

Abstract
There is a growing list of examples where perturbed mitochondrial function is associated with increased longevity, yet the exact mechanisms have remained elusive. This phenomenon was first documented, and has been studied most extensively, in C. elegans. One prominent model proposed that lifespan extension resulting from electron transport chain inhibition is due to induction of the mitochondrial unfolded protein response. This model requires revision in light of recent data showing that the mitochondrial unfolded protein response, as defined by the field, is neither necessary nor sufficient for lifespan extension in C. elegans. Several additional factors have been proposed to underlie this lifespan extension, which is likely to be multifactorial and complex.
Keywords: aging, ATFS-1, electron transport chain, hsp-6, longevity, lifespan, mitochondrial unfolded protein response
Introduction: Mitochondrial Stress and the Mitochondrial Unfolded Protein Response
Inhibition of mitochondrial function was first associated with longevity in C. elegans when it was discovered that clk-1 mutants with reduced coenzyme Q production are long-lived.1,2 Soon after this, animals defective for function of the electron transport chain complex III component, ISP-1, were also found to have increased lifespan.3 Genome-wide screens for RNAi clones that extend lifespan then identified dozens of mitochondrial factors that modulate aging, confirming the generality of these observations.4,5 To date, lifespan extension has been reported following RNAi knockdown of components of complex I, III, IV, and V of the electron transport chain (ETC), as well as mitochondrial ribosomal proteins, proteases, and other mitochondrial proteins.6
Much of the work aimed at understanding the relationship between mitochondrial inhibition and lifespan extension has focused on the quality control mechanisms that regulate mitochondrial homeostasis and respond to damaged proteins and complexes. During aging there is a decreased ability of cells to cope with misfolded, damaged, and aggregated proteins.7,8 This deterioration of protein quality control mechanisms contributes to negative effects on cellular function. Compartment specific responses have evolved to increase expression of proteins the promote proteostasis, including chaperones and proteases, when misfolded proteins are detected. Such compartment specific responses include the cytoplasmic heat shock response, the endoplasmic reticulum unfolded protein response (UPRER) and the mitochondrial unfolded protein response (UPRmt). Each of these has been proposed to promote increased lifespan in C. elegans by preserving protein homeostasis with age.
The UPRmt was first characterized in mammalian cells lacking mtDNA, which results in an imbalance of the nuclear and mitochondrial encoded subunits of the ETC.9 In this context, it was shown that several nuclear encoded mitochondrial chaperones are coordinately induced, akin to the well-characterized induction of ER chaperones in response to ER stress. In addition to depletion of mitochondrial DNA, targeting an unstable transgenic protein to the mitochondria was also sufficient to activate the UPRmt in mammalian cells.10
This pathway has since been extensively studied in C. elegans, primarily using GFP reporters for hsp-6 (mitochondrial Hsp70) and hsp-60, which have allowed for the identification of a handful of factors required for their full induction in response to mitochondrial stress.11 These include the ubiquitin-like protein UBL-5,12 the homeobox transcription factor DVE-1, the mitochondrial protease ClpP,13 the ATP-dependent peptide transporter HAF-1,14 and the basic-leucine zipper transcription factor ATFS-1.15
ATFS-1 is the best-characterized regulator of the UPRmt in C. elegans, and has reproducibly been shown to be required for UPRmt induction resulting from a variety of stressors including paraquat, ethidium bromide, and knockdown of and mutations in ETC components.15,16 ATFS-1 contains both nuclear and mitochondrial localization sequences allowing it to control mitochondrial homeostasis by shuttling to either subcellular compartment. When imported into the mitochondria, it is degraded by the Lon protease in the mitochondrial matrix.15 Under conditions of mitochondrial stress, ATFS-1 is excluded from the mitochondria and imported into the nucleus where it upregulates expression of mitochondrial stress response factors such as chaperones and proteases. Both hsp-6 and hsp-60 are among genes regulated in an ATFS-1-dependent manner.
