

Abstract
Homeostatic synaptic plasticity (HSP) has been implicated in the development of hyperexcitability and epileptic seizures following traumatic brain injury (TBI). Our in vivo experimental studies in cats revealed that the severity of TBI-mediated epileptogenesis depends on the age of the animal. To characterize mechanisms of these differences, we studied the properties of the TBI-induced epileptogenesis in a biophysically realistic cortical network model with dynamic ion concentrations. After deafferentation, which was induced by dissection of the afferent inputs, there was a reduction of the network activity and upregulation of excitatory connections leading to spontaneous spike-and-wave type seizures. When axonal sprouting was implemented, the seizure threshold increased in the model of young but not the older animals, which had slower or unidirectional homeostatic processes. Our study suggests that age-related changes in the HSP mechanisms are sufficient to explain the difference in the likelihood of seizure onset in young versus older animals.
SIGNIFICANCE STATEMENT Traumatic brain injury (TBI) is one of the leading causes of intractable epilepsy. Likelihood of developing epilepsy and seizures following severe brain trauma has been shown to increase with age. Specific mechanisms of TBI-related epileptogenesis and how these mechanisms are affected by age remain to be understood. We test a hypothesis that the failure of homeostatic synaptic regulation, a slow negative feedback mechanism that maintains neural activity within a physiological range through activity-dependent modulation of synaptic strength, in older animals may augment TBI-induced epileptogenesis. Our results provide new insight into understanding this debilitating disorder and may lead to novel avenues for the development of effective treatments of TBI-induced epilepsy.
Keywords: epileptic seizures, homeostatic plasticity, ion concentration dynamics, network models, synaptic plasticity
Introduction
Penetrating brain wounds or other forms of brain trauma commonly lead to epileptogenesis (Kollevold, 1976; Dinner, 1993; Temkin et al., 1995; Annegers et al., 1998; Topolnik et al., 2003b; Jin et al., 2006; Nita et al., 2007; Avramescu and Timofeev, 2008). Partial cortical deafferentation triggers the development of electrographic seizures within hours of the initial insult (Topolnik et al., 2003a; Nita et al., 2006). Following partial cortical deafferentation, epileptiform discharges were observed in vitro (Prince and Tseng, 1993; Xiong et al., 2011). In vivo studies in undercut cat cortex have revealed synaptic strength modulation and changes in intrinsic excitability (Avramescu and Timofeev, 2008), but exact mechanisms leading to seizure onset following brain trauma remain to be understood.
Homeostatic synaptic plasticity (HSP) has been implicated in epileptogenesis following severe brain trauma (Houweling et al., 2005; Echegoyen et al., 2007; Trasande and Ramirez, 2007; Fröhlich et al., 2008a; O'Leary et al., 2010; Volman et al., 2011a, b). HSP is a slow negative feedback mechanism that maintains neural activity within a physiological range through activity-dependent modulation of synaptic strength and intrinsic excitability (Burrone and Murthy, 2003; Turrigiano, 2008; Pozo and Goda, 2010). It has been proposed that this is achieved through the use of activity-dependent Ca2+ sensors, TNFα and BDNF levels, as well as changes in Arc/Arg3.1 expression (Burrone and Murthy, 2003; Turrigiano, 2008; Pozo and Goda, 2010). In a normal network, HSP works to counterbalance Hebbian plasticity preventing runaway excitation, and restricts the network activity to maintain network stability (Pozo and Goda, 2010). However, for chronic activity deprivation, as occurs after partial cortical deafferentation, HSP may fail to correctly compensate for the loss of network activity promoting hyperexcitability and may potentially lead to paroxysmal synchronized bursting events (Houweling et al., 2005; Fröhlich et al., 2008a; Volman et al., 2011a, b).
Recent studies of epileptogenesis in animals of different age revealed that older animals are more prone to epileptic seizures after traumatic brain injury (TBI) than the younger ones (Timofeev et al., 2013). It was suggested that, following recovery from injury, HSP may fail to control synaptic strength in deafferented areas leading to “overshooting” of synaptic strength and promoting epileptogenesis. In this new study, we explored the hypothesis that HSP regulation is age-dependent, and its changes may lead to epileptogenesis, using a biophysically realistic cortical network model in which synaptic strengths are under homeostatic regulation, and the dynamics of intracellular and extracellular ion concentrations are implemented to achieve realistic “in vivo-type” network dynamics. Following network deafferentation, we observed a decrease in the seizure threshold that was dependent on the severity of trauma. Implementing axonal sprouting rescued the normal physiological activity, but only when HSP was able to precisely control excitability. Impairment of the homeostatic downregulation, as observed in older animals, increased severity of epileptogenesis and led to persistent epileptiform discharges as observed experimentally in the older, but not in the young animals.
Materials and Methods
In vivo experiments.
All experiments were performed in accordance with the guideline of the Canadian Council on Animal Care and approved by the Université Laval Committee on Ethics and Animal Protection. Experiments were performed on cats of either sex. To create conditions that occur in penetrating wounds, the white matter underneath the suprasylvian gyrus (parietal cortex, area 5 and 7) was transected under isoflurane anesthesia. The details of this cortical undercut procedure were described previously (Topolnik et al., 2003b, Nita et al., 2007). The details of surgery and simultaneous wireless local field potential (LFP) behavioral recordings are described previously (Grand et al., 2013). In these experiments, the intracortical LFP electrodes were inserted to a depth of 1 mm in undercut cortex (left suprasylvian gyrus), left posterior marginal gyrus (secondary visual cortex, area 18), and right and left postcruciate gyri (primary somatosensory cortex, area 6). To control for states of vigilance and to record seizure-associated movements, we implanted electro-oculogram electrodes in the inferior surface of the orbital plate of the frontal bone, electromyogram electrodes into the neck muscles, and an accelerometer (ADXL-330, Analog Devices) was attached to the head bones. Continuous wireless recordings of electrographic and movement activities were achieved with NeuroWare W16-series system (Triangle Biosystems). The wireless amplifier and battery were attached to the head and protected with plastic housing. Head-restrained experiments were performed identically to previous description (Nita et al., 2007). The experiments lasted for 3–6 months.
Analysis.
All recordings were analyzed off-line using custom-written routines in IgorPro 4.
Pyramidal cell and interneuron models.
Pyramidal cells (PYs) and inhibitory interneurons (INs) were modeled as two-compartment neurons with dendritic and axosomatic compartments as described previously in detail (Mainen and Sejnowski, 1996; Kager et al., 2000; Bazhenov et al., 2004; Fröhlich and Bazhenov, 2006; Krishnan and Bazhenov, 2011). The change in voltage for each compartment can be described by the following equations:
 |
 |
where VD is the voltage of the dendritic compartment, IDleak and ISleak are the sum of the ionic leak currents, IDpump and ISpump are the sum of the Na+ and K+ currents through the Na+/K+ pump, and IDInt and ISInt are the intrinsic currents for the dendritic and axosomatic compartments, respectively.
The intrinsic current present in the dendritic compartment (IDInt) includes the voltage-gated sodium current (INa), persistent sodium current (INaP), high-threshold calcium current (ICa), calcium-activated potassium current (IKCa), slowly activating potassium current (IKm), hyperpolarization-activated depolarizing mix cationic currents (Ih), and leak conductances. The axosomatic compartment (ISInt) consisted of the delayed-rectifier potassium current (IKv), voltage-gated sodium current (INa), the persistent sodium current (INaP), and the sodium-activated potassium current (IKNa). All of these currents have been described in detail previously (Krishnan and Bazhenov, 2011).
Ion concentration dynamics.
This model exhibits changes in concentrations of [K+]o, [K+]i, [Na+]o, [Na+]i, [Ca2+]i, and [Cl−]i. The reversal potentials for each current calculated through the use of the Nernst equation for the specific ion or ions passing through the channel of interest. The concentration dynamics of these ions was modeled similar to previous work (Kager et al., 2000; Bazhenov et al., 2004; Fröhlich and Bazhenov, 2006; Krishnan and Bazhenov, 2011) and are described as follows:
 |
 |
 |
 |
 |
 |
 |
where F = 96,489 C/mol, the conversion factor k = 10, d = 0.15 determined the ratio of the extracellular compartment volume to the surface area, [K+]oc and [Na+]oc are the K+ and Na+ concentrations in the adjacent compartments, and [K+]o−1, [K+]o+1, [Na+]o−1, and [Na+]o+1 are the concentrations of K+ and Na+ in neighboring cells respectively. In this model, [K+]o represents the concentration of the extracellular potassium immediately surrounding a given neuron. Additionally, extracellular K+ was allowed to diffuse between the two compartments in the model and between the space volumes associated with neighboring neurons. G represents the glial K+ buffer. For the chloride dynamics, [Cl−]i∞ = 5 mM, τCl∞ = 2 × 104, and τKoCl = 0.08 s. As discussed previously (Krishnan and Bazhenov, 2011), these values were chosen to match experimental data with regards to the role of [K+]o on KCC2 cotransporter efficiency. Values for τCa and DCa were set to 300 ms and 0.85 ms, respectively.
