

Abstract
Brain edema formation occurs after dysfunctional control of extracellular volume partly through impaired astrocytic ion and water transport. Here, we show that such processes might involve synergistic cooperation between the glial water channel aquaporin 4 (AQP4) and the transient receptor potential isoform 4 (TRPV4), a polymodal swelling-sensitive cation channel. In mouse retinas, TRPV4 colocalized with AQP4 in the end feet and radial processes of Müller astroglia. Genetic ablation of TRPV4 did not affect the distribution of AQP4 and vice versa. However, retinas from Trpv4−/− and Aqp4−/− mice exhibited suppressed transcription of genes encoding Trpv4, Aqp4, and the Kir4.1 subunit of inwardly rectifying potassium channels. Swelling and [Ca2+]i elevations evoked in Müller cells by hypotonic stimulation were antagonized by the selective TRPV4 antagonist HC-067047 (2-methyl-1-[3-(4-morpholinyl)propyl]-5-phenyl-N-[3-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide) or Trpv4 ablation. Elimination of Aqp4 suppressed swelling-induced [Ca2+]i elevations but only modestly attenuated the amplitude of Ca2+ signals evoked by the TRPV4 agonist GSK1016790A [(N-((1S)-1-{[4-((2S)-2-{[(2,4-dichlorophenyl)sulfonyl]amino}-3-hydroxypropanoyl)-1-piperazinyl]carbonyl}-3-methylbutyl)-1-benzothiophene-2-carboxamide]. Glial cells lacking TRPV4 but not AQP4 showed deficits in hypotonic swelling and regulatory volume decrease. Functional synergy between TRPV4 and AQP4 during cell swelling was confirmed in the heterologously expressing Xenopus oocyte model. Importantly, when the swelling rate was osmotically matched for AQP4-positive and AQP4-negative oocytes, TRPV4 activation became independent of AQP4. We conclude that AQP4-mediated water fluxes promote the activation of the swelling sensor, whereas Ca2+ entry through TRPV4 channels reciprocally modulates volume regulation, swelling, and Aqp4 gene expression. Therefore, TRPV4–AQP4 interactions constitute a molecular system that fine-tunes astroglial volume regulation by integrating osmosensing, calcium signaling, and water transport and, when overactivated, triggers pathological swelling.
SIGNIFICANCE STATEMENT We characterize the physiological features of interactions between the astroglial swelling sensor transient receptor potential isoform 4 (TRPV4) and the aquaporin 4 (AQP4) water channel in retinal Müller cells. Our data reveal an elegant and complex set of mechanisms involving reciprocal interactions at the level of glial gene expression, calcium homeostasis, swelling, and volume regulation. Specifically, water influx through AQP4 drives calcium influx via TRPV4 in the glial end foot, which regulates expression of Aqp4 and Kir4.1 genes and facilitates the time course and amplitude of hypotonicity-induced swelling and regulatory volume decrease. We confirm the crucial facets of the signaling mechanism in heterologously expressing oocytes. These results identify the molecular mechanism that contributes to dynamic regulation of glial volume but also provide new insights into the pathophysiology of glial reactivity and edema formation.
Keywords: aquaporin 4, Muller cell, osmoregulation, retina, swelling, TRPV4
Introduction
Osmolality in the brain is an important physiological parameter that is under stringent homeostatic control. Activity-dependent fluxes of osmolytes and the volume of the extracellular space (ECS) are disproportionately regulated by astrocytes, which take up neuroactive substances (glutamate, K+, protons) and redistribute water among neuronal, glial, vascular, and ECS compartments. Osmotic gradients, caused by even relatively low levels of neuronal activity, cause sizeable increases in astrocyte volume as a result of influx of K+ and water (Risher et al., 2009; MacAulay and Zeuthen, 2010; Florence et al., 2012). Pathological swelling of astrocytes, caused by augmented rates of water flow into and out of the brain, occurs in many CNS diseases (Manley et al., 2000; Papadopoulos and Verkman, 2013). Therefore, investigations into the molecular mechanisms through which astrocytes sense osmotic imbalance and regulate their water and ionic homeostasis represent an important facet of translational neuroscience. However, it is unclear whether glial swelling is sensed by a single type of sensor molecule or whether it involves complex mechanisms composed of multiple components (Pasantes-Morales et al., 2006).
