

Abstract
Background
Plasmodium falciparum is responsible for the majority of global malaria deaths. During the pathogenic blood stages of infection, a rapid increase in parasitaemia threatens the survival of the host before transmission of slow-maturing sexual parasites to the mosquito vector to continue the life cycle. Programmed cell death (PCD) may provide the parasite with the means to control its burden on the host and thereby ensure its own survival. Various environmental stress factors encountered during malaria may induce PCD in P. falciparum. This study is the first to characterize parasite cell death in response to natural sunlight.
Methods
The 3D7 strain of P. falciparum was cultured in vitro in donor erythrocytes. Synchronized and mixed-stage parasitized cultures were exposed to sunlight for 1 h and compared to cultures maintained in the dark, 24 h later. Mixed-stage parasites were also subjected to a second one-hour exposure at 24 h and assessed at 48 h. Parasitaemia was measured daily by flow cytometry. Biochemical markers of cell death were assessed, including DNA fragmentation, mitochondrial membrane polarization and phosphatidylserine externalization.
Results
Sunlight inhibited P. falciparum growth in vitro. Late-stage parasites were more severely affected than early stages. However, some late-stage parasites survived exposure to sunlight to form new rings 24 h later, as would be expected during PCD whereby only a portion of the population dies. DNA fragmentation was observed at 24 and 48 h and preceded mitochondrial hyperpolarization in mixed-stage parasites at 48 h. Mitochondrial hyperpolarization likely resulted from increased oxidative stress. Although data suggested increased phosphatidylserine externalization in mixed-stage parasites, results were not statistically significant.
Conclusion
The combination of biochemical markers and the survival of some parasites, despite exposure to a lethal stimulus, support the occurrence of PCD in P. falciparum.
Keywords: Plasmodium falciparum, Programmed cell death, Sunlight
Background
During the recurring erythrocytic stages of Plasmodium falciparum malaria, each mature schizont-stage parasite produces around 20 new infective merozoites [1], resulting in an exponential increase in parasite load every 48 h. Severe parasitaemia threatens the survival of the human host before transmission of the slow-maturing gametocytes to the Anopheles mosquito, and thus careful regulation of parasite density may be advantageous to both the parasite and its host. Several mechanisms are postulated to allow the parasite to regulate its own parasitaemia, including altering the rate of division, the efficiency of invasion and the rate of cell death [2]. Programmed cell death (PCD) may provide the parasite with the most effective means to regulate parasitaemia [2]. In multicellular organisms PCD fulfils essential roles in development, immunity and the maintenance of homeostasis [3–5], but it has also been shown in unicellular protozoa [6, 7], including P. falciparum (reviewed in [8]). Although the exact phenotype remains debated, a growing body of evidence suggests that P. falciparum exhibits one or several PCD phenotypes.
PCD may be induced by a number of environmental stress factors encountered by the parasite during malaria illness, including high parasite population density [9] and febrile episodes [10–12]. However, the effect of sunlight is almost entirely overlooked. The considerable cardiac output delivered to cutaneous circulation [13] means that at any one time a significant number of intra-erythrocytic parasites are located in the superficial blood vessels, and are thus exposed to penetrating solar radiation. This proportion may further increase as a result of vasodilation during fever paroxysms [14].
Solar radiation is a potent inducer of apoptosis in various eukaryotic cell types. Natural sunlight is composed of UV-A (320–400 nm), UV-B (280–320 nm) and UV-C (200–280 nm) radiation [15], although UV-C radiation is filtered out by the atmosphere [16, 17]. UV-B radiation directly damages nuclear DNA by causing lesions such as cyclobutane pyrimidine dimers and pyrimidine 6–4 pyrimidone photoproducts [18–20]. However, UV-B may also indirectly induce apoptosis through the generation of reactive oxygen species (ROS) that in turn triggers mitochondrial cytochrome-c release [20–22]. UV-B radiation may also cause apoptosis via other cytoplasmic or membrane targets, such as direct activation of membrane-bound death receptors (reviewed in [22]). UV-A radiation causes oxidative stress that damages and permeabilizes lipid membranes (reviewed in [23]). In murine lymphoma cells, UV-A radiation was shown to induce apoptosis in less than four hours, while the apoptotic effects of UV-B and UV-C wavelengths were delayed (reviewed in [23]).
The in vitro photosensitivity of P. falciparum has previously been noted in the authors’ laboratory [24], but data on Plasmodium spp. are otherwise limited to the effect of UV radiation on the murine host [25]. In other protists, UV-B exposure did not affect the viability of Leishmania major parasites either in vitro or in vivo [26] and had no impact on parasite infectivity [26, 27].
The present study is the first to investigate biochemical markers of PCD in P. falciparum parasites in response to natural sunlight, a physiologically relevant stress factor. It should be noted that the present study focused on the effect of natural sunlight, rather than a specific wavelength spectrum. The use of culture flasks that were not UV transparent may have prevented light in the UV spectrum from reaching parasites, whereas physiological exposure would include UV light. However, even with the exclusion of the UV spectrum, the effects observed in this study derived from natural sunlight. Exposure to sunlight caused growth inhibition and induced PCD in a sub-population of parasites. Late-stage parasites—trophozoites and schizonts—were far more affected than ring-stage parasites. Sunlight caused DNA fragmentation that preceded mitochondrial hyperpolarization, suggesting a unique form of PCD in P. falciparum that is not initiated by the mitochondrion. These findings provide important new information on cell death mechanisms that are utilized by the parasite to limit its population and thereby prevent premature death of the human host and thus ensure its transmission to the mosquito vector.
Methods
Reagents
The APO-DIRECT TUNEL kit and FITC Annexin V Apoptosis Detection Kit II were obtained from Becton–Dickinson (BD Pharmingen, San Diego, CA, USA). Thiazole orange (TO), hydroethidine (HE), 3,3′-dihexyloxacarbocyanine iodide [DiOC6(3)], carbonyl cyanide m-chlorophenylhydrazone (CCCP) were obtained from Sigma-Aldrich (St Louis, MO, USA). Albumax II was obtained from Gibco (Gran Island, NY, USA).