Network and synaptic properties.
The cortical network was modeled as a one-dimensional network, which consisted of 120 PYs and 24 INs. Each PY neuron made local excitatory connections to five other PY neurons on both sides such that each PY neuron projected to 10 PYs with AMPA conductance strength of 9 nS and NMDA conductance of 0.9 nS. Each PY neuron also formed synaptic connections onto IN neurons with AMPA and NMDA conductance strengths of 3 nS and 0.3 nS, respectively. Each IN neuron, in turn, projected to five local PY neurons, forming GABAA connections with conductance strengths of 9 nS. Additionally, each PY and IN neuron received individual afferent excitatory input modeled as a Poisson process of 140 Hz. This network configuration, although simplified, reproduced realistic electrical activity found in cortical network during epileptic seizures and is similar to those used in previous studies (Bazhenov et al., 2002; Fröhlich and Bazhenov, 2006; Fröhlich et al., 2008b; Krishnan and Bazhenov, 2011).
Trauma to the network, in the form of cortical deafferentation, was modeled as a 50% reduction of the afferent input to a given set of PY neurons. The PYs that underwent deafferentation were selected at random and were within the 80 neurons in the center of the network. This provided a region of 40 PYs, 20 on both ends of the network, which would remain intact and allow for observation of seizure propagation and control for boundary effects. The fraction of deafferented PYs was varied from 0% to 100% within the aforementioned regions of 80 PY neurons, where 0% meant no PY was deafferented, and 100% meant that all 80 PYs underwent deafferentation.
The firing rate of the network was calculated every 5 s and was determined by averaging over all number of PY spikes in the 5 s interval. AMPA conductance between PYs were then adjusted, through homeostatic scaling, to maintain a target network firing rate of 5 Hz. We used a similar homeostatic rule as previous studies (Houweling et al., 2005; Fröhlich et al., 2008a) and given by the following:
where WiPY−PY is the AMPA conductance between two excitatory neurons at ith 5 s interval, v0 is the target firing rate (5 Hz), v̄ is the current network averaged firing rate, and αHSP is the rate of homeostatic scaling (αHSP = 0.01). Because it was not possible to simulate the network model on the longer time scale of the HSP, we applied HSP at a much faster rate (minutes) than observed in vivo (hours to days). We determined the value of αHSP by controlling the network activity and ion concentrations, ensuring that they always stayed in the physiological range without requiring excessively long computational times.
Seizures in humans and animal models of TBI generally last for only a brief period (30 s to a few minutes). Homeostatic changes in network excitability in cell cultures, in vitro and in vivo conditions, require several hours (>4 h) to days of chronically blocked activity (Topolnik et al., 2003a, b; Nita et al., 2006, 2007; Echegoyen et al., 2007; Rich and Wenner, 2007; Trasande and Ramirez, 2007; Avramescu and Timofeev, 2008; Ibata et al., 2008; O'Leary et al., 2010). Therefore, we assumed that the timescales for HSP and seizure activity are very different and that HSP may not have much of an effect on synaptic weights during a single seizure event. As such, to prevent unrealistic changes of synaptic weights during seizures in the model, where the rate of HSP was increased to obtain reasonable simulation times, we turned HSP off during seizure-like events.
In addition to the homeostatic scaling, we added synaptic recovery processes to some of the simulations to model trauma-induced axonal sprouting (Kuśmierczak et al., 2015) and to allow the deafferented PY neurons to recover the lost afferent inputs. This was implemented by the following:
 |
where AiPY is the strength of the current afferent input to the deafferented PY neurons, A0 is the original strength of the afferent input, and γSyn is the sprouting rate. The sprouting rate was varied in our simulations to test the hypothesis that the differences in axonal sprouting may be sufficient to explain observation that older cats are more susceptible to seizure than younger cats following partial cortical deafferentation (Timofeev et al., 2013). In our simulations, the homeostatic rule was applied globally, and axonal sprouting process was applied to all deafferented PY neurons.
Estimation of seizure thresholds.
The seizure threshold was determined using a binary search method using an iterative procedure. Initially, a high input that led to seizure and a low input that did not result in seizure were set as high (PUp) and low (PLow) bounds. At every iteration, the input to the network was set to be the average (<P>) of PUp and PLow. If the <P> resulted in a seizure, then the new high value was set as the average (PUp = <P>), otherwise the new low value was set as the average (PLow = <P>). The next iteration followed the same process as above with the new bounds until the difference between high and low bounds was less than 0.1, at which point the threshold was taken to be the average of these final bounds.
Results
Trauma-induced epileptogenesis in vivo
Our previous studies have demonstrated that cortical undercut induces acute seizures stopping within several hours (Topolnik et al., 2003b). In head-restrained adult cats, the seizures were also developed in chronic conditions, weeks and months from the undercut in 70% of animals (Nita et al., 2007). Although cats are very tolerant to head-restrained conditions, some physiological artifacts, due to unusual environment could be a factor that contributed to seizure generation. Therefore, in this study, we performed continuous wireless electrographic and behavioral recordings for 2–4 months in four young (10–12 months old at the time of surgery) and one adult cat (older than 8 years).
The acute seizures were recorded for 8–10 h after the end of isoflurane anesthesia in all animals. The amplitude of electric activity in the undercut suprasylvian area was lower compared with other sites of intracortical recordings. The general dynamics of pathological 3–5 Hz rhythmic activities around undercut cortex were not different from our previous study in terms of their spatial and temporal distribution (Nita et al., 2007). These electrical activities were recorded in the 8-year-old cat only. However, we observed new phenomena that have not been seen in head-restrained animals. The undercut cortex produced isolated large amplitude (∼0.5 mV) slow waves. There were 18–20 periods of these slow waves per day in the first week following the undercut, and then their occurrence increased to ∼50 periods per day. Within each period, there were 2–40 slow waves, none of which were rhythmic and they occurred at most once every 10 s. We have not observed these events in the head-restrained conditions in our previous experiments (Nita et al., 2007).
After 6 weeks, rare large paroxysmal electrographic activities were developed in the adult cat only. The full-blown seizures accompanied with motor jerks were developed at the end of the third month. Most of the seizures were detected in waking state, within a minute of transition from either slow-wave sleep or rapid eye movement sleep (Fig. 1A). In this animal, the highest amplitude of slow waves during slow-wave sleep were recorded in both right and left somatosensory cortices (Fig. 1B1,B2, green traces), and the lowest amplitude was recorded in the undercut cortex (Fig. 1B1,B2, black traces). During seizure, the maximal LFP amplitude was recorded in marginal gyrus (Fig. 1B1,B2, red trace) followed by left postcruciate gyrus (Fig. 1B1,B2, top green trace). Both these recording sites were surrounding the undercut cortex, suggesting that the areas surrounding the undercut cortex play a leading role in the seizure generation. After the end of the seizure, the cat awakened as demonstrated by activated LFP pattern in visual cortex and was at physical rest as evidenced by an absence of movements, low neck muscle tone, and strong mu rhythm recorded over somatosensory cortical areas (Fig. 1D1,D2).
Figure 1.
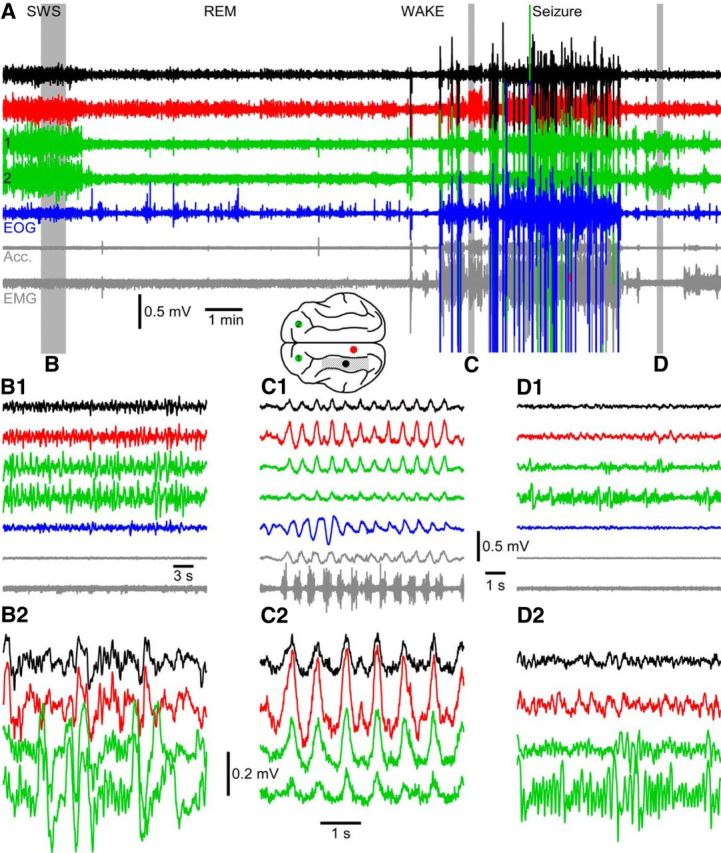
Cortical undercut leads to electrographic seizures in vivo. A, Transition between slow-wave sleep (SWS), rapid eye movement sleep (REM), waking state, and seizure. Brain drawing represents location of intracortical LFP electrodes. The other traces include the following: electro-oculogram, EMG, and accelerometer (Acc.). Segments of recordings indicated by the gray area are expanded in time in B1 (SWS), C1 (seizure), and D1 (postictal recovery). Further expansion in time and amplitude is shown in B2, C2, and D2.