Swelling in primary, cultured astrocytes and astrocytes in vivo is associated with dynamic changes in [Ca2+]i that can have multiple effects on cell physiology, including stimulation of Ca2+-dependent ion channels, glycogen synthesis, release of osmolytes, gliotransmitters, and arachidonic acid. Swelling in astrocytes can also lead to activation of regulatory volume decrease (RVD; an adaptive decrease in cell volume in the continued presence of hypotonicity; Kimelberg et al., 1992; Schliess et al., 1996; Fischer et al., 1997; Hoffmann et al., 2009). In addition, Ca2+ signals were associated with reactive gliosis, a graded progression of molecular, cellular, and functional changes in astrocytes that represents a hallmark of nearly every brain pathology (Huang et al., 2011; Kanemaru et al., 2013). Elimination of aquaporin 4 (AQP4) channels abolished hypotonically induced swelling-mediated Ca2+ signals, altered activity-dependent changes in ECS volume, and compromised glial RVD (Pannicke et al., 2010; Benfenati et al., 2011; Haj-Yasein et al., 2015). The impermeability of AQP4 to ions suggests that other channels must subserve swelling-induced Ca2+ entry. A strong candidate is transient receptor potential isoform 4 (TRPV4), a polymodal nonselective cation channel that was proposed to bind and/or functionally interact with multiple AQP isoforms (Liu et al., 2006; Benfenati et al., 2011; Galizia et al., 2012). The mechanism by which AQP4 might activate TRPV4 is unclear and the functional significance of AQP–TRPV4 interactions for astrocyte swelling, volume regulation, and intracellular signaling remains to be determined. Because TRPV4 expression is confined to a subset (∼30%) of cortical astrocytes (Shibasaki et al., 2014), we studied the effect of TRPV4–AQP4 interactions in Müller glia, which show close to 100% penetrance for both channels (Nagelhus et al., 1998; Ryskamp et al., 2014). Taking advantage of Trpv4−/− and Aqp4−/− animal models, we demonstrate that TRPV4 and AQP4 cooperatively participate in swelling-induced Ca2+ signaling and retinal glial cell volume regulation under hypotonic challenges. We confirmed the functional significance of AQP4–TRPV4 interactions in the Xenopus laevis expression system, leading us to conclude that the two structurally highly dissimilar channels form a functional symbiotic unit that mediates swelling-induced signaling and volume regulation in the retina. Part of this paper have been published previously in abstract form (Križaj et al., 2013).
Materials and Methods
Animals.
For mice, experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research, and the Institutional Animal Care and Use Committees at the University of Utah. C57BL/6J mice, Trpv4−/− mice with excised exon 12-encoding transmembrane pore domains 5 and 6 (Liedtke and Friedman, 2003), and Aqp4−/− mice with part of exon 1 replaced with a selection cassette causing transcript truncation (Ma et al., 1997) were maintained in a pathogen-free facility with a 12 h light/dark cycle and ad libitum access to food and water. Data were gathered from male and female mice, but no gender differences were noted. For Xenopus laevis, experiments complied with the European Community guidelines for the use of experimental animals and were approved by the Danish National Committee for Animal Studies.
Reagents.
The TRPV4 antagonist HC-067047 (2-methyl-1-[3-(4-morpholinyl)propyl]-5-phenyl-N-[3-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide) (HC-06) was purchased from Calbiochem or Sigma. Arachidonic acid and phospholipase A2 (PLA2) antagonists were purchased from Cayman Chemical or Sigma. All other salts and reagents were purchased from Sigma. GSK1016790A [(N-((1S)-1-{[4-((2S)-2-{[(2,4-dichlorophenyl)sulfonyl]amino}-3-hydroxypropanoyl)-1-piperazinyl]carbonyl}-3-methylbutyl)-1-benzothiophene-2-carboxamide] (GSK101; 1–10 mm) and HC-06 (4 mm) stock in DMSO were diluted 1:1000 in extracellular saline just before use and placed into reservoirs connected to gravity-fed perfusion systems (Warner Instruments).
