

Abstract
The last major advance in the treatment of glioblastoma multiforme (GBM) was the introduction of temozolomide in 1999. Treatment with temozolomide following surgical debulking extends survival rate compared to radiotherapy and debulking alone. However, virtually all glioblastoma patients experience disease progression within 7 to 10 months. Although many salvage treatments, including bevacizumab, rechallenge with temozolomide, and other alkylating agents, have been evaluated, none of these clearly improves survival. Monoamine oxidase B (MAOB) is highly expressed in glioblastoma cell mitochondria, and mitochondrial function is intimately tied to treatment-resistant glioblastoma progression. These glioblastoma properties provide a strong rationale for pursuing a MAOB-selective pro-drug treatment approach that, upon drug activation, targets glioblastoma mitochondria, especially mitochondrial DNA. MP-MUS is the lead compound in a family of pro-drugs designed to treat GBM that is converted into the mature, mitochondria-targeting drug, P+-MUS, by MAOB. We show that MP-MUS can successfully kill primary gliomas in vitro and in vivo mouse xenograft models.
Keywords: Glioblastoma multiforme, Monoamine oxidase B, Chemotherapy, Pro-drug, Xenograft, MP-MUS
Highlights
-
•
Monoamine oxidase B (MAOB) is upregulated in glioblastoma.
-
•
MAOB is used to convert a nitrogen mustard pro-drug into a mtDNA targeting drug.
-
•
In vitro data show MAOB dependent, mitochondrial-based, toxicity.
-
•
In vivo primary glioblastoma mouse models show efficacy.
Glioblastoma multiforme (GBM) is an inevitably fatal disease with inadequate treatment. MP-MUS, the lead compound in a family of pro-drugs specifically designed to treat GBM, is converted to the mature drug P+-MUS by the enzyme MAOB. This lipophilic cation accumulates in mitochondria, attacking mitochondrial DNA and promoting oxidative stress. GBM has markedly increased levels of MAOB compared to that in other cells; thus, mitochondrial and cell toxicity is restricted to GBM cells. MP-MUS significantly reduced tumor size in flank and intracranial xenograft models of GBM and induced permanent changes in the phenotype of surviving cells without causing systemic or organ-specific toxicities.
1. Introduction
Glioblastoma multiforme (GBM) is the most common and aggressive malignant primary brain tumor in humans, accounting for one-fifth of all intracranial tumors (Stupp et al., 2005). The most effective therapies for GBM are surgical resection (debulking), chemoradiation, and anti-angiogenic agents (Omuro and DeAngelis, 2013). However, despite the availability of these treatments, prognosis remains dismal with a mean survival of approximately 15 months (Henriksson et al., 2011). The search for more effective treatment regimens has been met with limited success during the past 50 years. In the present study, we describe a chemotherapeutic agent, MP-MUS, which specifically targets glioblastoma mitochondria, and we demonstrate its efficacy in cultured human primary GBM cells.
Mitochondria have three cellular functions: providing energy via oxidative phosphorylation, synthesizing cellular components (e.g., heme, cardiolipin, and pyrimidine), and regulating apoptosis. The metabolic function of mitochondria in cancer cells differs from that of healthy cells (Warburg, 1956). Otto Warburg noted that, in contrast to normal cells, which derive energy through the oxidative breakdown of pyruvate, cancer cells produce energy primarily through the anaerobic breakdown of glucose. Although Warburg mistakenly thought that this metabolic shift caused cancer, he correctly concluded that ATP synthesis in the absence of mitochondrial respiration was an inefficient use of energy resources (Moreno-Sánchez et al., 2014). Nonetheless, cancer cells remain dependent on mitochondria for survival because mitochondrial dihydroorotate dehydrogenase is required for pyrimidine synthesis (Lu et al., 2014). Based on their critical cellular functions, mitochondria are attractive anti-cancer drug targets (Ubah and Wallace, 2014).
Targeting mitochondria is also appealing due to the limited DNA repair mechanisms in these organelles (Alexeyev, 2009). Mitochondria repair damaged DNA through a simple base-excision repair mechanism. However, this repair mechanism is not active in cells containing damaged mitochondrial DNA (mtDNA), which effectively reduces the development of chemoresistance in these cells (Stupp et al., 2005). Thus, selective targeting of the mitochondrial genome is an attractive anti-cancer strategy; mitochondria may be the “Achilles' heel” of actively proliferating cancer cells, including stem cells (Loureiro et al., 2013). Glioblastoma mitochondria may be required for the maintenance and function of invasive pseudopodia (Arismendi-Morillo et al., 2012), which mediate the infiltration of cancer cells into the brain. Thus, a chemotherapeutic agent that targets glioblastoma mitochondria may block disease progression.
Monoamine oxidase type B (MAOB) is localized on the inner-face of the outer mitochondrial membrane. MAOB is mainly expressed in glial astrocytes in humans and mice (Ekblom et al., 1993) and oxidizes neuroactive amines, such as dopamine (Wang and Edmondson, 2011). MAOB activity levels have been reported to be significantly higher in GBMs, low-grade astrocytomas, and anaplastic astrocytomas compared to those of postmortem control brain (p < 0.01) (Gabilondo et al., 2008) (Callado et al., 2011). Glioma cells have 5–10 times fewer mitochondria than those of normal human astrocytes (Wolf et al., 2011) and far less complex I–IV (Kiebish et al., 2009). Mitochondrial cytochrome oxidase protein levels are five-fold lower in gliomas compared to those of normal brain tissue obtained from epileptic patients (Griguer et al., 2013). In addition, the state-3 respiration rates of glioblastoma mitochondria are 10-fold lower than those in normal human astrocytic mitochondria (Oliva et al., 2011). However, the MAOB levels per cell are higher in gliomas compared to those of normal astrocytes and so levels of MAOB per mitochondria are higher in glioma than in astrocytes and other cells of the brain.
The neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) crosses the blood–brain barrier and is bioactivated by MAOB oxidation (‘O’) to produce 1-methyl-4-phenyl-2,3-dihydropyridinium (MPDP+). MPDP+ is further oxidized (‘O’) by coenzyme Q to 1-methyl-4-phenylpyridinium (MPP+) (Shi et al., 1999) [summarized in Fig. 1A (I–III)]. The conversion and neuronal toxicity of MPTP, which causes symptoms similar to those associated with Parkinson's Disease, can be blocked by MAOB inhibitors, such as selegiline (Visanji and Brotchie, 2001).
Fig. 1.
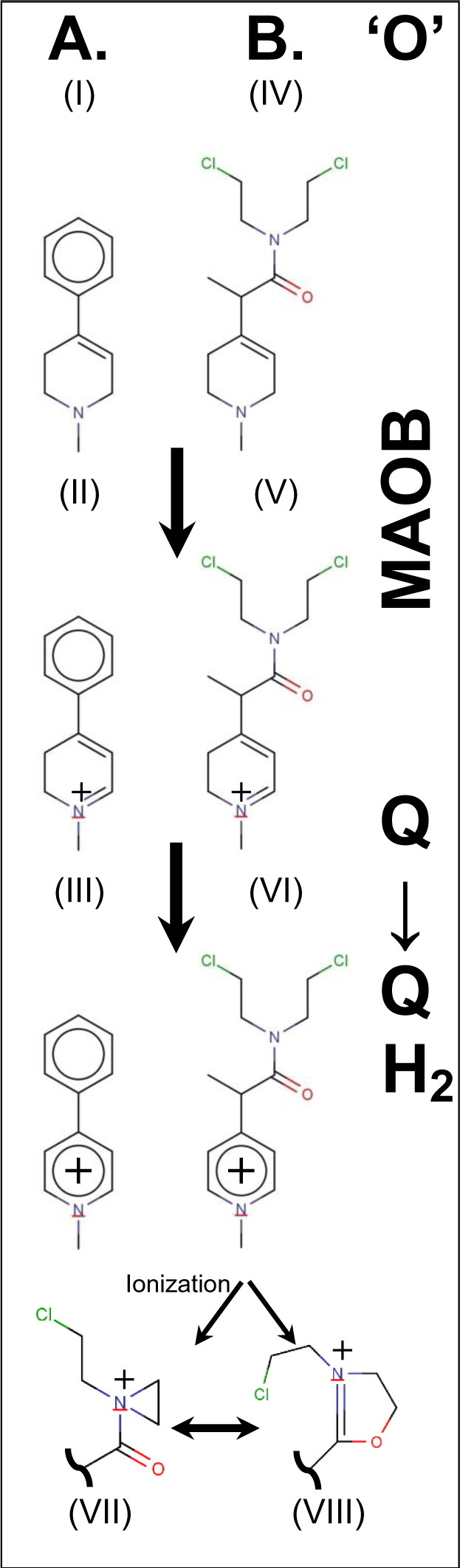
MPTP/MPMUS bioactivation pathways.
(A) Conversion of MPTP (I) to MPP+ (III) via MPDP+ (II) by sequential oxidation (‘O’); oxidation of MPTP is via MAOB and oxidation of MPDP+ is typically via the mitochondrial quinone pool.