Collection of human blood and ethics clearance
Human erythrocytes (RBC) were collected from healthy volunteers as whole blood into Vacutainer® ACD collection tubes (Becton–Dickinson, Franklin Lakes, NJ, USA). The plasma and buffy coat were removed after centrifugation and RBC were washed with RPMI to remove residual white blood cells. Ethics clearance for the collection of blood from volunteers by venepuncture was provided by the University of the Witwatersrand Human Research Ethics Committee (Medical) under the protocol number M130569.
Plasmodium falciparum culture
The 3D7 strain of P. falciparum was maintained according to established methods [28] with some modifications [29]. Briefly, parasites were maintained at 37 °C in malaria culture medium (RPMI 1640, 0.5 % Albumax II and 0.21 % sodium bicarbonate, supplemented with 50 mg/l gentamycin and 50 mg/l hypoxanthine) at 5 % haematocrit in RBC. Medium was changed daily. Optimal culture pH was maintained by gassing cultures daily for 60 s with a mixture of 2 % O2, 5 % CO2 and 93 % N2. Parasite morphology was monitored by Giemsa-stained smears and parasitaemia and staging were assessed by thiazole orange (TO) flow cytometry.
Synchronization of parasites to ring stages
For studies involving synchronized parasites, synchronization was performed similar to an established method [30]. Briefly, P. falciparum-infected red blood cells (pRBC) were centrifuged for 5 min at 1000×g and 25 °C. The resulting cell pellet was incubated in ten volumes of 5 % D-sorbitol for 5 min at 37 °C. Centrifugation was repeated and the cell pellet was resuspended to 5 % haematocrit with medium, returned to a clean 25 sq cm culture flask and incubated at 37 °C. This method synchronized parasites to ring stages. For studies involving late-stage parasites, pRBC cultures were allowed to mature for 20–24 h.
Exposure to natural sunlight
pRBC were seeded as 5 ml cultures in 25 sq cm, optically clear, sealed tissue culture flasks. Sunlight exposure was performed for one hour in the middle of the day (between 10.00 and 14.00 h) on clear, sunny days in Johannesburg, South Africa, with an elevation of 1753 m above sea level. Culture flasks were maintained at 37 °C by a Lauda B Circulator (temperature uniformity: ±0.01 °C) in a Lauda MA6 water bath (Lauda-Brinkmann, Delran, NJ, USA) throughout exposure. Temperature consistency was additionally monitored by a mercury-in-glass thermometer. Previous control experiments confirmed that culture medium inside the flasks exposed to sunlight in this manner remained constant at 37 °C. Control cultures were covered with thick cardboard to prevent exposure to sunlight, while experimental cultures were left uncovered in direct sunlight. Following exposure, all cultures were returned to the dark in an incubator at 37 °C.
Flow cytometry
Flow cytometric analyses were performed on a Beckman Coulter Gallios flow cytometer (Beckman Coulter Inc, Miami, FL, USA). Excitation for all assays was by 488 nm blue laser. Emission was detected with the use of 545/40BP (525 ± 20 nm, FL1) and, where indicated, 575/30BP (575 ± 15 nm, FL2) filters. Optical alignment was monitored daily with Beckman Coulter Flow Check Pro fluorospheres (Beckman Coulter Inc, Brea, CA, USA). Post-acquisition analyses were performed with Beckman Coulter Kaluza (v1.1) software.
TO flow cytometry for parasitaemia
Parasitaemia was measured daily by flow cytometry with the DNA-binding dye TO, similar to a previous method [31]. Whole culture samples (10 µl) were diluted 100-fold to 1 ml in Sorenson’s phosphate buffer (47 mM Na2HPO4, 20 mM KH2PO4, pH 7.2) with 1 µM TO final concentration (diluted from a 10 mM stock in methanol) and incubated at room temperature in the dark for 20 min. Stained cells were analysed by flow cytometry within 1 hour. Erythrocytes were gated on a forward- versus side-scatter dot plot and analysed on a FL1 integral (log) histogram, with regions for uninfected, ring-infected and trophozoite- or schizont-infected erythrocytes delineated. Regions had previously been confirmed by microscopy of Giemsa-stained smears of synchronized cultures. Approximately 50,000 events in the erythrocyte gate were counted.
TUNEL assay for DNA fragmentation
The terminal deoxynucleotidyltransferase (TdT)-mediated nick end labelling (TUNEL) assay was performed according to manufacturer’s recommendations, with modifications similar to a previous study [11]. Briefly, pRBC were pelleted and fixed on ice for 60–90 min in 4 % formaldehyde/phosphate-buffered saline (PBS: 10 mM Na2HPO4, 1.5 mM KH2PO4, 137 mMNaCl, 2.7 mMKCl, pH 7.4), followed by permeabilization with 0.1 % tri-sodium citrate (w/v) and 0.1 % Triton X-100 (v/v) in PBS for 3 min on ice. Labelling with DNA-staining solution (including TdT enzyme and FITC-dUTP) was performed according to manufacturer’s recommendations for 60–90 min at 37 °C, followed by staining with propidium iodide (PI) for 30 min at room temperature. Labelled cells were analysed by flow cytometry within three hours. PI-positive parasites were acquired on a FL2 time-of-flight (lin) versus FL2 integral (lin) dot plot, with gated parasites analysed on a FL1 integral (log) histogram for DNA fragmentation, measured as FITC-dUTP fluorescence. At least 10,000 PI-positive events were counted. DNase-treated, non-treated and unlabelled parasites were used as positive, negative and staining controls, respectively.