Semiquantitative real-time PCR.
Retinas were collected from two to four mice per group [wild type (WT), Trpv4−/−, and Aqp4−/− cohorts]. Retinal RNA was isolated with the TRI Reagent (Sigma), and total RNA concentrations (5 μg) were converted to cDNA using the SuperScript III First-Strand Synthesis kit (Invitrogen). RT-PCR was performed using the Cyber Green PCR master mix (Applied Biosystems) and was run on the CFX Connect Real-Time PCR System (Bio-Rad) following the protocols of the manufacturer (Takara; 95°C for 15 s, 95°C for 5 s, 60°C for 30 s, 40 cycles). After amplification, the ratio of gene-of-interest mRNA to the housekeeping gene α-tubulin was calculated for each sample. Four to eight independent biological replicates (retinas) were used. The primers are listed in Table 1.
Table 1.
Primers used in the study
| Primer | Sequence (5′ to 3′) | GenBank accession numbers |
|---|---|---|
| mAqp4 sense | AGCAATTGGATTTTCCGTTG | NM_009700 |
| mAqp4 antisense | TGAGCTCCACATCAGGACAG | |
| mKir4.1 sense | GCCCTAGGACTGATGAGCTG | NM_001039484 |
| mKir4.1 antisense | AACAGTAGGGAGCAGCCAGA | |
| mTrpv4 sense | TCCTGAGGCCGAGAAGTACA | NM_022017.3 |
| mTrpv4 antisense | TCCCCCTCAAACAGATTGGC | |
| mTuba1a sense | AAGCAGCAACCATGCGTGA | NM_011653.2 |
| mTuba1a antisense | CCTCCCCCAATGGTCTTGTC | |
| mGlul sense | TTGGTCTCTGCAGGTTTCCG | NM_008131.4 |
| mGlul antisense | TTGGTCTCTGCAGGTTTCCG | |
| mActb sense | CCACCATGTACCCAGGCATT | NM_007393.4 |
| mActb antisense | AGGGTGTAAAACGCAGCTCA | |
| mGapdh sense | GGTTGTCTCCTGCGACTTCA | NM_001289726.1 |
| mGapdh antisense | TAGGGCCTCTCTTGCTCAGT | |
| 18S rRNA sense | GCAATTATTCCCCATGAACG | NR_003278.3 |
| 18S rRNA antisense | GGCCTCACTAAACCATCCAA |
Immunofluorescence.
Enucleated eyes were punctured and fixed in 4% paraformaldehyde in PBS for 10 min. Eyes were hemisected, and the eyecup was fixed for another 50–60 min. After three washes for 10 min with PBS, eyecups were cryoprotected by incubation in 15% sucrose for 45 min at room temperature and in 30% sucrose overnight at 4°C (Witkovsky et al., 2008; Ryskamp et al., 2011). Eyecups were transferred to OCT (Ted Pella), frozen at −80°C, sliced with a cryostat (16 μm sections), and mounted onto Superfrost Plus slides. Slices were dried for 1 h at room temperature or overnight at 4°C before processing for immunohistochemistry or storage at −80°C. After washing with PBS, slices were blocked for 30 min in blocking buffer (10 ml of PBS, 30 μl of Triton X-100, 100 mg of BSA, and 200 μl of 5% w/v Na azide solution). Primary antibodies were diluted in PBS and applied overnight at 4°C. After rinsing, slices were incubated with secondary antibodies diluted to 1:1000 in PBS 1 h at room temperature. Unbound antibodies were rinsed away, and conjugated fluorophores were protected with Fluoromount-G (Southern Biotech) before mounting coverslips and confocal imaging. Primary antibodies included the following: rabbit anti-TRPV4 (1:100–1:1000; Lifespan Biosciences), rabbit anti-Kir4.1 (1:500; catalog #APC-035; Alomone), guinea pig anti-Kir4.1 (1:200; catalog #AGP-012; Alomone), rabbit anti-glial fibrillary acidic protein (GFAP; 1:500; catalog #Z0334; Dako), mouse anti-glutamine synthetase (GS; 1:1000–1:2000; catalog #610517; BD Biosciences), and mouse anti-AQP4 (1:1000; catalog #SC-20812; Santa Cruz Biotechnology). Goat anti-mouse, goat anti-rabbit, and goat anti-guinea pig IgG (heavy and light chain) secondary antibodies were conjugated to fluorophores (Alexa Fluor 488 or Alexa Fluor 594; Life Technologies). The primary antibodies did not label knock-out (KO) tissues (Da and Verkman, 2004; Ryskamp et al., 2011). Secondary antibodies did not label retinal tissue when primary antibodies were omitted. Immunostaining was imaged with confocal microscopes (Zeiss LSM 510 or Olympus FV1200) using argon (488 nm) and helium/neon (543 nm) lasers, suitable emission filters, and 40×/1.2 numerical aperture (NA) oil-immersion or 40×/0.9 NA water-immersion objectives.