(B) Analogous conversion by MAOB of MP-MUS (IV) to MD+-MUS (V). Again the dihydro species is converted to the pyridium, P+-MUS (VI). One of the chloroethyl groups of the mustard can ionize to give rise to either a 3-membered aziridinium (VII) or a 5-membered dihydrooxazolium (VIII) reactive ring system.
Our strategy for the design of a glioblastoma chemotherapeutic agent takes advantage of the high concentration of MAOB in gliomas by synthesizing and testing the pro-drug substrate, MP-MUS, which has a higher specificity for MAOB than MAO-A, and like the MPTP/MPP+ pair generates a lipophilic cation (Fig. 1B). MP-MUS is a chimeric MPTP-nitrogen mustard that accumulates in mitochondria through the MAOB-dependent mechanism of MPTP/MPDP+/MPP+. Mitochondrial accumulation allows the alkylating agent MP-MUS to target mtDNA and mitochondrial complexes. The MP-MUS pro-drug, MP-MUS (IV), is oxidized by MAOB to form the intermediate MD-MUS+ (V), which is then oxidized by quinones to the mature drug, P+-MUS (VI). The mitochondrial membrane potentials (ΔΨ) of cancer cells are typically ≈ 180 mV. Thus, the lipophilic cation P+-MUS will accumulate in the mitochondrial matrix and reach concentrations that are approximately three orders of magnitude greater than those in the cytosol, as with similar lipophilic cations (Reily et al., 2013; Pathak et al., 2014; Rin Jean et al., 2014). Nitrogen mustards alkylate DNA through a highly reactive, three-membered aziridinium ring (VII). Based on this alkylating function, nitrogen mustards have been used as chemotherapeutic agents since 1943 (DeVita and Chu, 2008) (Polavarapu et al., 2012). In addition to generating alkylating aziridinium groups, the presence of an amide in MP-MUS/P+-MUS allows the formation of dihydrooxazolium (VIII) alkylating agents. P+-MUS accumulates in the mitochondrial matrix and alkylates mtDNA and mitochondrial complexes, resulting in mitochondrial dysfunction and cell death. Based on the high levels of MAOB in the mitochondria of glioma cells, MP-MUS is expected to have increased specificity for glioblastoma cells. We have recently shown that MP-MUS is a MAOB specific substrate and that the oxidized product, P+-MUS, is indeed toxic toward cultured glioblastoma cells, but not normal human astrocytes (Sharpe et al., 2015).
A possible disadvantage of our MAOB-catalyzed MP-MUS/P+-MUS approach is that P+-MUS may cause similar effects as MPTP/MPP+, potentially leading to Parkinson's disease. The dopaminergic neuronal toxicity of MPTP results from efficient uptake of astrocytic MPP+ generated by MAOB by the dopamine transporter. Other dopamine mimetics that serve as dopamine transporter substrates include the closely related 4-(4-dimethylamino)phenyl-1-methylpyridinium (APP+) (Karpowicz et al., 2013) and N,N′-dimethyl-4,4′-bipyridinium dichloride or paraquat (Rappold et al., 2011). The crystal structure of the Drosophila melanogaster dopamine transporter in the nortriptyline-inhibited form, 4M48 (Penmatsa et al., 2013), provides hints regarding the possible actions of MPP+, APP+, and paraquat as dopamine mimetics. The substrate-binding pocket is highly constrained. The substrates dopamine, MPP+, APP+, and paraquat fit into this pocket; however, the parasol-like pair of phenyl rings in the inhibitor nortriptyline blocks movement. In silico modeling indicated that P+-MUS will be a very poor substrate of the dopamine transporter (Fig. S1).
2. Materials and Methods
2.1. Primary Human GBM
Glioma cells were isolated within 10 min of tumor excision. Tumors were homogenized with a pipette, and cells were grown for two weeks in DMEM with 20% fetal bovine serum, GlutaMax-I, sodium pyruvate, and penicillin/streptomycin. Unless otherwise specified, the same growth medium was used in the following experiments. Glioma cells were grown to confluence 24 h after treatment with an identical volume of drug (in DMSO) or DMSO alone (maximum 0.04% v/v of DMSO/medium); cells were cultured in either Costar 96-well plates (Corning, NYC, NY, USA) or 16-well Lab-Tek slide chambers (Nalge Nunc, Rochester, NY, USA). After treatment, cells were grown for 24 h in the absence or presence of all effectors (total volume of 250 μL).
The human glioma primary culture, BT150, was used at a low passage number in all of the in vitro experiments presented in Figs. 2, 3, and 4 and Supplementary Figs. 2 and 3.
Fig. 2.

Effect of MP-MUS on mitochondrial membrane potential, mtDNA, and MAOB.
(A) Glioma incubated ± MP-MUS and ± selegiline for 24 h show mitochondrial damage and upregulation of MAOB only when uninhibited MAOB is incubated with MP-MUS. MP-MUS/MAOB induces a drop in ΔΨ, an increase in fpg-sensitive mtDNA lesions, and an elevation of MAOB levels.
(B) Effects of MP-MUS with respect to time; the first changes in mitochondrial function, ΔΨ, and damage are observed after 30 min and continue up to 24 h.
(C) The data presented in A and B is shown as a plot. All changes reported are at p < 0.05, except fpg-sensitive DNA lesions at 2 h (‡), mean ± SEM, n = 8.
Fig. 3.

LD50 of MP-MUS in primary human glioma.
(A) Changes in live cell numbers and %dead cells following 24 hour incubation with increasing concentrations of MP-MUS indicate an LD50 of ≈ 80 μM MP-MUS. Mean ± SEM, n = 4 individual wells.
(B) LDH and XTT formazan assays performed on other half of the 96-well plate appear to demonstrate an increase in levels of both XTT reduction and LDH, per living cell. Mean ± SEM, n = 8 individual wells.
Fig. 4.

MP-MUS induces ROS and causes LDHA and mitochondrial upregulation.
(A) Increase in oxidative stress; representative images of glioma labeled for hydrazine reactive aldehydes/ketones (I), ddTUNEL (II), peroxides (III) hydroxyl radical (IV) all labeled green, with MAOB labeled red. The final panel shows the levels of LDHA (V). The arrows point to cells with massive oxidative stress.
(B) MP-MUS increases levels of oxidative stress, DNA breaks, mitochondrial enzymes, and LDHA.
(C) 3′OH DNA lesions caused by MP-MUS are colocalized with mitochondrial MAOB, and are exclusively cytosolic and non-nuclear.
2.2. Fixation and Permeabilization
Cells in 96-well plates and 16-well slide tanks were fixed in ice-cold 4% paraformaldehyde for at least one hour and then washed in phosphate-buffered saline solution (PBS) (Thermo Fisher Scientific, Rockford, IL, USA). MitoTracker™, MitoSox™ Red, H2-DCF, and Hoe assays were performed in non-permeabilized cells. Hydroxyl radical, aldehyde & ketone, antibody labeling, ddTUNEL, Fpg-ddTUNEL, and blunt-ended ligation experiments were performed with cells that were permeabilized with 0.1% Triton X-100 in PBS (Thermo Fisher Scientific, Rockford, IL, USA).
2.3. Probes
Mitochondrial membrane potential was assayed with the MitoTracker™ (MT, M22425) probe. Mitochondrial superoxide generation required the MitoSox Red (M36008) probe. Probes or dyes used for evaluation and visualization included hydroxyphenyl fluorescein (HPF, H36004) for hydroxyl radicals, 2′,7′-dichlorodihydrofluorescein diacetate (H2-DCFDA/H2-DCF, D399) for peroxides, N-(aminooxyacetyl)-N′-(D-Biotinoyl) hydrazine (ARP, A10550) for aldehydes & ketones, either Hoechst 33342 (Hoe, H21492) or 4′,6-Diamidino-2-Phenylindole (DAPI, D3571) for DNA, either FITC-Avidin (A2662) or Texas Red-Avidin (A2665) for biotinylated probes, goat anti-mouse IgG antibody labeled with Alexa Fluor®594 (A11032) or Alexa Fluor®488 (A10680) for primary mouse antibodies, and Alexa Fluor®594 (A11012) or Alexa Fluor®488 (A11034) for goat anti-rabbit IgG antibody. All reagents were purchased from Invitrogen/Molecular Probes, Eugene, Oregon, USA.
2.3.1. ddTUNEL
A reaction buffer for terminal deoxynucleotidyl transferase (Tdt) was prepared by diluting a stock solution of TUNEL buffer (125 mM Tris–HCl, 1 M sodium cacodylate, 1.25 mg/mL BSA, pH 6.6) at a 1:5 ratio and a 25 mM cobalt chloride stock solution at a 1:25 ratio. Each well was washed twice with 0.1% Triton X-100 in PBS and then twice in reaction buffer. Cells were then incubated for one hour in 50 μL of the reaction buffer containing 20 units/mL of Tdt and 250 nM of Biotin-16-ddUTP (Roche, IN, USA) (Baskin et al., 2010b).
