

Abstract
Background
Marketing authorization holders (MAHs) are obligated to report adverse events (AEs) within 15 days (some cases 30 days) to the Pharmaceuticals and Medical Devices Agency (PMDA) of Japan.
Methods
To analyze the timeliness of AE reporting to the PMDA, 6610 reports for five categories of cardiovascular devices were retrieved. Two durations were calculated: (1) time from the date the AE occurred to that when the MAH captured it (DOC: days); and (2) time from the date of MAH capture to that of MAH report (DCR: days). Number of DOC > 15 days (DOC15) and delayed reports (DCR > 15 or 30 days) were also calculated.
Results
AEs included 9.2% deaths and 7.5% non-recoveries. DOC15 and delayed reports were 51.0% and 10.9%, respectively. By multivariate analysis, DOC15 was associated with foreign AE, device category, MAH, patient outcome, event category, and AE that had to be reported within 15 or 30 days (AE15/30). Delayed report was associated with device category, MAH, patient outcome, event category, and AE15/30.
Comments
Although Japanese MAHs complied with the obligation to report AEs, they often failed to share AEs with healthcare providers. Registry may be a potential solution, although the cooperation of healthcare providers to input data is essential.
Keywords: Adverse event report, Cardiovascular device, Registry, Pharmaceuticals and Medical Devices Agency
Highlights
-
•
Japanese marketing authorization holders (MAHs) complied with the obligation to report adverse events within the legally determined duration.
-
•
MAHs often failed to share adverse events with healthcare providers in a timely fashion.
-
•
Registry may be a potential solution, although the cooperation of healthcare providers to input data is essential.
With the recent progress of medical technology, an increasing number of new or modified medical devices that require special control has been developed and approved for clinical use (List of approved products: new & improved medical devices). The Pharmaceuticals and Medical Devices Agency (PMDA) of Japan works on the principle that efficacy and safety must be adequately evaluated until approval is granted (Our philosophy. Reviews and related services). However, it is also important to balance this with patient access to such medical devices (Curfman and Redberg, 2011). As the submission files for approval consist of a limited number of cases in pre-approval clinical trials, they are insufficient to reveal the entire safety profile before approval. Adverse event (AE) reporting is thus an important safety measure in combination with detailed case investigation in a certain number of cases as a form of post-marketing surveillance (PMS) in Japan (Post-marketing measures for drug/medical devices; Kramer et al., 2013). Most AEs are provided by the marketing authorization holder (MAH), while a very small number of them come directly from healthcare professionals. Under the provision of the Japanese PMD ACT,1 article 68–10, Japanese MAHs are obligated to report AEs of their own accord within either 15 or 30 days, depending on the nature of the event (Fig. 1a) (Brief overview of the Draft Amendment of Pharmaceutical Affairs Law (PAL)). It is crucial that such AEs be reported promptly so that hazard signals can be detected immediately. The purpose of the present study was to identify the factors that affect the duration of time from AE occurrence to a PMDA report, and discuss how to reduce such durations so that appropriate safety measures can be taken in a timely manner.
Fig. 1.
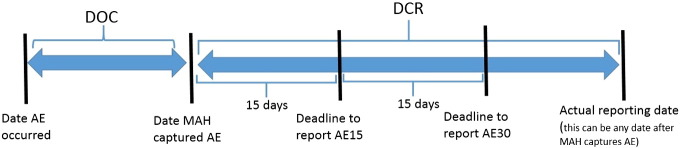
Definition of DOC and DCR.
DOC: duration from the date when AE occurred to the date when MAH captured it. DCR: duration from the date when MAH captured AE to the date MAH reported it. AE: adverse event, AE15: DCR of AE must be within 15 days, AE15 consists of a death or any serious health damage that has not been listed in labeling, and any domestic death event that has been already listed in labeling. AE30: DCR of AE must be within 30 days. AE30 consists of any serious health damage or foreign death event that is listed in labeling.
1. Materials and Methods
The present study was approved by the PMDA's review board and was managed by the Department of Regulatory Science.
AEs that must be reported to the PMDA are separated into the following five categories: AEs that result in death, AEs that are life threatening, AEs that require inpatient hospitalization or prolongation of existing hospitalization, AEs that result in persistent or significant disability or incapacity, and AEs that are a congenital anomaly or birth defect.
From a variety of medical devices, five categories of implantable cardiovascular devices that require surgical intervention were selected for the current analysis because they are implanted permanently, and because the failed products may require replacement by re-do surgery. From the AE database at the PMDA, the initial reports of AEs were retrieved from fiscal year 2004 through 2013. Detailed reports following the initial one were excluded from the analysis, and a total of 6610 AE reports were identified. The five device categories consisted of bio-prosthetic heart valve, mechanical heart valve, prosthetic vascular graft, stent graft, and valved conduit for aortic root replacement.
