

Abstract
The unfolded protein response of the endoplasmic reticulum (UPRER) is a conserved signaling circuit that ensures ER protein homeostasis (proteostasis). In the UPRER of higher eukaryotes, multiple sensors cooperatively perceive proteostatic disturbances in the ER lumen and induce downstream adaptive changes. Besides direct proteotoxic insults, altered lipid profiles can also lead to UPRER activation, evidently because abnormal lipid composition impairs protein folding. However, 2 recent studies propose an alternative mechanism of UPRER sensor activation. In one report, UPRER activation occurred in cells expressing UPRER sensors lacking the very domains that sense unfolded proteins; the other study found that Caenorhabditis elegans worms displayed UPRER activation without apparent proteostatic imbalance in the ER lumen. Collectively, these studies suggest that lipid disequilibrium-activated UPRER is not strictly accompanied by compromised ER proteostasis and hint at a lipid membrane-monitoring role of the UPRER. These discoveries raise several important questions: does the UPRER monitor and maintain homeostasis of the ER membrane and/or its lipids? In turn, does the UPRER initiate downstream regulatory events that specifically alleviate lipid or proteostatic imbalance? And what is the physiological significance of proteostasis-independent UPRER activation? In this commentary, we will discuss these issues and highlight the utility of C. elegans as an in vivo model to study lipid disequilibrium-induced UPRER and related pathways.
Keywords: endoplasmic reticulum, mediator complex, phosphatidylcholine, phospholipid, unfolded protein response
Abbreviations
- ER
endoplasmic reticulum
- UPR
unfolded protein response
- IRE-1
Inositol-Requiring-Enzyme 1
- PERK
protein kinase RNA-like ER kinase
- ATF-6
Activating Transcription Factor 6
- PC
phosphatidylcholine
- SAM
S-Adenosyl methionine
- SAMS-1
SAM synthetase 1
Introduction
The unfolded protein response of the endoplasmic reticulum (UPRER) is an evolutionarily conserved regulatory mechanism that allows the ER to adapt to proteostatic imbalance.1 Specifically, the accumulation of un- or misfolded proteins in the ER lumen is perceived directly and indirectly by ER-membrane anchored sensors. In higher eukaryotes, 3 parallel UPRER signaling pathways detect disturbed proteostasis and implement appropriate downstream response programs: the Inositol-Requiring-Enzyme 1 (IRE-1) branch, the protein kinase RNA-like ER kinase (PERK) branch, and the Activating Transcription Factor 6 (ATF-6) branch.1,2 Together, these factors reprogram transcription and translation, thus allowing cells, tissues, or organisms to adapt to the stress or initiate programmed cell death.1,3 The evolutionary conservation of the key sensor and effector genes and their downstream signaling pathways illuminates the critical need of ER integrity for cell and organism function and survival.
The UPRER is not only activated by proteostatic imbalance, but also by perturbations of other processes that contribute to ER homeostasis, e.g. calcium storage and membrane lipid composition.4-11 For example, decreased synthesis of phosphatidylcholine (PC), altered fatty acid desaturation, abnormal sterol levels, and organismal obesity all induce the UPRER. In most of these scenarios, UPRER induction appears to reflect underlying proteostatic imbalance, as it can be alleviated by exogenous chemical chaperones that improve protein folding. However, recent studies suggest that the UPRER may additionally sense and signal disturbances of the ER membrane lipids themselves, in manners that are independent of ER proteostasis. First, Volmer et al. showed that cultured cells and reconstituted vesicles experiencing abnormal membrane lipid saturation retain the ability to induce downstream signals even when expressing IRE1 and PERK mutants that lack unfolded protein sensing domains.8 Subsequently, we showed that, in the nematode worm Caenorhabditis elegans, the UPRER is activated by abnormal lipid saturation or reduced PC levels in the absence of unfolded proteins.12 Thus, in vivo states apparently exist whereby the UPRER is activated despite unfolded proteins either being absent or not being detectable, suggesting that lipid disequilibrium may directly activate the UPRER. Here, we discuss the implications of these findings, focusing on a possible role of lipids as inputs or outputs of the UPRER.
