emergency granulopoiesis. Nat Rev Immunol. 2014;14(5):302–314. doi: 10.1038/nri3660. [DOI] [PubMed] [Google Scholar]
- 13.Gomez Perdiguero E, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518(7540):547–551. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hashimoto D, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38(4):792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoeffel G, et al. C-myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity. 2015;42(4):665–678. doi: 10.1016/j.immuni.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okabe Y, Medzhitov R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell. 2014;157(4):832–844. doi: 10.1016/j.cell.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kohyama M, et al. Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature. 2009;457(7227):318–321. doi: 10.1038/nature07472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8(8):618–631. doi: 10.1038/nrc2444. [DOI] [PubMed] [Google Scholar]
- 19.Gosselin D, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell. 2014;159(6):1327–1340. doi: 10.1016/j.cell.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lavin Y, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159(6):1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bain CC, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol. 2014;15(10):929–937. doi: 10.1038/ni.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franklin RA, et al. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344(6186):921–925. doi: 10.1126/science.1252510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galon J, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 24.Cortez-Retamozo V, et al. Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci U S A. 2012;109(7):2491–2496. doi: 10.1073/pnas.1113744109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shand FH, et al. Tracking of intertissue migration reveals the origins of tumor-infiltrating monocytes. Proc Natl Acad Sci U S A. 2014;111(21):7771–7776. doi: 10.1073/pnas.1402914111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rossner S, Voigtlander C, Wiethe C, Hanig J, Seifarth C, Lutz MB. Myeloid dendritic cell precursors generated from bone marrow suppress T cell responses via cell contact and nitric oxide production in vitro. Eur J Immunol. 2005;35(12):3533–3544. doi: 10.1002/eji.200526172. [DOI] [PubMed] [Google Scholar]
- 27.Kusmartsev S, Gabrilovich DI. Effect of tumor-derived cytokines and growth factors on differentiation and immune suppressive features of myeloid cells in cancer. Cancer Metastasis Rev. 2006;25(3):323–331. doi: 10.1007/s10555-006-9002-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marigo I, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPβ transcription factor. Immunity. 2010;32(6):790–802. doi: 10.1016/j.immuni.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 29.Ugel S, et al. Immune tolerance to tumor antigens occurs in a specialized environment of the spleen. Cell Rep. 2012;2(3):628–639. doi: 10.1016/j.celrep.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 30.Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. 2015;125(9):3356–3364. doi: 10.1172/JCI80005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dolcetti L, et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur J Immunol. 2010;40(1):22–35. doi: 10.1002/eji.200939903. [DOI] [PubMed] [Google Scholar]
- 32.Movahedi K, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111(8):4233–4244. doi: 10.1182/blood-2007-07-099226. [DOI] [PubMed] [Google Scholar]
- 33.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181(8):5791–5802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corzo CA, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207(11):2439–2453. doi: 10.1084/jem.20100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Colegio OR, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513(7519):559–563. doi: 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hammami I, Chen J, Murschel F, Bronte V, De Crescenzo G, Jolicoeur M. Immunosuppressive activity enhances central carbon metabolism and bioenergetics in myeloid-derived suppressor cells in vitro models. BMC Cell Biol. 2012;13:18. doi: 10.1186/1471-2121-13-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Movahedi K, et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010;70(14):5728–5739. doi: 10.1158/0008-5472.CAN-09-4672. [DOI] [PubMed] [Google Scholar]