In our previous experiments, ∼70% of cats developed seizure activities in chronic conditions after undercut (Nita et al., 2007). These animals were of unknown age. Here we reanalyzed these data. Of 6 animals, the two that did not develop epilepsy within 3–4 months of observation were the cats that weighed 1.9–2.2 kg. The cats that developed epilepsy within the first several months from the undercut weighed 2.3–3.9 kg; veterinary examination did not consider these cats to be obese. The cats that weighed >2.3 kg were most likely fully adult. This further suggests that cortical undercuts lead to the development of epilepsy only in adult cats.
Partial cortical deafferentation triggers seizure-like activity in the network model
Following our previous studies (Timofeev et al., 2000; Topolnik et al., 2003a, b; Houweling et al., 2005; Fröhlich and Bazhenov, 2006; Fröhlich et al., 2008a, b; Timofeev et al., 2010; Volman et al., 2011a, b), partial network deafferentation was used as a computational model of TBI. The network model incorporated populations of synaptically coupled excitatory pyramidal cells, and inhibitory interneurons. In addition, realistic ion concentration dynamics were implemented to simulate in vivo-like conditions (for details, see Materials and Methods). HSP was implemented to maintain a target firing rate by upregulation or downregulation of synaptic strength. To achieve feasible simulation times, the HSP scaling rate was significantly increased to achieve much faster than physiological scaling.
In an intact, nondeafferented network, application of a low amplitude 1 s current pulse stimulus to all pyramidal neurons was unable to elicit seizure activity (Fig. 2A–C). The brief stimulus caused a small transient increase in network activity reflected in the rise of the mean firing rate of the network (Fig. 3A, red trace). Before the stimulus, the network firing rate was fairly constant around the target firing rate of 5 Hz. At the onset of the stimulus (Fig. 3A, 300 s), the firing rate of the network increased to ∼10 Hz, but quickly returned to the baseline following termination of current pulse. Increase in the network mean firing rate was accompanied by small transient increases in extracellular potassium and intracellular sodium concentrations, which then returned to the baseline shortly after the stimulus ended (Fig. 3D, red traces). The firing pattern of this intact network remained unperturbed, showing spontaneous activity across the entire network (Fig. 2A–C).
Figure 2.

Network deafferentation leads to reduction of seizure threshold. A, Raster plot of activity in control network without deafferentation. Brief 1 s stimulus was applied at 300 s (red bar). B, Single-cell activity from control network in A (location indicated by arrow). C, Zoom-in of activity from B showing spontaneous firing pattern. D, Raster plot of activity of a network with 50% deafferentation applied at 100 s (green triangle). Stimulation was applied at 300 s (red bar) of equal duration and strength as that applied in A. E, Single-cell activity from the deafferented network in D. F, Left, synchronized bursting events with spike inactivation during spike and wave seizure-like activity in E. Right, Background bursting firing pattern generated between seizures. G, Left, LFP of the network corresponding to the spike and wave epileptiform activity shown in F (left). Right, the LFP corresponding to background bursting in F (right).
Figure 3.

Synaptic weights and ion concentration dynamics. A, Average firing rate of a network without deafferentation (red) and network with 50% deafferentation (black). B, Average synaptic weight dynamics. HSP scaling was blocked during seizure state to avoid nonphysiologically fast changes of synaptic weights. C, Phase space projection shows dynamics of the averaged network firing rate and synaptic weight for intact (red) and deafferented (black) networks. D, Left (Right), Evolution of the extracellular potassium (intracellular sodium) concentrations near (from) a single cortical pyramidal neuron. E, F, Raster plots of extracellular potassium and intracellular sodium concentrations, respectively, for the network with deafferentation. Green triangle represents time of deafferentation. Red bar represents stimulus application.
Inducing 50% deafferentation (afferent input was reduced by half) (Fig. 2D, green triangle) to a middle region of 80 neurons (of a total of 120 neurons) led to immediate decrease of activity in the region affected by deafferentation (Figs. 2D, 3A, black). This triggered homeostatic upregulation of excitatory synaptic connectivity leading to the slow recovery of the network firing rate (Fig. 3A, see the time interval 100–200 s). Figure 2E shows the voltage trace of a single representative neuron within the deafferented region and the reduction in activity of the neuron following deafferentation. Extracellular potassium concentration (Fig. 3D, black trace, and Fig. 3E) was also slightly reduced as a result of the deafferentation. Synaptic weights between pyramidal neurons progressively increased (Fig. 3B, black trace) through HSP regulation in response to the reduction of firing rate.
Upon application of external stimulus to deafferented network (Fig. 2D, red bar; strength same as Fig. 2A), the network generated seizure-like response. We found a significant reduction in the seizure threshold: seizure-like activity could be generated in the deafferented network using only 4% of the stimulus strength needed to induce seizure-like activity in the intact network. Pathological activity showed characteristic state transitions between fast runs and spike-and-wave complexes, followed by spontaneous termination and postictal depression (Fig. 2D,E) resembling in vivo recordings of electrographic seizures (Topolnik et al., 2003a). The LFPs during the fast runs and spike-and-wave complexes in the model (Fig. 2G) resemble the LFPs recorded in vivo in Figure 1. Following postictal depression, the baseline firing rate of the deafferented network fluctuated around the target firing rate of 5 Hz. However, there was a marked difference in the network baseline firing pattern after deafferentation. Unlike asynchronous spontaneous firing observed in the intact network (Fig. 2A–C), the neurons showed isolated bursting events following deafferentation (Fig. 2D–F, right). Bursting was observed independently of the episodes of seizure, suggesting that the origin of this pattern was purely a consequence of network deafferentation.
The dynamics of network activity can be illustrated by following the trajectory in a reduced phase space of the average synaptic weights plotted against the average firing rate (Fig. 3C). Without deafferentation (Fig. 3C, red), the state of the network was in a fixed point-like attractor and stayed there for the duration of the simulation. Following 50% deafferentation, the network state drifted away from the fixed point (Fig. 3C, black) due to HSP-mediated increase in synaptic weights. It approached a limit cycle-like attractor corresponding to the slow oscillatory dynamics of the baseline activity. In this state, the network was sensitive to small perturbations; thus, a small external stimulus could trigger a seizure. The seizure-like activity resulted in a rapid rise in the firing frequency followed by return of oscillations (Fig. 3C, spike in black plot).
Analysis of the extracellular potassium concentration (Fig. 3D,E) suggests that the seizure onset was triggered by a positive feedback mechanism. Following external stimulation, [K+]o in the region of the network affected by deafferentation reached a level high enough to increase excitability and neuronal firing; that further increased [K+]o leading to seizure onset (Fig. 3 D,E). During seizure-like activity, [K+]o reached as high as 7 mm, while the intracellular sodium concentration ([Na+]i) reached 22.5 mm; in contrast, the levels of [K+]o and [Na+]i in the intact network remained at ∼4 and 20 mm, respectively (Fig. 3D, red traces). [K+]o and [Na+]i levels remained high in the intact regions of the deafferented network compared with the baseline levels in the nondeafferented network (Fig. 3E,F). Dynamics of [K+]o and [Na+]i determined spike-and-wave pattern of epileptic seizure and its termination. As previously shown, [K+]o increases during fast runs and decreases during spike and wave phase mediating periodic transitions between two dynamic states in a presence of bistability between fast runs and spike-and-wave complexes (Fröhlich et al., 2005; Fröhlich and Bazhenov, 2006). Progressive increase of [Na+]i over the course of seizure eliminated bistability, leading to termination of seizure and postictal depression state (Krishnan and Bazhenov, 2011).
We found that stimulation of the deafferented network during either the peak or trough of the slow baseline [K+]o oscillation did not significantly affect seizure threshold. Although we previously reported dependence of the threshold for seizure onset on the level of [K+]o (Fröhlich et al., 2010), fluctuations of the baseline [K+]o reported here were too small (<1 mm) to produce a significant change of the seizure threshold.
In this model, seizure-like activity started first near the center of the network. This was likely because there were enough intact neurons within the deafferented region to trigger a seizure. In networks with more severe deafferentation (e.g., 90% deafferentation; see Fig. 5), there were fewer active neurons within the deafferented region, but neurons from the intact part of the network stimulated activity near the border of the intact and deafferented regions; that area became a primary site for seizure initiation.
Figure 5.