Acute dissociation of Müller cells.
After the animals were killed, mouse eyes were enucleated, and retinas were isolated by dissection in ice-cold Leibovitz 15 (L-15) medium (Life Technologies) containing 11 mg/ml L-15 powder, 20 mm d-glucose, 10 mm Na-HEPES, 2 mm Na-pyruvate, 0.3 mm Na-ascorbate, and 1 mm glutathione. Retinas were incubated at room temperature in L-15 medium containing papain (7 U/ml; Worthington) to digest the extracellular matrix. Retinas were rinsed with cold L-15 medium and diced into ∼500 μm pieces. One to two pieces of retina were dissociated mechanically by trituration and plated on coverslips pretreated with concanavalin A (1 mg/ml). Plated cells were loaded with the ratiometric Ca2+ indicator fura-5F AM for 30–40 min and were washed for 10–20 min in dye-free L-15 medium. Ca2+ homeostasis and stimulus responsiveness is maintained for several hours under these experimental conditions (Ryskamp et al., 2014). Müller cells were identified by their distinctive morphology and prominent Ca2+ signals during TRPV4 activation. Cellular identity was confirmed in a subset of experiments by post-imaging immunocytochemistry (mouse anti-GS) as in the study by Ryskamp et al. (2014).
Superfusion of Müller cells and cell swelling assays.
Volume regulation in dissociated Müller glia was investigated following published protocols (Ryskamp et al., 2011, 2014). Briefly, saline solutions were delivered through a gravity-fed eight-reservoir system (Warner Instruments) that converged toward a manifold tube inserted into the experimental chamber. The control saline contained the following (in mm): 133 NaCl, 2.5 KCl, 1.5 NaH2PO4, 1.5 MgCl2 (6H20), 2 CaCl2, 10 glucose, 10 HEPES hemisodium salt, 1 pyruvic acid, 1 lactic acid, 0.5 l-glutamine, 0.5 glutathione, and 0.3 Na-ascorbate, pH 7.4 (osmolarity at 300 mOsm). With NaCl kept at 57.5 mm, anisosmotic solutions were prepared by addition or removal of mannitol, a procedure that maintains the ionic strength of the extracellular solution. Mannitol may reduce the contribution of volume-activated channels to RVD, whereas the reduction in NaCl could disinhibit volume-activated chloride channels (Fernández et al., 2013; Minieri et al., 2015). Osmolarity was checked thermometrically using a vapor pressure osmometer (Wescor). Cell volume was measured by calcein fluorescence and by adapting a protocol developed by the Putney laboratory (Chiavaroli et al., 1994). Fluorescence emissions from 340 and 380 nm stimuli were normalized to baseline fluorescence and algebraically summed (Fvol = F340 + F380/x, where x = 1–3, a value derived empirically for each preparation to equalize the magnitude of the Ca2+-dependent and opposing changes in F340 and F380). Thus, at the appropriate x value, the intensity of the summed fluorescence was calcium insensitive (i.e., the summed trace showed no response to GSK101 or glutamate). In a subset of experiments using hypotonic stimulation (HTS), cells were coloaded with fura-5F AM and calcein AM (1 μm; Invitrogen). Calcein fluorescence was evoked using 490 nm excitation attenuated as described previously (Ryskamp et al., 2011). The x–y cross-sectional area was determined offline using NIS-Elements AR 3.2 or Olympus image analysis software. We confirmed with confocal z stacks over time that swelling occurs uniformly in all directions (Ryskamp et al., 2014), indicating that cell volume is proportional to √area3. The percentage RVD was calculated as 100 × (peak cell area in hypotonicity − cell area at the end of hypotonicity)/(peak cell area in hypotonicity − cell area before hypotonicity).