The Relationship Between Membrane Lipids and the UPRER
The ER synthesizes, folds, and secretes proteins. In line with this important role, an ER resident machinery has evolved that monitors and adapts proteostasis, the UPRER.1,2,13 However, the ER is also a major site of lipid synthesis, particularly of phospholipids and sterols. Taken together with the discovery of UPRER activation without accompanying proteostatic imbalance,8,12 this hints at a broader potential regulatory role for the UPRER (Fig. 1). In such a view, the UPRER would integrate 2 inputs, namely protein and lipid homeostasis, and generate separable outputs to alleviate or eliminate only the specific and relevant insult. In line with this hypothesis, proteotoxic conditions trigger adaptive changes that enable organisms to cope with this type of stress, including the attenuation of general translation, and the induction of chaperones that assist protein folding.1,13,14 As the UPRER is apparently capable of sensing lipid disequilibrium independently of sensing proteostatic imbalance, it will be interesting to assess whether the compensatory changes to lipid disequilibrium similarly serve to alleviate the specific upstream stressors, namely abnormal lipid compositions. For example, in our worm models of increased fatty acid saturation (leading to increased PC saturation) or reduced PC production,12 are relevant biosynthetic pathways modulated to alleviate this membrane stress? Studies using metabolic labeling approaches to quantitate the flux through lipid synthesis, modification, and turnover pathways15 should be particularly insightful in this context.
Figure 1.
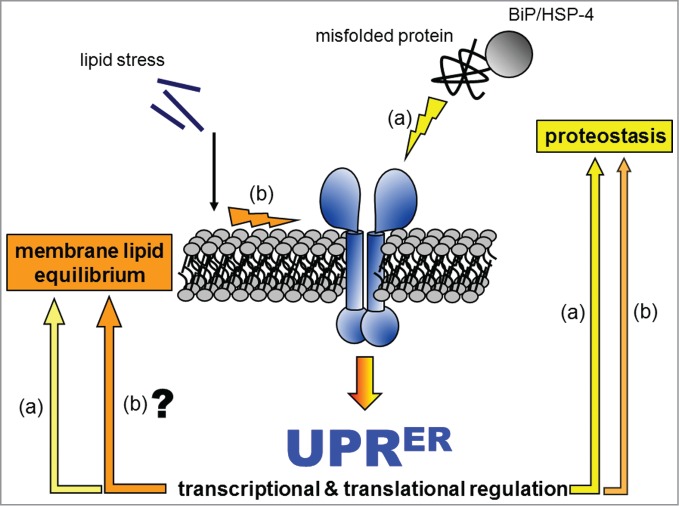
The UPRER engages different downstream outputs in response to distinct upstream inputs. Our hypothetical model of the UPRER incorporates independent sensing of proteostatic or lipid stress, which leads to distinctive downstream outputs that aim to alleviate the specific input stress. In one scenario (A), the UPRER sensors directly and/or indirectly sense the accumulation of misfolded protein in the ER. The resulting downstream outputs promote protein quality control by inducing the expression of chaperones and of the protein degradation machinery, by optimizing the redox environment for protein folding, and by attenuating general translation. In the other scenario (B), lipid stress imposed by free saturated fatty acids, decreased fatty acid desaturation, or reduced PC biosynthesis activates the UPRER sensors through its impact on the ER membrane. This type of UPRER input can occur either with or without misfolded-proteins, suggesting that there may be downstream adaptive outputs dedicated specifically to membrane lipid homeostasis. Simultaneously, lipid disequilibrium also activates pathways promoting protein quality control, as indicated by the activation of conventional UPRER markers (e.g., the chaperone BiP).