Dissociating the Mitochondrial UPR from Longevity
The Dillin lab first introduced the hypothesis that the UPRmt promotes longevity during mitochondrial dysfunction with the report that inhibition of ETC function specifically in neurons was sufficient to induce the UPRmt in non-neuronal cells, including intestine, as well as extend lifespan.17 Cell non-autonomous induction of the UPRmt in this study was shown only using the hsp-6p::gfp reporter (Fig. 1). Thus, it remains unclear whether this represents a general induction of the UPRmt, induction of only a subset of UPRmt targets, or induction of only the reporter construct. In the same study, the authors concluded that the UPRmt promoted lifespan extension based on their observation that lifespan extension from mutation of isp-1 required factors previously reported to be involved in UPRmt signaling.17
Figure 1.
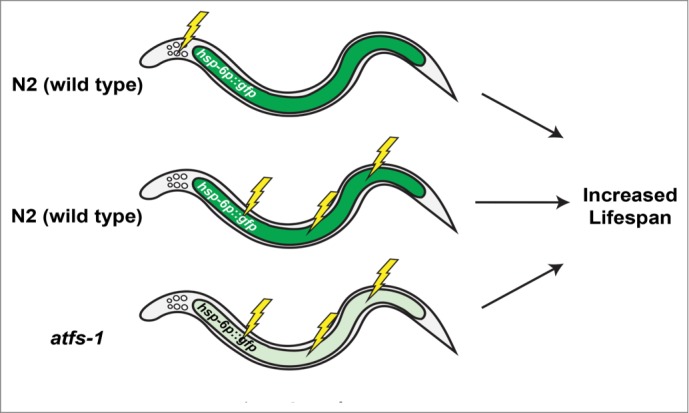
Mitochondrial dysfunction induces the UPRmt in a cell autonomous and non-autonomous fashion, but does not require the UPRmt for longevity. The C. elegans UPRmt can be monitored by the transcriptional upregulation of mitochondrial chaperone genes including hsp-6 and hsp-60. Electron transport chain inhibition by RNAi knockdown of cco-1 (indicated by lightning bolts) in neurons or the intestine is sufficient for lifespan extension in worms and causes obvious induction of the hsp-6p::gfp reporter in intestinal cells (Panels 1 and 2). Loss of function in the UPRmt transcription factor atfs-1 prevents induction of hsp-6p::gfp following cco-1(RNAi) but does not prevent lifespan extension (Panel 3).
Based on this model, we set out to identify novel longevity factors by performing an RNAi screen for genes that negatively regulate the UPRmt. Specifically, we looked for RNAi clones that induced both the hsp-6p::gfp reporter and the hsp-60p::gfp reporter. Although conceptually similar screens had been performed previously, we identified dozens of novel genetic modifiers of UPRmt signaling using this approach.18 We anticipated that a majority of the RNAi clones found to induce the hsp-6p::gfp and hsp-60p::gfp reporters should also extend lifespan. In contrast to this prediction, about half of the clones tested had no effect or significantly shortened lifespan. This led us to speculate that the UPRmt may not be a direct regulator of longevity.
To further explore the role of the UPRmt in aging, we utilized gain and loss of function alleles of atfs-1. As previously reported for RNAi knockdown of the nuclear-encoded mitochondrial metalloprotease spg-7,15 we observed that loss of function in atfs-1 suppressed induction of the UPRmt following knock-down or mutation of cco-1 or isp-1.18 This was determined both by reduced expression of GFP reporter for hsp-6, as well as endogenous expression of several UPRmt target genes. Despite blocking induction of the UPRmt by these measures, we still observed increased lifespan in every case tested. We also found that 2 gain of function alleles of atfs-1 failed to extend lifespan relative to wild type N2 animals, despite the fact that these alleles did cause increased expression of UPRmt target genes. Based on these observations, we concluded that the UPRmt, as currently defined by the field, is neither necessary nor sufficient for lifespan extension in C. elegans.
The UPRmt and Aging: Where did Things Go Wrong?
