

Abstract
A major question in mapping protein-ligand or protein-protein interactions in solution is to distinguish direct-binding interactions from long-range conformational changes at allosteric sites. We describe here the applicability of amide hydrogen deuterium exchange mass spectrometry (HDXMS) in addressing this important question using the bacteriophage HK97 capsid proteins’ interactions with their processing protease. HK97 is a lambda-like dsDNA bacteriophage that is ideal for studies of particle assembly and maturation. Its capsid precursor protein is composed of two main regions, the scaffolding protein (δ-domain, residues 2-103), and the coat subunit (residues 104-385), which spontaneously forms a mixture of hexamers and pentamers upon association. Activation of the viral protease, which occurs after particle assembly, is initiated by the protease mediated digestion of the scaffolding domains to yield Prohead-2. This irreversible step is obligatory for activation of the virus maturation pathway. Here we provide an “addendum” to our previous study of Prohead I and Prohead I+pro (a transient complex of Prohead I and the protease) where we investigated the interactions between the δ domain and the packaged protease using HDXMS. Our results revealed two sites on the δ domain: one set of contiguous peptides that showed decreased exchange at the protease binding site at early time points of deuterium labeling and another separate set of continuous peptides that showed decreased exchange at later time points. Even though this cannot reveal the time scales of molecular processes governing binding and allostery, we believe this differential pattern of exchange across deuteration times can allow spatial distinction between binding sites and long range conformational changes with allosteric implications. This partitioning can be discerned from the lag between noncontiguous regions on a protein showing maximal changes in deuterium exchange and highlights a powerful application of HDXMS in distinguishing direct binding in transient protein-protein interactions from allosteric changes.
Keywords: allostery, binding, HDXMS, protein-protein interactions, viral protein dynamics
A major question in mapping protein-ligand or protein-protein interactions in solution is to distinguish direct binding and allosteric sites. We describe here the applicability of HDXMS in addressing this important question using the bacteriophage HK97 capsid proteins’ interaction with their processing protease. HK97 is a lambda-like dsDNA bacteriophage that is exceptionally amenable for studies of particle assembly and maturation.1 Prohead I−pro is the first assembly product when the capsid protein gene (gp5) is expressed in E. coli. It is formed by 420 copies of the subunit arranged on a T = 7l quasi-equivalent surface lattice. HK97 gp5 is a 384 amino acid protein with residues 2–103 (called the δ domain) required for proper assembly of the capsid. Expression of the viral protease (gp4) with gp5 results in a transient particle, Prohead I+pro, that undergoes proteolysis of the δ domain when the particle is fully assembled, followed by gp4 auto-digestion resulting in small polypeptides that diffuse out of the particles.2 The resulting assembly product is Prohead II and can be readily purified from the expression system. Prohead I+pro is stable if gp4 is made inactive by mutation.3 Here we provide an “addendum” to our previous study of Prohead I−pro and Prohead I+pro where we investigated the structural relationship between the δ domain and the packaged protease.4 We established that the δ domain in the absence or presence of protease was found to be largely disordered in EM or crystallographic studies suggesting that it adopted multiple conformations. We therefore used amide hydrogen deuterium exchange mass spectrometry (HDXMS) to provide additional insights regarding the dynamic character of the δ domain and capsid protein in Prohead I−pro and Prohead I+pro particles. Here we further discuss the implications of the data reported in Veesler et al.4 and provide a working model to explain the deuterium exchange profiles measured for different regions of the viral coat protein. This analysis has implications not discussed in the original paper.
Deuterium exchange at backbone amide hydrogens is highly dependent on the environment and is directly affected by the secondary, tertiary and quaternary structure of a protein5. Amides are thus powerful probes for monitoring both solvent accessibility as well as intrinsic H-bonding and neighborhood effects. Consequently, it offers a powerful tool for mapping protein-protein interactions in solution. The observed rate of deuterium exchange (kobs) is a function of the association (ka) and dissociation (kd) rates as well as of the concentrations of interacting partners (protein or ligand). It is expressed as follows:
(Mandell et al.6) kex represents the intrinsic rate of deuterium exchange in solution.