There are two kinds of AE (Fig. 1). The first type should be reported to the PMDA within 15 days of MAH capture (AE15); whereas the other type should be reported within 30 days (AE30) (Fig. 1). AE15s include fatal events, any serious health damage not listed in the labeling, and any domestic fatal event that has already been listed in the labeling. AE30s consist of any serious health damage that has been listed in the labeling or fatal foreign event that is listed in the labeling. Each AE was classified into one of five event categories: device infection, device related, patient related, procedure related, and under investigation at the time of initial report. Two durations were calculated: duration from the date when the AE occurred to the date when the MAH captured it (DOC: days), and duration from the date when the MAH captured the AE to the date when the MAH reported it to the PMDA (DCR: days) (Fig. 1). Number of DOC15 (DOC > 15 days) and delayed reports (DCR > 15 days in AE15 or DCR > 30 days in AE30) were also calculated.
This study was supported by internal funding from the PMDA.
1.1. Statistical Analysis
Statistical analysis was conducted using the statistical software package JMP version 4.1 (SAS Institute, Inc.). The narrative paragraph of each report was excluded, and the following seven variables, which potentially affect DOC and DCR, were identified and utilized as explanatory variables: (1) location where the event occurred, either domestic or foreign (LOC); (2) known or unknown event (KNW); (3) device category; (4) Japanese MAH; (5) event category; (6) patient outcome; and (7) AE15/30. Two groups were compared using chi-square tests for categorical variables and student t-test for continuous variables. Comparison among multiple groups was conducted using a likelihood ratio test for categorical variables and analysis of variance (ANOVA) for continuous variables. Associations between DOC15 and the seven variables, as well as between the variables and delayed report, were analyzed by multiple logistic regression analysis. The multivariate adjusted odds ratios (ORs) are expressed with 95% confidence intervals (CI). P values of less than 0.05 were considered statistically significant.
2. Results
A total of 6610 AEs were reported over the 10-year period. The number of reports per year dramatically increased over the period (Fig. 2). Median numbers of the two durations were 17 days (range: 0–6271) in DOC, and 19 days (range: 0–1110) in DCR. Median of DOC and DCR over the 10 years is shown in Fig. 3. Of the total AE reports, 51.0% were DOC15, while 10.9% were delayed reports. Proportions of device category, event category, patient outcome, LOC and KNW are listed in the second left column of Table 1. The proportion of DOC15 and delayed reports are detailed in the second column on the right side of Table 1. It is noted that 94.1% of AEs were known events and 5.9% were unknown. Japanese MAHs failed to capture 56.5% of unknown AEs within 15 days, while 13.9% of unknown events had delayed reports. About half of AEs occurred in foreign countries. Japanese MAHs failed to capture 74.7% of AEs from foreign countries within 15 days, and 18.6% of AEs to the PMDA were delayed.
Fig. 2.
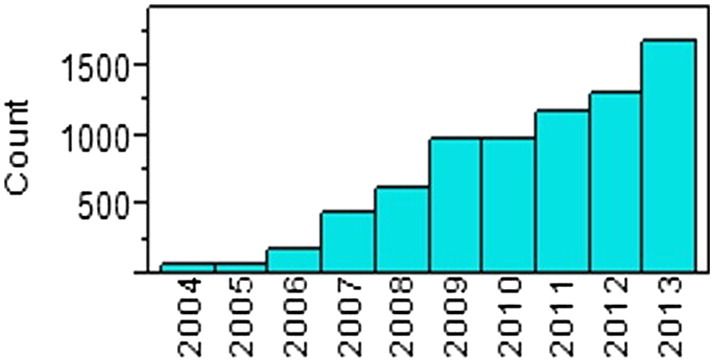
Number of AE reports over 10 years.
AE: adverse event.
Fig. 3.
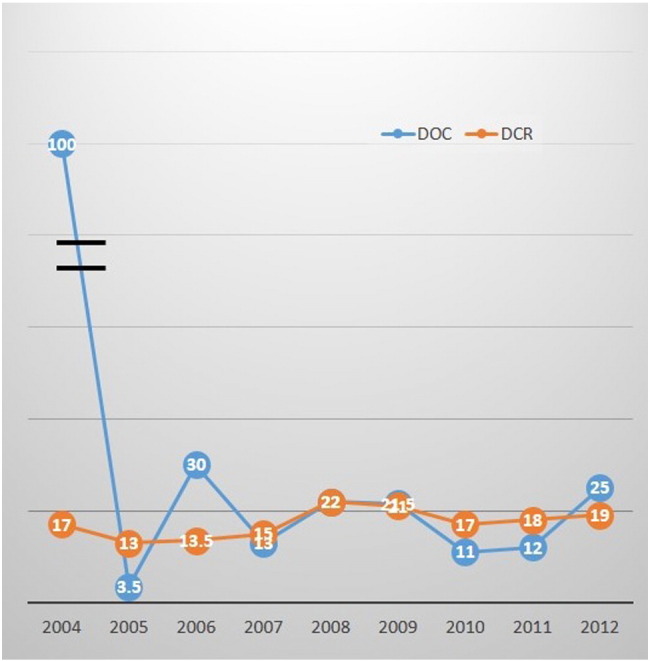
Medians of DOC and DCR over 10 years.