2.3.2. Fpg-ddTUNEL Assay
Permeabilized cells were washed, incubated with NEBuffer 3 for 30 min, and then treated with 50 μL of the same buffer containing 100 units/mL of calf intestinal alkaline phosphatases (CAIP, Sigma) for ≥ 2 h to convert 3′PO4 to 3′OH ends. Cells were washed twice with Tdt reaction buffer before capping the 3′OH ends with biotinylated-ddUTP, blocking with avidin and biotin. Samples were washed twice in 10 mM HEPES, 10 mM NaCl, 2 mM EDTA, and 0.1% BSA. The same buffer (50 μL) containing 100 units/mL of formamidopyrimidine DNA glycosylase (Fpg) (USB, Cleveland, OH, USA) was added to each well, and cells were incubated in a humidified chamber for ≥ 2 h. Each sample was washed twice in 1 × PBS in NEBuffer 3, and 50 μL of the same buffer containing 100 units/mL of CIAP was applied to each section before incubating for ≥ 2 h. Samples were treated for ddTUNEL labeling (Baskin et al., 2010b).
2.3.3. Blunt-ended DNA Breaks
A blunt-ended oligonucleotide probe (biotinylated, hairpin-structured) was used as previously described (Baskin et al., 2010a). The wells were pre-incubated with ligation buffer without probe (66 mM Tris HCl, pH 7.5, 5 mM MgCl2, 0.1 mM dithioerythritol, 1 mM ATP, and 15% polyethylene glycol-8000) and then aspirated. The ligation mix containing probe (35 μg/μL) and T4 DNA ligase (0.5 U/μL) (New England Biolabs, Ipswich, MA, USA) was added prior to an overnight incubation in a humidified chamber. The biotinylated probes were visualized using either FITC-Avidin or Texas Red-Avidin.
2.3.4. Epifluorescence Microscopy
Signals were acquired with the Nikon Eclipse TE2000-E fluorescence microscope. The microscope was equipped with a CoolSnap ES digital camera system (RoperScientific) containing a CCD-1300-Y/HS 1392 × 1040 imaging array cooled by Peltier. Images were recorded and analyzed using Nikon NIS-Elements software.
2.3.5. Transmission Microscopy
Images were acquired using the Olympus IX71 microscope, which was equipped with an Olympus DP72 camera and Olympus TH4-100 light source. Images were recorded using the DP2-BSW software (Olympus).
2.4. Assay for XTT, LDH, and Cell Viability
We grew glioma cells in 96-well plates and 16-well Lab-Tek slide chambers (total volume of 240 μL). At t = 0, 10 μL DMSO was added with or without effectors (titrated along plate columns) and accompanied by equivalent concentrations of MP-MUS. At t = 23 h, columns 9–12 were incubated with Hoechst 33342 (final concentration of 10 μM), and columns 1–8 were washed with XTT buffer (3 mM Tris–HCl, 30 mM HEPES, 10 mM NaCl, pH 7.4, 37 °C).
2.4.1. XTT Assay
A solution of 1.5 mM 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT, Invitrogen) and 10 μM phenazine methosulfate (PMS, Sigma) XTT buffer (Roehm et al., 1991) was freshly prepared and placed in a pre-warmed BioTek Synergy HT plate reader. The first 64 wells were read at 575–675 nm every two and a half minutes. After formazan generation became non-linear (> 10 min), the XTT/PMS was removed, wells were washed twice, and the first 64 wells were used for LDH measurement. XTT generation rate was calculated from the 575–675-nm reading taken at 10 min minus the reading taken at 2.5 min.
2.4.2. LDH Assay
A 100 μL solution [110 mM lactate, 3.35 mM NAD+, 350 μM resazurin, and 2.2 units/mL of diaphorase in 3 mM Tris, 30 mM HEPES, 10 mM NaCl buffer, and 0.45% Triton X-100], pH 7.4, 37 °C (Sharpe et al., 2012) was added to each well. The resorufin that eventually formed was measured over 20 min in a plate reader using 530/25 nm excitation and 590/35 nm emission. The rate of resorufin formation was calculated over the 20-minute period when the curve was linear. A background reading of resorufin generation was then measured on a fresh plate (n = 16 readings from individual wells). LDH levels were calculated from t = 20 min minus t = 5 min, subtracting the background readings recorded during the same time period.
2.4.3. Cell Counts
At t = 24 h and after two-thirds of the plate had been used for XTT/LDH assays, the remaining cells were fixed with ice-cold paraformaldehyde (PFA). We counted cells in the center field at 20 × magnification. There was an average of 150 cells per image when cells were confluent and between 1 and 8 dead/dying cells in the same field. Dead/dying cells were identified by condensed nuclei with signal intensities greater than three times those of the median cell nuclei. Dividing cells were identified by paired and brightly labeled nuclei and were counted as two living cells.
2.4.4. Labeling Glioma in 16-well Slide Tanks
At t = 23.5 h, cells were incubated with 10 μM Hoe and either 1 μM MitoTracker™, 10 μM Mitosox, 10 μM H2-DCFAM, 10 μM HPF, or DMSO for 30 min prior to fixing in ice-cold PFA. After washing in PBS, the levels of fluorophore emission from the fixed cells were measured. Cells were subsequently permeabilized and labeled with probes or antibodies for DNA lesions.
2.4.5. Primary Antibodies
Antibodies included rabbit anti-MAOB (clone EPR7103, Cat#ab125010, Abcam), Armenian Hamster monoclonal Anti-CD3 epsilon antibody (clone 145-2C11, Cat#ab24947, Abcam), mouse anti-cytochrome c (clone 7H8, Cat#sc-13560, Santa Cruz Biotechnology, CA), mouse anti-vimentin antibody clone V9 (M0725, Dako, North America, Carpinteria, CA), mouse anti-LDHA (E-9 clone, Cat#sc-137243, Santa Cruz Biotechnology) and mouse monoclonal tyrosine hydroxylase (F-11 clone, Cat#sc − 25269). Dako antibody diluent (S3022) and protein blocking solution (X0909) were used.
2.5. Animal Models of Primary Brain Glioma
2.5.1. Study Approval
The animal research in mice was conducted according to Institutional Animal Care and Use Committee (IACUC) Protocol AUP-0315-0016, approved by the IACUC of Methodist Hospital. All animal care procedures conformed to the Guide for the Care and Use of Laboratory Animals (National Research Council, National Academy Press, Washington DC 1996, USA). Gliomal cells used in laboratory studies do not meet the definition of human subject research per 45 CFR 46.102 since cells were isolated from deidentified patient tissues and have no identifiable private information.Initial toxicology model. 10 nude mice (NU-FoxnNu) were injected into the tail vein with 200 μL saline or 0.2 mg MP-MUS (in saline) on days 0, 12, and 23. The following day they were sacrificed and their major organs were fixed. After fixation, immunohistochemical slides of 10 μM thickness were prepared. Laterally sliced brain sections of all ten animals were investigated for the levels and the localization of tyrosine hydroxylase; the gold standard marker for the identification of dopaminergic neurons, which is the rate limiting enzyme in dopamine synthesis. A mouse monoclonal antibody (F-11, Santa Cruz, sc-25269) was used to identify dopaminergic neurons and nuclei were counter stained with hematoxylin.
2.5.2. Flank Model
Glioblastoma cell line BT111 was derived from a high-grade GBM. BT111 contains an unmethylated MGMT promoter region and lacks the EGFR oncogene (i.e., no demonstrable gene amplification). In the flank model of GBM using BT11, 20 nude mice (NU-FoxnNu) were injected with 1 × 106 primary human glioblastoma cells at the 6th passage in media:Matrigel (1:1). Animals were monitored for changes in weight and tumor volume based on caliper measurements. The greatest longitudinal diameter (length) and greatest transverse diameter (width) were recorded. Volume was calculated by the modified ellipsoidal formula (Euhus et al., 1986) as tumor volume = 1/2(length × width2). Mice with an average tumor volume of 45 mm3 after four weeks were categorized into two groups of seven. Either saline or 0.2 mg MP-MUS (in saline) was injected into the tail vein on days 0, 12, and 23; mice were monitored until day 36 (the last injection time point). At this time, two of the control animals and all of the MP-MUS-treated animals were sacrificed. Tumors were excised, halved, and either preserved in PFA or grown in cell culture media. Tumor volumes (87 mm3) were determined for the remaining five control animals. Each of these mice was given one tail-vein injection of 0.2 mg MP-MUS in saline, and tumor volume was re-assessed 24 h later.