HDXMS is especially suited to mapping protein-protein interactions at peptide resolution where every backbone amide within a protein is a potential reporter for monitoring protein dynamics, mapping protein-protein interactions and allostery. In HDXMS experiments, deuterium exchange is localized to specific regions of the protein by proteolysis of the target protein with the enzyme, pepsin and mass spectrometry is then applied to measure the increase in mass upon deuterium exchange.7 The interactions between the capsid protein (gp5) and the protease (gp4) were examined using HDXMS. In these mapping studies, use of a mutant protease stably maintained the Prohead I+pro intermediate and was used to map capsid protein-protease interactions, focusing on the capsid protein.4 A total of 140 molecules of protease per virus particle were used, resulting in a 3-fold molar excess of viral capsid protein (420 capsid proteins/virus particle) to protease. HDXMS of the free viral capsid protein provided a relative dynamic profile of the protein at peptide resolution, wherein certain regions were observed to be more dynamic overall as evidenced by their faster deuterium exchange rates (Fig. 1). These can be considered “adaptable regions” with greater relative intrinsic flexibility or disorder in the absence of protease and represented putative sites for protease-capsid protein interactions. In the presence of protease, our studies revealed differential kinetics of exchange at 2 clusters of peptides within these ‘adaptable regions’.
Figure 1.
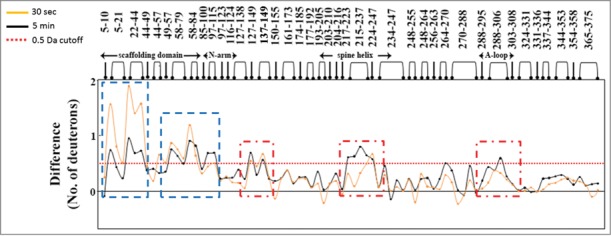
Difference in deuterium exchange between protease-free procapsids and protease containing procapsids at peptide resolution highlight the direct and indirect effects of protease binding. Specific pepsin-digest fragment peptides of procapsid protein generated are listed from the N- to C-terminus (x-axis). The 2 plots represent the difference in deuterium exchange (y-axis) at 2 time points of deuterium on-labeling (t = 30 sec (orange) and t = 5 min (black)) with each point representing a single pepsin-digest fragment peptide. Positive differences represent regions in the procapsid showing reduced deuterium exchange in presence of protease. Peptides showing differences at early time points represent direct binding sites (blue dashed boxes) and those regions showing larger differences in deuterium exchange (t = 5 min) represent long range stabilization/allosteric sites (red dashed boxes). Deuterium exchange of 0.5 Da is considered significant and is represented by a red dashed line in the plot.
One set of peptides (residues 22–44) in Prohead I+pro, spanning the N-terminal δ domain of the capsid protein, showed decreased deuterium exchange compared to Prohead I−pro at early time points (Deuterium exchange time, t = 0.5 min, summarized in Fig. 1). We interpret this as the direct binding site within the δ domain of the capsid protein for the gp4 protease (schematic is in Fig. 2). At early time points, direct binding of the protease lowers deuterium exchange by sequestering H-bond contacts or by limiting solvent accessibility. This deuterium exchange protection disappears with increasing deuteration time. This is likely attributed to the rapid dissociation rates of the gp4 protease from the target capsid protein together with the molar excess of deuterium in the experiment. It must be pointed out that these mapping studies were carried out under sub-stoichiometric ratios of protease to capsid protein (molar ratio of 1:3). The deuterium exchange observed in the presence of protease being an average of both the protease-bound and free conformations, we predict that the magnitude difference in deuterium exchange would be greater if increased protease to capsid protein ratios are used under comparable experimental conditions. A slow dissociation rate relative to the deuterium exchange time would have shown up as multimodal deuterium exchange profiles for the binding site regions, reflecting the relative populations of protease-bound and free capsid protein. There was no evidence for any multimodal profiles under our experimental conditions.