DOC: duration from the date when adverse event (AE) occurred to the date when marketing authorization holder captured it. DCR: duration from the date when MAH captured AE to the date MAH reported it.
Table 1.
Proportion of DOC15 and delayed report.
| Proportion (%) | DOC15 (%) | P value | Delayed report (%) | P value | |
|---|---|---|---|---|---|
| All AE reports | 6610 (100%) | 3305 (51.0%) | 72 (10.9%) | ||
| Known or unknown | |||||
| Known event | 6213 (94.1%) | 315 (50.8%) | 0.1189 | 67 (10.8%) | 0.3555 |
| Unknown event | 397 (5.9%) | 224 (56.5%) | 55 (13.9%) | ||
| Location of event | |||||
| Domestic AE | 3252 (49.2%) | 741 (22.8%) | < 0.0001 | 16 (4.9%) | 0.5316 |
| Foreign AE | 3358 (50.8%) | 2508 (74.7%) | 625 (18.6%) | ||
| AE15/30 | |||||
| Within 15 days | 1752 (26.5%) | 336 (19.2%) | < 0.0001 | 482 (27.5%) | < 0.0001 |
| Within 30 days | 4858 (73.5%) | 3089 (63.6%) | 209 (4.3%) | ||
| MAH | |||||
| A | 2132 (32.2%) | 772 (36.2%) | < 0.0001 | 14 (0.66%) | < 0.0001 |
| B | 1477 (22.3%) | 940 (63.6%) | 570 (38.6%) | ||
| C | 1223 (18.5%) | 410 (33.5%) | 58 (4.74%) | ||
| D | 641 (9.7%) | 335 (52.3%) | 18 (2.81%) | ||
| E | 595 (9.0%) | 506 (85.0%) | 25 (4.20%) | ||
| F | 185 (2.8%) | 120 (64.9%) | 16 (8.65%) | ||
| G | 152 (2.3%) | 74 (48.7%) | 7 (4.61%) | ||
| H | 106 (1.6%) | 23 (21.7%) | 4 (3.77%) | ||
| Others | 99 (1.5%) | 59 (59.6%) | 14 (14.1%) | ||
| Event category | |||||
| Device infection | 336 (5.1%) | 186 (55.4%) | < 0.0001 | 12 (3.6%) | 0.0215 |
| Device related | 2932 (44.4%) | 1358 (46.3%) | 343 (11.7%) | ||
| Patient related | 667 (10.1%) | 446 (66.9%) | 39 (5.8%) | ||
| Procedure related | 2193 (33.2%) | 1097 (50.0%) | 270 (12.3%) | ||
| Under investigation | 482 (7.3%) | 305 (63.3%) | 32 (6.6%) | ||
| Patient outcome | |||||
| No health damage | 588 (8.9%) | 255 (43.4%) | < 0.0001 | 91 (15.5%) | < 0.0001 |
| Recovered | 2571 (38.9%) | 1327 (51.6%) | 247 (9.6%) | ||
| Non-recovery | 496 (7.5%) | 192 (38.7%) | 77 (15.5%) | ||
| Death | 608 (9.2%) | 424 (69.7%) | 58 (9.5%) | ||
| Observation or under treatment | 985 (14.9%) | 171 (17.4%) | 93 (9.4%) | ||
| Unknown or under investigation | 1362 (20.6%) | 801 (58.8%) | 151 (11.1%) | ||
| Device category | |||||
| Mechanical heart valve | 793 (12.0%) | 416 (52.5%) | < 0.0001 | 29 (3.7%) | 0.0350 |
| Tissue heart valve | 945 (14.3%) | 229 (24.2%) | 45 (4.8%) | ||
| Prosthetic vascular graft | 304 (4.6%) | 124 (40.8%) | 32 (10.5%) | ||
| Stent graft | 4383 (66.3%) | 2533 (57.8%) | 587 (13.4%) | ||
| Valved conduit | 185 (2.8%) | 62 (33.5%) | 20 (10.8%) |
Two groups were compared using chi-square tests for categorical variables and student t-test for continuous variables. Comparison among multiple groups was conducted using a likelihood ratio test for categorical variables and analysis of variance (ANOVA) for continuous variables. DOC15: duration more than 15 days from the date the adverse event occurred to that when the MAH captured it, AE: adverse event, AE15/30: adverse event which should be reported within 15 days versus within 30 days, and MAH: marketing authorization holder.