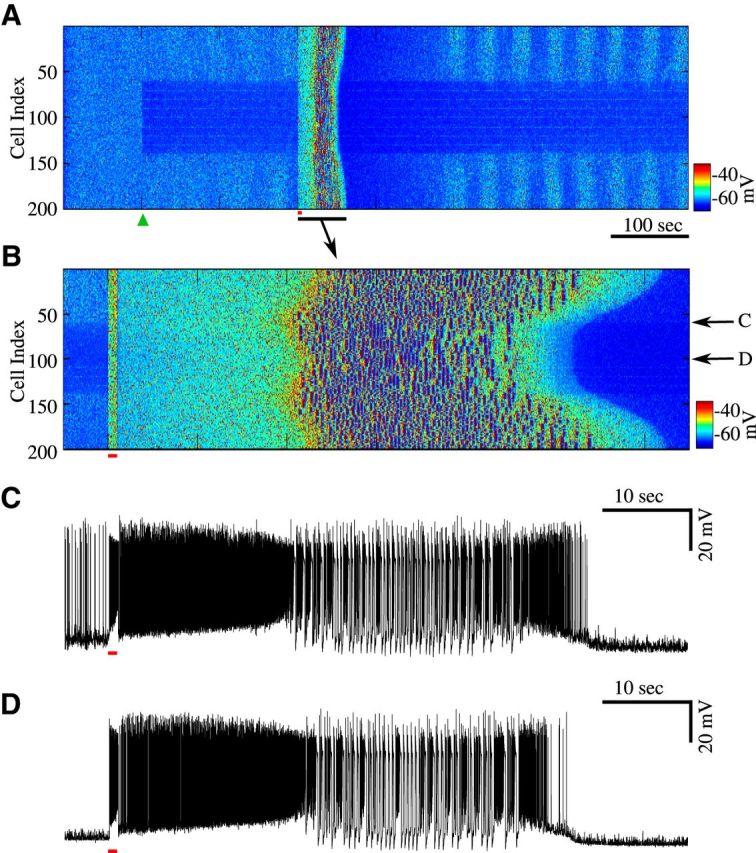
Seizures initiate at the boundaries and propagate toward intact and deafferented regions. A, Raster plot of activity from a network with 90% deafferentation applied at 100 s (green triangle) followed by stimulus at 300 s (red bar). B, Zoom-in of the raster plot of activity during one episode of seizure in A. C, D, Single-cell activity from the network in B. Arrow indicate locations. Note bursting with spike inactivation followed by tonic spiking (fast run) just before termination of seizure.
Severity of deafferentation determines seizure threshold
We next explored effects of the severity of deafferentation on seizure threshold. We began by keeping the size of the deafferented region constant while varying the percentage of deafferented neurons within the affected region. In a 1D network of 120 PY neurons, an affected region of 80 contiguous neurons was chosen, and within these 80 neurons the percentage of deafferented neurons was varied. Starting with a network with 0% deafferentation (all neurons were left intact) and increasing the severity of the trauma to 100% deafferentation (all 80 neurons experienced partial deafferentation), there was a significant decrease in seizure threshold (Fig. 4A). In intact nondeafferented networks, seizure-like activity could still be induced by a sufficiently strong current pulse (100% in Fig. 4A,C). Similar to the seizures in the deafferented network, the intact network exhibited characteristic fast run and spike-and-wave complex state transitions, and large increases in [K+]o as well as increases in [Na+]i. With a small deafferentation (<10%), the seizure threshold was reduced but remained relatively high (Fig. 4A). Deafferentation of 10% of the neural population (Fig. 4B) resulted in a seizure threshold of 70% of the stimulus strength necessary to evoke seizure in intact network. In that case, reduction in the network baseline activity following deafferentation (green triangle) was relatively small. Nevertheless, even such minor “trauma” led to appearance of isolated bursting events (similar to that shown in Fig. 2F, right).
Figure 4.

Severity of deafferentation affects seizure threshold. A, Threshold values for different degrees of deafferentation. Red line indicates the mean threshold (±SD) for a given degree of deafferentation. Black bars represent a range of thresholds from individual simulations. B, Raster plot of activity from a network with 10% deafferentation (green triangle). Bottom, Activity of a single neuron from the network (top panel). Arrow indicates location. C, Threshold values for different sizes of deafferented area. D, Raster plot of activity from a network with a block of 70 neurons undergoing 50% deafferentation (green triangle). Bottom, Activity of a single neuron from the network.
The seizure threshold quickly reduced with deafferentation of 20% and higher; 50% deafferentation produced the lowest seizure threshold of ∼4% of that in the intact network (Fig. 4A). This condition (50% deafferentation) was chosen in simulations shown in Figures 2 and 3. Increasing deafferentation to 100% led to a small increase in seizure threshold compared with that in the model with 50% deafferentation. Remaining intact (and therefore, more active) neurons at the intermediate levels of deafferentation (e.g., 50%) could likely help initiate seizure; this can explain the minor threshold increase when an entire population was deafferented. In these simulations, although seizure initiation threshold became very low with increasing size of deafferentation, spontaneous seizures were not generated. Independent of the severity of the trauma, all seizure-like events exhibited characteristic fast run and spike-and-wave complex state transitions and increases in [K+]o. Additionally, all but the fully intact network developed isolated bursting following deafferentation.
We then tested the effect of the size of deafferented region on the susceptibility of the network for seizure generation by changing the number of neurons in the affected region (i.e., 80 neurons for the previous condition) and applying 50% deafferentation (∼40 neurons were deafferented of 80 neuron for the previous condition). Increasing the size of the affected region from 0 neurons to a block of 100 neurons also showed a significant change in seizure threshold (Fig. 4C). Affected regions of 10–20 neurons resulted in a small decrease of threshold (>60% of that in intact network), very similar to the small change in seizure threshold observed in the 10% deafferented condition (Fig. 4A). Deafferented regions of 30–100 neurons, however, led to much higher decrease of the seizure threshold. When 100 neurons were deafferented, a seizure threshold reduced to only 3% of that in the intact condition. Figure 4D shows a network with a deafferented region of 70 neurons. Surprisingly, this network produced seizure pattern that was very similar to that in the network with only 10% deafferentation (Fig. 4B). Unlike the result from varying the percentage of deafferented neurons (Fig. 4A), we did not observe an increase in threshold as the size of the affected region approached 100 neurons. Although both of the trauma conditions resulted in very low seizure initiation thresholds, varying the size of the deafferented region showed a smoother drop in threshold (Fig. 4C) as opposed to the abrupt drop in Figure 4A. As was the case with varying the percentage of neurons being deafferented, varying the size of the affected region did not result in the generation of spontaneous seizures.
Seizure-like events start near the boundary between intact and deafferented regions
To study spatiotemporal properties of initiation and spread of seizure-like activity, we increased the size of the network to 200 neurons leaving an intact region of 60 neurons on either side of the deafferented area of 80 neurons in the center of the network (Fig. 5A). We then applied 90% deafferentation at the time 100 s (Fig. 5A, green triangle). As in the previous experiments, before the “trauma” the network displayed an asynchronous firing pattern. After deafferentation, we observed an immediate and strong drop in activity of the deafferented region. A brief, 1 s stimulus applied to entire population of neurons (red bar) evoked seizure-like activity. Figure 5B provides a closer look at the spatiotemporal pattern of activity near the time of stimulation. The stimulus (red bar) triggered tonic firing, which lasted ∼20 s before transitioning to spike-and-wave bursting activity. In all models of epileptiform activity reported in this study, [K+]o increased during fast runs and decreased during spike-and-wave bursting phase (Fröhlich et al., 2005; Fröhlich and Bazhenov, 2006). This created bistability between two dynamic network states for a range of [K+]o mediating transitions from tonic firing to spike-and-wave bursting.
We observed that epileptic bursts initiated at the boundaries between intact and deafferented regions (Fig. 5B) and then propagated outward to the rest of the network. A closer look at a single neuron near the boundary and a neuron from the center of the deafferented region revealed the delay in the onset of transition from fast runs to spike-and-wave complexes (Fig. 5C,D), thus indicating that the synchronized activity propagated away from the boundaries and well inside the intact and deafferented regions. Seizure-like activity in the intact regions of the network appeared to last longer than that in the deafferented region. These results are consistent with our new experimental study (Fig. 1) reported that the seizure amplitude was highest near the boundary of the undercut area, and with the previous experimental studies showing that seizure initiation in vivo occurs near the boundary between intact and deafferented regions (Nita et al., 2007). Unlike the neurons located well inside deafferented region, neurons near the border receive synaptic input from active intact neurons while some of their excitatory connections are homeostatically upregulated. This combination of upregulated synaptic strength and strong synaptic input made these neurons hyperexcitable and primed for seizure (Volman et al., 2011b).