DiOC6(3) flow cytometry for mitochondrial transmembrane potential
Plasmodium falciparum whole culture samples (50 µl) were diluted to 1 ml in PBS containing 10 nM DiOC6(3) (diluted from a 100 mM stock in DMSO) and 50 µM hydroethidine HE (diluted from a 10 mM stock in DMSO) and incubated at 37 °C for 45 min in the dark. Following incubation, cells were washed and suspended in 1 ml PBS and analysed immediately by flow cytometry. Erythrocytes were gated on a forward- versus side-scatter dot blot. HE-positive pRBC counted on a FL2 integral (log) histogram were analysed for DiOC6(3) fluorescence on a FL1 integral (log) histogram. At least 10,000 HE-positive events were counted. Positive controls were treated with 200 nM CCCP for one hour before staining. Unstained cells, cells stained with only HE or DiOC6(3) and non-parasitized erythrocytes were used as staining controls.
Annexin V-FITC for PS externalization
Plasmodium falciparum culture samples (20 µl) were diluted 50-fold to 1 ml in PBS and stained with a final concentration of 50 µM HE (diluted from a 10 mM stock in DMSO) in PBS for 15 min at 37 °C in the dark. Cells were then pelleted, suspended to 100 µl in 1X annexin-binding buffer (provided with the kit) and stained with annexin V-FITC for 15 min in the dark according to manufacturer’s recommendations with modifications similar to a previous study [32]. Stained cells were diluted to 1 ml in 1X annexin-binding buffer and analysed by flow cytometry within 1 h. Erythrocytes were gated on a forward- versus side-scatter dot blot and pRBC were discriminated on a FL 2 integral (log) histogram for HE fluorescence. Gated pRBC were analysed for annexin V-FITC fluorescence on a FL1 integral (log) histogram. At least 50,000 events in the erythrocyte gate were counted. pRBC treated with recombinant annexin V (BD Pharmingen, San Diego, CA, USA) before staining were used as a negative control and unstained parasite cultures and parasite cultures stained with only HE or annexin V-FITC were used as staining controls.
Statistical analysis
Bar graphs were compiled using GraphPad Prism 5, with raw data values exported from analyses by Beckman Coulter Kaluza (v.1.1) software. Student’s unpaired t tests were performed with Microsoft Office Excel 2010 to test for significance between treated and control groups. Data distributions were not tested for normality.
Results
Sunlight inhibited Plasmodium falciparum growth in vitro
Mixed and synchronized parasite cultures were exposed to one hour of sunlight and assessed for growth 24 h later. Mixed-stage cultures were exposed to another 1 h of sunlight at 24 h and assessed again 24 h later, at 48 h. Sunlight decreased the in vitro growth of both mixed (Fig. 1) and synchronized (Fig. 2) parasite cultures.
Fig. 1.
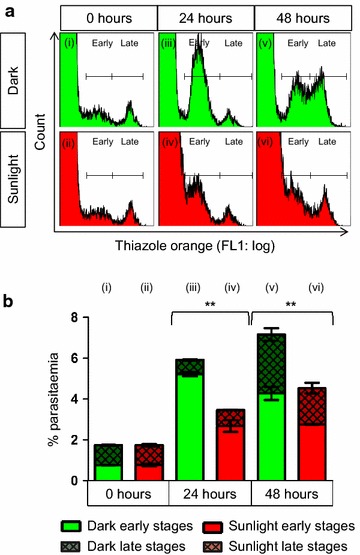
Mixed-stage Plasmodium falciparum growth decreased after exposure to sunlight in vitro. Flow cytometry histograms (a) and statistical analyses (b) showed that late-stage parasites (Late) were more affected by exposure to solar radiation than early stage parasites (Early). When compared to experimental cultures exposed to sunlight for 1 h (Sunlight), control cultures (Dark) showed a large population of new early-stage parasites (iii), whereas the formation of early stage parasites was significantly reduced in experimental cultures (iv) indicating that late-stage parasites exposed to sunlight produced fewer new rings. Statistical comparisons were made between total parasitaemia values and were significant at P < 0.01 (double asterisk). For all comparisons, n = 2
Fig. 2.
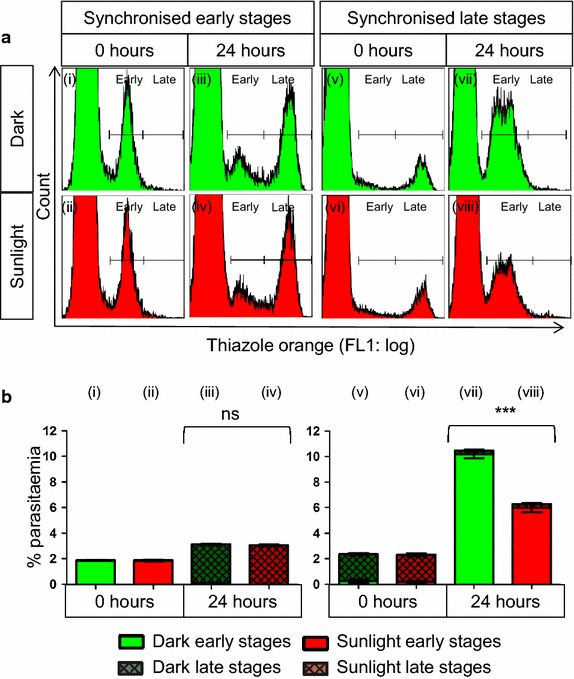
Sunlight decreased the growth of late-stage Plasmodium falciparum, but early stages were unaffected. Flow cytometry histograms (a) and statistical analyses (b) showed that early-stage parasites (i – iv) were unaffected by exposure to solar radiation, whereas late-stage parasites (v – viii) were significantly affected. Synchronized early-stage parasites showed no difference in development between cultures maintained in the dark (Dark) and those exposed to sunlight for one hour (Sunlight), while synchronized late-stage parasites showed significantly reduced growth (viii). Statistical comparisons were made between total parasitaemia values and were not significant (ns) or significant at P < 0.001 (triple asterisk). For all comparisons, n = 4
Early-stage parasites were unaffected by sunlight. Twenty-four hours after a single exposure, ring stage parasites in both mixed stage (Fig. 1aii, bii) and synchronized (Fig. 2aii, bii) parasites progressed to trophozoite stages (Figs. 1aiv, biv, 2aiv, biv, respectively) similar to control cultures maintained in the dark (Figs. 1aiii, biii, 2aiii, biii, respectively).