Calcium imaging.
Epifluorescence images were acquired using an inverted Nikon Ti or an upright Nikon E600 FN microscope with 20× (0.75 NA oil-immersion), 40× (1.3 NA oil-immersion and 0.8 NA water-immersion), and 60× (1.0 NA water-immersion) objectives. Excitation light from a xenon arc lamp was delivered and filtered using a Lambda DG-4 (Sutter Instruments). Emissions were detected with 14-bit CoolSNAP HQ2 cameras and analyzed using NIS-Elements AR 3.2 and MS Excel. [Ca2+]i was detected using the ratiometric fluorescent dyes fura-2 (Kd at room temperature = 225 nm) and fura-5F (Kd at room temperature = 400 nm; Invitrogen Handbook) as described previously (Molnar et al., 2012; Ryskamp et al., 2014). Although cells were loaded with 5–10 μm of the AM dyes, trapping by de-esterification was assumed to accumulate them to ∼100 μm (Križaj and Copenhagen, 1998). ΔR/R (peak F340/F380 ratio − baseline/baseline) was used to quantify the amplitude of Ca2+ signals. In a subset of experiments using extreme HTS (≤30% tonicity), the duration of 380 nm excitation was increased to decrease signal noise. Results represent averages of Müller cell responses from at least three animals.
Electrophysiology in Müller glia.
Retinal pieces were incubated in L-15 medium, and cells were dissociated, plated, and identified as described previously (Szikra et al., 2009; Ryskamp et al., 2014). The extracellular solution contained the following (in mm): 51 NaCl, 0.6 Na2HPO4, 0.75 MgCl2, 0.5 sodium pyruvate, 5 lactic acid, 5 HEPES hemisodium salt, 0.5 glutathione, 0.3 sodium l-ascorbate, 10 d-glucose, 0.5 l-glutamine, 2 CaCl2, and 135 mannitol, pH 7.3. The hypotonic saline had the same composition, without mannitol. Experiments were performed at room temperature (20–22°C). Solution exchange was achieved through rapid switching of solenoid valves regulating gravity-driven flow through a tubing manifold placed ∼5 cm from the perfusion chamber.
The recording pipettes were pulled using a P-2000 horizontal puller (Sutter Instruments) from borosilicate glass (1.5 mm outer diameter, 0.84 mm inner diameter) to obtain tips with resistance between 8 and 10 MΩ. We patched Müller end feet, recognized as semicircular protuberances at the end of the proximal stalk. Membrane currents were acquired after application of ramp pulses assessing from −100 to 100 mV for 1 s (applied at 0.2 Hz) using pClamp 10.5 acquisition software (Molecular Devices) and a Digidata 1422 interface (Molecular Devices). Data were sampled at 10 kHz and filtered at 5 kHz with an eight-pole Bessel filter and analyzed using Clampfit 10.5 (Molecular Devices) and Origin Pro8 software. The time dependence of HTS-induced currents was quantified as duration of HTS-induced whole-cell current changes from 5% of the maximum response to the maximum response.
Heterologous expression in Xenopus oocytes.
Xenopus laevis frogs were obtained from Nasco or the National Center for Scientific Research (France). Oocytes were removed surgically from anesthetized frogs and prepared as described previously (Fenton et al., 2010). cDNA encoding rat Trpv4 and Aqp4.M23 in the oocyte expression vector pXOOM (TRPV4 and AQP4.M23) were linearized downstream from the poly(A) segment and in vitro transcribed using T7 mMessage Machine (Ambion). cRNA was extracted with MEGAclear (Ambion) and microinjected into defolliculated Xenopus laevis oocytes (4 ng of TRPV4 and/or 10 ng of AQP4 RNA/oocyte). Oocytes were kept at 19°C in Kulori medium (in mm: 90 NaCl, 1 KCl, 1 CaCl2, 1 MgCl2, and 5 HEPES, pH 7.4) for 3–4 d before experiments. Because of basal activity of TRPV4 and associated cell lysis resulting from the toxicity of overexpressed channels, the oocytes were kept in 1 μm Ruthenium Red (Sigma) but rinsed before experiments (Loukin et al. 2010).