Disproving hypotheses is an important and necessary part of the scientific process. In this case, we did not set out to disprove the model proposed by Durieux et al.17; however, in the course of our experiments, it became apparent to us that important details were missing from the initial and subsequent publications in this area. Among those subsequent publications are 2 papers from the Auwerx lab that have propagated this model, arguing the C. elegans lifespan extension from knockdown of mitochondrial ribosomal proteins, rapamycin, resveratrol, antibiotics, overexpression of sirtuins, and NAD+ precursors all require the UPRmt.19,20 As enticing as this idea may be, the data supporting these claims is not entirely consistent, and key controls are missing in some cases.21
One major limitation in this area is that the field has not carefully defined the UPRmt in C. elegans, which has resulted in the near exclusive use of the hsp-6p::gfp reporter to measure this response. Indeed, this was the only measure of UPRmt activity reported in the papers proposing a mechanistic link between the UPRmt and longevity in C. elegans.17,19,20 Aside from the obvious limitations associated with using a single readout for a coordinated stress response involving dozens of genes, this is also a questionable choice given that the hsp-6 ortholog, mtHsp70, is not generally considered a component of the UPRmt in mammals.10 The Haynes lab has recently identified numerous genes that are transcriptionally altered in C. elegans in response to mitochondrial stress induced by RNAi knockdown of spg-7.15 Induction of the canonical UPRmt genes was dependent upon ATFS-1; however, there were also numerous additional ATFS-1-dependent transcriptional changes as well as genes whose expression was modulated in an ATFS-1-independent manner (Fig. 2). It would be quite valuable going forward if some consensus could be reached as to which subset of genes regulated in response to mitochondrial stress make up the UPRmt.
Figure 2.
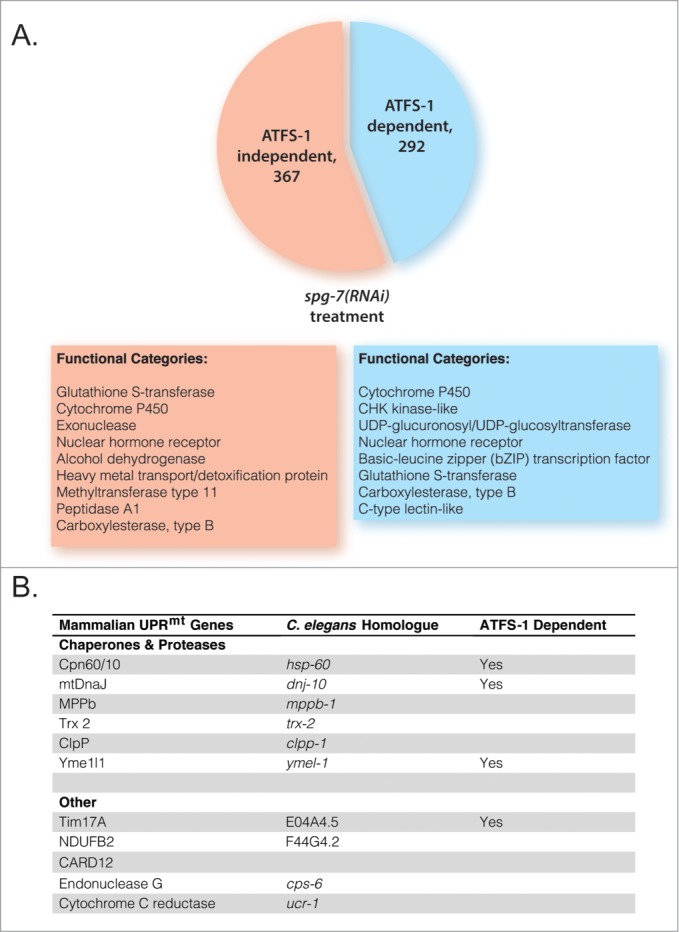
ATFS-1 regulates expression of several functional categories of genes, besides mitochondrial chaperones and proteases. (A) ATFS-1 regulates expression of at least 292 genes during mitochondrial stress. These include a multitude of cytochrome P450 enzymes, kinases, and nuclear hormone transcription factors, among others. Based on data from Nargund et al.15 (B) The UPRmt appears to be highly conserved between mammals and worms with respect to target genes that function as mitochondrial chaperones, proteases, and protein import machinery. If a UPRmt gene is known to require ATFS-1 for induction, ‘Yes’ is marked in the third column. Mammalian UPRmt genes from Aldridge et al.42
A second limitation of prior studies of the UPRmt in C. elegans is the use of genetic modifiers of UPRmt signaling whose function and mechanisms of action are poorly characterized. For example, loss of function alleles or RNAi knockdown of ubl-5 and haf-1 have been 2 methods used to argue for UPRmt-mediated effects on longevity.17,19,20 UBL-5 is an ubiquitin-like protein that interacts with the homeobox protein DVE-1 to promote expression of hsp-6 and hsp-60 in response to mitochondrial stress. The yeast ortholog of UBL-5, Hub1, is an essential gene involved in cell cycle regulation and pre-mRNA splicing,22 and numerous phenotypes have been associated with ubl-5(RNAi) in C. elegans, most of which are not obviously linked to its role as a regulator of the UPRmt. Thus it seems likely that UBL-5 has functions in addition to its role