In contrast, sunlight decreased the growth of late stage parasites in both mixed (Fig. 1) and synchronized (Fig. 2) cultures. Fewer new rings were formed (Figs. 1aiv, biv, 2aiii, bviii) from late-stage parasites exposed to sunlight (Figs. 1ai, bi, 2avi, bvi) than from corresponding cultures maintained in the dark. However, growth inhibition was only partial, as evidenced by increased parasitaemia between 0 and 24 h, albeit less than in control cultures.
In mixed-stage cultures, a decrease in both early and late stage parasites was seen at 48 h (Fig. 1avi, bvi vs. Av and Bv), after a second dose of sunlight was delivered at 24 h. A decrease in late-stage parasites, compared to control cultures at 48 h, is attributed to the initial exposure of trophozoites at 0 h causing fewer rings at 24 h and correspondingly fewer late stage parasites at 48 h. As sunlight had no effect on the growth of early-stage parasites, only mixed-stage parasites and synchronized late-stage parasites were used for cell death assays.
Exposure to sunlight caused DNA fragmentation in Plasmodium falciparum
DNA fragmentation was quantified from fixed and isolated parasites by the flow cytometric TUNEL assay, 24 h after every sunlight exposure. DNA fragmentation was seen at 24 h in synchronized late stage parasites (Fig. 3b) and at 48 h in mixed-stage parasites (Fig. 3c).
Fig. 3.
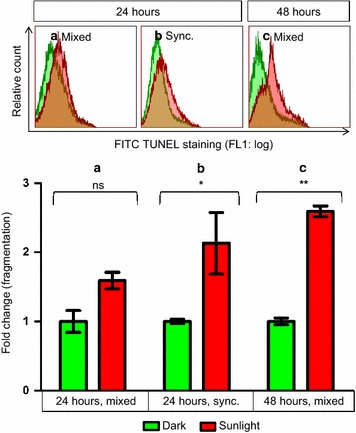
Exposure to sunlight caused DNA fragmentation in Plasmodium falciparum in vitro. Although DNA fragmentation was not observed in mixed-stage parasites after a single exposure to sunlight (a), synchronized late-stage parasites did show DNA fragmentation at 24 h (b). DNA fragmentation was also seen in mixed-stage parasites at 48 h, after two exposures to sunlight (c). Statistical comparisons were not significant (ns), or significant at P < 0.05 (asterisk) or at P < 0.01 (double asterisk). n = 2 for (a) and (b); n = 4 for (c)
Mixed-stage parasites showed suggestive but insignificant DNA fragmentation at 24 h (Fig. 3a), likely due to the negating effect of early-stage parasites that were unaffected by sunlight that reduced the fragmentation of the population as a whole. Therefore, it seems likely that late-stage parasites in mixed cultures exhibited DNA fragmentation in the same manner as synchronized late-stage parasites did.
Exposure to sunlight caused mitochondrial hyperpolarization in mixed-stage parasites
Mixed-stage parasites showed increased DiOC6(3) fluorescence, suggesting mitochondrial hyperpolarization, the day following a second exposure to sunlight (Fig. 4c). No mitochondrial dysregulation was observed in synchronized late-stage parasites or in mixed-stage parasites 24 h after the first exposure to sunlight (Fig. 4a, b). DNA fragmentation therefore preceded mitochondrial dysregulation, with DNA fragmentation observed in late stage parasites at 24 h, and mitochondrial hyperpolarization only observed at 48 h in mixed-stage parasites. Mitochondrial hyperpolarization was likely due to increased reactive oxygen species (ROS) [10, 24], but it is not clear whether there is any causative relationship between DNA fragmentation and the appearance of mitochondrial hyperpolarization.
Fig. 4.
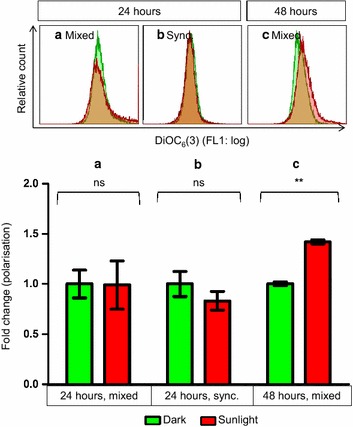
Sunlight caused changes in mitochondrial polarization in Plasmodium falciparum only after 48 h. No change was seen in the mitochondrial polarization of mixed-stage parasites (a) or synchronized late-stage parasites (b) exposed to sunlight for 1 h and maintained in the dark for the remainder of 24 h, compared to control parasites kept in the dark. After a second exposure to solar radiation, mixed-stage parasites showed an apparent increase in mitochondrial polarization at 48 h, compared to parasites maintained in the dark (c). Statistical comparisons were not significant (ns) or significant at P < 0.01 (double asterisk). n = 2 for (a) and (b); n = 4 for (c)
Exposure to sunlight caused no change in phosphatidylserine externalization
Flow cytometry data showed increased PS externalization in mixed-stage pRBC on the day following both the first (Fig. 5a) and second exposure (Fig. 5c) to natural sunlight, but the difference was not statistically significant. No increase in PS externalization was observed in synchronized late-stage pRBC exposed to sunlight (Fig. 5b).
Fig. 5.