2.5.3. Brain Model
Recently, Iwami and co-workers (Iwami et al., 2012) developed an innovative methodology for injecting human GBM cultures into mouse brain via the postglenoid foramen. Percutaneous injection into the adult mouse brain via the postglenoid foramen is technically simpler, more time efficient, and less stressful for mice than traditional stereotaxic methodologies. GBM cells (5 × 104) in a solution of Matrigel™ media (4:6) were injected into the postglenoid foramen of 40 SCID mice. The primary GBM culture BT116 was from an aggressive, grade IV GBM that was fatal in less than two years. An excessive number of SCID mice died following the glioma injections due to brain hemorrhage; this complication or high death rate was not observed with NU-FoxnNu or NIHIII mice using the same brain injection methodology (data not shown). After more than three months, 24 animals were asymptomatic. Two animals were sacrificed on day 115; brains were fixed/embedded in wax and archived as pre-treatment controls. The remaining 22 animals were split into MP-MUS-treated and saline-treated groups. The animals received tail-vein injections of saline or 0.2 mg MP-MUS in saline on days 115, 123, and 131 and monitored until day 307, which was the end of the study. When mice became symptomatic, they were sacrificed, and their brains were removed and fixed in 2% PFA.
2.6. Histological Analysis of GBM in Mouse Brain
To detect human glioblastoma cells in mouse brain, we used a 1:100 dilution of the mouse anti-vimentin antibody clone V9 (Dako, M0725), which has no cross-reactivity with mouse vimentin (Valadez et al., 2014). We also determined the levels of immune cells positive for CD3-Ɛ using a 1:10 dilution of Armenian Hamster anti-CD3-Ɛ antibody-FITC followed by a 1:100 dilution of the secondary mouse anti-FITC antibody (clone FL-D6, Cat# A1812, Sigma). The HiDefTM HRP-polymer system was used for detection (Cell Marque, Rocklin, CA). Endogenous peroxidase activity was eliminated with treatment under mild conditions as follows: 1.8% H2O2 for 5 min, 1% periodate for 5 min, and 0.02% NaBH4 for 2 min (Polak and Van Noorden, 2003). Signal detection was based on the Dako DAB chromogen kit according to the manufacturer's guidelines. We found that hot citric acid and slow cooling of the slide/citrate solution to room temperature (> 20 min) was the best method to retrieve the epitope, but lost epitope if we cooled quickly. The complete methodology can be found in Supplemental Experimental Procedure 1.
2.7. Chemical Synthesis of MP-MUS
A full description of the synthesis and characterization of MP-MUS has been recently published by our laboratory (Sharpe et al., 2015). The synthetic route taken for the synthesis of MP-MUS that was used in our initial work is found in Supplemental Experimental Procedure 2. It should be noted that the BOP used in this synthesis generates a carcinogenic hexamethylphosphoramide product. We advise that our ChemMedChem route which does not use BOP, be used instead.
2.8. Statistical Analysis
Statistical analyses were performed with Microsoft Office Excel. All numerical data are presented as mean ± SEM. An n number is a unique experiment, typically an image or enzyme-linked assay performed in a single well. Grouped analysis was performed by ANOVA. Pairwise analysis was performing using the Student's t-test with p < 0.05 as the threshold for statistical significance following a Bonferroni correction for multiple analyses.
3. Results
3.1. Inhibition of MAOB Attenuates MP-MUS-Induced Mitochondrial Toxicity
We demonstrated that MP-MUS targets the mitochondria of primary human glioma cells in a MAOB-dependent manner (Fig. 2). Human glioblastoma cells were incubated with or without MP-MUS for 24 h in the absence or presence of the MAOB-specific inhibitor selegiline (Fig. 2A). We examined mitochondrial ΔΨ, formamidopyrimidine-DNA glycosylase or 8-oxoguanine DNA glycosylase (fpg)-sensitive DNA lesions, and MAOB by fluorescence microscopy. The MitoTracker™ ΔΨ signal per cell was not significantly affected in cells treated with 2 μM selegiline compared to DMSO vehicle. However, 90 μM MP-MUS resulted in a 50% reduction in ΔΨ (p < 0.0001 for n = 8 images from individual wells), which was completely blocked by co-treatment with selegiline. The MitoTracker™ signal is a proxy for both mitochondrial density and mitochondrial energization status. Low signal levels indicate that either the mitochondria are depolarized, thus incapable of generating significant levels of ATP by oxidative phosphorylation, or that the number of mitochondria per cell has dropped, or both. Oxidation and alkylation of mtDNA was evaluated by the fpg-ddTUNEL assay (Sharpe et al., 2012; Baskin et al., 2010b). MP-MUS increased fpg-sensitive oxidation/alkylation of DNA bases by 60% (p < 0.0001). Statistically significant changes in mitochondrial or nuclear DNA damage were not observed in cells treated with selegiline alone or with the combination of selegiline and MP-MUS. MP-PUS treatment caused a significant (p < 0.001) 220% increase in MAOB antibody labeling. Thus, MP-MUS damaged mtDNA through an MAOB-dependent mechanism that was blocked by selegiline.
The effects of MP-MUS treatment duration on ΔΨ, fpg-sensitive DNA lesions, and MAOB in glioblastoma cells are shown in Fig. 2B. After only 30 min, 20% of the ΔΨ signal was lost (p < 0.05); after two hours, MP-MUS-treated cells had 70% of the control mitochondrial ΔΨ (p < 0.001). We observed an initial 20% increase in fpg-sensitive DNA lesions after 30 min of treatment; these lesions continued to increase before damaged DNA levels reached a plateau at 30% above control levels at 24 h. There was a 40% increase in MAOB levels within 30 min of treatment and a 60% increase by one hour. MAOB levels showed a slight decline at 2 h after treatment, which is when cells began to die and detach. A time series is shown for three mitochondrial parameters in Fig. 2C. All data showed statistically significant changes beyond 30 min of treatment, with the exception of the fpg-assay at 120 min (‡). We did not observe statistically significant changes in mitochondrial phenotype at incubation times < 30 min, which may be due to MAOB-induced maturation of the pro-drug and mtDNA replication time (≈ 17 min) (Song et al., 2011).
3.2. MP-MUS is a Glioblastoma Toxin In Vitro
We examined the concentration range at which MP-MUS affected the viability of glioma cells. Cells were cultured in a 96-well plate for 24 h. Viability was assessed by counting cells using fluorescence microscopy, measuring the levels of mitochondrial complexes I and III using the XTT assay, and measuring the levels of detergent-permeabilized lactate dehydrogenase. Hoechst staining showed that the LD50 of MP-MUS was 77 μM after 24 h in primary human glioma cultures (Fig. 3A), whereas the LD50 was 50 μM at 48 h (data not shown).
Total cellular LDH levels are an indicator of glucose/lactate flux (‘anaerobic respiration’) and XTT reduction levels are an indicator of the respiratory flux through the respiratory chain (‘aerobic flux’), when combined with cell numbers these assays are informative as to changes in cellular energy changes. MP-MUS affected levels of both XTT formazan generation and glioblastoma lactate dehydrogenase levels (Fig. 3B) and the changes in the proxy's anaerobic and aerobic respiration are quite different from changes in cell counts (Fig. 3A); Treatment with MP-MUS increased the levels of mitochondrial complexes and lactate dehydrogenase in glioma cells. In Figs. 3A and B one can compare the levels of LDH and XTT in the control and in cells treated with 90 μM MP-MUS to be alerted to the fact that large-scale metabolic changes occur in response to MP-MUS. There are less than half the total number of cells that have been treated with 90 μM MP-MUS treated as in the controls (Fig. 3A). None the less, this much smaller cell population has almost the same level of LDH and XTT formazan production as the more numerous control cells (Fig. 3B). Thus, on a per cell basis, 90 μM MP-MUS induces a 2.2-fold increase in cellular mitochondrial levels and a 2-fold increase in lactate dehydrogenase activity.
Parallel control experiments were performed to examine the effects of blocking the activity of MAOB or the parental mustard compound. Mitochondrial ΔΨ, XTT reduction, LDH levels, cell viability, and the percentage of dead cells did not change in response to co-treatment with 210 μM MP-MUS and 2 μM selegiline. Similarly, 210 μM of the parental mustard compound, 2,2′-dichlorodiethylamine, did not significantly alter any of the measured parameters in glioma cells (Fig. S2). Thus, MP-MUS toxicity toward glioblastoma cells is dependent upon its conversion to P+-MUS, by MAOB and not due to MP-MUS itself nor the parental mustards toxicity. In Fig. 2 we showed that the MitoTracker™ signal collapses after 24 h incubation with 90 μM MP-MUS, while Fig. 3B shows that concurrent with this loss of mitochondrial signal there is a doubling, per cell, of the rate that XTT is reduced by mitochondrial complexes, which is mirrored by the increase in MAOB levels (Fig. 2A). Thus, in response to MP-MUS glioblastoma increase their mitochondrial levels, but these mitochondria are poorly energized and so are less able to maintain normal ATP production.
3.3. MP-MUS Induces Changes in Reactive Oxygen Species (ROS) Levels and Other Cellular Responses in vitro
In addition to damaging glioblastoma mtDNA, MP-MUS increased the levels of mitochondrial ROS (Fig. 4). In addition, exposure of cells to 90 μM MP-MUS increased mitochondrial MitoTracker™ labeling and lactate dehydrogenase A (LDHA) levels. The levels of mitochondrial proteins cytochrome c (columns I–II) and MAOB (columns III–IV) (Fig. 4A, labeled in red) increased by 70% and 130% (plotted in Fig. 4B). The increased mitochondrial levels were accompanied by a 4.8-fold increase in LDHA levels (column V, red). These changes in protein levels were consistent with the increased mitochondrial respiratory chain activity and increased LDH activity, per cell, following exposure to MP-MUS (Fig. 3).