Electrophysiology and volume measurements in oocytes.
Conventional two-electrode voltage-clamp studies were performed with a Dagan CA-1B High Performance oocyte clamp (Dagan) with Digidata 1322A interface controlled by pClamp 9.2 (Molecular Devices). The oocyte membrane potential was clamped at −30 mV, and the current–voltage (I–V) relationship was determined by stepping the clamp potential to test potentials ranging from +60 to −140 mV in 20 mV increments (100 ms pulses). The experimental setup for measuring water permeability of oocytes has been described in detail previously (Zeuthen et al., 2006). Briefly, the oocyte was placed in a small chamber with a glass bottom and perfused with a control solution (in mm: 40 NaCl, 2 KCl, 1 CaCl2, 1 MgCl2, 125 mannitol, and 10 HEPES, pH 7.4). Oocyte images were captured continuously from below at a rate of 25 images/s. To induce cell swelling, the oocytes were challenged with a hypotonic solution (composition as in control solution but with removal of 125 or 15 mm mannitol), and cell swelling was determined as the percentage increase in cross-sectional area of the oocyte.
Statistics.
Statistical analyses were performed with GraphPad Prism version 6.0. Data were acquired from at least three different retinas and three different batches of oocytes. Means are shown ± SEM. Unless indicated otherwise, an unpaired t test was used to compare two means, and a one-way or two-way ANOVA along with the Holm–Sidak test was used to compare three or more means. p > 0.05 is not significant, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Results
TRPV4 colocalizes with AQP4 in Müller glial end feet
Although TRPV4 and AQP channels were implicated in osmotic regulation as sensors and effectors for osmotically induced Müller glial swelling and water flux, respectively (Nagelhus and Ottersen, 2013; Ryskamp et al., 2014), the nature of their combined contribution to cellular osmoregulation is not well defined. Therefore, we tested whether selective ablation of either channel affects the production and localization of its putative partner and investigated their respective contributions to volume regulation and Ca2+ homeostasis. As shown previously (Nagelhus et al., 1998; Da and Verkman, 2004; Goodyear et al., 2009), immunostaining of WT mouse retinas with the AQP4 antibody labeled Müller cell somata within the inner nuclear layer, inner retinal processes, and end feet forming the pericapillary sheath within the inner limiting membrane (ILM; Fig. 1Ai). Likewise, consistent with Müller glial expression, TRPV4 immunoreactivity was observed in glial end feet and transverse processes in the proximal inner plexiform layer (IPL) in which it overlapped with Müller cell markers GS and the inwardly rectifying K+ channel (composed of Kir4.1 subunits) that has been suggested to regulate Müller cell resting membrane potential and underlie retinal K+ siphoning toward vascular sinks (Newman, 1985; Fig. 1Aii). The extensive colocalization between AQP4 and TRPV4 signals included pronounced overlap within the end foot compartment and radial processes traversing the IPL (Fig. 1Ai, top, arrows), whereas TRPV4–Kir4.1 colocalization was less pronounced, with the latter signal most concentrated in the most proximal portion of the ILM (Fig. 1Aiii). AQP4-immunonegative TRPV4-immunoreactive signals in the inner retina correspond to retinal ganglion cell (RGC) somata (Fig. 1A, arrowheads) and proximal dendrites. The colocalization of TRPV4, AQP4, and Kir4.1 channels in Müller end feet is consistent with a putative synergistic function of these channels in Müller cell volume regulation at the blood–retina barrier.
Figure 1.