Of the 6610 AE reports, about a quarter was AE15, while three quarters were AE30. The proportion of DOC15 was higher in AE30 than in AE15, while the proportion of delayed reports was higher in AE15 than in AE30. With respect to event category, device-related events were the most frequent, about 44.4% of AEs. AEs related to procedures were the second most common. DOC15 of device-related AEs was 46.3%, while delayed report of device-related AEs was 11.7%. Fig. 4 is a scatter diagram showing the proportions of DOC15 and delayed reports stratified by Japanese MAH. The figure shows that out of eight major MAHs in Japan, seven reported more than 90% of AEs to the PMDA within the legally determined duration. Meanwhile, the proportion of DOC15 was widely varied among the MAHs. Among patient outcomes, death and non-recovery events represented 9.2% and 7.5%, respectively.
Fig. 4.
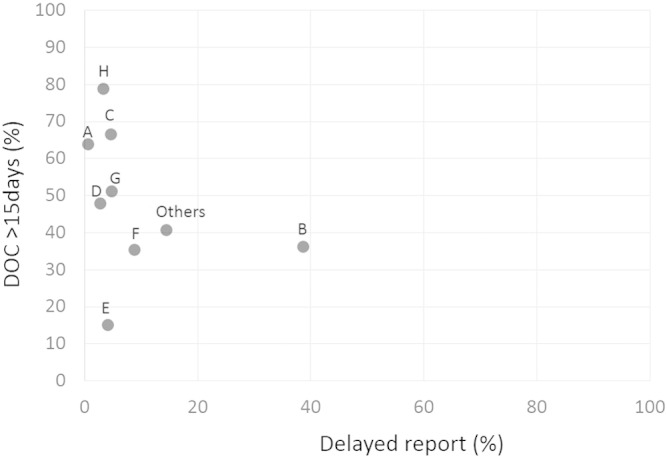
DOC15/delayed report stratified by Japanese MAH.
DOC: duration from the date the AE occurred to that when the MAH captured it and DOC15: DOC more than 15 days.
2.1. Multiple Logistic Regression Analysis for DOC15
Although the univariate analyses of the proportion of DOC15 for KNW did not show any significant differences (P = 0.1189), it can potentially affect DCR. Therefore, all seven variables were included as explanatory variables in the multiple logistic regression analysis of DOC15. The result demonstrated that DOC15 was associated with the seven explanatory variables as follows: LOC (P < 0.0001), KNW (P < 0.0001), device category (P < 0.0001), event category (P < 0.0001), MAH (P < 0.0001), patient outcome (P < 0.0001), and AE15/30 (P < 0.0001) (Table 2). The multivariate adjusted odds ratio was calculated with reference to the mechanical heart valve in the device category, device-related AE in the event category, company A in MAH, and no health damage in patient outcome. The results are shown in Table 2. Adjusted odds ratio of unknown to known AE regarding DOC15 was 3.35 (CI: 2.16–5.20, P < 0.0001). Adjusted odds ratio of foreign to domestic AE was 8.72 (CI: 7.24–10.54, P < 0.0001). Several MAHs showed higher odds ratio of DOC15 compared with company A. Out of the five event categories, DOC15 of device infection had a significantly higher odds ratio compared to that of device-related events. In terms of patient outcome, DOC15 of any AEs that caused any health damage to the patient had a higher odds ratio than that of an AE that caused no heath damage. In particular, DOC15 of a fatal event had the highest odds ratio of 5.84 (CI: 4.07–8.44).
Table 2.
Results of multiple logistic regression analysis for DOC15.