Figure 2.
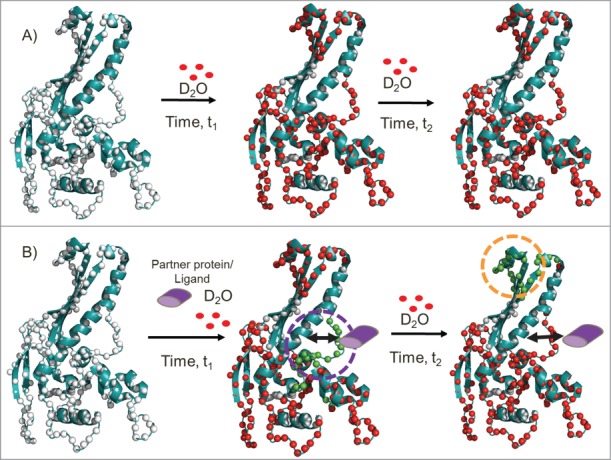
Comparative HDXMS analysis across multiple protein loci enables a distinguishing of direct protein/ligand binding from long range stabilization/allosteric sites on proteins. The schematic shows the application of HDXMS in probing protein-protein/ligand interactions. The HK-97 procapsid protein (PDB ID:3QPR) (cartoon in teal) with the backbone amide nitrogen atoms as white spheres. Deuterons (red) exchange preferentially at regions with greater solvent accessibility or that are more dynamic. Deuterium exchange of the protein at 2 different times of deuterium exchange (t2 > t1) in the absence (A) and presence of partner protein/ligand (purple) (B). In the presence of partner protein/ligand, differential decreases in deuterium exchange are observed at both proximal binding sites and distal sites. At early time points (represented by t1), reduced exchange (green) highlights the ‘direct’ binding site. With increased deuteration time (represented by t2), the binding site begins to show greater exchange arising from the reversible kinetics of association and dissociation of the partner protein/ligand, while a distal site shows reduced exchange (orange dashed lines). No differences in rates of deuterium exchange are observable between these 2 sites in the protein/ligand-free protein.
A second set of peptides, residues 215–237 of the spine helix, residues 264–270 and residues 288–306 of the A-loop, require a more sophisticated interpretation and correspond to capsid subunit regions distal from the δ domain. While there were no observable differences in deuterium exchange in these peptides between Prohead I−pro and Prohead I+pro at early time points (t = 0.5 min), the difference became evident with increasing deuterium exchange times (deuterium exchange across the protein at t = 5 min is displayed in Fig. 1). We predict once again that these magnitude differences across different deuterium exchange times would be greater if equimolar ratios of protease to capsid protein were present. However, the ratio (140 protease: 420 capsid proteins) used in our HDXMS studies is determined by assembly of Prohead I+pro intermediate and cannot be altered. There is a structure of the bacteriophage HK97 Prohead I+pro (PDB ID: 3QPR) but it lacks the N-terminal 118 residues, which we predict to span the protease binding site. Since the viral capsid would encapsulate the protease and render it invisible, for ease of visualization, we have mapped our results onto the hexameric unit of Prohead I+pro (Fig. 3). In this figure, the N-terminal 118 residues are in dashed lines and as illustrative art. The green regions highlight our predicted binding sites while the red regions highlight the regions reflecting the long range conformational changes induced by protease binding.
Figure 3.