as a regulator of hsp-6 and hsp-60 expression, which may complicate epistasis studies aimed at exploring its role in the UPRmt and aging. HAF-1 encodes an ATP-Binding Cassette (ABC) transporter in the mitochondrial inner membrane that transports peptide fragments from the mitochondrial matrix.14 There is some evidence that HAF-1 negatively regulates mitochondrial protein import, including that of ATFS-1, by an unknown mechanism; however, the role of HAF-1 in the UPRmt may be limited to low levels of mitochondrial stress, and HAF-1 has been shown to be dispensable for induction of UPRmt target genes in several cases.15,16
With respect to the role of the UPRmt in aging, it is noteworthy that neither HAF-1 nor UBL-5 has been shown to be required for induction of multiple UPRmt target genes under the conditions where lifespan is extended. In one case, Houtkooper et al.19 did examine expression of the hsp-6p::gfp reporter in response to mrps-5(RNAi) and cco-1(RNAi), both of which extend lifespan, and found that induction is only partially reduced by either haf-1 or ubl-5 knockdown or mutation. Interpretation of these experiments is somewhat further complicated by the use of double RNAi knockdown in some cases without the corresponding control of quantifying the level of knockdown for each targeted gene. Double RNAi knockdown is generally less efficient than the use of single RNAi clones,23 and properly controlling for this may be especially important in the case of electron transport inhibition, where there is a clear dose response with respect to efficacy of RNAi knockdown and longevity.24 Unfortunately, there is also currently very little information regarding the effects of haf-1 and ubl-5 knockdown or mutation on endogenous expression of UPRmt target genes. Thus, we feel strongly that the current data suggesting that UBL-5 or HAF-1 are sometimes required for lifespan extension cannot be rigorously interpreted due to the absence of necessary controls and lack of understanding regarding how and whether these factors do, indeed, generally regulate the UPRmt.
In contrast to other factors reported to be involved in the UPRmt in C. elegans, the function and mode of regulation for ATFS-1 is relatively well characterized. ATFS-1 is critical for induction of the UPRmt, as measured both by GFP reporters and endogenous expression of UPRmt target genes, across many different mitochondrial stressors including paraquat, ethidium bromide, and RNAi depletion of the mitochondrial AAA metalloprotease spg-7 and subunits of the TIM complex.14-16 We found that partial deletion and knockdown of atfs-1 greatly attenuated the UPRmt without causing any overt phenotypes including effects on lifespan. The atfs-1(tm4525) allele prevented induction of the hsp-6p::gfp reporter as well as increased expression of hsp-6, hsp-60, and timm-23 following RNAi knockdown of cco-1 or mutation of isp-1 without influencing the lifespan extension associated with these interventions.18 We note that this allele causes a truncated atfs-1 mRNA to be expressed,15 so it is possible that residual ATFS-1 activity could be present. Nevertheless we observed a complete block in UPRmt induction, based on expression of 3 different endogenous UPRmt target genes. Taken in combination with the failure of gain of function alleles in ATFS-1 to extend lifespan, as observed both by us and the Pilon lab,18,25 these data greatly weaken the model that the UPRmt plays a direct role in promoting longevity in C. elegans.
Mitochondrial Stress and Aging: Multiple Paths to Longevity?