Spontaneous versus evoked epileptiform events
To test the effect of HSP properties on seizure threshold, we varied the rate of homeostatic scaling (αHSP; which is the rate at which synaptic weights change during HSP; for details, see Materials and Methods); higher scaling rate manifested faster, but also less accurate adjustment of synaptic weights. Spontaneous seizures were generated after 50% deafferentation (similar to conditions presented in Fig. 2) when the rate of homeostatic scaling was increased from 0.01 to 0.06, and the update time for synaptic weights was increased from every 5 s to every 20 s (Fig. 6A,B). Less precise synaptic scaling at higher rates prevented fine adjustments of synaptic weights, thereby making the network more prone to overcompensate for the loss of activity and increasing propensity for seizure. Physiologically, this effect could result from the feedback delay between increase of synaptic strength and changes of the ambient glutamate level sufficient to stop synaptic upregulation. These spontaneous seizures exhibited characteristic fast runs and spike-and-wave complexes (Fig. 6B) and were accompanied by a large increase in the network mean firing rate, with a maximum rate reaching 65 Hz, and large increases of synaptic weights (Fig. 6C,D). The network generated the first spontaneous seizure ∼100 s after the deafferentation, with ∼250 s between seizure-like events (Fig. 6A). Postictal depression lasted for ∼100 s. Before the first seizure onset, synaptic weights reached higher level than that in the model with a slower and more precise HSP rate (compare Fig. 3B with Fig. 6D), which could explain the occurrence of spontaneous seizures. This experiment suggests that precise regulation of the HSP scaling may have an important impact on the network dynamics.
Figure 6.

Spontaneous epileptiform events. A, Raster plot of activity with spontaneous, recurrent seizure-like events. Deafferentation time is indicated by the green triangle (αHSP = 0.06 and check time = 20 s). B, Top, Single-cell activity from the network in A (black arrow). Bottom, Zoom-in shows individual bursts with spike inactivation during seizure. C, Average firing rate of the network in A. D, Mean synaptic weights dynamics as a function of time for the network in A. HSP scaling was blocked during seizure state to avoid nonphysiologically fast changes of synaptic weights.
Effects of axonal sprouting on seizure threshold
Local axonal sprouting has been observed in cortex following cortical deafferentation (Carmichael and Chesselet, 2002; Avramescu and Timofeev, 2008; Timofeev et al., 2013; Kuśmierczak et al., 2015). Connection probabilities progressively increased up to 6 weeks after cortical undercut was administered in cats (Avramescu and Timofeev, 2008). Axonal sprouting in response to cortical trauma was found to be regulated by synchronous network activity after the trauma (Carmichael and Chesselet, 2002). To study effect of trauma-induced axonal sprouting on seizure threshold and generation, we implemented a recovery process that modeled the axonal sprouting observed following brain trauma and that regulated the amount of external input received by deafferented population of neurons. This model of axonal sprouting was only applied to deafferented neurons, and the level of input was not allowed to exceed that in the intact network.
Because both homeostatic scaling and axonal sprouting may vary with age, we varied the rate of both processes in our model, and we tested effect of these changes on seizure threshold (Fig. 7A). In general, we found that as the sprouting rate increased, the threshold for seizure generation also increased. For high sprouting rates, increasing HSP rate, αHSP, led to threshold increase. For networks with very fast HSP rate (αHSP = 0.06) and sprouting rate (γSyn = 0.001), the seizure threshold reached the level observed in a completely intact network (Fig. 7A, top/right). Figure 7B shows examples of the seizure threshold dynamics over time for two combinations of HSP and sprouting rate parameters (Fig. 7A, see 1 and 2). We compared a “fast” network with faster HSP and faster sprouting rate (αHSP = 0.005; γSyn = 0.001) to a “slow” network that had a slower HSP and slower sprouting rate (αHSP = 0.001; γSyn = 0.0002). A total of 50% deafferentation was applied to both networks at the same time point (Fig. 7B, arrow). In Figure 7B, early and late HSP are defined by the time elapsed following the initial cortical insult, early HSP was the time almost immediately after deafferentation and late HSP was the time when the target firing rate was fully recovered. From a clinical perspective, early HSP can be considered the time when early onset seizures are prevalent, and late HSP is when the incidence of late onset seizures is increased. The threshold values presented before the onset of the deafferentation represent the threshold for intact networks.
Figure 7.

Effect of axonal sprouting rate on seizure threshold. A, Seizure threshold (color map) as a function of HSP αHSP and sprouting rate, γSyn. All thresholds were tested in late HSP condition representing the last data points in B. The tiles numbered 1 and 2 correspond to the thresholds of the fast and slow networks, respectively, in B. B, Threshold dynamics (±SD) for two sample networks. A total of 50% deafferentation was applied at 100 s (arrow). The “fast” network (red) has a fast HSP and fast sprouting rate (αHSP = 0.005, γSyn = 0.001), whereas the “slow” network (blue) has slow HSP and slow sprouting rate (αHSP = 0.001, γSyn = 0.0002). A total of 100% represents seizure threshold of an intact network.
During early stages of HSP, the fast network had a lower seizure threshold than the slow network (Fig. 7B; compare the first point after deafferentation). However, as the time progressed, the fast network displayed a significant increase in the threshold level (>70% of intact network), whereas the slow network's seizure threshold continued to decline. It should be noted that both networks, with the fast and slow HSP and sprouting rate in Figure 7B, were able to fully recover their seizure thresholds after a substantial amount of time; however, the overall duration of the time window when the threshold was reduced was much longer in the network with the slow sprouting rates. Experimental studies with cats (Timofeev et al., 2013) suggest that the age-related increase in seizure susceptibility following cortical deafferentation may be caused by the differences in homeostatic scaling and axonal sprouting. Indeed, our model predicts that it may be a plausible mechanism to explain reduced likelihood of late-onset seizure as seen in animal experiments, as well as in juvenile TBI patients compared with adult TBI patients.
Impact of impaired homeostatic downregulation on seizure threshold
In the previous section, we showed that the differences in the properties of HSP scaling and axonal sprouting rate may explain why younger cats are less likely to develop seizures than older animals following severe brain trauma (Timofeev et al., 2013). Below we tested several specific mechanisms that could explain the difference between TBI impact on young and adult animals.
Experimental data suggest that different biophysical mechanisms are responsible for homeostatic upregulation and downregulation and that they may be differentially impaired with aging. Recent data (Sun and Turrigiano, 2011) revealed that downregulation of synaptic strength in older animals may be impaired by increased expression levels of PSD-95 (see more in Discussion). We therefore proposed that homeostatic synaptic scaling may be less bidirectional in the older animal compared with the young ones. As such, we tested a possibility of age-related changes in seizure susceptibility by varying the downregulation of synaptic weights (αHSP−) while keeping upregulation constant (αHSP+ = 0.01) fixed. First, we set the sprouting rate to zero. Following the onset of deafferentation, the network generated spontaneous seizures for values of αHSP− ≤ 0.003 (Fig. 8A). Seizure threshold was reduced (compared with a control network) for all values of αHSP− ≤ 0.002 (Fig. 8A, inset). Reducing αHSP− to 0.001 or removing downscaling all together led to the generation of multiple recurrent spontaneous seizures in response to 50% deafferentation.
Figure 8.

Seizure susceptibility in the “young” versus “old” animal models. A total of 50% deafferentation was applied at 100 s (black arrow or green triangle). A, Number of spontaneous seizures versus HSP downregulation rate, αHSP−. Inset, Seizure thresholds for networks with different αHSP−. B, Top, Raster plot of activity of a network with αHSP− = 0.009. Bottom, Phase space projection shows dynamics of synaptic weights and averaged firing rate. Inset, Amplitude of the steady-state oscillation in the phase space projections for values of αHSP− = 0.003 − 0.01; these values did not lead to spontaneous seizures. C, Top, Raster plot of the network activity for αHSP− = 0.002. Bottom, Phase space projection shows dynamics of synaptic weights and averaged firing rate leading to seizure. D, Time evolution of seizure threshold for networks with varying αHSP−. All networks implemented synaptic sprouting. Vertical arrow indicates the time of deafferentation. E, Raster plots of the network activity with bidirectional HSP (left) and unidirectional HSP (right). F, Single-cell activity from the networks with bidirectional HSP (left) and unidirectional HSP (right).
Figure 8B, C shows examples of the networks with different degrees of impairment of downregulation processes. A network model with a slightly reduced αHSP− = 0.009, representing scenario of a “younger” animal (Fig. 8B), did not produce spontaneous seizures following deafferentation (Fig. 8B, green triangle). Phase trajectory of this network converged to a fixed point like attractor (Fig. 8B, bottom). In the network model representing an “older” animal and having a larger reduction of αHSP− = 0.002 (Fig. 8C), we observed recurrent spontaneous seizures (Fig. 8C, top, end of the simulation), and the phase trajectory converged to a limit cycle-like attractor in which the synaptic weights slowly oscillated along with the average firing rate (Fig. 8C, bottom).
Analysis of the network dynamics for αHSP− values, which did not lead to spontaneous seizures (αHSP− = 0.003 − 0.01), revealed that the amplitude of the steady-state oscillations in the reduced phase space of the average firing rate plotted against synaptic weights increased as the αHSP− value decreased (compare Fig. 8B with Fig. 8C; see Fig. 8B, inset), suggesting a dynamic mechanism of instability and seizure initiation in the models with imprecise synaptic scaling for lower αHSP− values.