| Odds ratio | 95% Lower limit | 95% Upper limit | P value | P value | |
|---|---|---|---|---|---|
| Known AE | 1.00 | Reference | < 0.0001 | ||
| Unknown AEa | 3.35 | 2.16 | 5.20 | ||
| AE30 | 1.00 | Reference | < 0.0001 | ||
| AE15b | 0.24 | 0.18 | 0.32 | ||
| Domestic AE | 1.00 | Reference | < 0.0001 | ||
| Foreign AEc | 8.72 | 7.24 | 10.54 | ||
| MHA | |||||
| A | 1.00 | Reference | < 0.0001 | ||
| B | 1.96 | 1.53 | 2.55 | < 0.0001 | |
| C | 3.48 | 2.71 | 4.48 | < 0.0001 | |
| D | 0.66 | 0.41 | 1.04 | 0.0749 | |
| E | 3.30 | 1.95 | 5.58 | < 0.0001 | |
| F | 0.61 | 0.29 | 1.29 | 0.1971 | |
| G | 1.37 | 0.75 | 2.48 | 0.3033 | |
| H | 12.10 | 7.06 | 21.64 | < 0.0001 | |
| Others | 1.38 | 0.70 | 2.65 | 0.3440 | |
| Event category | |||||
| Device related | 1.00 | Reference | < 0.0001 | ||
| Device infection | 2.45 | 1.68 | 3.58 | < 0.0001 | |
| Patient related | 1.27 | 0.96 | 1.67 | 0.0904 | |
| Procedure related | 0.70 | 0.59 | 0.84 | 0.0001 | |
| Under investigation | 1.13 | 0.85 | 1.51 | 0.3871 | |
| Patient outcome | |||||
| No health damage | 1.00 | Reference | < 0.0001 | ||
| Recovered | 1.85 | 1.42 | 2.39 | < 0.0001 | |
| Unrecovered | 2.03 | 1.45 | 2.84 | < 0.0001 | |
| Death | 5.84 | 4.07 | 8.44 | < 0.0001 | |
| Observation or under treatment | 0.61 | 0.39 | 0.95 | 0.0303 | |
| Unknown or under investigation | 1.78 | 1.36 | 2.33 | < 0.0001 | |
| Device category | |||||
| Mechanical heart valve | 1.00 | Reference | < 0.0001 | ||
| Tissue heart valve | 0.64 | 0.45 | 0.92 | 0.0152 | |
| Prosthetic vascular graft | 2.61 | 0031.37 | 4.98 | 0.0038 | |
| Stent graft | 1.85 | 1.14 | 2.99 | 0.0129 | |
| Valved conduit | 0.65 | 0.37 | 1.12 | 0.1189 |
AE: adverse event. MAH: marketing authorization holder. DOC15: duration more than 15 days from the date the AE occurred to that when the MAH captured it. DCR: duration from the date when the MAH captured the AE to the date when the MAH reported it to the Pharmaceutical and Medical Device Agency. DCR of AE must be within 15 days, AE15 consists of a death or any serious health damage that has not been listed in labeling, and any domestic death event that has been already listed in labeling. AE30: DCR of AE must be within 30 days. AE30 consists of any serious health damage or foreign death event that is listed in labeling.
Associations between DOC15 and the seven variables were analyzed by multiple logistic regression analysis. The multivariate adjusted odds ratios (ORs) are expressed with 95% confidence interval (CI). P values of less than 0.05 are considered statistically significant.
The odds ratio of unknown AE refer to Known AE.
The odds ration of AE15 refer to AE 30.
The odds ration of foreign AE refer to domestic AE.
2.2. Multiple Logistic Regression Analysis for Delayed Report
Although the univariate analyses of the proportion of delayed reports for KNW and LOC did not show any significant differences, they potentially affect DCR. Therefore, all seven variables were included in the multivariate analysis for delayed report. The results of multiple logistic regression analysis for delayed report demonstrated that it was associated with device category (P = 0.0350), MAH (P < 0.0001), patient outcome (P < 0.0001), event category (P = 0.0215) and AE15/30 (P < 0.0001) (Table 3). The multivariate adjusted odds ratio was calculated in reference to the mechanical heart valve in the device category, device-related AE in the event category, company A in MAH, and no health damage in patient outcome. Delayed report was more frequent in AE15 than in AE30. For certain MAHs, odds ratios of delayed report were significantly higher compared with that of company A. With respect to event category, delayed report of procedure-related event was less frequent than that of device-related AEs. In terms of patient outcome, reports of health damage in which the patient either recovered or did not recover were less frequent than events in which there was no health damage.
Table 3.
Results of multiple logistic regression analysis for delayed report.