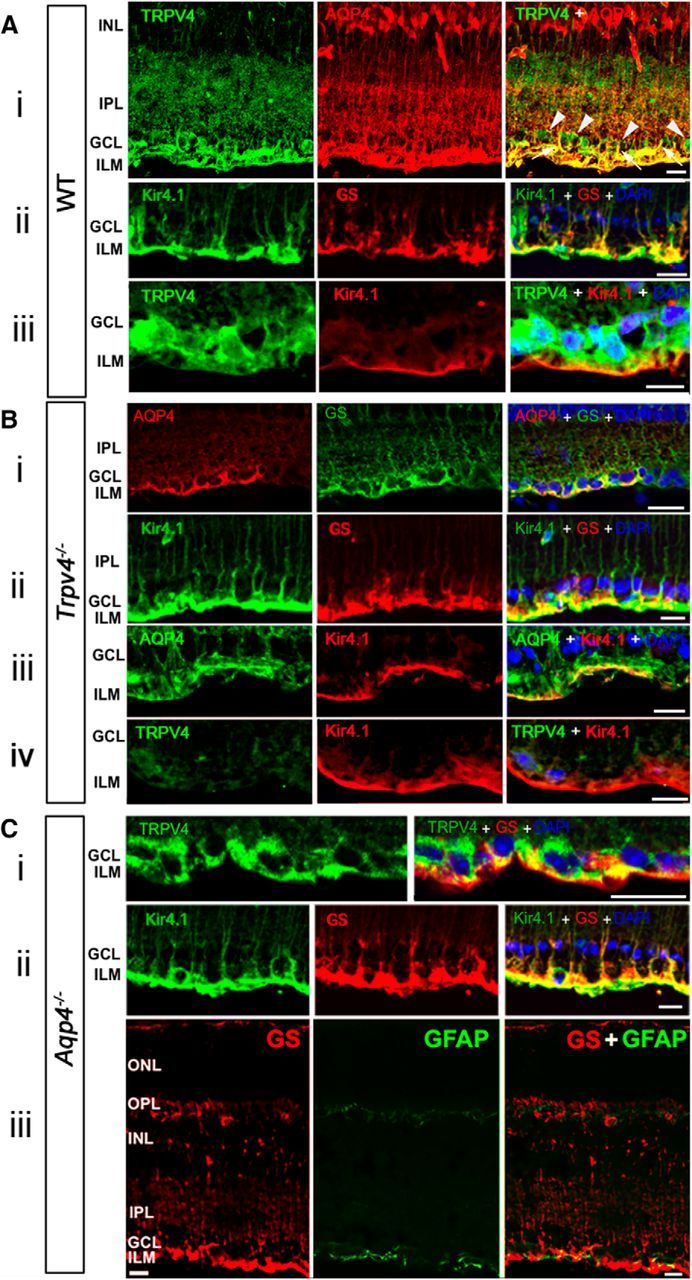
TRPV4 and AQP4 colocalize in the mouse inner retina, yet TRPV4 and AQP4 trafficking are independent of each other. Ai, Vertical cryosections of WT mouse retinas immunolabeled for TRPV4 (Alexa Fluor 488) and AQP4 (Alexa Fluor 594) show preferential colocalization in Müller end feet and proximal processes entering the IPL (arrows). RGC somata are TRPV4 immunopositive and do not express AQP4 (arrowheads). Aii, Aiii, Higher magnification of Müller end feet double labeled for Kir4.1 and GS, and TRPV4 and Kir4.1, respectively. Nuclei labeled with DAPI. GCL, Ganglion cell layer; INL, inner nuclear layer. Scale bars, 20 μm. Bi–Biii, Trpv4−/− retina. GS and Kir4.1 signals in KO tissue are indistinguishable from WT controls, whereas AQP4 signals appear to be less pronounced. Biv, Limited nonspecific TRPV4 immunoreactivity in Trpv4−/− retina costained for Kir4.1. C, Aqp4−/− retinas. Ci, TRPV4 and GS colocalize in glial end feet at the blood–retina barrier, whereas RGC somata are immunoreactive for TRPV4. Cii, Double labeling for GS and Kir4.1 shows normal targeting to glial processes and end feet. Ciii, GFAP is expressed in astrocyte processes but is absent from GS immunoreactivity in Müller processes and end feet, indicating absence of reactive gliosis. Scale bars, 20 μm.