Although the UPRmt has gained a level of prominence as a potential mediator of longevity due to the high profile papers mentioned above, a host of additional cellular factors have also been implicated in controlling longevity in response to mitochondrial dysfunction. These include GCN-2, CEP-1, HIF-1, CEH-23, TAF-4, AHA-1, CEH-18, JUN-1, NHR-27 and NHR-49.26-30 To date, however, no universal mechanism has been demonstrated that can account for all, or even most, of the cases where mitochondrial dysfunction is associated with longevity in C. elegans. This raises the possibility that there are multiple pathways to longevity mediated by mitochondrial stress (Fig. 3), which is supported by studies from the Hekimi lab suggesting that RNAi knockdown of the genes encoding the ETC components isp-1 or nuo-6 extends lifespan by a mechanism that is distinct from mutations in those same genes.31 Along these lines, there is growing evidence for at least 2 distinct general mechanisms by which mitochondrial dysfunction can extend lifespan in C. elegans: reactive oxygen species (ROS)-mediated signaling and metabolic adaptation.
Figure 3.
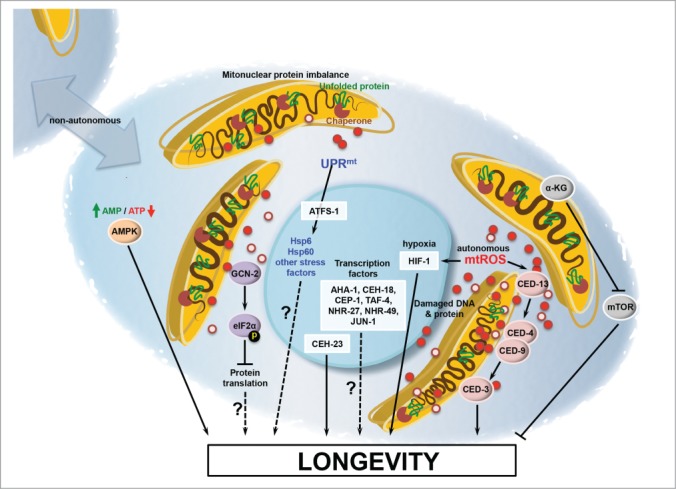
Current models for mitochondrial longevity. Inhibition of ETC function can lead to increased level of ROS (red circles), which can induce lifespan extension. One effect of this ROS production is stabilization of the hypoxia-inducible transcription factor HIF-1 and activation of the intrinsic apoptotic pathway. Protein misfolding in the mitochondria causes induction of the UPRmt and inhibition of translation by the kinase GCN-2. This concerted mechanisms promotes protein homeostasis in the mitochondria. Other factors have also been implicated in lifespan extension following inhibition of mitochondrial function including AMPK, the homebox protein CEH-23, the p53 homolog CEP-1, and other transcription and signaling factors.
One way in which ROS-mediated signaling can extend lifespan is through stabilization of the hypoxia inducible transcription factor HIF-1.27,32 HIF-1 is typically degraded under normoxic conditions by the proteasome. When oxygen levels are very low, however, HIF-1 becomes stabilized and induces transcription of a suite of genes involved in promoting survival under hypoxia.33 Interestingly, HIF-1 can also become stabilized under conditions where mitochondrial respiration is inhibited, and HIF-1 is required for lifespan extension from mutations in isp-1 or clk-1.27 Several studies have shown that stabilization of HIF-1 is sufficient to extend lifespan in animals with normally functioning mitochondria,34-36 providing additional compelling evidence in support of this model. HIF-1 is only partially required for lifespan extension from RNAi knockdown of electron transport chain components,27 however, indicating that additional mechanisms must also exist.
Recently, the intrinsic apoptotic pathway has been implicated in ROS-mediated lifespan extension.37 Lifespan extending mutations in isp-1 or nuo-6, as well as treatment with paraquat, can activate a protective program mediated by CED-3/Casp9, CED-4/Apaf1, and CED-9/Bcl2. Despite the requirement of apoptotic genes, this response does not induce apoptosis. Furthermore, the BH3-only protein CED-13 also functions in this pathway, conceivably upstream of mitochondrial ROS since it is required for full lifespan extension from mutations in nuo-6 and isp-1, but not paraquat. In addition to longevity, the phenotypes of decreased pumping rate, thrashing, and developmental rate from mitochondrial ETC mutations were partially dependent on CED-4 and CED-13. Thus, the intrinsic apoptotic pathway can function to promote organismal health and cellular survival in the presence of mitochondrial dysfunction. The importance of this type of response to mitochondrial dysfunction is plausible, since C. elegans is a largely post-mitotic adult organism with a small number of cells that cannot be lost without severe consequence. Therefore, it will be interesting to determine if a similar mechanism exists in higher eukaryotes, perhaps in cell types that have limited regenerative capacity.