- 39.Murray PJ, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cavnar MJ, et al. KIT oncogene inhibition drives intratumoral macrophage M2 polarization. J Exp Med. 2013;210(13):2873–2886. doi: 10.1084/jem.20130875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chittezhath M, et al. Molecular profiling reveals a tumor-promoting phenotype of monocytes and macrophages in human cancer progression. Immunity. 2014;41(5):815–829. doi: 10.1016/j.immuni.2014.09.014. [DOI] [PubMed] [Google Scholar]
- 42.Casbon AJ, et al. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc Natl Acad Sci U S A. 2015;112(6):E566–E575. doi: 10.1073/pnas.1424927112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kowanetz M, et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc Natl Acad Sci U S A. 2010;107(50):21248–21255. doi: 10.1073/pnas.1015855107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67(19):9518–9527. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- 45.Damuzzo V, et al. Complexity and challenges in defining myeloid-derived suppressor cells. Cytometry B Clin Cytom. 2015;88(2):77–91. doi: 10.1002/cyto.b.21206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khaled YS, Ammori BJ, Elkord E. Increased levels of granulocytic myeloid-derived suppressor cells in peripheral blood and tumour tissue of pancreatic cancer patients. J Immunol Res. 2014;2014:879897. doi: 10.1155/2014/879897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bayne LJ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21(6):822–835. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmidt K, Zilio S, Schmollinger JC, Bronte V, Blankenstein T, Willimsky G. Differently immunogenic cancers in mice induce immature myeloid cells that suppress CTL in vitro but not in vivo following transfer. Blood. 2013;121(10):1740–1748. doi: 10.1182/blood-2012-06-436568. [DOI] [PubMed] [Google Scholar]
- 49.Su S, et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell. 2014;25(5):605–620. doi: 10.1016/j.ccr.2014.03.021. [DOI] [PubMed] [Google Scholar]
- 50.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193(6):727–740. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4(7):540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- 54.Allavena P, Mantovani A. Immunology in the clinic review series; focus on cancer: tumour-associated macrophages: undisputed stars of the inflammatory tumour microenvironment. Clin Exp Immunol. 2012;167(2):195–205. doi: 10.1111/j.1365-2249.2011.04515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gray MJ, Poljakovic M, Kepka-Lenhart D, Morris SM, Morris SM., Jr Induction of arginase I transcription by IL-4 requires a composite DNA response element for STAT6 and C/EBPβ. Gene. 2005;353(1):98–106. doi: 10.1016/j.gene.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 56.Kohanbash G, et al. GM-CSF promotes the immunosuppressive activity of glioma-infiltrating myeloid cells through interleukin-4 receptor-α. Cancer Res. 2013;73(21):6413–6423. doi: 10.1158/0008-5472.CAN-12-4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gallina G, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest. 2006;116(10):2777–2790. doi: 10.1172/JCI28828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roth F, De La Fuente AC, Vella JL, Zoso A, Inverardi L, Serafini P. Aptamer-mediated blockade of IL4Rα triggers apoptosis of MDSCs and limits tumor progression. Cancer Res. 2012;72(6):1373–1383. doi: 10.1158/0008-5472.CAN-11-2772. [DOI] [PubMed] [Google Scholar]
- 59.Gocheva V, et al. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010;24(3):241–255. doi: 10.1101/gad.1874010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DeNardo DG, et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009;16(2):91–102. doi: 10.1016/j.ccr.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 62.Srivastava MK, Bosch JJ, Thompson JA, Ksander BR, Edelman MJ, Ostrand-Rosenberg S. Lung cancer patients’ CD4(+) T cells are activated in vitro by MHC II cell-based vaccines despite the presence of myeloid-derived suppressor cells. Cancer Immunol Immunother. 2008;57(10):1493–1504. doi: 10.1007/s00262-008-0490-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mandruzzato S, et al. IL4Rα+ myeloid-derived suppressor cell expansion in cancer patients. J Immunol. 2009;182(10):6562–6568. doi: 10.4049/jimmunol.0803831. [DOI] [PubMed] [Google Scholar]