Four different oxidative stress assays (all depicted in green) were also performed with the same glioma cultures labeled for mitochondrial proteins. The levels of cellular aldehydes and ketones, including protein carbonyls, were labeled with biotin-hydrazine and visualized with fluorescein isothiocyanate labeled avidin (FITC-avidin); Fig. 4A (I). No changes in the overall levels of these markers of oxidative stress were found in MP-MUS-treated gliomas. The levels of damaged DNA were significantly increased, as determined by labeling 3′-OH DNA breaks and nicks using ddTUNEL; Fig. 4A (II). Damaged DNA was identified as mtDNA based on the extra-nuclear location and co-localization with mitochondrial protein markers. Formation of 2′-7′dichlorofluorescein (DCF), a measure of steady-state cellular peroxides, increased by 75% after MP-MUS treatment and co-localized with mitochondria; Fig. 4A (III). The hydroxyl radical probe hydroxyphenyl fluorescein (HPF) showed a three-fold increase in hydroxyl radical production, Fig. 4A (IV), which was indicative of mitochondrial Fenton chemistry (Sharpe et al., 2012). Histograms and statistical significance of the changes in labeling due to incubation with MP-MUS are shown in Fig. 4B. We examined control- and MP-MUS-treated cells at high resolution with MAOB labeled in red, 3′OH DNA nicks/breaks labeled in green, and DAPI labeled in blue (Fig. 4C). DNA damage was only visible in the cytosol, indicating that mtDNA, rather than nuclear DNA, was the target of MP-MUS.
These data (Figs. 2–4) demonstrate that MP-MUS is highly damaging to mitochondria in glioblastoma cultures. In response to this damage the glioblastoma cells respond by increasing mitochondrial biogenesis and upregulate production of LDHA.
3.4. MP-MUS Decreases Tumor Volume in a Flank Model of GBM But Does Not Cause Parkinsonian Syndromes
We examined the effects of two MP-MUS treatment regimens on primary human glioma xenografts in a Nu/Nu nude mouse model (Fig. 5). Saline or 0.2 mg MP-MUS in saline was injected into the tail veins of mice (≈ 25 g), 8 mg/kg, on days 0, 12, and 23 with seven mice per group (Fig. 5A). None of the mice exhibited adverse symptoms or weight loss during the study period. Tumor volumes shrank in the MP-MUS treatment group after the first injection, whereas tumors in the saline controls grew steadily throughout the study. Tumor volumes were significantly different between groups (p < 0.0005) from day 22 onward.
Fig. 5.
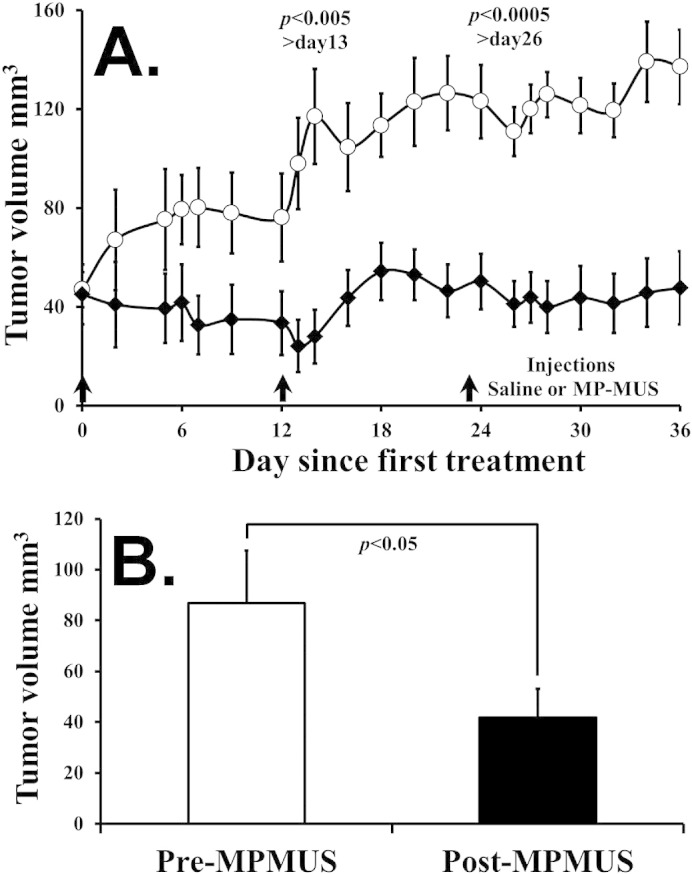
MP-MUS is a chemotherapeutic in a flank glioblastoma model.
(A) Three treatments of 8 mg/kg of MP-MUS were able to halt tumor growth compared with saline controls.
(B) A single treatment of 8 mg/kg MP-MUS was able to shrink tumor volume by 50% in 24 h.
The two control mice with the largest tumors and all seven mice treated with MP-MUS were sacrificed on day 36. No tumors were detected in two of the seven MP-MUS-treated mice. Half of the excised tumor tissue was used for histological studies, and the remaining portion was gently homogenized and grown in culture. In addition, the major organs were fixed, sectioned, and examined by H&E staining and ddTUNEL for identification of apoptotic nuclei. No abnormalities or differences were found in the major organs of control or treated animals. Necrotic areas and increased numbers of apoptotic, ddTUNEL-positive cells were found in MP-MUS-treated xenografts compared with those of controls. Representative images of ddTUNEL and DAPI labeled saline control and MP-MUS treated tumors are shown in Supplemental Fig. 3. Cells undergoing apoptotic cell death were present in both tumor types, but are more numerous in the MP-MUS treated tissue. In addition, ddTUNEL labeling is found in many non-apoptotic MP-MUS treated cells, with DNA damage localized within the cytosol, suggesting that these cells have mtDNA damage.
The five remaining saline-treated control mice were given a single injection of 0.2 mg MP-MUS. A greater than 50% decrease in tumor burden occurred after 24 h of treatment (Fig. 5B). Thus, a single treatment with MP-MUS was effective against large, well-established glioblastoma tumors.
Treatment of mice with MPTP gives rise to a phenotype indicating dysfunctional motor control resembling those in human Parkinson's disease, and symptoms include akinesia, rigidity, tremor, gait and posture disturbances (Sedelis et al., 2001). In preliminary toxicology studies, and in the following efficacy studies, we observed no evidence of symptoms consistent with the induction of Parkinsonian syndromes. To investigate whether MP-MUS usage had caused ablation of dopaminergic neurons, but at a level that was behaviorally asymptomatic, we performed a number of histological studies on the brains of mice used in preliminary toxicology studies. We probed saline control and MP-MUS treated brains for dopaminergic neuronal density, using a tyrosine hydroxylase specific monoclonal antibody and typical low and high magnified images are shown in Supplemental Fig. 4. We found no indication of loss of dopaminergic neurons in any of the MP-MUS treated mouse brain sections in this study. Treatment of mice with MPTP can also give rise to infiltration of surviving dopaminergic neurons by activated astrocytes (Ghosh et al., 2012) and to iron rich voids that are formed following dopaminergic neuronal cell death (Jiang et al., 2010). We labeled the brain slices of the same two cohorts for iron, using Perl's Prussian blue, and for astrocytic infiltrates, using anti-GFAP antibodies, and could not identify any differences between the MP-MUS treated and untreated groups (results not shown).
3.5. MP-MUS Treatment of Xenografts Induces Changes in the Glioblastoma Phenotype
We then examined if permanent changes to the mitochondrial phenotype were present in the MP-MUS-treated glioblastoma tumors. Cells grown from MP-MUS-treated or saline-treated xenografts were assessed for markers of mitochondrial function and oxidative stress. The levels of Mitotracker™ and MitoSox, which measure mitochondrial superoxide, were statistically identical between the two treatment groups. However, cells from the MP-MUS xenografts exhibited a 2.8-fold increase in the level of mitochondrial MAOB and 1.9-fold increase in the level of cytosolic 3′OH mtDNA nicks and breaks (Figs. 6A and 6B). We explored the type of mtDNA damage using blunt-ended ligation as previously described (Baskin et al., 2010a). MP-MUS-treated glioblastoma xenografts had ~ 50% more blunt-ended mtDNA breaks (Fig. 6C). These mitochondria increased cellular oxidative stress because there was a 50% greater level of hydroxyl radical generation and a 3.5-fold increase in the steady state levels of peroxides in cells derived from MP-MUS-treated xenografts. MP-MUS treatment affects the glioblastoma phenotype, with subsequent daughter cells having altered mitochondrial function, presumably due to heritable mtDNA damage.
Fig. 6.

Alteration in glioblastoma phenotype after MP-MUS treatment.
(A) Representative images of the two cell lineages treated ± MP-MUS show an increase in steady state of MAOB levels, mtDNA lesions and permanent changes in the generation of reactive oxygen species.