Significant metabolic adaptations to mitochondrial stress have also been recently described and there is growing evidence that they are likely to be important for longevity. Metabolomic profiling of mitochondrial mutants revealed that specific α-ketoacids and α-hydroxyacids are produced a high levels by long-lived mutants, due to inhibition of α-ketoacid dehydrogenases.38 Such metabolites could act as signaling factors or inhibit/activate specific enzymes important for longevity. Along these lines, certain α-ketoacids are known to inhibit the prolyl hydroxylase EGL-9, a negative regulator of HIF-1 stability.39 It was also recently reported that the α-ketoacid α-ketoglutarate is sufficient to extend lifespan in C. elegans by binding to and inhibiting the mitochondrial ATP synthase.40 This was associated with inhibition of the mechanistic target of rapamycin (mTOR) pathway and induction of autophagy,40 which has previously been shown to extend lifespan in C. elegans as well as several other species.41 The importance of α-ketoglutarate and the mTOR pathway, if any, in lifespan extension from mitochondrial dysfunction remains to be determined.
Conclusion
The basis for C. elegans lifespan extension from mitochondrial stress is still not fully understood and appears to be mediated by multiple downstream processes. Whether these processes act by truly distinct mechanisms or as part of a coordinated metabolic and genetic network, and whether the UPRmt plays any role in such a longevity network, will require further study. It will be important for the field to arrive at some consensus as to what defines the UPRmt. We suggest that endogenous expression of multiple UPRmt target genes, perhaps along with in the currently widely utilized GFP reporters, should minimally be required in future studies of the UPRmt in C. elegans and that similar approaches should be used when studying this pathway in other model organisms.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
Studies related to this topic in the Kaeberlein Lab are funded by NIH Grant R01AG039390 to MK. CFB was supported by NIH training grants T32GM07270 and T32ES007032. HC was supported by a grant from the Samsung Electronics Company.
References
- 1. Wong A, Boutis P, Hekimi S. Mutations in the clk-1 gene of Caenorhabditis elegans affect developmental and behavioral timing. Genetics 1995; 139:1247-59; PMID:7768437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Felkai S, Ewbank JJ, Lemieux J, Labbé JC, Brown GG, Hekimi S. CLK-1 controls respiration, behavior and aging in the nematode Caenorhabditis elegans. EMBO J 1999; 18:1783-92; PMID:10202142; http://dx.doi.org/ 10.1093/emboj/18.7.1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Feng J, Bussière F, Hekimi S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev Cell 2001; 1:633-44; PMID:11709184; http://dx.doi.org/ 10.1016/S1534-5807(01)00071-5 [DOI] [PubMed] [Google Scholar]
- 4. Dillin A, Hsu AL, Arantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, Kenyon C. Rates of behavior and aging specified by mitochondrial function during development. Science 2002; 298:2398-401; PMID:12471266; http://dx.doi.org/ 10.1126/science.1077780 [DOI] [PubMed] [Google Scholar]
- 5. Lee SS, Lee RY, Fraser AG, Kamath RS, Ahringer J, Ruvkun G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet 2003; 33:40-8; PMID:12447374; http://dx.doi.org/ 10.1038/ng1056 [DOI] [PubMed] [Google Scholar]
- 6. Yanos ME, Bennett CF, Kaeberlein M. Genome-Wide RNAi Longevity Screens in Caenorhabditis elegans. Curr Genomics 2012; 13:508-18; PMID:23633911; http://dx.doi.org/ 10.2174/138920212803251391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Labbadia J, Morimoto RI. Proteostasis and longevity: when does aging really begin? F1000Prime Rep 2014; 6:7; PMID:24592319; http://dx.doi.org/ 10.12703/P6-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Prahlad V, Morimoto RI. Integrating the stress response: lessons for neurodegenerative diseases from C. elegans. Trends Cell Biol 2009; 19:52-61; PMID:19112021; http://dx.doi.org/ 10.1016/j.tcb.2008.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martinus RD, Garth GP, Webster TL, Cartwright P, Naylor DJ, Høj PB, Hoogenraad NJ. Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur J Biochem 1996; 240:98-103; PMID:8797841; http://dx.doi.org/ 10.1111/j.1432-1033.1996.0098h.x [DOI] [PubMed] [Google Scholar]