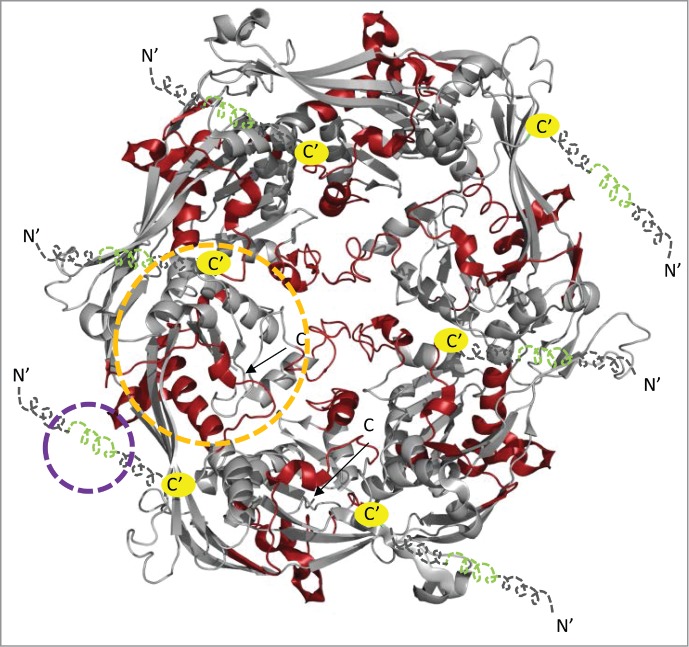
Distinguishing direct binding of processing protease from allosteric effects in the hexameric unit of bacteriophage HK97 Prohead I. The structure of the viral capsid (PDB ID: 3QPR)3 lacks the N-terminal 118 residues and shows the viral capsid as it would encapsidate the virally encoded protease. In order to map our predicted binding sites and distinguish it from allostery, we have represented our HDXMS results mapped onto a hexameric unit of the prohead I viral capsid. For illustrative purposes, we have represented the N-terminal 118 residues for which no structural information is available in dashed lines. The terminii of the illustrative art are represented by N′ and C′ and yellow spheres indicate the junction between the illustrative art and the structural coordinates of the hexameric unit of the viral capsid (PDB ID: 3QPR). A subset of the N-terminal region in green showed decreased deuterium exchange at early time points and reflects the protease binding site, while the regions in red at distal regions indicate the regions showing maximal decreases in deuterium exchange at later points and these indicate the allosteric changes upon protease binding. The binding and allosteric sites for one representative monomer are shown in blue and orange dashed lines respectively.
We believe the lag in deuterium exchange times when the greatest protection is seen across the above 2 sites on the capsid protein offer important insights into binding and allostery. Under the optimum experimental conditions, binding sites would show deuterium exchange protection at earlier times, whereas distal non-contiguous regions, characteristic of long range stabilizing effects would show the greatest shifts in deuterium exchange at later times. This identifies the capsid protein δ domain as the binding site of the protease and identifies possible allosteric implications across the entire capsid. These observable long range effects can be distinguished from direct binding from their kinetic profiles as they occur over longer time scales and involve structural stabilization of the dynamic unbound state. Stabilized regions include both fast exchanging amides and slower exchanging reporter amides. When remote interactions occur, the faster exchanging amides continue to exchange, but the slower exchanging amides are further stabilized, which translates into a gradual decrease in their intrinsic exchange rate. Thus, at the earlier time points the differences in protection between the unbound and protease-bound states are too low to be observable, but as labeling time increases, the decreased exchange rates are magnified to be detectable as significant differences in deuterium exchange (Schematic shown in Fig. 2). This behavior is consistent with a model proposed by Gertsman et al.8 when the Prohead II crystal structure was determined and further elaborated by Johnson.9 It was proposed that interactions between the δ domains allosterically affect the tertiary structure of the rest of the capsid protein creating distortions and instability. This conformational stress is trapped by the quaternary structure when the δ domains are removed by proteolysis, making Prohead II metastable and poised for exothermic expansion and further maturation. The fact that interactions between the δ domains and inactive protease allosterically affect the structure of the subunit at a distance is consistent with this hypothesis. Moreover, other studies have shown that the Prohead I+pro is more stable than Prohead I−pro, making the decrease in HDX after 10 minutes consistent with these observations.3
In conclusion, we report that careful kinetic analysis of HDXMS across time scales can allow for distinguishing direct binding sites with partner proteins/ligands from long range conformational changes. Our results, with the capsid protein, allow us to predict the direct binding site for the protease to be localized entirely within the δ domain. Furthermore we propose that the entire Prohead I+pro complex is highly dynamic and perturbation of the δ domains by direct protease interaction does affect the tertiary structure of the capsid subunit at a distance, supporting the previously proposed role of the δ domains in allosterically influencing the capsid subunit tertiary structure.