(B) Graphical representations of the altered phenotypes are shown with significant changes in levels of MAOB, the mtDNA breaks, peroxides and hydroxyl radical. ** p < 0.01 and *** p < 0.005 at the n = number of individual wells shown in the insert.
(C) High resolution images of blunt ended DNA breaks (green) and DNA (blue) of cells grown from xenografts. Blunt-ended DNA is non-nuclear, cytosolic mtDNA with some invaginations of cytosol into the nucleus.
3.6. MP-MUS is Successful in the Treatment of Intracranial Xenografts
To study the effects of MP-MUS on morbidity and body mass, we used an intracranial mouse model of primary human GBM (Fig. 7A and 7B). NOD/SCID mice were randomized into two treatment groups (n = 11 mice per group). Primary human GBM cells encased in a Matrigel matrix were injected into the right hemisphere of the brain of each mouse using the method developed by Iwami and co-workers (Iwami et al., 2012). The first of three tail-vein injections of saline or 0.2 mg MP-MUS, 8 mg/kg, was delivered on day 115. The saline-treated group exhibited morbidity in 50% of animals on day 248, whereas the MP-MUS treatment group had only one symptomatic mouse during the entire experimental period. Shortly after the final injection, animals in the MP-MUS-treated group steadily gained weight. In contrast, control mice showed a delayed weight gain after the final saline injection. Saline controls initially averaged 95% of the weight of MP-MUS-treated animals; however, the body weights of control animals declined consistently after day 250 until weights were about 88% of the final average weight of the MP-MUS-treated mice. In addition to a failure to thrive, most of the control mice lost clumps of body hair throughout the study, typically prior to the onset of neurological deficits or sudden weight loss; in contrast, only 40% of the MP-MUS-treated group exhibited these changes. Fig. S5 shows the surviving control mice on day 307 compared with four representative MP-MUS treated animals.
Fig. 7.

Glioblastoma successfully treated by MP-MUS in an intracranial xenograft model.
(A) Kaplan–Meier plot of mice after intracranial injection following 3 × Saline or 3 × MP-MUS (8 mg/kg) tail vein injections, n = 11.
(B) Alteration in mouse body mass during course of trial shows that MP-MUS treated gain and hold body weight better than saline treated.
(C) Labeling of uninjected hemisphere of saline treated mouse with (I) anti-vimentin (human glioblastoma cells) IgG, (III) CD3-ε (mouse immune cells) and (II) no-primary IgG control (background peroxidase activity).
(D) Images of C. (I) taken as increasing magnification demonstrating the pattern of infiltration of the brain by the glioblastoma cells.
(E) Unambiguous demonstration that vimentin labeling co-localized with DAPI/nuclear DNA can be seen by attenuation of fluorescent nuclei within glioblastoma DAB staining.
(F) Levels of glioma and CD3-ε in MP-MUS treated brain (right hemispheres).
MP-MUS #1 displays vimentin in human glioblastoma, found only on a single slide, but CD3-ε labeling is found throughout hemisphere.
MP-MUS #8 has the most vimentin signal, hence glioblastoma cells, of any of the MP-MUS treated mice.
After the onset of symptoms that indicated an ethical endpoint or at the end of the study, we performed a sagittal brain dissection into two hemispheres and prepared sections for microscopy. We identified infiltrating human cells with a mouse anti-vimentin antibody derived from V9, a clone that does not bind to mouse vimentin (Caretti et al., 2011). This antibody recognizes all non-mouse forms of vimentin in a mouse vimentin background. We also used an antibody against CD3-Ɛ to label another set of hemispheric pairings. CD3-Ɛ is found on the surface of tumor-associated macrophages and T-cells (Borroto et al., 2014) and serves as an indicator of increased immune response to glioma xenografts in rodents (Kong et al., 2012; Strojnik et al., 2010).
DAB-stained images of human vimentin (I), a no-primary internal control (II), and CD3-Ɛ staining (III) of sequential slices of the left hemisphere of a saline control brain, all mounted on a single slide, are shown on in Fig. 7C. The same vimentin-labeled section of individual human glioblastoma cells in the right hemisphere infiltrating the left hemisphere is also shown at an increased magnification [Fig. 7D (I)]. To confirm that we had labeled human cells and were not observing artifacts, we also labeled DNA with DAPI. We took an image of the DAB-vimentin cells using transmitted black and white light [Fig. 7E (I)] and an image of DAPI fluorescence (II). The overlaid image [Fig. 7E (III)] shows that nuclear DNA co-localized with DAB staining and that DAB staining attenuates DAPI fluorescence. The DAPI signal was inside DAB-labeled cells, indicating that the antibodies were recognizing the correct cellular antigen.
3.7. MP-MUS Treatment Lowers Glioma Burden in Intracranial Xenografts
Glioma xenografts were developed in the right hemisphere of the brain of each mouse; thus, a comparison of right & left hemisphere glioblastoma load is an index of glioblastoma infiltration. The glioblastoma load and levels of CD3-Ɛ-positive cells in each hemisphere of all 22 animals are shown in Supplementary Table 1. We found human gliomas in 100% of the right hemispheres of the brains of control mice, with infiltration into the left hemispheres of 10 of 11 control mice. In contrast, no cells of human origin were detected in 45% of the brains of mice treated with MP-MUS. There were glioblastoma cells in the right hemispheres of 6 of 11 MP-MUS-treated mice, but there were no glioblastoma infiltrates into the left hemispheres. The only symptomatic MP-MUS-treated animal that reached an ethical endpoint (MP-MUS #1) had a very low glioblastoma burden; positive cells were observed in only one 5-μM section on one slide of a sample near the injection point (Fig. 7F). Although we observed very few human glioma cells, we found large numbers of activated immune cells positive for CD3-Ɛ. The highest glioblastoma burden in an MP-MUS-treated mouse (MP-MUS #8) occurred near the injection site and glioblastoma cells co-localized with CD3-Ɛ-positive cells, Fig. 7F. CD3-Ɛ-positive cells were located in both brain hemispheres of all saline-treated controls. CD3-Ɛ-positive cells were found in the injected and uninjected hemispheres of 7 of 11 MP-MUS-treated animals. We were unable to identify any CD3-Ɛ-positive immune cells or gliomas in the remaining four MP-MUS-treated mouse brains.
4. Discussion
Human gliomas exhibit significant and selective increases in MAOB activity compared to other types of common brain tumors, such as meningiomas (Callado et al., 2011), or non-tumor-bearing brain tissues (Gabilondo et al., 2008). This characteristic presents a distinct opportunity for rational drug design using MAOB as a catalyst for converting a non-toxic pro-drug into a mature chemotherapeutic, as demonstrated here with MP-MUS. The ability of MAO to irreversibly oxidize the neutral 1,2,3,6-tetrahydropyridine into a cationic pyridinium species is the basis of the mitochondria-mediated neurotoxicity of MPTP (Visanji and Brotchie, 2001; Shi et al., 1999; Sullivan and Tipton, 1990); this mechanism was the starting point for our pro-drug design. An ideal MAOB-activated pro-drug should cross the blood–brain barrier and have high MAOB flux and high specificity for MAOB versus MAO-A. The design of the “warhead” attached to the tetrahydropyridine “seeker”, N,N‐bis(2‐chloroethyl) propanamide, was partly based on the size, shape, and LogP of the indolyl and 4-(1-methylpyrrol-2-yl) analogues of MPTP, which have very good MAOB kinetic properties (Sullivan and Tipton, 1990; Nimkar et al., 1996, 1999).
Sub-lethal levels of MP-MUS induce increased levels of mitochondria and mitochondrial MAOB in glioma cells in vitro and in vivo. Cells derived from MP-MUS-treated glioma xenografts harbored dysfunctional mitochondria many generations after treatment. The mitochondria in these cells had increased MAOB expression and increased ROS generation. Thus, glioma cells that survive after one or more cycles of MP-MUS treatment express increased levels of MAOB, rendering these glioma cells more sensitive to MP-MUS therapy. Unlike most chemotherapeutic drugs, which lose their efficacy during repetitive rechallenge, giving rise to drug resistance, serial challenge with MP-MUS appears to have the opposite effect; MP-MUS treated cells upregulate mitochondrial levels, and in doing so further upregulate their levels of MAOB, making them sensitive to further MP-MUS treatment.
We have recently shown the kinetics of rhMAO-A/-B acting on MP-MUS and known MAO substrates (Sharpe et al., 2015). MP-MUS is a very poor rhMAO-A substrate, but MP-MUS has a similar km and Vmax to benzylamine in rhMAOB. We also show that in vitro, MP-MUS is more toxic to primary GBM than to normal human astrocytes and that this difference in toxicity at least partly due to the higher levels of MAOB in glioma.