- 10. Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J 2002; 21:4411-9; PMID:12198143; http://dx.doi.org/ 10.1093/emboj/cdf445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Haynes CM, Ron D. The mitochondrial UPR - protecting organelle protein homeostasis. J Cell Sci 2010; 123:3849-55; PMID:21048161; http://dx.doi.org/ 10.1242/jcs.075119 [DOI] [PubMed] [Google Scholar]
- 12. Benedetti C, Haynes CM, Yang Y, Harding HP, Ron D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 2006; 174:229-39; PMID:16816413; http://dx.doi.org/ 10.1534/genetics.106.061580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell 2007; 13:467-80; PMID:17925224; http://dx.doi.org/ 10.1016/j.devcel.2007.07.016 [DOI] [PubMed] [Google Scholar]
- 14. Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell 2010; 37:529-40; PMID:20188671; http://dx.doi.org/ 10.1016/j.molcel.2010.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012; 337:587-90; PMID:22700657; http://dx.doi.org/ 10.1126/science.1223560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Runkel ED, Liu S, Baumeister R, Schulze E. Surveillance-activated defenses block the ROS-induced mitochondrial unfolded protein response. PLoS Genet 2013; 9:e1003346; PMID:23516373; http://dx.doi.org/ 10.1371/journal.pgen.1003346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 2011; 144:79-91; PMID:21215371; http://dx.doi.org/ 10.1016/j.cell.2010.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bennett CF, Vander Wende H, Simko M, Klum S, Barfield S, Choi H, Pineda VV, Kaeberlein M. Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nat Commun 2014; 5:3483; PMID:24662282; http://dx.doi.org/ 10.1038/ncomms4483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013; 497:451-7; PMID:23698443; http://dx.doi.org/ 10.1038/nature12188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Cantó C, Mottis A, Jo YS, Viswanathan M, Schoonjans K, et al. . The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013; 154:430-41; PMID:23870130; http://dx.doi.org/ 10.1016/j.cell.2013.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bennett CF, Kaeberlein M. The mitochondrial unfolded protein response and increased longevity: cause, consequence, or correlation? Exp Gerontol 2014; 56:142-6; PMID:24518875; http://dx.doi.org/ 10.1016/j.exger.2014.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mishra SK, Ammon T, Popowicz GM, Krajewski M, Nagel RJ, Ares M, Jr., Holak TA, Jentsch S. Role of the ubiquitin-like protein Hub1 in splice-site usage and alternative splicing. Nature 2011; 474:173-8; PMID:21614000; http://dx.doi.org/ 10.1038/nature10143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J. Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol 2001; 2: 1-10; PMID:11178279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rea SL, Ventura N, Johnson TE. Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol 2007; 5:e259; PMID:17914900; http://dx.doi.org/ 10.1371/journal.pbio.0050259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rauthan M, Ranji P, Aguilera Pradenas N, Pitot C, Pilon M. The mitochondrial unfolded protein response activator ATFS-1 protects cells from inhibition of the mevalonate pathway. Proc Natl Acad Sci U S A 2013; 110:5981-6; PMID:23530189; http://dx.doi.org/ 10.1073/pnas.1218778110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ventura N, Rea SL, Schiavi A, Torgovnick A, Testi R, Johnson TE. p53/CEP-1 increases or decreases lifespan, depending on level of mitochondrial bioenergetic stress. Aging Cell 2009; 8:380-93; PMID:19416129; http://dx.doi.org/ 10.1111/j.1474-9726.2009.00482.