Crosstalk Between Proteostatic and Lipid Metabolic Inputs and Outputs of the UPRER
Although the model presented in the previous paragraph is attractive at first glance, it also fails to reflect many observations made in vivo. That is, there may be overlap and/or crosstalk between the downstream regulatory outputs regardless of the precise nature of the upstream input (i.e., protein or lipid imbalance). Indeed, several studies have reported that proteotoxic insults elevate the expression of genes involved in lipid metabolism. For example, in C. elegans, acute ER stress imposed by the widely used protein glycosylation inhibitor tunicamycin caused adaptive changes in the expression of numerous genes.14 These include phospholipid metabolism genes such as choline kinases, which catalyze de novo synthesis of PC, the most abundant ER membrane lipid.16 These inductions may reflect altered demands of the ER, which the UPRER accommodates by specifically adjusting ER membrane lipid composition and hence properties of the ER membrane. Indeed, ER volume expands substantially in yeast under protein folding stress,17 and overall ER morphology also changes.9,17 Similarly, ER expansion also occurs in human B-cells undergoing proliferation and differentiation, evidently to provide an enlarged ER lumen during times of active protein synthesis and secretion.18 Overexpression of spliced XBP1 (i.e., mimicking the activation of IRE1) is sufficient to drive PC production and ER membrane proliferation,19,20 but whether increased PC biosynthesis and/or ER membrane proliferation is a universal response to proteostatic stress remains unclear. Nevertheless, the potential for a general functional role of membrane lipid adaptation in the response to disturbed proteostasis is also suggested by the fact that depletion of certain lipid metabolism genes results in tunicamycin sensitivity in C. elegans.21
Juxtaposed to the induction of lipid metabolism genes by proteostatic imbalance is the induction of protein quality control factors by lipid disequilibrium. For instance, in our models of lipid imbalance, we observed activation of hsp-4 (encoding the ER-resident chaperone BiP) and phosphorylation of the eukaryotic translation initiation factor eIF2α, an event that attenuates general protein translation;12 similar events were reported during the activation of UPRER in cultured cells expressing sensor mutants lacking unfolded proteins-sensing domains.8 If the insult on the ER in these scenarios were indeed limited to lipid alterations, why would compensatory changes adapt proteostasis? To some extent, these findings may reflect the limitation of the reporters currently available to study the UPRER. For example, induction of HSP-4/BiP is usually interpreted as an indicator of compromised protein folding in the ER, yet Volmer et al. and we showed that such activation need not strictly be accompanied by compromised proteostasis.8,12 Gene expression profiling in worms under lipid disequilibrium might help identify genes that are specifically activated by such changes but not by altered proteostasis. Alternatively, protein folding and processing may be adapted even if the initial insult affected lipid levels or composition, potentially to preemptively prevent further aggregation of overall ER health.
Interestingly, yeast strains with decreased membrane lipid desaturation akin to our worm models present an altered ER morphology that is distinct from the structural ER changes induced by proteostatic stress.9 Importantly, the UPRER was required for these adaptive changes. These data suggest a selective adaptation of lipid metabolism and/or ER structure dependent on the specific upstream input, albeit modulated in both cases by the UPRER. In addition to these studies on ER morphology, experiments on the lipid composition in worms experiencing various types of insults leading to UPR activation (e.g., proteostatic stress by tunicamycin treatment, chaperone depletion, or interference with calcium homeostasis, and lipid disequilibrium induced by depletion of fatty acid desaturases or PC biosynthesis enzymes) should help clarify whether selective adaptation of lipid synthesis is an active regulatory step to alleviate particular types of ER stress.
What is the Physiological Role of Lipid Disequilibrium Induced UPRER?
In our C. elegans models of lipid disequilibrium, we observed activation of at least 2 branches of the UPRER, the IRE-1 and the PERK branch.12 At least for the IRE-1 branch, this activation reflected canonical signaling, as xbp-1 – the key effector of activated IRE-1 – was required for the activation of the downstream target hsp-4. Such canonical activation might reflect a necessity to activate a protective response upon the physiological disturbances in the models we employed, but surprisingly, ire-1, pek-1/PERK, and atf-6 inactivation all failed to synergize with lipid disequilibrium. This raises the question as to why these pathways would be activated when not providing any evident benefit? A simple explanation would be that the individual branches of the UPRER are redundant in the protection they provide against lipid disequilibrium, or act redundantly with other, unidentified pathways to respond to such insults. Alternatively, a requirement for canonical UPRER signaling may exist in a physiological context different from the one we studied (embryonic development). For example, ire-1, pek-1, or atf-6 might be required to protect developing larvae from lipid disequilibrium induced stress, perhaps in combination, in line with redundant requirements for these factors for larval development of C. elegans.14,22 Given that ire-1 and xbp-1 are required for the longevity of a long-lived mutant,23 and given the emerging role of lipid metabolism in the regulation of longevity,24 it is also possible that lipid metabolism and the UPRER functionally cooperate to assure normal health and lifespan of adult worms.