Next, we explored the effects of impaired homeostatic downregulation on seizure threshold in the presence of synaptic recovery. We tested this by determining the seizure thresholds of four different networks. Each network had the same sprouting rate but varied in the rate of homeostatic downregulation of synaptic weights, αHSP− (Fig. 8D). Following deafferentation, all four networks had similar seizure thresholds at the early stages of the HSP. However, a clear-cut difference was found at the late stages of synaptic homeostasis. The networks with less impaired αHSP− were able to recover some of the seizure threshold and prevent spontaneous seizure generation (Fig. 8D, blue and green lines). In contrast, the networks with more impaired αHSP− produced recurrent spontaneous seizures (Fig. 8D, black and red lines overlap in most of the plot). Even for very long simulation times, the unidirectional network, αHSP− = 0, was unable to recovery its seizure threshold.
Representative examples of the network dynamics and activity of one pyramidal cell from a bidirectional αHSP− = αHSP+ and unidirectional αHSP− = 0 networks are shown in Figure 8E, F. The green triangles in both plots indicate the time of deafferentation. Figure 8E (left, red bar) indicates the stimulus used to induce seizure-like activity. The first spontaneous seizure generated in the unidirectional HSP network occurred 100 s before the evoked seizures in the bidirectional network. Bidirectional network never generated spontaneous seizures.
Discussion
Previous theoretical studies predicted that synaptic upregulation after severe brain trauma may lead to a hyperexcitable network where susceptibility for seizures is dramatically increased (Houweling et al., 2005; Fröhlich et al., 2008a; Volman et al., 2011a, b). Reduction of the seizure threshold after severe deafferentation is consistent with experimental studies where it was observed that cortical undercut increased local neuronal connectivity, which eventually increased network excitability and promoted seizures (Salin et al., 1995; Jin et al., 2006, 2011; Avramescu and Timofeev, 2008).
In this study, we present in vivo experimental data and results of computer simulations to test the hypothesis that age-related changes in HSP may be sufficient to explain the difference in the likelihood of seizure onset in young versus older animals. Our study, based on detailed modeling of the interaction between homeostatic synaptic scaling and ion dynamics, revealed that following deafferentation the threshold for initiation of seizures recovers to a state close to that in an intact network for models of young animals, but remains low for models implementing HSP changes that may occur during aging.
Evidence for age dependence of trauma-induced epileptogenesis
Our new in vivo data suggest a strong correlation between seizure susceptibility after brain trauma and the age of an animal. Adult cats in our study developed trauma-induced epilepsy, but none of the young cats developed epilepsy. Previous studies in rats revealed age-dependent differences in susceptibility to pharmacologically induced seizure following cortical injury. FeCl3 injection induced post-traumatic seizures in young (4 months) and old (18 months) rats; however, older rats were more susceptible to post-traumatic epilepsy than younger animals and exhibited both faster seizure spread and faster seizure onset (Jyoti et al., 2009).
Age-dependent differences in the development of TBI-induced epilepsy have been observed in human patients. One study reported that patients 65 years of age and older at the time of injury were more susceptible to the development of epilepsy than younger patients (Annegers et al., 1998). More recent study (Christensen et al., 2009) assessed the risk of developing epilepsy in children and young adults suffering from TBI. Taking into consideration age, severity of trauma, sex, and family history, it found that the risk of epilepsy was dependent on the severity of the trauma and the age of patient at the time of injury.
Role of HSP in epileptogenesis
Reduced network activity, through bath application of TTX in vitro or through cortical deafferentation in vivo, leads to changes in synaptic strengths and increases in the excitability of the network (Topolnik et al., 2003a, b; Wierenga et al., 2005; Jin et al., 2006; Nita et al., 2006; Echegoyen et al., 2007; Trasande and Ramirez, 2007; Avramescu and Timofeev, 2008; Ibata et al., 2008; Lemieux et al., 2014). It is generally accepted that these changes are caused by homeostatic plasticity, a regulatory mechanism, which maintains a target firing rate through upregulation of excitatory intrinsic and synaptic factors and downregulation of inhibitory factors (Burrone and Murthy, 2003; Rich and Wenner, 2007; Turrigiano, 2008; Pozo and Goda, 2010).
Our previous modeling studies explored the hypothesis that HSP may fail to control “normal” excitability in heterogeneous networks, where there are subpopulations of neurons with severely different levels of activity, conditions found in traumatized cortex (Houweling et al., 2005; Fröhlich et al., 2008a; Volman et al., 2011a, b). In these models, HSP-dependent epileptogenesis was manifested by periodic bursting events; however, realistic patterns of spike-and-wave seizures was impossible to reproduce because of the model simplicity. Changes of the ion concentrations can have profound effects on the network dynamics and may be responsible for the characteristic patterns of electrical activity observed during seizures. In particular, an increase of [K+]o during epileptic seizures functions as a positive feedback loop to depolarize the neurons and to make the network more excitable leading to further increase of [K+]o (Somjen, 2002; Fröhlich et al., 2008b; Krishnan and Bazhenov, 2011). The model presented in our new study included dynamics of intracellular and extracellular concentrations of K+ and Na+ ions, as well as intracellular concentrations of Cl− and Ca2+. In this model, seizure-like activity closely resembled in vivo intracellular recordings of spike-and-wave electrographic seizures in cats (Avramescu and Timofeev, 2008), which spontaneously terminate and are followed by an interictal period.
Our model predicts the role of the severity of brain trauma in determining the susceptibility to seizure onset. It has been reported that cats that have undergone partial cortical deafferentation are much more likely to have seizures when anesthetized with ketamine/xylazine than cats that do not have cortical trauma (Topolnik et al., 2003b; Nita et al., 2006). Changing the degree of deafferentation in our model resulted in different thresholds for seizure generation such that larger deafferentation produced lower seizure thresholds; this is consistent with previously reported results in simplified models of the cortical network (Volman et al., 2011b). We found that the seizure activity could also be induced in control, nondeafferented network by sufficiently strong stimulation. This result supports an idea of bistability of the cortical network with physiological and pathological attractors coexisting and having different basins of attraction in epileptic and nonepileptic brains (Fröhlich et al., 2010).
In vivo data presented in our study suggest that state of vigilance may affect the transition from physiological activity to epileptic seizures. Neocortical seizures commonly occur during slow-wave sleep. One possible explanation depends on the presence of repetitive hyperpolarized down states in the cortex found during slow-wave sleep. It has been suggested that such periods of silence may lead to an increase in cortical excitability and may facilitate seizure onset (Timofeev et al., 2000, 2010, 2013; Topolnik et al., 2003a, b; Nita et al., 2006, 2007; Avramescu and Timofeev, 2008). Transition from sleep to waking is associated with the activation of the cholinergic system and overall increase in excitability (Brown et al., 2012). Together, it suggests that the time just after the sleep–wake transition, when the sleep-related increase in excitability is still in place and increase in excitability associated with cholinergic changes just occurred, would constitute ideal conditions for seizures to occur. Indeed, in our in vivo study, seizures always occurred shortly after transitions from sleep to waking states.
Mechanisms of age dependence in epileptogenesis
Our in vivo study revealed that the outcome of trauma-induced epileptogenesis significantly depends on the age of the animal, with older animals being much more prone to seizures. The multiplicative nature of HSP is of the special interest in terms of age-related differences. The multiplicativity of HSP depends on the modulation of AMPA receptors that occurs at all synapses on a given neuron. This type of HSP is different from synapse-specific HSP expression (Echegoyen et al., 2007; Goel and Lee, 2007; Rich and Wenner, 2007; Turrigiano, 2008; Lee et al., 2014). Age-dependent differences in multiplicative versus synapse-specific HSP expression in response to dark rearing were previously reported (Goel and Lee, 2007).
The properties of homeostatic scaling or/and the rate of axonal sprouting of the traumatized connectivity may be different in young and adult animals, and may lead to the different final states of the cortical network excitability. Impaired homeostatic downregulation in the older animals will prevent full restoration of intact synaptic connectivity after recovery of synaptic input to affected areas (e.g., by axonal sprouting) (Timofeev et al., 2013). Indeed, studies on spinal cord injury-induced sprouting and hippocampal deafferentation-induced sprouting have shown age-dependent differences (Schauwecker et al., 1995; Jaerve et al., 2011). Following trauma, increases in growth-associated proteins occurred in the hippocampus young rats (3 months old), but not in adult rats (24 months old).
In our model, we tested this mechanism by introducing a model of axonal sprouting processes, which worked to compensate for the afferent input lost during partial deafferentation. Our results revealed that the network with faster sprouting rate and homeostatic scaling displayed a rapid increase of seizure thresholds after initial decline, whereas the network with slow axonal sprouting and synaptic scaling displayed prolonged periods of seizure-like activity. Although the later network eventually restored its initial high seizure threshold, we can speculate that older animals may have limited abilities for axonal sprouting; and, therefore, synaptic projections to the undercut area may never fully recover, leading to chronically reduced seizure threshold.