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee SJ, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol 2010; 20:2131-6; PMID:21093262; http://dx.doi.org/ 10.1016/j.cub.2010.10.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Khan MH, Ligon M, Hussey LR, Hufnal B, Farber R, 2nd, Munkácsy E, Rodriguez A, Dillow A, Kahlig E, Rea SL. TAF-4 is required for the life extension of isp-1, clk-1 and tpk-1 Mit mutants. Aging (Albany NY) 2013; 5:741-58; PMID:24107417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Walter L, Baruah A, Chang HW, Pace HM, Lee SS. The homeobox protein CEH-23 mediates prolonged longevity in response to impaired mitochondrial electron transport chain in C. elegans. PLoS Biol 2011; 9:e1001084; PMID:21713031; http://dx.doi.org/ 10.1371/journal.pbio.1001084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Baker BM, Nargund AM, Sun T, Haynes CM. Protective coupling of mitochondrial function and protein synthesis via the eIF2α kinase GCN-2. PLoS Genet 2012; 8:e1002760; PMID:22719267; http://dx.doi.org/ 10.1371/journal.pgen.1002760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang W, Hekimi S. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell 2010; 9:433-47; PMID:20346072; http://dx.doi.org/ 10.1111/j.1474-9726.2010.00571.x [DOI] [PubMed] [Google Scholar]
- 32. Hwang AB, Jeong DE, Lee SJ. Mitochondria and organismal longevity. Curr Genomics 2012; 13:519-32; PMID:23633912; http://dx.doi.org/ 10.2174/138920212803251427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Leiser SF, Kaeberlein M. The hypoxia-inducible factor HIF-1 functions as both a positive and negative modulator of aging. Biol Chem 2010; 391:1131-7; PMID:20707608; http://dx.doi.org/ 10.1515/bc.2010.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leiser SF, Begun A, Kaeberlein M. HIF-1 modulates longevity and healthspan in a temperature-dependent manner. Aging Cell 2011; 10:318-26; PMID:21241450; http://dx.doi.org/ 10.1111/j.1474-9726.2011.00672.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mehta R, Steinkraus KA, Sutphin GL, Ramos FJ, Shamieh LS, Huh A, Davis C, Chandler-Brown D, Kaeberlein M. Proteasomal regulation of the hypoxic response modulates aging in C. elegans. Science 2009; 324:1196-8; PMID:19372390; http://dx.doi.org/ 10.1126/science.1173507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Y, Shao Z, Zhai Z, Shen C, Powell-Coffman JA. The HIF-1 hypoxia-inducible factor modulates lifespan in C. elegans. PLoS One 2009; 4:e6348; PMID:19633713; http://dx.doi.org/ 10.1371/journal.pone.0006348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yee C, Yang W, Hekimi S. The intrinsic apoptosis pathway mediates the pro-longevity response to mitochondrial ROS in C. elegans. Cell 2014; 157:897-909; PMID:24813612; http://dx.doi.org/ 10.1016/j.cell.2014.02.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Butler JA, Mishur RJ, Bhaskaran S, Rea SL. A metabolic signature for long life in the Caenorhabditis elegans Mit mutants. Aging Cell 2013; 12:130-8; PMID:23173729; http://dx.doi.org/ 10.1111/acel.12029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lu H, Dalgard CL, Mohyeldin A, McFate T, Tait AS, Verma A. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J Biol Chem 2005; 280:41928-39; PMID:16223732; http://dx.doi.org/ 10.1074/jbc.M508718200 [DOI] [PubMed] [Google Scholar]
- 40. Chin RM, Fu X, Pai MY, Vergnes L, Hwang H, Deng G, Diep S, Lomenick B, Meli VS, Monsalve GC, et al. . The metabolite α-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 2014; 510:397-401; PMID:24828042; http://dx.doi.org/ 10.1038/nature13264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature 2013; 493:338-45; PMID:23325216; http://dx.doi.org/ 10.1038/nature11861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aldridge JE, Horibe T, Hoogenraad NJ. Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS One 2007; 2:e874; PMID:17849004; http://dx.doi.org/ 10.1371/journal.pone.0000874 [DOI] [PMC free article] [PubMed] [Google Scholar]