Alternatively, no separate system may have evolved to specifically deal with lipid disequilibrium. Such a scenario might imply a lack of physiological significance of the UPRER in lipid homeostasis. Perhaps, the conditions that induce lipid disequilibrium related stress responses are relatively rare in natural settings, although starvation, obesity, and low-temperature induced changes in lipid membranes represent naturally occurring conditions that are potentially accompanied by lipid disequilibrium. Nevertheless, the lipid stresses observed in our experimental systems may not parallel an evolutionary pressure imposed on the UPRER during evolution, in contrast to e.g., the UPR induction upon massively increased synthesis and processing of secreted proteins (e.g., B-cell proliferation, see above). The mechanism proposed by Volmer et al. could support this view: in this model, differential packing of ER membrane lipids is thought to affect the activity of UPRER sensors, and therefore, the upregulation of UPRER markers (e.g., BiP) simply reflects non-specific UPRER activation.8 To test whether lipid stress-induced UPRER activation is indeed physiologically significant, it will be important to examine regulatory events downstream of the UPRER in response to various upstream inputs. For example, a comparison of UPRER-dependent genes under exogenous lipid vs. proteostatic stresses in C. elegans could be informative. Similarly, genetic screens for factors required for nematode survival under lipid imbalance-triggered ER stress might reveal the genes and pathways linking lipid and ER homeostasis.
Lipids and the Mitochondrial UPR
Besides the ER, the mitochondria also synthesize lipids and proteins. Accordingly, mitochondria possess an unfolded protein response that is mechanistically distinct from the UPRER, the UPRmito.25 Given that lipid disequilibrium can directly activate the UPRER, it is tempting to speculate that it might also activate the UPRmito, especially as the UPRmito relies on membrane-anchored sensors, like the UPRER.25 Moreover, phospholipids are asymmetrically partitioned in the inner and outer mitochondrial membranes, and phospholipids can influence the biophysical and biochemical properties of embedded mitochondrial proteins including the electron transport chain. As such, it appears possible that a mechanism monitoring the 2 mitochondrial lipid membranes exists.
In our study, changes in lipid desaturation did not induce the UPRmito.12 In contrast, the model we used as a proxy of reduced PC synthesis (worms depleted of the S-Adenosyl methionine synthetase sams-1, which are unable to synthesize the universal methyl-group donor SAM)26 displayed an activated UPRmito.12 Mutation or depletion of sams-1 reduces PC synthesis, and this defect as well as UPRER and UPRmito activation can be rescued by dietary choline, pinpointing altered PC metabolism, not methylation in general as the culprit of these phenotypes.12,26 Thus, normal PC levels are apparently essential for mitochondria and ER homeostasis, which is supported by the fact that PC is the most abundance PL in both organelles.16 Interestingly, as mitochondria do not synthesize PC and obtain their PC content from other sites such as the ER, it is likely that there is a monitoring pathway (e.g., the UPRmito) to ensure mitochondrial membrane lipid balance.
In our study, we also examined the overall levels and the fatty acid profiles of cardiolipin,12 a mitochondria-specific lipid,27 though it is unclear whether reduction in cardiolipin synthesis truly affects mitochondrial function of C. elegans. Whether cardiolipin abundance or profiles are affected in sams-1 worms has not yet been determined. A detailed analysis of the lipid profiles of these worms, and of the relationship between altered lipid profiles and UPRmito activation would be insightful.