Homeostatic scaling is a bidirectional process that aims to maintain a target firing rate through the strengthening and weakening of synaptic connections. Most of the synaptic strength modulation involved in this process can be attributed to changes in synaptic AMPA receptor densities (Rich and Wenner, 2007; Hou et al., 2008; Pozo and Goda, 2010; Remme and Wadman, 2012). It was recently demonstrated that the scaffolding protein PSD-95 is necessary for synaptic scaling, and its role in scaling the synaptic strength was dependent on age and directionality of the scaling (Sun and Turrigiano, 2011). Expression levels of PSD-95 increased as the neurons aged and PSD-95 affected mainly the downregulation of synaptic strength, whereas upregulation was much less affected. These results support our hypothesis that the downregulation of synaptic strength in older animals may be more difficult to regulate, leading to higher susceptibility for seizures (Timofeev et al., 2013). We found that impairing the sensitivity of the homeostatic scaling sensor not only reduced seizure threshold, but also led to the generation of spontaneous seizures.
Age-dependent changes of the homeostatic upregulation may contribute to seizure susceptibility. BDNF has been shown to be involved in the increase of AMPA receptor insertion following decrease in synaptic activity, but not to the removal of receptors in response to increased activity (Rutherford et al., 1998; Leslie et al., 2001; Turrigiano, 2008; Pozo and Goda, 2010). Additionally, BDNF expression levels have been shown to decrease with age (Lommatzsch et al., 2005). Together, these results suggest that reduced levels of BDNF in older animals could reduce the dynamic range of BDNF regulation of homeostatic upscaling. This may lead to larger increases in AMPA receptor insertion in response to the smaller changes in BDNF expression in older animals compared with younger ones and may provide a complimentary mechanism for age-dependent changes of the seizure susceptibility.
Strategies to prevent epileptogenesis
Trauma-induced epilepsy is poorly controlled by antiepileptic drugs (Hernandez, 1997; Chang et al., 2003; Temkin, 2003, 2009; Agrawal et al., 2006). Administration of antiepileptic drugs immediately following TBI reduces incidence of early seizure onset within the first week following brain insult (Annegers et al., 1998; Agrawal et al., 2006; Szaflarski et al., 2014), but rarely controls late seizures. Our study predicts that reduction of the neuronal excitability, a common target of the antiepileptic drugs (e.g., levetiracetam) (Szaflarski et al., 2014), in some conditions can enhance HSP-mediated synaptic upregulation and increase severity of epileptogenesis. Thus, our study provides new insight into the development of interventions that can be used to treat TBI, which would target to maintain physiological levels of activity in deafferented areas and thus reduce likelihood of epileptic seizures.
Footnotes
This work was supported by National Institutes of Health Grants R01EB009282 and R01NS081243, Office of Naval Research Multidisciplinary University Research Initiative Grant N000141310672, and Canadian Institutes of Health Research Grants 201403MOP-325213 and 201403MOP-324941. O.C.G. was supported by the National Science Foundation Graduate Research Fellowship Grant DGE-1326120.
The authors declare no competing financial interests.
References
- Agrawal A, Timothy J, Pandit L, Manju M. Post-traumatic epilepsy: an overview. Clinical Neurol Neurosurg. 2006;108:433–439. doi: 10.1016/j.clineuro.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Annegers JF, Hauser WA, Coan SP, Rocca WA. A population-based study of seizures after traumatic brain injuries. N Engl J Med. 1998;338:20–24. doi: 10.1056/NEJM199801013380104. [DOI] [PubMed] [Google Scholar]
- Avramescu S, Timofeev I. Synaptic strength modulation after cortical trauma: a role in epileptogenesis. J Neurosci. 2008;28:6760–6772. doi: 10.1523/JNEUROSCI.0643-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ. Model of thalamocortical slow-wave sleep oscillations and transitions to activated states. J Neurosci. 2002;22:8691–8704. doi: 10.1523/JNEUROSCI.22-19-08691.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ. Potassium model for slow (2–3 Hz) in vivo neocortical paroxysmal oscillations. J Neurophysiol. 2004;92:1116–1132. doi: 10.1152/jn.00529.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Basheer R, McKenna JT, Strecker RE, McCarley RW. Control of sleep and wakefulness. Physiol Rev. 2012;92:1087–1187. doi: 10.1152/physrev.00032.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrone J, Murthy VN. Synaptic gain control and homeostasis. Curr Opin Neurobiol. 2003;13:560–567. doi: 10.1016/j.conb.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Carmichael ST, Chesselet MF. Synchronous neuronal activity is a signal for axonal sprouting after cortical lesions in the adult. J Neurosci. 2002;22:6062–6070. doi: 10.1523/JNEUROSCI.22-14-06062.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang BS, Lowenstein DH Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: antiepileptic drug prophylaxis in severe traumatic brain injury: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2003;60:10–16. doi: 10.1212/01.WNL.0000031432.05543.14. [DOI] [PubMed] [Google Scholar]
- Christensen J, Pedersen MG, Pedersen CB, Sidenius P, Olsen J, Vestergaard M. Long-term risk of epilepsy after traumatic brain injury in children and young adults: a population-based cohort study. Lancet. 2009;373:1105–1110. doi: 10.1016/S0140-6736(09)60214-2. [DOI] [PubMed] [Google Scholar]
- Dinner D. Posttraumatic epilepsy. In: Wyllie E, editor. The treatment of epilepsy: principles. Philadelphia: Lea & Fibinger; 1993. pp. 654–658. [Google Scholar]
- Echegoyen J, Neu A, Graber KD, Soltesz I. Homeostatic plasticity studied using in vivo hippocampal activity-blockade: synaptic scaling, intrinsic plasticity and age-dependence. PLoS One. 2007;2:e700. doi: 10.1371/journal.pone.0000700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fröhlich F, Bazhenov M. Coexistence of tonic firing and bursting in cortical neurons. Phys Rev E Stat Nonlin Soft Matter Phys. 2006;74 doi: 10.1103/PhysRevE.74.031922. 031922. [DOI] [PubMed] [Google Scholar]
- Fröhlich F, Bazhenov M, Timofeev I, Sejnowski TJ. Maintenance and termination of neocortical oscillations by dynamic modulation of intrinsic and synaptic excitability. Thalamus Relat Syst. 2005;3:147–156. doi: 10.1017/S1472928807000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fröhlich F, Bazhenov M, Sejnowski TJ. Pathological effect of homeostatic synaptic scaling on network dynamics in diseases of the cortex. J Neurosci. 2008a;28:1709–1720. doi: 10.1523/JNEUROSCI.4263-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fröhlich F, Bazhenov M, Iragui-Madoz V, Sejnowski TJ. Potassium dynamics in the epileptic cortex: new insights on an old topic. Neuroscientist. 2008b;14:422–433. doi: 10.1177/1073858408317955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fröhlich F, Sejnowski TJ, Bazhenov M. Network bistability mediates spontaneous transitions between normal and pathological brain states. J Neurosci. 2010;30:10734–10743. doi: 10.1523/JNEUROSCI.1239-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel A, Lee HK. Persistence of experience-induced homeostatic synaptic plasticity through adulthood in superficial layers of mouse visual cortex. J Neurosci. 2007;27:6692–6700. doi: 10.1523/JNEUROSCI.5038-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grand L, Ftomov S, Timofeev I. Long-term synchronized electrophysiological and behavioral wireless monitoring of freely moving animals. J Neurosci Methods. 2013;212:237–241. doi: 10.1016/j.jneumeth.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez TD. Preventing post-traumatic epilepsy after brain injury: weighing the costs and benefits of anticonvulsant prophylaxis. Trends Pharmacol Sci. 1997;18:59–62. doi: 10.1016/S0165-6147(97)89801-X. [DOI] [PubMed] [Google Scholar]
- Hou Q, Zhang D, Jarzylo L, Huganir RL, Man HY. Homeostatic regulation of AMPA receptor expression at single hippocampal synapses. Proc Natl Acad Sci U S A. 2008;105:775–780. doi: 10.1073/pnas.0706447105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houweling AR, Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ. Homeostatic synaptic plasticity can explain post-traumatic epileptogenesis in chronically isolated neocortex. Cereb Cortex. 2005;15:834–845. doi: 10.1093/cercor/bhh184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57:819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Jaerve A, Schiwy N, Schmitz C, Mueller HW. Differential effect of aging on axon sprouting and regenerative growth in spinal cord injury. Exp Neurol. 2011;231:284–294. doi: 10.1016/j.expneurol.2011.07.002. [DOI] [PubMed] [Google Scholar]