| Odds ratio | 95% Lower limit | 95% Upper limit | P value | P value | |
|---|---|---|---|---|---|
| Known AE | 1.00 | Reference | 0.3555 | ||
| Unknown AEa | 1.34 | 0.72 | 2.43 | ||
| AE30 | 1.00 | Reference | < 0.0001 | ||
| AE15b | 3.51 | 2.26 | 5.50 | ||
| Domestic AE | 1.00 | Reference | 0.5316 | ||
| Foreign AEc | 0.87 | 0.57 | 1.34 | ||
| MHA | |||||
| A | 1.00 | Reference | < 0.0001 | ||
| B | 50.8 | 28.1 | 101.9 | < 0.0001 | |
| C | 3.35 | 1.53 | 7.60 | 0.0025 | |
| D | 1.87 | 0.64 | 5.55 | 0.2558 | |
| E | 0.84 | 0.30 | 2.43 | 0.7415 | |
| F | 0.38 | 0.43 | 12.39 | 0.3113 | |
| G | 2.71 | 0.72 | 10.09 | 0.1405 | |
| H | 5.57 | 1.20 | 19.17 | 0.0307 | |
| Others | 5.22 | 11.47 | 18.14 | 0.0106 | |
| Classification of event | |||||
| Device related | 1.00 | Reference | 0.0004 | ||
| Device infection | 0.83 | 0.34 | 1.77 | 0.6584 | |
| Patient related | 1.11 | 0.62 | 1.90 | 0.725 | |
| Procedure related | 0.66 | 0.52 | 0.85 | 0.0014 | |
| Under investigation | 0.73 | 0.43 | 1.19 | 0.2148 | |
| Patient outcome | |||||
| No health damage | 1.00 | Reference | < 0.0001 | ||
| Recovered | 0.44 | 0.30 | 0.66 | < 0.0001 | |
| Unrecovered | 0.37 | 0.23 | 0.60 | < 0.0001 | |
| Death | 0.71 | 0.42 | 1.21 | 0.2152 | |
| Observation or under treatment | 0.93 | 0.49 | 1.77 | 0.836 | |
| Unknown or under investigation | 0.86 | 0.57 | 1.30 | 0.4714 | |
| Device category | |||||
| Mechanical heart valve | 1.00 | Reference | < 0.0001 | ||
| Tissue heart valve | 1.17 | 0.51 | 2.71 | 0.7184 | |
| Prosthetic vascular graft | 2.28 | 0.65 | 7.66 | 0.194 | |
| Stent graft | 0.56 | 0.23 | 1.45 | 0.2275 | |
| Valved conduit | 1.73 | 0.68 | 4.45 | 0.251 |
AE: adverse event. MAH: marketing authorization holder. DCR: duration from the date when the MAH captured the AE to the date when the MAH reported it to the Pharmaceutical and Medical Device Agency. AE15: AE15 consists of a death or any serious health damage that has not been listed in labeling, and any domestic death event that has been already listed in labeling. AE30: DCR of AE must be within 30 days. AE30 consists of any serious health damage or foreign death event that is listed in labeling.
Associations between the variables and delayed report were analyzed by multiple logistic regression analysis. The multivariate adjusted odds ratios (ORs) are expressed with 95% confidence intervals (CI). P values of less than 0.05 are considered statistically significant.
The odds ratio of unknown AE refer to known AE.
The odds ration of AE15 refer to AE 30.
The odds ration of foreign AE refer to domestic AE.
2.3. The PMDA's Reaction to AE Reports
Out of the 6610 AEs, the PMDA took safety measures, such as recall, for 44 events (0.67%) and modification of package inserts for 51 events (0.77%). More than half of AE reports (3940/6610) did not require any additional safety measures, and 28.6% (1888/6610) only required reminders to medical professionals of alerts or instructions for use already noted in the package inserts.
3. Discussion
The PMDA was founded in April 2004. Since then, AE reports of medical devices have been collected in the database of the PMDA Safety Department. It was evident that the number of AEs increased dramatically over the 10-year period considered here (Fig. 1b). This must be related to the rapid increase in new and modified medical devices, in particular the approval of specially controlled medical devices (List of approved products: new & improved medical devices). In the present study, in order to analyze in how timely a manner the PMDA has been provided with AE reports, two durations were calculated, DOC and DCR. DOC is related to how Japanese MAHs collect AE reports from healthcare providers or the central headquarters of manufacturers in foreign countries. Although healthcare professionals or medical institutions reported a very small number of AEs directly to the PMDA, the overwhelming majority of AEs were reported by Japanese MAHs (Kramer et al., 2013). In the details of the study results, MAHs appeared to struggle to capture AEs characterized as unknown event, device infection, and certain types of prostheses such as prosthetic vascular graft or stent graft. In addition, it took more than 15 days for MAHs to capture about 70% of fatal events, and about 60% of events not listed in labeling or still under investigation at the time of the initial report. Unknown AEs and death events should be reported to the PMDA immediately so that the hidden health hazard can be determined. Overall, only 49% of AEs were recognized by MAHs within 15 days. Although most MAHs reported more than 90% of AEs within the legally determined duration, the proportion of DOC15 varied widely among Japanese MAHs (Fig. 2). This was probably due to varying attitudes of MAHs as to how aggressively to collect AEs, or to the philosophy of foreign manufacturers as to how to share AEs with Japanese MAHs.