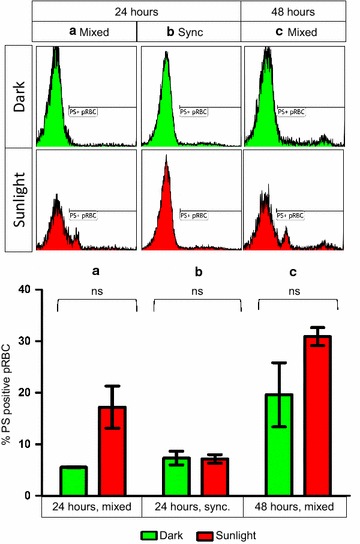
No significant change was seen in the phosphatidylserine externalization of parasitized erythrocytes after exposure sunlight. Flow cytometry histograms (upper panels) showed an apparent increase in PS externalization in mixed-stage pRBC at both 24 (a) and 48 (c) hours after one and two exposures to sunlight, respectively. However, statistical analyses of bar graphs (bottom panels) showed no significant differences (ns). Synchronized late-stage pRBC exposed to solar radiation for 1 h and maintained in the dark for the remainder of 24 h also showed no change in PS externalization, compared to control cultures maintained in the dark (b). n = 2 for (a) and (b); n = 4 for (c)
Discussion
Shedding light on a killer: sunlight inhibits Plasmodium falciparum growth
Since ancient times, humans have linked sunlight and malaria and one of the myths was that staying out in the sun would cause malaria [33]. However, sun worship in early cultures may have in fact conferred a survival advantage to its practitioners, by reducing the burden of disease [Mendelow and Coetzer, unpublished observations]. The data from this study showed that exposure to sunlight inhibited the in vitro growth of intra-erythrocytic P. falciparum. The effect of sunlight was tested in both mixed and synchronous parasites. In vivo malaria often involves multiple, asynchronous infections [34]. Therefore, the use of mixed parasite populations with multiple sunlight exposures was essential to determine the effect of sunlight in conditions that mimic in vivo disease. However, the use of synchronous parasites with a single exposure allows further narrowing of the effects of sunlight on different asexual developmental stages. Early-stage parasites were resistant to damage caused by sunlight and although late-stage parasites were more severely affected, some late-stage parasites survived sunlight exposure. These may have been early trophozoites at the time of exposure, rather than late trophozoites or schizonts. The conservation of some parasites, despite exposure to a lethal stress stimulus, supports the occurrence of PCD, which requires a portion of the population to survive. It is not clear whether decreased parasite growth was due to fewer schizonts being produced or if some of the newly released merozoites were non-viable and hence could not invade new erythrocytes. Another possibility for reduced parasite growth that also conserves a portion of the population may involve some quorum sensing-like mechanism, with parasite-derived signalling molecules resulting in the induction of PCD. However, such a mechanism has not been identified in P. falciparum and furthermore it is not clear why only a portion of the parasite population would react to such signalling molecules (reviewed in [8]). It seems more likely that the conservation of some parasites relates to the specific asexual developmental stage of those parasites. The details of this mechanism require further elucidation.
To further investigate the decline in parasitaemia after exposure to sunlight, biochemical markers of cell death were evaluated. DNA fragmentation is one of the characteristics of PCD and was observed in late-stage parasites, confirming that the reduced growth resulted from the death of some parasites, rather than the reduced reproduction of the population as a whole. DNA fragmentation has been widely reported in P. falciparum in response to anti-malarial drugs [2, 35–38], heat stress [10, 11], high parasite density [9], bilirubin [39], etoposide [36], and staurosporine [37]. Some studies have attributed the occurrence of DNA fragmentation, without other biochemical markers of cell death, to apoptosis in P. falciparum [2, 11, 36], although it has also been attributed to an undefined pathway [40].
The effect of sunlight on another key element of PCD, the polarization of the mitochondrial membrane, was also investigated. Mitochondrial hyperpolarization was observed in mixed-stage parasites the day following a second sunlight exposure. Although mitochondrial depolarization, resulting from permeabilization of the outer mitochondrial membrane, is expected during apoptosis [41–46], both mitochondrial depolarization and hyperpolarization may form part of an apoptosis response in other protozoa [47–49]. The asexual stages of P. falciparum contain only a single mitochondrion that is chiefly involved in pyrimidine synthesis [50]; however, previous studies have shown the importance of mitochondria in parasite programmed cell death [51]. It should also be noted that transient hyperpolarization in metazoans may occur as an early checkpoint in deciding cell fate [48]; however, hyperpolarization in this case was not observed immediately after exposure to sunlight, but only a day after the second exposure and two days after the first. Mitochondrial hyperpolarization was likely the result of increased accumulation of reactive oxygen species (ROS), which has previously been suggested to be the cause of parasite death after sunlight exposure [24]. The previous study showed that P. falciparum growth is reduced and the developmental cycle of parasites delayed by 30-min daily exposures to natural sunlight for 4 days. The observed effects were ascribed to photo-activation of ROS, possibly free erythrocyte protoporphyrin [24].
The externalization of PS in pRBC was evaluated as a third marker of PCD. PS externalization has previously been noted to result from parasite maturation or stress [32, 52]. Although results suggested increased PS externalization in mixed-stage cultures, the results were not statistically significant. The absence of increased PS externalization, particularly in late-stage pRBC, supports the notion of a unique PCD pathway in P. falciparum.
These combined data therefore showed that sunlight exposure induced DNA fragmentation followed by mitochondrial hyperpolarization in the absence of PS externalization. This unusual manifestation of biochemical markers suggests a cell death phenotype that does not entirely fit with any metazoan PCD models and might be unique to P. falciparum.
Sunlight and heat stress: cooperative effect
Plasmodium falciparum utilizes a unique method of erythrocyte modification to allow adhesion of late-stage asexual and immature sexual parasites to endothelium, thereby sparing parasites from splenic clearance [1]. Heat stress, such as febrile episodes characteristic of malaria [1], increases cardiac output to cutaneous circulation to as much as 60 % of total vascular conductance [13] and further increases the cyto-adherence of mature P. falciparum-infected RBC [53]. Therefore, a large number of late-stage parasites would be present in the microvasculature near the skin and thus be exposed to sunlight. Heat stress reduces P. falciparum numbers in vitro [10–12, 34, 54, 55] and has been suggested to cause PCD in the parasite [10, 11]. Sunlight and heat stress may therefore have a cooperative effect in parasite clearance.