- Jin X, Prince DA, Huguenard JR. Enhanced excitatory synaptic connectivity in layer v pyramidal neurons of chronically injured epileptogenic neocortex in rats. J Neurosci. 2006;26:4891–4900. doi: 10.1523/JNEUROSCI.4361-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Huguenard JR, Prince DA. Reorganization of inhibitory synaptic circuits in rodent chronically injured epileptogenic neocortex. Cereb Cortex. 2011;21:1094–1104. doi: 10.1093/cercor/bhq181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jyoti A, Sethi P, Sharma D. Aging accelerates the progression and manifestation of seizures in post-traumatic model of epilepsy. Neurosci Lett. 2009;453:86–91. doi: 10.1016/j.neulet.2009.01.082. [DOI] [PubMed] [Google Scholar]
- Kager H, Wadman WJ, Somjen GG. Simulated seizures and spreading depression in a neuron model incorporating interstitial space and ion concentrations. J Neurophysiol. 2000;84:495–512. doi: 10.1152/jn.2000.84.1.495. [DOI] [PubMed] [Google Scholar]
- Kollevold T. Immediate and early cerebral seizures after head injuries, I. J Oslo City Hosp. 1976;26:99–114. [PubMed] [Google Scholar]
- Krishnan GP, Bazhenov M. Ionic dynamics mediate spontaneous termination of seizures and postictal depression state. J Neurosci. 2011;31:8870–8882. doi: 10.1523/JNEUROSCI.6200-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuśmierczak M, Lajeunesse F, Grand L, Timofeev I. Changes in long-range connectivity and neuronal reorganization in partial cortical deafferentation model of epileptogenesis. Neuroscience. 2015;284:153–164. doi: 10.1016/j.neuroscience.2014.09.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KF, Soares C, Béïque JC. Tuning into diversity of homeostatic synaptic plasticity. Neuropharmacology. 2014;78:31–37. doi: 10.1016/j.neuropharm.2013.03.016. [DOI] [PubMed] [Google Scholar]
- Lemieux M, Chen JY, Lonjers P, Bazhenov M, Timofeev I. The impact of cortical deafferentation on the neocortical slow oscillation. J Neurosci. 2014;34:5689–5703. doi: 10.1523/JNEUROSCI.1156-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie KR, Nelson SB, Turrigiano GG. Postsynaptic depolarization scales quantal amplitude in cortical pyramidal neurons. J Neurosci. 2001;21:RC170. doi: 10.1523/JNEUROSCI.21-19-j0005.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lommatzsch M, Zingler D, Schuhbaeck K, Schloetcke K, Zingler C, Schuff-Werner P, Virchow JC. The impact of age, weight and gender on BDNF levels in human platelets and plasma. Neurobiol Aging. 2005;26:115–123. doi: 10.1016/j.neurobiolaging.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Mainen ZF, Sejnowski TJ. Influence of dendritic structure on firing pattern in model neocortical neurons. Nature. 1996;382:363–366. doi: 10.1038/382363a0. [DOI] [PubMed] [Google Scholar]
- Nita DA, Cissé Y, Timofeev I, Steriade M. Increased propensity to seizures after chronic cortical deafferentation in vivo. J Neurophysiol. 2006;95:902–913. doi: 10.1152/jn.00742.2005. [DOI] [PubMed] [Google Scholar]
- Nita DA, Cissé Y, Timofeev I, Steriade M. Waking-sleep modulation of paroxysmal activities induced by partial cortical deafferentation. Cereb Cortex. 2007;17:272–283. doi: 10.1093/cercor/bhj145. [DOI] [PubMed] [Google Scholar]
- O'Leary T, van Rossum MC, Wyllie DJ. Homeostasis of intrinsic excitability in hippocampal neurones: dynamics and mechanism of the response to chronic depolarization. J Physiol. 2010;588:157–170. doi: 10.1113/jphysiol.2009.181024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozo K, Goda Y. Unraveling mechanisms of homeostatic synaptic plasticity. Neuron. 2010;66:337–351. doi: 10.1016/j.neuron.2010.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince DA, Tseng GF. Epileptogenesis in chronically injured cortex: in vitro studies. J Neurophysiol. 1993;69:1276–1291. doi: 10.1152/jn.1993.69.4.1276. [DOI] [PubMed] [Google Scholar]
- Remme MW, Wadman WJ. Homeostatic scaling of excitability in recurrent neural networks. PLoS Comput Biol. 2012;8:e1002494. doi: 10.1371/journal.pcbi.1002494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich MM, Wenner P. Sensing and expressing homeostatic synaptic plasticity. Trends Neurosci. 2007;30:119–125. doi: 10.1016/j.tins.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Rutherford LC, Nelson SB, Turrigiano GG. BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron. 1998;21:521–530. doi: 10.1016/S0896-6273(00)80563-2. [DOI] [PubMed] [Google Scholar]
- Salin P, Tseng GF, Hoffman S, Parada I, Prince DA. Axonal sprouting in layer V pyramidal neurons of chronically injured cerebral cortex. J Neurosci. 1995;15:8234–8245. doi: 10.1523/JNEUROSCI.15-12-08234.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauwecker PE, Cheng HW, Serquinia RM, Mori N, McNeill TH. Lesion-induced sprouting of commissural/associational axons and induction of GAP-43 mRNA in hilar and CA3 pyramidal neurons in the hippocampus are diminished in aged rats. J Neurosci. 1995;15:2462–2470. doi: 10.1523/JNEUROSCI.15-03-02462.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somjen GG. Ion regulation in the brain: implications for pathophysiology. Neuroscientist. 2002;8:254–267. doi: 10.1177/1073858402008003011. [DOI] [PubMed] [Google Scholar]
- Sun Q, Turrigiano GG. PSD-95 and PSD-93 play critical but distinct roles in synaptic scaling up and down. J Neurosci. 2011;31:6800–6808. doi: 10.1523/JNEUROSCI.5616-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szaflarski JP, Nazzal Y, Dreer LE. Post-traumatic epilepsy: current and emerging treatment options. Neuropsychiatr Dis Treat. 2014;10:1469–1477. doi: 10.2147/NDT.S50421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temkin NR. Risk factors for posttraumatic seizures in adults. Epilepsia. 2003;44(Suppl 10):18–20. doi: 10.1046/j.1528-1157.44.s10.6.x. [DOI] [PubMed] [Google Scholar]
- Temkin NR. Preventing and treating posttraumatic seizures: the human experience. Epilepsia. 2009;50(Suppl 2):10–13. doi: 10.1111/j.1528-1167.2008.02005.x. [DOI] [PubMed] [Google Scholar]
- Temkin NR, Haglund MM, Winn HR. Causes, prevention, and treatment of post-traumatic epilepsy. New Horizons. 1995;3:518–522. [PubMed] [Google Scholar]
- Timofeev I, Grenier F, Bazhenov M, Sejnowski TJ, Steriade M. Origin of slow cortical oscillations in deafferented cortical slabs. Cereb Cortex. 2000;10:1185–1199. doi: 10.1093/cercor/10.12.1185. [DOI] [PubMed] [Google Scholar]
- Timofeev I, Bazhenov M, Avramescu S, Nita DA. Posttraumatic epilepsy: the roles of synaptic plasticity. Neuroscientist. 2010;16:19–27. doi: 10.1177/1073858409333545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timofeev I, Sejnowski TJ, Bazhenov M, Chauvette S, Grand LB. Age dependency of trauma-induced neocortical epileptogenesis. Front Cell Neurosci. 2013;7:154. doi: 10.3389/fncel.2013.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topolnik L, Steriade M, Timofeev I. Partial cortical deafferentation promotes development of paroxysmal activity. Cereb Cortex. 2003a;13:883–893. doi: 10.1093/cercor/13.8.883. [DOI] [PubMed] [Google Scholar]
- Topolnik L, Steriade M, Timofeev I. Hyperexcitability of intact neurons underlies acute development of trauma-related electrographic seizures in cats in vivo. Eur J Neurosci. 2003b;18:486–496. doi: 10.1046/j.1460-9568.2003.02742.x. [DOI] [PubMed] [Google Scholar]
- Trasande CA, Ramirez JM. Activity deprivation leads to seizures in hippocampal slice cultures: is epilepsy the consequence of homeostatic plasticity? J Clin Neurophysiol. 2007;24:154–164. doi: 10.1097/WNP.0b013e318033787f. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volman V, Sejnowski TJ, Bazhenov M. Topological basis of epileptogenesis in a model of severe cortical trauma. J Neurophysiol. 2011a;106:1933–1942. doi: 10.1152/jn.00458.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volman V, Bazhenov M, Sejnowski TJ. Pattern of trauma determines the threshold for epileptic activity in a model of cortical deafferentation. Proc Natl Acad Sci U S A. 2011b;108:15402–15407. doi: 10.1073/pnas.1112066108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga CJ, Ibata K, Turrigiano GG. Postsynaptic expression of homeostatic plasticity at neocortical synapses. J Neurosci. 2005;25:2895–2905. doi: 10.1523/JNEUROSCI.5217-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, Ping X, Gao J, Jin X. Preparing undercut model of posttraumatic epileptogenesis in rodents. J Vis Exp. 2011;55:pii2840. doi: 10.3791/2840. [DOI] [PMC free article] [PubMed] [Google Scholar]