The Japanese Pharmaceutical Affairs Law was amended to the PMD ACT in November 2013, which enforces the safety of drugs, medical devices and regenerative products (Act on Securing Quality, Efficacy and Safety of Pharmaceuticals, Medical Devices, Regenerative and Cellular Therapy Products, Gene Therapy Products, and Cosmetic). The Japanese PMD ACT imposes AE reports and PMS, as well as clinical evaluation and inspection on MAHs that sell Class III or IV devices. It mandated that Japanese MAHs report AEs directly to the PMDA (Article 68–10) (Kramer et al., 2013). Regarding foreign AEs, Japanese MAHs have a strict obligation to obtain such information from the manufacturer or medical healthcare professionals abroad and report them to the PMDA immediately. Unfortunately, the present study suggested that AEs from foreign countries were significantly delayed, probably because foreign manufacturers failed to share AEs with healthcare providers, or because these manufacturers delayed informing the Japanese MAHs about the AE. Healthcare providers and foreign manufacturers are asked to make an effort to cooperate with Japanese MAHs. After the PMDA analyzes an individual AE report, it may conclude that further action is required, and impose additional safety measures such as recalls or changes in labeling. Although AEs that required additional measures totaled only 1.4% in this study, timely reporting of AEs is fundamental to the success of PMS. As a safety measure, the Japanese PMD ACT requires MAHs to provide the most recent evidence regarding the effectiveness and safety profile in the package inserts of drugs, medical devices, and regenerative products (Brief overview of the Draft Amendment of Pharmaceutical Affairs Law (PAL)). The modification and new evidence must then be registered in the PMDA database and disclosed to the public (Act on Securing Quality, Efficacy and Safety of Pharmaceuticals, Medical Devices, Regenerative and Cellular Therapy Products, Gene Therapy Products, and Cosmetic).
The efficacy and safety profile of new drugs and devices should be reasonably demonstrated in the pre-market review process before approval. However, the limited number of cases in the submission file, limited combination therapy, and limited follow-up period leaves unanswered questions of effectiveness and safety. The PMS in Japan includes systems for reporting foreign and domestic AEs, identifying safety signals emerging from international markets, and re-filing applications 3–7 years after initial market approval (Kramer et al., 2013; Brief overview of the Draft Amendment of Pharmaceutical Affairs Law (PAL)). Japan has a unique system to reexamine approved devices that carry a high risk by surveying all cases in a pre-specified period or pre-determined number of subjects. Sponsors must aggregate information from healthcare providers, clinical trials, and published studies such as foreign and domestic observational research or experience from registries to demonstrate that the device is providing the expected safety and effectiveness results (Kramer et al., 2013; Brief overview of the Draft Amendment of Pharmaceutical Affairs Law (PAL)). Although a brief report is submitted to the PMDA annually, a final report to re-file that includes full assessment of effectiveness and safety is usually submitted 3–7 years after approval. By then, a modified device may have been approved so that that the original device may have disappeared from the market. It is obvious that life-cycle management of medical devices has shortened due to frequent modifications in response to requests from the clinical field (Act on Securing Quality, Efficacy and Safety of Pharmaceuticals, Medical Devices, Regenerative and Cellular Therapy Products, Gene Therapy Products, and Cosmetic; Levesque et al., 2014). Early detection of new hazards is essential in the short life cycle of device modification. In addition, if the modification of the device is small, leaving the modified device practically equivalent to the original one, no clinical data or re-assessment is necessary for approval. In such cases, AE reports are the only source of information in detecting hidden hazard signals. Accordingly, AE reports play a central role in detecting hazard signals quickly after approval.
The PMDA address two conflicting goals: the first is to assure the public that the devices are reasonably safe and effective; and the second is to avoid overregulation of manufacturers in the development of Class III or IV devices (Post-marketing measures for drug/medical devices). The PMDA shifted the regulatory standards to a less burdensome approach in all areas of medical devices and reduced the terms of device development. As a part of the implementation of reducing such terms for certain devices with little clinical data in the Japanese population, the PMDA has accepted clinical trials conducted in the US, European Union, or other countries. In these cases, reports of foreign as well as domestic AEs are an extremely important source of information.
Registry, in collaboration with academic societies as well as manufacturers, is a promising potential solution for PMS (Resnic and Normand, 2012). For example, Interagency Registry for Mechanically Assisted Circulatory Support (INTERMACS) was very successful in monitoring the results of a left ventricular assist device in the real world and detecting hazard signals (Grady et al., 2015 Feb; Kirklin et al., 2014). J-MACS, the Japanese version of INTERMACS, has a system that sends an alert mail to the device company when healthcare professionals input an AE into the system (Japanese Registry for Mechanically Assisted Circulatory Support). AEs in combination with registry of follow-up data would provide both the number of events and number of devices implanted for a patient-year follow-up period. One possible drawback of registry is that the more patients that register with the device, the more difficult it would be to collect follow-up data (Smith et al., 2012). However, in the advent of trans-catheter aortic valve replacement (TAVR) in Japan, PMS with an AE alert mail system was embedded in a national TAVR registry formed by academic societies; and the system is designed to collect 5 years of follow-up data (Japanese Registry of Trans-Catheter Aortic Valve Replacement).