Sunlight and heat stress: different stress, different phenotype
In contrast to the effect of sunlight on late-stage pRBC, PCD induced by heat stress was characterized by mitochondrial hyperpolarization and PS externalization without any DNA fragmentation [10]. Interestingly, heat stress also induced PCD in some early stage parasites that exhibited DNA fragmentation and mitochondrial depolarization [10], whereas sunlight did not affect ring stages. Therefore, P. falciparum shows unique combinations of cell death markers that differ depending on the type of stress.
Early-stage parasites appear to be well adapted to surviving lethal stress stimuli. In vivo, the rupture of pRBC and the egress of merozoites are responsible for the onset of fever, but the merozoites would have re-invaded and be present as ring-stage parasites by the time heat stress occurs and will thus not be affected [10, 56]. In the case of sunlight, early-stage parasites are exposed as a result of continuous circulation, but since they do not sequester as late-stage parasites do, they presumably receive less sunlight than late-stage parasites.
Conclusion
In vitro exposure to natural sunlight reduced the growth of P. falciparum due to the death of a portion of the parasite population. The occurrence of DNA fragmentation prior to mitochondrial depolarization suggests a cell death mechanism that is not initiated by the mitochondrion. This combination of biochemical markers may offer clues to a unique phenotype of PCD in P. falciparum. Although conflicting data concerning the phenotype of PCD in P. falciparum might be attributed to the use of various strains and cell death markers, it is also apparent that P. falciparum may vary its response to different potentially fatal stimuli. However, whether this should be attributed to diverse underlying mechanisms or simply different facets of the same pathway remains unclear.
Authors’ contributions
DE participated in the study design and conception as well as data interpretation, and was responsible for all data collection, statistical analyses and drafted the manuscript. TLC participated in the design, conception and coordination of the study, as well as data interpretation and helped to draft the manuscript. Both authors read and approved the final manuscript.
Acknowledgements
We thank Professor Lesley E Scott and Beckman Coulter South Africa for the support provided in the use of the Beckman Coulter Gallios flow cytometer installed at the Department of Molecular Medicine and Haematology, University of the Witwatersrand and National Health Laboratory Service, Johannesburg, South Africa. We further express our gratitude to laboratory members of the Wits Research Institute for Malaria and Plasmodium Molecular Research Unit for helpful discussions. Funding was provided by the National Research Foundation (NRF)—Grants 66072 and 73703, the University of the Witwatersrand and National Health Laboratory Service. DE is funded by the National Research Foundation (NRF) Scarce Skills Scholarship—Grant no. 81556. The funders were not involved in the study design, experimental aspects, data interpretation or manuscript preparation.
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Abbreviations
- CCCP
carbonyl cyanide m-chlorophenylhydrazone
- DiOC6(3)
3,3′-dihexyloxacarbocyanine iodide
- HE
hydroethidine
- PBS
phosphate-buffered saline
- PCD
programmed cell death
- PI
propidium iodide
- pRBC
Plasmodium falciparum-infected red blood cells
- PS
phosphatidylserine
- ROS
reactive oxygen species
- TdT
terminal deoxynucleotidyltransferase
- TO
thiazole orange
- TUNEL
TdT-mediated nick end labelling
Contributor Information
Dewaldt Engelbrecht, Email: dewaldtengelbrecht@gmail.com.
Thérèsa Louise Coetzer, Email: theresa.coetzer@nhls.ac.za.
References
- 1.Miller LH, Baruch DI, Marsh K, Doumbo OK. The pathogenic basis of malaria. Nature. 2002;415:673–679. doi: 10.1038/415673a. [DOI] [PubMed] [Google Scholar]
- 2.Deponte M, Becker K. Plasmodium falciparum—do killers commit suicide? Trends Parasitol. 2004;20:165–169. doi: 10.1016/j.pt.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 3.Ameisen JC. On the origin, evolution, and nature of programmed cell death: a timeline of four billion years. Cell Death Differ. 2002;9:367–393. doi: 10.1038/sj.cdd.4400950. [DOI] [PubMed] [Google Scholar]
- 4.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 6.Van Zandbergen G, Lüder CGKK, Heussler V, Duszenko M. Programmed cell death in unicellular parasites: a prerequisite for sustained infection? Trends Parasitol. 2010;26:477–483. doi: 10.1016/j.pt.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 7.Lüder CG, Campos-Salinas J, Gonzalez-Rey E, van Zandbergen G. Impact of protozoan cell death on parasite-host interactions and pathogenesis. Parasit Vectors. 2010;3:116–126. doi: 10.1186/1756-3305-3-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engelbrecht D, Durand PM, Coetzer TL. On programmed cell death in Plasmodium falciparum: status quo. J Trop Med. 2012;2012:1–15. doi: 10.1155/2012/646534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mutai BK, Waitumbi JN. Apoptosis stalks Plasmodium falciparum maintained in continuous culture condition. Malar J. 2010;9(Suppl 3):S6. doi: 10.1186/1475-2875-9-S3-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engelbrecht D, Coetzer TL. Turning up the heat: heat stress induces markers of programmed cell death in Plasmodium falciparum in vitro. Cell Death Dis. 2013;4:e971. doi: 10.1038/cddis.2013.