It is possible that like MPP+, P+-MUS could be a dopamine transporter substrate and so can induce Parkinsonian syndromes due to DAT-mediated toxicity. We designed MP-MUS so that the active compound, P+-MUS, would be too large to serve as a DAT substrate, based on the known kinetics of MPP+ analogues. We observed no Parkinsonian symptoms in any of the MP-MUS treated animals, nor did an examination of treated brains reveal any toxicity toward the dopaminergic neurons. As human and mouse DAT share very similar sequence homology and substrate kinetics (Han and Gu, 2006) the inability of MP-MUS to target mouse dopaminergic neurons via DAT-mediated toxicity should be replicated in humans.
Treatment of glioblastoma has been a challenge for decades. The Radiation Therapy Oncology Group (RTOG) study in 1993 demonstrated the limited value of surgery (Simpson et al., 1993). Patients who underwent radical surgery and radiotherapy vs. biopsy only and radiation therapy had an increase in survival from only 6.6 to 11.3 months. The addition of radiation therapy and chemotherapy, including the use of temozolomide, has only extended median survival to 15 months over 20 years later. Furthermore, the final 3 months of life generally have a low quality attached to them. Thus, a novel approach is desperately needed for these patients.
MP-MUS may be such a therapy. It targets an essential organelle of the cell, the mitochondria, which cancer cells must have to survive. The up regulation of MAOB in response to treatment is a maladaptive response of the glioma cell, which may work to the advantage of treatment.
The next steps for this construct is to perform detailed toxicological studies, pharmacodynamic testing, and to fine-tune the details of the manufacturing process. This work is under way, and we are hopeful to see this drug in clinical trials in the next 18 months.
Author Contributions
MAS did the labeling/imaging and worked with AL on the flank model and with TG on the brain model of glioma. MAS and DSB were responsible for the design of MP-MUS. MAS did the in silico modeling and designed the initial synthetic route. PG did the original MP-MUS synthesis. JH improved the synthesis and purified and chemically characterized the MP-MUS used in our reported studies.
Conflicts of Interest
The authors have no conflicting financial interests. Baskin and Sharpe have applied for intellectual property rights for MAO-based prodrugs in Patent application PCT/US2012/062850.
Acknowledgments
We thank Dr. Michael Cooper, M.D., Department of Neurology, Vanderbilt Medical Center, for his helpful advice as to the identification of infiltrating glioma in mouse brain.
Sophie Lopez handled all our tissue culture work and maintained our cultures.
This work was funded by, the Donna and Kenneth R. Peak Foundation, the Taub Foundation, the Blanche Green Estate Fund of the Pauline Sterne Wolff Memorial Foundation, the Veralan Foundation, the Methodist Hospital Foundation, the John S. Dunn Foundation, the American Brain Tumor Association, and was made possible by the many patients and families who have been affected by the devastating effects of brain tumors and central nervous system disease.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.ebiom.2015.08.013.
Contributor Information
Martyn A. Sharpe, Email: masharpe@houstonmethodist.org.
David S. Baskin, Email: dbaskin@houstonmethodist.org.
Appendix A. Supplementary Data
Supplementary material.
References
- Alexeyev M.F. Is there more to aging than mitochondrial DNA and reactive oxygen species? FEBS J. 2009;276:5768–5787. doi: 10.1111/j.1742-4658.2009.07269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arismendi-Morillo G., Hoa N.T., Ge L., Jadus M.R. Mitochondrial network in Glioma's Invadopodia displays an activated state both in situ and in vitro: potential functional implications. Ultrastruct. Pathol. 2012;36:409–414. doi: 10.3109/01913123.2012.694582. [DOI] [PubMed] [Google Scholar]
- Baskin D.S., Widmayer M.A., Sharpe M.A. Quantification and calibration of images in fluorescence microscopy. Anal. Biochem. 2010;404:118–126. doi: 10.1016/j.ab.2010.05.029. [DOI] [PubMed] [Google Scholar]
- Baskin D.S., Widmayer M.A., Sharpe M.A. Quantification of DNase type I ends, DNase type II ends, and modified bases using fluorescently labeled ddUTP, terminal deoxynucleotidyl transferase, and formamidopyrimidine-DNA glycosylase. Biotechniques. 2010;49:505–512. doi: 10.2144/000113439. [DOI] [PubMed] [Google Scholar]
- Borroto A., Arellano I., Blanco R., Fuentes M., Orfao A., Dopfer E.P., Prouza M., Suchanek M., Schamel W.W., Alarcon B. Relevance of Nck-CD3 epsilon interaction for T cell activation in vivo. J. Immunol. 2014;192:2042–2053. doi: 10.4049/jimmunol.1203414. [DOI] [PubMed] [Google Scholar]
- Callado L., Garibi J., Meana J.J. Gliomas: role of monoamine oxidase B in diagnosis. In: Hayat M.A., editor. Tumors of the Central Nervous System, Volume 1. Springer; Netherlands: 2011. [Google Scholar]
- Caretti V., Zondervan I., Meijer D.H., Idema S., Vos W., Hamans B., Bugiani M., Hulleman E., Wesseling P., Vandertop W.P., Noske D.P., Kaspers G., Molthoff C.F., Wurdinger T. Monitoring of tumor growth and post-irradiation recurrence in a diffuse intrinsic pontine glioma mouse model. Brain Pathol. 2011;21:441–451. doi: 10.1111/j.1750-3639.2010.00468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVita V.T., Chu E. A history of cancer chemotherapy. Cancer Res. 2008;68:8643–8653. doi: 10.1158/0008-5472.CAN-07-6611. [DOI] [PubMed] [Google Scholar]
- Ekblom J., Jossan S.S., Bergstrüm M., Oreland L., Walum E., Aquilonius S.-M. Monoamine oxidase-B in astrocytes. Glia. 1993;8:122–132. doi: 10.1002/glia.440080208. [DOI] [PubMed] [Google Scholar]
- Euhus D.M., Hudd C., LaRegina M.C., Johnson F.E. Tumor measurement in the nude mouse. J. Surg. Oncol. 1986;31:229–234. doi: 10.1002/jso.2930310402. [DOI] [PubMed] [Google Scholar]
- Gabilondo A.M., Hostalot C., Garibi J.M., Meana J.J., Callado L.F. Monoamine oxidase B activity is increased in human gliomas. Neurochem. Int. 2008;52:230–234. doi: 10.1016/j.neuint.2007.05.015. [DOI] [PubMed] [Google Scholar]
- Ghosh A., Kanthasamy A., Joseph J., Anantharam V., Srivastava P., Dranka B.P., Kalyanaraman B., Kanthasamy A.G. Anti-inflammatory and neuroprotective effects of an orally active apocynin derivative in pre-clinical models of Parkinson's disease. J. Neuroinflammation. 2012;9:241. doi: 10.1186/1742-2094-9-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griguer C.E., Cantor A.B., Fathallah-Shaykh H.M., Gillespie G.Y., Gordon A.S., Markert J.M., Radovanovic I., Clement-Schatlo V., Shannon C.N., Oliva C.R. Prognostic relevance of cytochrome C oxidase in primary glioblastoma multiforme. PLoS One. 2013;8:e61035. doi: 10.1371/journal.pone.0061035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D.D., Gu H.H. Comparison of the monoamine transporters from human and mouse in their sensitivities to psychostimulant drugs. BMC Pharmacol. 2006;6:6. doi: 10.1186/1471-2210-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksson R., Asklund T., Poulsen H.S. Impact of therapy on quality of life, neurocognitive function and their correlates in glioblastoma multiforme: a review. J. Neuro-Oncol. 2011;104:639–646. doi: 10.1007/s11060-011-0565-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwami K., Momota H., Natsume A., Kinjo S., Nagatani T., Wakabayashi T. A novel method of intracranial injection via the postglenoid foramen for brain tumor mouse models. J. Neurosurg. 2012;116:630–635. doi: 10.3171/2011.10.JNS11852. [DOI] [PubMed] [Google Scholar]
- Jiang H., Song N., Xu H., Zhang S., Wang J., Xie J. Up-regulation of divalent metal transporter 1 in 6-hydroxydopamine intoxication is IRE/IRP dependent. Cell Res. 2010;20:345–356. doi: 10.1038/cr.2010.20. [DOI] [PubMed] [Google Scholar]