Funding
We acknowledge funding from the NIH (R01 AI040101) to JEJ and from a FP7 Marie-Curie IOF fellowship (273427) awarded to DV. We also acknowledge funding from Ministry of Education (MOE2012-T3–1–008), Singapore and Mechanobiology Institute, NUS, Singapore to GSA. Part of this work was carried out at the National Resource for Automated Molecular Microscopy which is supported by the National Institutes of Health though the National Center for Research Resources (2P41RR017573–11) and the National Institute of General Medical Sciences (9 P41 GM103310–11). J.S and A.J.R.H. are supported by the Netherlands Proteomics Centre.
References
- 1. Hendrix RW, Johnson JE. Bacteriophage HK97 capsid assembly and maturation. Adv Exp Med Biol 2012; 726:351-63; PMID:22297521; http://dx.doi.org/ 10.1007/978-1-4614-0980-9_15 [DOI] [PubMed] [Google Scholar]
- 2. Duda RL, Oh B, Hendrix RW. Functional domains of the HK97 capsid maturation protease and the mechanisms of protein encapsidation. J Mol Biol 2013; 425:2765-81; PMID:23688818; http://dx.doi.org/ 10.1016/j.jmb.2013.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang RK, Khayat R, Lee KK, Gertsman I, Duda RL, Hendrix RW, Johnson JE. The Prohead-I structure of bacteriophage HK97: implications for scaffold-mediated control of particle assembly and maturation. J Mol Biol 2011; 408:541-54; PMID:21276801; http://dx.doi.org/ 10.1016/j.jmb.2011.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Veesler D, Khayat R, Krishnamurthy S, Snijder J, Huang RK, Heck AJ, Anand GS, Johnson JE. Architecture of a dsDNA viral capsid in complex with its maturation protease. Structure 2014; 22:230-7; PMID:24361271; http://dx.doi.org/ 10.1016/j.str.2013.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Englander SW, Kallenbach NR. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q Rev Biophys 1983; 16:521-655; PMID:6204354; http://dx.doi.org/ 10.1017/S0033583500005217 [DOI] [PubMed] [Google Scholar]
- 6. Mandell JG, Baerga-Ortiz A, Akashi S, Takio K, Komives EA. Solvent accessibility of the thrombin-thrombomodulin interface. J Mol Biol 2001; 306:575-89; PMID:11178915; http://dx.doi.org/ 10.1006/jmbi.2000.4416 [DOI] [PubMed] [Google Scholar]
- 7. Hoofnagle AN, Resing KA, Ahn NG. Protein analysis by hydrogen exchange mass spectrometry. Annu Rev Biophys Biomol Struct 2003; 32:1-25; PMID:12598366; http://dx.doi.org/ 10.1146/annurev.biophys.32.110601.142417 [DOI] [PubMed] [Google Scholar]
- 8. Gertsman I, Gan L, Guttman M, Lee K, Speir JA, Duda RL, Hendrix RW, Komives EA, Johnson JE. An unexpected twist in viral capsid maturation. Nature 2009; 458:646-50; PMID:19204733; http://dx.doi.org/ 10.1038/nature07686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Johnson JE. Virus particle maturation: insights into elegantly programmed nanomachines. Curr Opin Struct Biol 2010; 20:210-6; PMID:20149636; http://dx.doi.org/ 10.1016/j.sbi.2010.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]