- 64.Bennett JA, Rao VS, Mitchell MS. Systemic bacillus Calmette-Guerin (BCG) activates natural suppressor cells. Proc Natl Acad Sci U S A. 1978;75(10):5142–5144. doi: 10.1073/pnas.75.10.5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Savage ND, et al. Human anti-inflammatory macrophages induce Foxp3+ GITR+ CD25+ regulatory T cells, which suppress via membrane-bound TGFβ-1. J Immunol. 2008;181(3):2220–2226. doi: 10.4049/jimmunol.181.3.2220. [DOI] [PubMed] [Google Scholar]
- 66.Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 2008;68(13):5439–5449. doi: 10.1158/0008-5472.CAN-07-6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Curiel TJ, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 68.Liu J, et al. Tumor-associated macrophages recruit CCR6+ regulatory T cells and promote the development of colorectal cancer via enhancing CCL20 production in mice. PLoS One. 2011;6(4):e19495. doi: 10.1371/journal.pone.0019495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schlecker E, et al. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J Immunol. 2012;189(12):5602–5611. doi: 10.4049/jimmunol.1201018. [DOI] [PubMed] [Google Scholar]
- 70.Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. 2007;179(2):977–983. doi: 10.4049/jimmunol.179.2.977. [DOI] [PubMed] [Google Scholar]
- 71.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117(5):1155–1166. doi: 10.1172/JCI31422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol. 2012;22(4):275–281. doi: 10.1016/j.semcancer.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11(10):889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 74.Ruffell B, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. 2014;26(5):623–637. doi: 10.1016/j.ccell.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5(8):641–654. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- 76.Munn DH, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22(5):633–642. doi: 10.1016/j.immuni.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 77.Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010;70(1):68–77. doi: 10.1158/0008-5472.CAN-09-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mazzoni A, et al. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol. 2002;168(2):689–695. doi: 10.4049/jimmunol.168.2.689. [DOI] [PubMed] [Google Scholar]
- 79.Macphail SE, Gibney CA, Brooks BM, Booth CG, Flanagan BF, Coleman JW. Nitric oxide regulation of human peripheral blood mononuclear cells: critical time dependence and selectivity for cytokine versus chemokine expression. J Immunol. 2003;171(9):4809–4815. doi: 10.4049/jimmunol.171.9.4809. [DOI] [PubMed] [Google Scholar]
- 80.Corzo CA, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009;182(9):5693–5701. doi: 10.4049/jimmunol.0900092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schmielau J, Nalesnik MA, Finn OJ. Suppressed T-cell receptor zeta chain expression and cytokine production in pancreatic cancer patients. Clin Cancer Res. 2001;7(3):933s–939s. [PubMed] [Google Scholar]
- 82.Hardy LL, Wick DA, Webb JR. Conversion of tyrosine to the inflammation-associated analog 3′-nitrotyrosine at either TCR- or MHC-contact positions can profoundly affect recognition of the MHC class I-restricted epitope of lymphocytic choriomeningitis virus glycoprotein 33 by CD8 T cells. J Immunol. 2008;180(9):5956–5962. doi: 10.4049/jimmunol.180.9.5956. [DOI] [PubMed] [Google Scholar]
- 83.Nagaraj S, Schrum AG, Cho HI, Celis E, Gabrilovich DI. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol. 2010;184(6):3106–3116. doi: 10.4049/jimmunol.0902661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.De Sanctis F, et al. The emerging immunological role of post-translational modifications by reactive nitrogen species in cancer microenvironment. Front Immunol. 2014;5:69. doi: 10.3389/fimmu.2014.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Molon B, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. 2011;208(10):1949–1962. doi: 10.1084/jem.20101956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Duraiswamy J, Freeman GJ, Coukos G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res. 2013;73(23):6900–6912. doi: 10.1158/0008-5472.CAN-13-1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U S A. 2003;100(9):5336–5341. doi: 10.1073/pnas.0931259100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Noman MZ, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211(5):781–790. doi: 10.1084/jem.20131916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kennedy BC, et al. Tumor-associated macrophages in glioma: friend or foe? J Oncol. 2013;2013:486912. doi: 10.1155/2013/486912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Germano G, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23(2):249–262. doi: 10.1016/j.ccr.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 91.LeMaoult J, Krawice-Radanne I, Dausset J, Carosella ED. HLA-G1-expressing antigen-presenting cells induce immunosuppressive CD4+ T cells. Proc Natl Acad Sci U S A. 2004;101(18):7064–7069. doi: 10.1073/pnas.0401922101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Di Mitri D, et al. Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature. 2014;515(7525):134–137. doi: 10.1038/nature13638. [DOI] [PubMed] [Google Scholar]