- Karpowicz R.J., Dunn M., Sulzer D., Sames D. APP +, a fluorescent analogue of the neurotoxin MPP +, is a marker of catecholamine neurons in brain tissue, but not a fluorescent false neurotransmitter. ACS Chem. Neurosci. 2013;4:858–869. doi: 10.1021/cn400038u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiebish M.A., Han X., Cheng H., Seyfried T.N. In vitro growth environment produces lipidomic and electron transport chain abnormalities in mitochondria from non-tumorigenic astrocytes and brain tumours. ASN Neuro. 2009;1 doi: 10.1042/AN20090011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong S., Sengupta S., Tyler B., Bais A.J., Ma Q., Doucette S., Zhou J., Sahin A., Carter B.S., Brem H., Junghans R.P., Sampath P. Suppression of human glioma xenografts with second-generation IL13R-specific chimeric antigen receptor–modified T cells. Clin. Cancer Res. 2012;18:5949–5960. doi: 10.1158/1078-0432.CCR-12-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loureiro R., Mesquita K.A., Oliveira P.J., Vega-Naredo I. Mitochondria in cancer stem cells: a target for therapy. Recent Pat. Endocr., Metab. Immune Drug Discovery. 2013;7:102–114. doi: 10.2174/18722148113079990006. [DOI] [PubMed] [Google Scholar]
- Lu P., Wang Y., Ma B., She J., Zhang Q., He M., Liu Y. Pharmacophore-based discovery of new human dihydroorotate dehydrogenase inhibitor. Med. Chem. 2014;10:402–408. doi: 10.2174/15734064113096660047. [DOI] [PubMed] [Google Scholar]
- Moreno-Sánchez R., Marín-Hernández A., Saavedra E., Pardo J.P., Ralph S.J., Rodríguez-Enríquez S. Who controls the ATP supply in cancer cells? Biochemistry lessons to understand cancer energy metabolism. Int. J. Biochem. Cell Biol. 2014;50:10–23. doi: 10.1016/j.biocel.2014.01.025. [DOI] [PubMed] [Google Scholar]
- Nimkar S.K., Anderson A.H., Rimoldi J.M., Stanton M., Castagnoli K.P., Mabic S., Wang Y.X., Castagnoli N., Jr. Synthesis and monoamine oxidase B catalyzed oxidation of C-4 heteroaromatic substituted 1,2,3,6-tetrahydropyridine derivatives. Chem. Res. Toxicol. 1996;9:1013–1022. doi: 10.1021/tx960063o. [DOI] [PubMed] [Google Scholar]
- Nimkar S.K., Mabic S., Anderson A.H., Palmer S.L., Graham T.H., de Jonge M., Hazelwood L., Hislop S.J., Castagnoli N., Jr. Studies on the monoamine oxidase-B-catalyzed biotransformation of 4-azaaryl-1-methyl-1,2,3,6-tetrahydropyridine derivatives. J. Med. Chem. 1999;42:1828–1835. doi: 10.1021/jm9900319. [DOI] [PubMed] [Google Scholar]
- Oliva C.R., Moellering D.R., Gillespie G.Y., Griguer C.E. Acquisition of chemoresistance in gliomas is associated with increased mitochondrial coupling and decreased ROS production. PLoS One. 2011;6:e24665. doi: 10.1371/journal.pone.0024665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omuro A., DeAngelis L.M. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310:1842–1850. doi: 10.1001/jama.2013.280319. [DOI] [PubMed] [Google Scholar]
- Pathak R.K., Marrache S., Harn D.A., Dhar S. Mito-DCA: a mitochondria targeted molecular scaffold for efficacious delivery of metabolic modulator dichloroacetate. ACS Chem. Biol. 2014;9:1178–1187. doi: 10.1021/cb400944y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penmatsa A., Wang K.H., Gouaux E. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature. 2013;503:85–90. doi: 10.1038/nature12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak J., Van Noorden S. Bios Scientific Publishers/Royal Microscopical Society; Oxford, England: 2003. Introduction to Immunocytochemistry. [Google Scholar]
- Polavarapu A., Stillabower J.A., Stubblefield S.G.W., Taylor W.M., Baik M.-H. The mechanism of guanine alkylation by nitrogen mustards: a computational study. J. Org. Chem. 2012;77:5914–5921. doi: 10.1021/jo300351g. [DOI] [PubMed] [Google Scholar]
- Rappold P.M., Cui M., Chesser A.S., Tibbett J., Grima J.C., Duan L., Sen N., Javitch J.A., Tieu K. Paraquat neurotoxicity is mediated by the dopamine transporter and organic cation transporter-3. Proc. Natl. Acad. Sci. 2011;108:20766–20771. doi: 10.1073/pnas.1115141108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reily C., Mitchell T., Chacko B.K., Benavides G.A., Murphy M.P., Darley-Usmar V.M. Mitochondrially targeted compounds and their impact on cellular bioenergetics. Redox Biol. 2013;1:86–93. doi: 10.1016/j.redox.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rin Jean S., Tulumello D.V., Wisnovsky S.P., Lei E.K., Pereira M.P., Kelley S.O. Molecular vehicles for mitochondrial chemical biology and drug delivery. ACS Chem. Biol. 2014;9:323–333. doi: 10.1021/cb400821p. [DOI] [PubMed] [Google Scholar]
- Roehm N.W., Rodgers G.H., Hatfield S.M., Glasebrook A.L. An improved colorimetric assay for cell proliferation and viability utilizing the tetrazolium salt XTT. J. Immunol. Methods. 1991;142:257–265. doi: 10.1016/0022-1759(91)90114-u. [DOI] [PubMed] [Google Scholar]
- Sedelis M., Schwarting R.K., Huston J.P. Behavioral phenotyping of the MPTP mouse model of Parkinson's disease. Behav. Brain Res. 2001;125:109–125. doi: 10.1016/s0166-4328(01)00309-6. [DOI] [PubMed] [Google Scholar]
- Sharpe M.A., Livingston A.D., Baskin D.S. Thimerosal-derived ethylmercury is a mitochondrial toxin in human astrocytes: possible role of Fenton chemistry in the oxidation and breakage of mtDNA. J. Toxicol. 2012;2012:373678. doi: 10.1155/2012/373678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe M.A., Han J., Baskin A.M., Baskin D.S. Design and synthesis of a MAO-B-selectively activated prodrug based on MPTP: a mitochondria-targeting chemotherapeutic agent for treatment of human malignant gliomas. ChemMedChem. 2015;10:621–628. doi: 10.1002/cmdc.201402562. [DOI] [PubMed] [Google Scholar]
- Shi H., Noguchi N., Xu Y., Niki E. 1-Methyl-4-phenyl-2,3-dihydropyridinium is transformed by ubiquinone to the selective nigrostriatal toxin 1-methyl-4-phenylpyridinium. FEBS Lett. 1999;461:196–200. doi: 10.1016/s0014-5793(99)01444-1. [DOI] [PubMed] [Google Scholar]
- Simpson J.R., Horton J., Scott C., Curran W.J., Rubin P., Fischbach J., Isaacson S., Rotman M., Asbell S.O., Nelson J.S., Weinstein A.S., Nelson D.F. Influence of location and extent of surgical resection on survival of patients with glioblastoma multiforme: results of three consecutive radiation therapy oncology group (RTOG) clinical trials. Int. J. Radiat. Oncol. Biol. Phys. 1993;26:239–244. doi: 10.1016/0360-3016(93)90203-8. [DOI] [PubMed] [Google Scholar]
- Song Z., Cao Y., Samuels D.C. Replication pauses of the wild-type and mutant mitochondrial DNA polymerase gamma: a simulation study. PLoS Comput. Biol. 2011;7:e1002287. doi: 10.1371/journal.pcbi.1002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strojnik T., Kavalar R., Barone T.A., Plunkett R.J. Experimental model and immunohistochemical comparison of U87 human glioblastoma cell xenografts on the chicken chorioallantoic membrane and in rat brains. Anticancer Res. 2010;30:4851–4860. [PubMed] [Google Scholar]
- Stupp R., Mason W.P., van den Bent M.J., Weller M., Fisher B., Taphoorn M.J.B., Belanger K., Brandes A.A., Marosi C., Bogdahn U., Curschmann J., Janzer R.C., Ludwin S.K., Gorlia T., Allgeier A., Lacombe D., Cairncross J.G., Eisenhauer E., Mirimanoff R.O. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- Sullivan J.P., Tipton K.F. The interactions of monoamine oxidase with some derivatives of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) J. Neural Transm. Suppl. 1990;29:269–277. doi: 10.1007/978-3-7091-9050-0_26. [DOI] [PubMed] [Google Scholar]
- Ubah O.C., Wallace H.M. Cancer therapy: targeting mitochondria and other sub-cellular organelles. Curr. Pharm. Des. 2014;20:201–222. doi: 10.2174/13816128113199990031. [DOI] [PubMed] [Google Scholar]
- Valadez J.G., Sarangi A., Lundberg C.J., Cooper M.K. Primary orthotopic glioma xenografts recapitulate infiltrative growth and isocitrate dehydrogenase I mutation. J. Vis. Exp. 2014 doi: 10.3791/50865. e50865. http://www.jove.com/video/50865/primary-orthotopic-glioma-xenografts-recapitulate-infiltrative-growth. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visanji N.P., Brotchie J.M. John Wiley & Sons, Inc.; 2001. MPTP-induced models of parkinson's disease in mice and non-human primates. (Current Protocols in Pharmacology). [DOI] [PubMed] [Google Scholar]
- Wang J., Edmondson D.E. Topological probes of monoamine oxidases A and B in rat liver mitochondria: inhibition by TEMPO-substituted pargyline analogues and inactivation by proteolysis. Biochemistry. 2011;50:2499–2505. doi: 10.1021/bi101722b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Wolf A., Agnihotri S., Micallef J., Mukherjee J., Sabha N., Cairns R., Hawkins C., Guha A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011;208:313–326. doi: 10.1084/jem.20101470. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material.