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oakley MSM, Kumar S, Anantharaman V, Zheng H, Mahajan B, Haynes JD, et al. Molecular factors and biochemical pathways induced by febrile temperature in intraerythrocytic Plasmodium falciparum parasites. Infect Immun. 2007;75:2012–2025. doi: 10.1128/IAI.01236-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Porter H, Gamette MJ, Cortes-Hernandez DG, Jensen JB. Asexual blood stages of Plasmodium falciparum exhibit signs of secondary necrosis, but not classical apoptosis after exposure to febrile temperature (40 C) J Parasitol. 2008;94:473–480. doi: 10.1645/GE-1343.1. [DOI] [PubMed] [Google Scholar]
- 13.Minson CT. Hypoxic regulation of blood flow in humans. Skin blood flow and temperature regulation. Adv Exp Med Biol. 2003;543:249–262. doi: 10.1007/978-1-4419-8997-0_18. [DOI] [PubMed] [Google Scholar]
- 14.Starr C, McMillan B. Human Biology. 10. Belmont: Cengage Learning; 2013. pp. 82–83. [Google Scholar]
- 15.Latonen L, Laiho M. Cellular UV damage responses—functions of tumor suppressor p53. Biochim Biophys Acta. 2005;1755:71–89. doi: 10.1016/j.bbcan.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 16.Norval M. Sun Protection in Man. The Netherlands: Elsevier; 2001. pp. 91–113. [Google Scholar]
- 17.Afaq F. Natural agents: cellular and molecular mechanisms of photoprotection. Arch Biochem Biophys. 2011;508:144–151. doi: 10.1016/j.abb.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sinha RP, Häder D-P. UV-induced DNA damage and repair: a review. Photochem Photobiol Sci. 2002;1:225–236. doi: 10.1039/b201230h. [DOI] [PubMed] [Google Scholar]
- 19.Rastogi RP, Richa, Kumar A, Tyagi MB, Sinha RP. Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J Nucleic Acids. 2010;2010:592980. doi: 10.4061/2010/592980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He Y-Y, Hader D-P. Reactive oxygen species and UV-B: effect on cyanobacteria. Photochem Photobiol Sci. 2002;1:729–736. doi: 10.1039/b110365m. [DOI] [PubMed] [Google Scholar]
- 21.Kulms D, Zeise E, Pöppelmann B, Schwarz T. DNA damage, death receptor activation and reactive oxygen species contribute to ultraviolet radiation-induced apoptosis in an essential and independent way. Oncogene. 2002;21:5844–5851. doi: 10.1038/sj.onc.1205743. [DOI] [PubMed] [Google Scholar]
- 22.Kulms D, Schwarz T. Independent contribution of three different pathways to ultraviolet-B-induced apoptosis. Biochem Pharmacol. 2002;64:837–841. doi: 10.1016/S0006-2952(02)01146-2. [DOI] [PubMed] [Google Scholar]
- 23.Pourzand C, Tyrrell RM. Apoptosis, the role of oxidative stress and the example of solar UV radiation. Photochem Photobiol. 1999;70:380–390. doi: 10.1111/j.1751-1097.1999.tb08239.x. [DOI] [PubMed] [Google Scholar]
- 24.Coetzer TL, Lauterbach SB, Gulumian M, Jenkins T, Grimmer H, Mendelow BV. Skin colour variation and susceptibility to malaria. In: 11th biennial congress of The Southern African Society for Human Genetics combined with the 2nd annual meeting of The African Society of Human Genetics; 2005, p. 52.
- 25.Yamamoto K, Ito R, Koura M, Kamiyama T. UV-B irradiation increases susceptibility of mice to malarial infection. Infect Immun. 2000;68:2353–2355. doi: 10.1128/IAI.68.4.2353-2355.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giannini MS. Suppression of pathogenesis in cutaneous leishmaniasis by UV irradiation. Infect Immun. 1986;51:838–843. doi: 10.1128/iai.51.3.838-843.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giannini SH. Effects of ultraviolet B irradiation on cutaneous leishmaniasis. Parasitol Today. 1992;8:44–48. doi: 10.1016/0169-4758(92)90083-E. [DOI] [PubMed] [Google Scholar]
- 28.Trager W, Jensen J. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 29.Mphande F, Nilsson S, Bolad A. Culturing of erythrocytic asexual stages of Plasmodium falciparum and P. vivax. In: Moll K, Ljungström I, Perlmann H, Scherf A, Wahlgren M, editors. Methods in Malaria Research, 5th edn. Virginia: MR4/ATCC; 2008. p. 1–3.
- 30.Lambros C, Vanderberg JP. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol. 1979;65:418–420. doi: 10.2307/3280287. [DOI] [PubMed] [Google Scholar]
- 31.Schulze DLC, Makgatho EM, Coetzer TL, Louw AI, Van Rensburg CEJ, Visser L. Development and application of a modified flow cytometric procedure for rapid in vitro quantitation of malaria parasitaemia. S Afr J Sci. 1997;93:156–158. [Google Scholar]
- 32.Pattanapanyasat K, Sratongno P, Chimma P, Chitjamnongchai S, Polsrila K, Chotivanich K. Febrile temperature but not proinflammatory cytokines promotes phosphatidylserine expression on Plasmodium falciparum malaria-infected red blood cells during parasite maturation. Cytom A. 2010;77:515–523. doi: 10.1002/cyto.a.20879. [DOI] [PubMed] [Google Scholar]
- 33.Bawa JT. Malaria: from myths to management. In: From the field. Malaria Vaccine Initiative 2014. http://www.malariavaccine.org/from-the-field-Bawa-myths-to-mgmt.php. Accessed 18 August 2015.