- 93.Cui TX, et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity. 2013;39(3):611–621. doi: 10.1016/j.immuni.2013.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Panni RZ, et al. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol Immunother. 2014;63(5):513–528. doi: 10.1007/s00262-014-1527-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mitchem JB, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73(3):1128–1141. doi: 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schwitalla S, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell. 2013;152(1):25–38. doi: 10.1016/j.cell.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 97.Wan S, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. 2014;147(6):1393–1404. doi: 10.1053/j.gastro.2014.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lu H, et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat Cell Biol. 2014;16(11):1105–1117. doi: 10.1038/ncb3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jinushi M, et al. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Natl Acad Sci U S A. 2011;108(30):12425–12430. doi: 10.1073/pnas.1106645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mukhopadhyay D, Tsiokas L, Zhou XM, Foster D, Brugge JS, Sukhatme VP. Hypoxic induction of human vascular endothelial growth factor expression through c-Src activation. Nature. 1995;375(6532):577–581. doi: 10.1038/375577a0. [DOI] [PubMed] [Google Scholar]
- 101.Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood. 2004;104(8):2224–2234. doi: 10.1182/blood-2004-03-1109. [DOI] [PubMed] [Google Scholar]
- 102.Schmidt T, Carmeliet P. Blood-vessel formation: bridges that guide and unite. Nature. 2010;465(7299):697–699. doi: 10.1038/465697a. [DOI] [PubMed] [Google Scholar]
- 103.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shojaei F, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25(8):911–920. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- 105.Shojaei F, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature. 2007;450(7171):825–831. doi: 10.1038/nature06348. [DOI] [PubMed] [Google Scholar]
- 106.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124(2):263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 107.Wyckoff JB, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67(6):2649–2656. doi: 10.1158/0008-5472.CAN-06-1823. [DOI] [PubMed] [Google Scholar]
- 108.Kitamura T, Qian BZ, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol. 2015;15(2):73–86. doi: 10.1038/nri3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. 2008;181(7):4666–4675. doi: 10.4049/jimmunol.181.7.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hiratsuka S, et al. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol. 2008;10(11):1349–1355. doi: 10.1038/ncb1794. [DOI] [PubMed] [Google Scholar]
- 111.Qian BZ, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sceneay J, et al. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res. 2012;72(16):3906–3911. doi: 10.1158/0008-5472.CAN-11-3873. [DOI] [PubMed] [Google Scholar]
- 113.Coffelt SB, et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522(7556):345–348. doi: 10.1038/nature14282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Toh B, et al. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 2011;9(9):e1001162. doi: 10.1371/journal.pbio.1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bonde AK, Tischler V, Kumar S, Soltermann A, Schwendener RA. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer. 2012;12:35. doi: 10.1186/1471-2407-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fu XT, et al. Macrophage-secreted IL-8 induces epithelial-mesenchymal transition in hepatocellular carcinoma cells by activating the JAK2/STAT3/Snail pathway. Int J Oncol. 2015;46(2):587–596. doi: 10.3892/ijo.2014.2761. [DOI] [PubMed] [Google Scholar]