Conclusions
The recent studies highlighted here8,12 have expanded our view of UPRER function, in that it may control proteostasis and lipid homeostasis independently as well as in combination (Fig. 1). While attractive, our hypothetical model needs to be validated, and the putative physiological in vivo role of lipid disequilibrium activated UPRER needs to be clarified. For these endeavors, C. elegans should provide a powerful organism to dissect the various inputs and outputs of the UPRER, an important homeostatic signaling circuit in eukaryotes.
Disclosure of Potential Conflict of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Amy K. Walker for insightful comments on the manuscript.
Funding
Research support was from CIHR (MOP-93713), NSERC (RGPIN 386398–13), CFI, CMMT, and CFRI (to ST). ST holds the Canada Research Chair in Transcriptional Regulatory Networks. The funding organizations had no role in the decision to publish or in the preparation of the manuscript.
References
- 1. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007; 8:519-29; PMID:17565364; http://dx.doi.org/ 10.1038/nrm2199 [DOI] [PubMed] [Google Scholar]
- 2. Gardner BM, Pincus D, Gotthardt K, Gallagher CM, Walter P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb Perspect Biol 2013; 5:a013169; PMID:23388626; http://dx.doi.org/ 10.1101/cshperspect.a013169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol 2012; 197:857-67; PMID:22733998; http://dx.doi.org/ 10.1083/jcb.201110131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, Hotamisligil GS. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011; 473:528-31; PMID:21532591; http://dx.doi.org/ 10.1038/nature09968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Görgün CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006; 313:1137-40; PMID:16931765; http://dx.doi.org/ 10.1126/science.1128294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pineau L, Ferreira T. Lipid-induced ER stress in yeast and β cells: parallel trails to a common fate. FEMS Yeast Res 2010; 10:1035-45; PMID:20738405; http://dx.doi.org/ 10.1111/j.1567-1364.2010.00674.x [DOI] [PubMed] [Google Scholar]
- 7. Pineau L, Colas J, Dupont S, Beney L, Fleurat-Lessard P, Berjeaud J-M, Bergès T, Ferreira T. Lipid-induced ER stress: synergistic effects of sterols and saturated fatty acids. Traffic 2009; 10:673-90; PMID:19302420; http://dx.doi.org/ 10.1111/j.1600-0854.2009.00903.x [DOI] [PubMed] [Google Scholar]
- 8. Volmer R, van der Ploeg K, Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci USA 2013; 110:4628-33; PMID:23487760; http://dx.doi.org/ 10.1073/pnas.1217611110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Surma MA, Klose C, Peng D, Shales M, Mrejen C, Stefanko A, Braberg H, Gordon DE, Vorkel D, Ejsing CS, et al. . A lipid E-MAP identifies Ubx2 as a critical regulator of lipid saturation and lipid bilayer stress. Mol Cell 2013; 51:519-30; PMID:23891562; http://dx.doi.org/ 10.1016/j.molcel.2013.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab 2006; 291:E275-81; PMID:16492686; http://dx.doi.org/ 10.1152/ajpendo.00644.2005 [DOI] [PubMed] [Google Scholar]
- 11. Ariyama H, Kono N, Matsuda S, Inoue T, Arai H. Decrease in membrane phospholipid unsaturation induces unfolded protein response. J Biol Chem 2010; 285:22027-35; PMID:20489212; http://dx.doi.org/ 10.1074/jbc.M110.126870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hou NS, Gutschmidt A, Choi DY, Pather K, Shi X, Watts JL, Hoppe T, Taubert S. Activation of the endoplasmic reticulum unfolded protein response by lipid disequilibrium without disturbed proteostasis in vivo. Proc Natl Acad Sci USA 2014; 111:E2271-80; PMID:24843123; http://dx.doi.org/ 10.1073/pnas.1318262111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lagace TA, Ridgway ND. The role of phospholipids in the biological activity and structure of the endoplasmic reticulum. Biochim Biophys Acta 2013; 1833:2499-510; PMID:23711956; http://dx.doi.org/ 10.1016/j.bbamcr.2013.05.018 [DOI] [PubMed] [Google Scholar]