4. Conclusions
The present study suggested that multiple factors influenced the delay of AE reports. Japanese MAHs had difficulty in sharing AE appropriately with healthcare providers or foreign manufacturers, particularly for critical events such as death or unknown event. Registry, in combination with a system to alert the MAH, may be a potential solution for functional AE reporting. Even so, the cooperation of healthcare providers to input the data into the registry will be essential. Once a medical device is approved for the Japanese market, it is strongly encouraged that MAHs develop a functional AE reporting system in collaboration with healthcare providers and academic societies, particularly given the current short life cycle of new or modified medical devices.
Author Contributions
Study concept and design: Handa and Ishii.
Acquisition of data: Handa and Matsui.
Statistical analysis: Ando and Handa.
Critical revision of the manuscript: Ishii, Ando and Matsui.
Analysis and interpretation of data: Matsui, Ishii, and Handa.
Financial Disclosure
This study was supported by internal funding from the Pharmaceuticals and Medical Devices Agency of Japan.
Acknowledgment
The views expressed in this article are those of the authors and do not necessarily reflect the official views of the Pharmaceuticals and Medical Devices Agency of Japan.
Footnotes
Disclosures: all authors assert that they have no conflict of interest to disclose.
References
- List of approved products: new & improved medical devices. http://www.pmda.go.jp/english/service/list_s.htm Accessed March 11, 2015, at.
- Our philosophy. Reviews and related services. http://www.pmda.go.jp/english/about/pdf/profile_of_services.pdf Accessed March 11, 2015, at.
- Brief overview of the Draft Amendment of Pharmaceutical Affairs Law (PAL) http://www.puntofocal.gov.ar/notific_otros_miembros/jpn428_t.pdf
- Act on Securing Quality, Efficacy and Safety of Pharmaceuticals, Medical Devices, Regenerative and Cellular Therapy Products, Gene Therapy Products, and Cosmetic. http://www.mhlw.go.jp/stf/seisakunitsuite/bunya/0000045726.html Accessed March 11, 2015, at. (Japanese)
- Japanese Registry for Mechanically Assisted Circulatory Support. http://www.pmda.go.jp/english/service/pdf/j-macs/j-macs_brochure.pdf Accessed March 11, 2015, at.
- Japanese Registry of Trans-Catheter Aortic Valve Replacement. http://j-tavr.com/about.html Accessed March 11, 2015, at. (Japanese)
- Curfman G.D., Redberg R.F. Medical devices—balancing regulation and innovation. N. Engl. J. Med. 2011;365:975–977. doi: 10.1056/NEJMp1109094. [DOI] [PubMed] [Google Scholar]
- Grady K.L., Naftel D.C., Myers S. Change in health-related quality of life from before to after destination therapy mechanical circulatory support is similar for older and younger patients: analyses from INTERMACS. J. Heart Lung Transplant. 2015;34(2):213–221. doi: 10.1016/j.healun.2014.10.001. (Feb) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirklin J.K., Naftel D.C., Pagani F.D. Sixth INTERMACS annual report: a 10,000-patient database. J. Heart Lung Transplant. 2014;33:555–564. doi: 10.1016/j.healun.2014.04.010. [DOI] [PubMed] [Google Scholar]
- Kramer D.B., Tan Y.T., Sato C., Kesselheim A.S. Postmarket surveillance of medical devices: a comparison of strategies in the US, EU, Japan, and China. PLoS Med. 2013;10:e1001519. doi: 10.1371/journal.pmed.1001519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque K., Coqueblin C., Guillot B. Post-approval studies in France, challenges facing medical devices. Therapie. 2014;69:303–321. doi: 10.2515/therapie/2014051. [DOI] [PubMed] [Google Scholar]
- Post-marketing measures for drug/medical devices. http://www.mhlw.go.jp/english/wp/wp-hw5/dl/23010232e.pdf Accessed March 11, 2015, at.
- Resnic F.S., Normand S.L. Postmarketing surveillance of medical devices—filling in the gaps. N. Engl. J. Med. 2012;366(10):875–877. doi: 10.1056/NEJMp1114865. (Mar 8) [DOI] [PubMed] [Google Scholar]
- Smith A.J., Dieppe P., Vernon K., Porter M., Blom A.W. Failure rates of stemmed metal-on-metal hip replacements: analysis of data from the National Joint Registry of England and Wales. Lancet. 2012;379:1199–1204. doi: 10.1016/S0140-6736(12)60353-5. [DOI] [PubMed] [Google Scholar]