- 117.Gao D, et al. Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer Res. 2012;72(6):1384–1394. doi: 10.1158/0008-5472.CAN-11-2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Solito S, Marigo I, Pinton L, Damuzzo V, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann N Y Acad Sci. 2014;1319:47–65. doi: 10.1111/nyas.12469. [DOI] [PubMed] [Google Scholar]
- 119.Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. 2013;13(10):739–752. doi: 10.1038/nrc3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58(1):49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yuan XK, Zhao XK, Xia YC, Zhu X, Xiao P. Increased circulating immunosuppressive CD14(+)HLA-DR(–/low) cells correlate with clinical cancer stage and pathological grade in patients with bladder carcinoma. J Int Med Res. 2011;39(4):1381–1391. doi: 10.1177/147323001103900424. [DOI] [PubMed] [Google Scholar]
- 122.Porembka MR, et al. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol Immunother. 2012;61(9):1373–1385. doi: 10.1007/s00262-011-1178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shen P, Wang A, He M, Wang Q, Zheng S. Increased circulating Lin(–/low) CD33(+) HLA-DR(–) myeloid-derived suppressor cells in hepatocellular carcinoma patients. Hepatol Res. 2014;44(6):639–650. doi: 10.1111/hepr.12167. [DOI] [PubMed] [Google Scholar]
- 124.Arihara F, et al. Increase in CD14+HLA–DR–/low myeloid-derived suppressor cells in hepatocellular carcinoma patients and its impact on prognosis. Cancer Immunol Immunother. 2013;62(8):1421–1430. doi: 10.1007/s00262-013-1447-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang L, Chang EW, Wong SC, Ong SM, Chong DQ, Ling KL. Increased myeloid-derived suppressor cells in gastric cancer correlate with cancer stage and plasma S100A8/A9 proinflammatory proteins. J Immunol. 2013;190(2):794–804. doi: 10.4049/jimmunol.1202088. [DOI] [PubMed] [Google Scholar]
- 126.Huang A, Zhang B, Wang B, Zhang F, Fan KX, Guo YJ. Increased CD14(+)HLA-DR (–/low) myeloid-derived suppressor cells correlate with extrathoracic metastasis and poor response to chemotherapy in non-small cell lung cancer patients. Cancer Immunol Immunother. 2013;62(9):1439–1451. doi: 10.1007/s00262-013-1450-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Vasquez-Dunddel D, et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J Clin Invest. 2013;123(4):1580–1589. doi: 10.1172/JCI60083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lin Y, Gustafson MP, Bulur PA, Gastineau DA, Witzig TE, Dietz AB. Immunosuppressive CD14+HLA-DRlow/– monocytes in B-cell non-Hodgkin lymphoma. Blood. 2011;117(3):872–881. doi: 10.1182/blood-2010-05-283820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ko JS, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15(6):2148–2157. doi: 10.1158/1078-0432.CCR-08-1332. [DOI] [PubMed] [Google Scholar]
- 130.Liu CY, et al. Population alterations of L-arginase- and inducible nitric oxide synthase-expressed CD11b+/CD14–/CD15+/CD33+ myeloid-derived suppressor cells and CD8+ T lymphocytes in patients with advanced-stage non-small cell lung cancer. J Cancer Res Clin Oncol. 2010;136(1):35–45. doi: 10.1007/s00432-009-0634-0. [DOI] [PubMed] [Google Scholar]
- 131.Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. 2011;60(10):1419–1430. doi: 10.1007/s00262-011-1028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kitano S, et al. Computational algorithm-driven evaluation of monocytic myeloid-derived suppressor cell frequency for prediction of clinical outcomes. Cancer Immunol Res. 2014;2(8):812–821. doi: 10.1158/2326-6066.CIR-14-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Solito S, et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood. 2011;118(8):2254–2265. doi: 10.1182/blood-2010-12-325753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Markowitz J, et al. Patients with pancreatic adenocarcinoma exhibit elevated levels of myeloid-derived suppressor cells upon progression of disease. Cancer Immunol Immunother. 2015;64(2):149–159. doi: 10.1007/s00262-014-1618-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Walter S, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med. 2012;18(8):1254–1261. doi: 10.1038/nm.2883. [DOI] [PubMed] [Google Scholar]
- 136.Page DB, Postow MA, Callahan MK, Allison JP, Wolchok JD. Immune modulation in cancer with antibodies. Annu Rev Med. 2014;65:185–202. doi: 10.1146/annurev-med-092012-112807. [DOI] [PubMed] [Google Scholar]