- 14. Shen X, Ellis RE, Sakaki K, Kaufman RJ. Genetic interactions due to constitutive and inducible gene regulation mediated by the unfolded protein response in C. elegans. PLoS Genet 2005; 1:e37; PMID:16184190; http://dx.doi.org/ 10.1371/journal.pgen.0010037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Perez CL, Van Gilst MR. A 13C isotope labeling strategy reveals the influence of insulin signaling on lipogenesis in C. elegans. Cell Metabolism 2008; 8:266-74; PMID:18762027; http://dx.doi.org/ 10.1016/j.cmet.2008.08.007 [DOI] [PubMed] [Google Scholar]
- 16. van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 2008; 9:112-24; PMID:18216768; http://dx.doi.org/ 10.1038/nrm2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol 2006; 4:e423; PMID:17132049; http://dx.doi.org/ 10.1371/journal.pbio.0040423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brewer JW, Jackowski S. UPR-mediated membrane biogenesis in B cells. Biochem Res Int 2011; 2012; 738471; PMID:22110962; http://dx.doi.org/ 10.1155/2012/738471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sriburi R, Bommiasamy H, Buldak GL, Robbins GR, Frank M, Jackowski S, Brewer JW. Coordinate regulation of phospholipid biosynthesis and secretory pathway gene expression in XBP-1(S)-induced endoplasmic reticulum biogenesis. J Biol Chem 2007; 282:7024-34; PMID:17213183; http://dx.doi.org/ 10.1074/jbc.M609490200 [DOI] [PubMed] [Google Scholar]
- 20. Sriburi R, Jackowski S, Mori K, Brewer JW. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol 2004; 167:35-41; PMID:15466483; http://dx.doi.org/ 10.1083/jcb.200406136z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Struwe WB, Hughes BL, Osborn DW, Boudreau ED, Shaw KMD, Warren CE. Modeling a congenital disorder of glycosylation type I in C. elegans: a genome-wide RNAi screen for N-glycosylation-dependent loci. Glycobiology 2009; 19:1554-62; PMID:19729382; http://dx.doi.org/ 10.1093/glycob/cwp136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shen X, Ellis RE, Lee K, Liu CY, Yang K, Solomon A, Yoshida H, Morimoto R, Kurnit DM, Mori K, et al. . Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 2001; 107:893-903; PMID:11779465; http://dx.doi.org/ 10.1016/S0092-8674(01)00612-2 [DOI] [PubMed] [Google Scholar]
- 23. Henis-Korenblit S, Zhang P, Hansen M, McCormick M, Lee S-J, Cary M, Kenyon C. InsulinIGF-1 signaling mutants reprogram ER stress response regulators to promote longevity. Proc Nat Acad Sci 2010; 107:9730-5; PMID:20460307; http://dx.doi.org/ 10.1073/pnas.1002575107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hou NS, Taubert S. Function and regulation of lipid biology in caenorhabditis elegans Aging. Front Physiol 2012; 3:143; PMID:22629250; http://dx.doi.org/ 10.3389/fphys.2012.00143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pellegrino MW, Nargund AM, Haynes CM. Signaling the mitochondrial unfolded protein response. Biochim Biophys Acta 2013; 1833:410-6; PMID:22445420; http://dx.doi.org/ 10.1016/j.bbamcr.2012.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Walker AK, Jacobs RL, Watts JL, Rottiers V, Jiang K, Finnegan DM, Shioda T, Hansen M, Yang F, Niebergall LJ, et al. . A conserved SREBP-1phosphatidylcholine feedback circuit regulates lipogenesis in metazoans. Cell 2011; 147:840-52; PMID:22035958; http://dx.doi.org/ 10.1016/j.cell.2011.09.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Horvath SE, Daum G. Lipids of mitochondria. Prog Lipid Res 2013; 52:590-614; PMID:24007978; http://dx.doi.org/ 10.1016/j.plipres.2013.07.002 [DOI] [PubMed] [Google Scholar]