- 34.Kwiatkowski D. Febrile temperatures can synchronize the growth of Plasmodium falciparum in vitro. J Exp Med. 1989;169:357–361. doi: 10.1084/jem.169.1.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Picot S, Burnod J, Bracchi V, Chumpitazi BFF, Ambroise-Thomas P. Apoptosis related to chloroquine Plasmodium falciparum sensitivity of the human malaria parasite. Trans R Soc Trop Med Hyg. 1997;91:590–591. doi: 10.1016/S0035-9203(97)90039-0. [DOI] [PubMed] [Google Scholar]
- 36.Meslin B, Barnadas C, Boni V, Latour C, De Monbrison F, Kaiser K, et al. Features of apoptosis in Plasmodium falciparum erythrocytic stage through a putative role of PfMCA1 metacaspase-like protein. J Infect Dis. 2007;195:1852–1859. doi: 10.1086/518253. [DOI] [PubMed] [Google Scholar]
- 37.Ch’ng J-H, Kotturi SR, Chong AG-L, Lear MJ, Tan KS-W. A programmed cell death pathway in the malaria parasite Plasmodium falciparum has general features of mammalian apoptosis but is mediated by clan CA cysteine proteases. Cell Death Dis. 2010;1:e26. doi: 10.1038/cddis.2010.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.López ML, Vommaro R, Zalis M, de Souza W, Blair S, Segura C. Induction of cell death on Plasmodium falciparum asexual blood stages by Solanum nudum steroids. Parasitol Int. 2010;59:217–225. doi: 10.1016/j.parint.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 39.Kumar S, Guha M, Choubey V, Maity P, Srivastava K, Puri SK, et al. Bilirubin inhibits Plasmodium falciparum growth through the generation of reactive oxygen species. Free Radic Biol Med. 2008;44:602–613. doi: 10.1016/j.freeradbiomed.2007.10.057. [DOI] [PubMed] [Google Scholar]
- 40.Nyakeriga AM, Perlmann H, Hagstedt M, Berzins K, Troye-Blomberg M, Zhivotovsky B, et al. Drug-induced death of the asexual blood stages of Plasmodium falciparum occurs without typical signs of apoptosis. Microbes Infect. 2006;8:1560–1568. doi: 10.1016/j.micinf.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 41.Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Skulachev VP. Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis. 2006;11:473–485. doi: 10.1007/s10495-006-5881-9. [DOI] [PubMed] [Google Scholar]
- 43.Banki K, Hutter E, Gonchoroff NJ, Perl A. Elevation of mitochondrial transmembrane potential and reactive oxygen intermediate levels are early events and occur independently from activation of caspases in Fas signaling. J Immunol. 1999;162:1466–1479. [PMC free article] [PubMed] [Google Scholar]
- 44.Joshi B, Li L, Taffe BG, Zhu Z, Wahl S, Tian H, et al. Apoptosis induction by a novel anti-prostate cancer compound, BMD188 (a fatty acid-containing hydroxamic acid), requires the mitochondrial respiratory chain. Cancer Res. 1999;59:4343–4355. [PubMed] [Google Scholar]
- 45.Sánchez-Alcázar JA, Ault JG, Khodjakov A, Schneider E. Increased mitochondrial cytochrome c levels and mitochondrial hyperpolarization precede camptothecin-induced apoptosis in Jurkat cells. Cell Death Differ. 2000;7:1090–1100. doi: 10.1038/sj.cdd.4400740. [DOI] [PubMed] [Google Scholar]
- 46.Scarlett JL, Sheard PW, Hughes G, Ledgerwood EC, Ku HH, Murphy MP. Changes in mitochondrial membrane potential during staurosporine-induced apoptosis in Jurkat cells. FEBS Lett. 2000;475:267–272. doi: 10.1016/S0014-5793(00)01681-1. [DOI] [PubMed] [Google Scholar]
- 47.Alzate JF, Arias AA, Moreno-Mateos D, Álvarez-Barrientos A, Jiménez-Ruiz A. Mitochondrial superoxide mediates heat-induced apoptotic-like death in Leishmania infantum. Mol Biochem Parasitol. 2007;152:192–202. doi: 10.1016/j.molbiopara.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 48.Genes C, Baquero E, Echeverri F, Maya JD, Triana O. Mitochondrial dysfunction in Trypanosoma cruzi: the role of Serratia marcescens prodigiosin in the alternative treatment of Chagas disease. Parasit Vectors. 2011;4:8. doi: 10.1186/1756-3305-4-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smirlis D, Duszenko M, Ruiz AJ, Scoulica E, Bastien P, Fasel N, et al. Targeting essential pathways in trypanosomatids gives insights into protozoan mechanisms of cell death. Parasit Vectors. 2010;3:107. doi: 10.1186/1756-3305-3-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vaidya AB. The mitochondrion. In: Sherman IW, editor. Molecular approaches to malaria. Washington: ASM Press; 2005. pp. 234–252. [Google Scholar]
- 51.Rathore S, Jain S, Sinha D, Gupta M, Asad M, Srivastava A, et al. Disruption of a mitochondrial protease machinery in Plasmodium falciparum is an intrinsic signal for parasite cell death. Cell Death Dis. 2011;2:e231. doi: 10.1038/cddis.2011.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sherman IW, Eda S, Winograd E. Erythrocyte aging and malaria. Cell Mol Biol. 2004;50:159–169. [PubMed] [Google Scholar]
- 53.Udomsangpetch R, Pipitaporn B, Silamut K, Pinches R, Kyes S, Looareesuwan S, et al. Febrile temperatures induce cytoadherence of ring-stage Plasmodium falciparum-infected erythrocytes. Proc Natl Acad Sci USA. 2002;99:11825–11829. doi: 10.1073/pnas.172398999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Long HY, Lell B, Dietz K, Kremsner PG. Plasmodium falciparum: in vitro growth inhibition by febrile temperatures. Parasitol Res. 2001;87:553–555. doi: 10.1007/s004360100374. [DOI] [PubMed] [Google Scholar]
- 55.Joshi B, Biswas S, Sharma YD. Effect of heat-shock on Plasmodium falciparum viability, growth and expression of the heat-shock protein `PFHSP70-I’ gene. FEBS Lett. 1992;312:91–94. doi: 10.1016/0014-5793(92)81417-K. [DOI] [PubMed] [Google Scholar]
- 56.Kwiatkowski D, Greenwood BM. Why is malaria fever periodic? A hypothesis. Parasitol Today. 1989;5:264–266. doi: 10.1016/0169-4758(89)90261-5. [DOI] [PubMed] [Google Scholar]