- 137.Meyer C, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. 2014;63(3):247–257. doi: 10.1007/s00262-013-1508-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tarhini AA, et al. Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PLoS One. 2014;9(2):e87705. doi: 10.1371/journal.pone.0087705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196(3):254–265. doi: 10.1002/path.1027. [DOI] [PubMed] [Google Scholar]
- 140.Pyonteck SM, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Vincent J, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70(8):3052–3061. doi: 10.1158/0008-5472.CAN-09-3690. [DOI] [PubMed] [Google Scholar]
- 142.Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11(18):6713–6721. doi: 10.1158/1078-0432.CCR-05-0883. [DOI] [PubMed] [Google Scholar]
- 143.Klug F, et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. 2013;24(5):589–602. doi: 10.1016/j.ccr.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 144.Watson GA, Fu YX, Lopez DM. Splenic macrophages from tumor-bearing mice co-expressing MAC-1 and MAC-2 antigens exert immunoregulatory functions via two distinct mechanisms. J Leukoc Biol. 1991;49(2):126–138. doi: 10.1002/jlb.49.2.126. [DOI] [PubMed] [Google Scholar]
- 145.De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. 2013;23(3):277–286. doi: 10.1016/j.ccr.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 146.Dijkgraaf EM, et al. Chemotherapy alters monocyte differentiation to favor generation of cancer-supporting M2 macrophages in the tumor microenvironment. Cancer Res. 2013;73(8):2480–2492. doi: 10.1158/0008-5472.CAN-12-3542. [DOI] [PubMed] [Google Scholar]
- 147.Filipazzi P, et al. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J Clin Oncol. 2007;25(18):2546–2553. doi: 10.1200/JCO.2006.08.5829. [DOI] [PubMed] [Google Scholar]
- 148.Diaz-Montero CM, Finke J, Montero AJ. Myeloid-derived suppressor cells in cancer: therapeutic, predictive, and prognostic implications. Semin Oncol. 2014;41(2):174–184. doi: 10.1053/j.seminoncol.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ries CH, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25(6):846–859. doi: 10.1016/j.ccr.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 150.Guiducci C, Vicari AP, Sangaletti S, Trinchieri G, Colombo MP. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res. 2005;65(8):3437–3446. doi: 10.1158/0008-5472.CAN-04-4262. [DOI] [PubMed] [Google Scholar]
- 151.Weiss JM, et al. Macrophage-dependent nitric oxide expression regulates tumor cell detachment and metastasis after IL-2/anti-CD40 immunotherapy. J Exp Med. 2010;207(11):2455–2467. doi: 10.1084/jem.20100670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Priceman SJ, et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: combating tumor evasion of antiangiogenic therapy. Blood. 2010;115(7):1461–1471. doi: 10.1182/blood-2009-08-237412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Pienta KJ, et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest New Drugs. 2013;31(3):760–768. doi: 10.1007/s10637-012-9869-8. [DOI] [PubMed] [Google Scholar]
- 154.Sandhu SK, et al. A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer Chemother Pharmacol. 2013;71(4):1041–1050. doi: 10.1007/s00280-013-2099-8. [DOI] [PubMed] [Google Scholar]
- 155.Ortiz ML, et al. Immature myeloid cells directly contribute to skin tumor development by recruiting IL-17-producing CD4+ T cells. J Exp Med. 2015;212(3):351–367. doi: 10.1084/jem.20140835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cassier PA, et al. Efficacy of imatinib mesylate for the treatment of locally advanced and/or metastatic tenosynovial giant cell tumor/pigmented villonodular synovitis. Cancer. 2012;118(6):1649–1655. doi: 10.1002/cncr.26409. [DOI] [PubMed] [Google Scholar]
- 157.Beatty GL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Rolny C, et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell. 2011;19(1):31–44. doi: 10.1016/j.ccr.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 159.Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011;32(1):19–25. doi: 10.1016/j.it.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Brown HK, Holen I. Anti-tumour effects of bisphosphonates--what have we learned from in vivo models? Curr Cancer Drug Targets. 2009;9(7):807–823. doi: 10.2174/156800909789760339. [DOI] [PubMed] [Google Scholar]
- 161.Veltman JD, et al. Zoledronic acid impairs myeloid differentiation to tumour-associated macrophages in mesothelioma. Br J Cancer. 2010;103(5):629–641. doi: 10.1038/sj.bjc.6605814. [DOI] [PMC free article] [PubMed] [Google Scholar]