If AQP4 channels in Müller glia form an obligatory complex with TRPV4, the distribution and/or expression of the osmosensor might be influenced by AQP4 elimination (Benfenati et al., 2011). However, evaluation of Müller glial morphology and TRPV4 immunolocalization in Aqp4−/− retinas (Fig. 1Ci) failed to reveal substantial differences from WT retinas (Fig. 1A). Aqp4−/− retinas did not show a discernible phenotype in terms of layering, cytoarchitecture, and Kir4.1 immunoreactivity (Fig. 1Cii). To test the possibility that targeting of AQP4/Kir4.1 channels to the plasma membrane might depend on TRPV4 channels, we examined their respective immunoreactivities in Trpv4−/− retinas. Trpv4−/− retinas were morphologically normal, evincing the typical Kir4.1 localization patterns within Müller cell processes and end feet (Fig. 1B). Further suggestive of normal Müller glial morphological and molecular characteristics in KO tissue was the typical expression pattern of GS staining in Trpv4−/− retinas and Aqp4−/− retinas (Fig. 1Aii,Bii,Cii/Ciii). Consistent with downregulation of Aqp4 mRNA (Fig. 2), the intensity of AQP4 labeling in Trpv4−/− retinas appeared to be lower (Fig. 1Bi,Biii) compared with WT tissue (Fig. 1Ai).
Figure 2.

Expression of Trpv4, Aqp4, and Kir4.1 transcripts is effected by genetic elimination of TRPV4 or AQP4. Semiquantitative RT-PCR analysis of mRNA levels in WT, Trpv4−/−, and Aqp4−/− mouse retinas. Fold change in mRNA expression was calculated relative to basal after normalization of expression to α-tubulin (Tuba1a) mRNA. The relative abundance of Trpv4 transcripts is decreased in Aqp4−/− samples and vice versa. Kir4.1 transcript levels are lower in both Trpv4−/− and Aqp4−/− retinas, whereas the abundance of presumed housekeeping genes Glul, Actb, Gapdh, and 18S rRNA (the 18S structural subunit of the small eukaryotic 40S ribosomal subunit, 18S-rRNA) mRNAs was unaffected by the deletion of Trpv4 or Aqp4.
Müller cells respond to injury with reactive gliosis, an adaptive response typified by astroglial hypertrophy and cytoskeletal reorganization that can be visualized by upregulation of the gliotic marker GFAP. In the retina, astrocytes on the vitreal surface constitutively express GFAP, whereas Müller glia only express GFAP during the reactive response. We noted previously that ablation of the Trpv4 gene and TRPV4 overactivation in vivo result in mild Müller glial reactivity indicated by de novo GFAP expression (Ryskamp et al., 2014). Here we examined whether the absence of physiological AQP4 functions might evoke a similar gliotic response. Although astrocytes on the vitreal surface were labeled clearly by the GFAP antibody, the marker was absent from the radial processes of Müller glia (Fig. 1Ciii). This absence of overt retinal distress is consistent with the previous report by Pannicke et al. (2010) and implies that AQP4 may have more subtle roles in normal retinal function (Li et al., 2002), whereas AQP4-mediated water fluxes may have more influence under pathophysiological conditions (Da and Verkman, 2004).
These results suggest the following: (1) TRPV4 expression is not a prerequisite for proper trafficking or localization of AQP4 and Kir4.1; (2) AQP4 expression is not required for trafficking or localization of TRPV4 and Kir4.1; and (3) unlike loss of TRPV4, which is associated with reactive gliosis (Ryskamp et al., 2014), loss of AQP4 function does not induce Müller glial expression of the gliotic marker GFAP.
Genetic deletion of TRPV4 and AQP4 alters the transcription of Trpv4, Aqp4, and Kir4.1 genes
Despite independent trafficking of TRPV4 and AQP4 to Müller end feet, the two channels might interact indirectly through their effects on gene expression, calcium homeostasis, cell swelling, and/or volume regulation. We analyzed the expression of Trpv4 and Aqp4 transcripts in KO retinas but also tested for expression of Kir4.1 mRNA, because these inwardly rectifying K+ channels were suggested to form a complex with AQP4 (Puwarawuttipanit et al., 2006) and TRPV4 (Lanciotti et al., 2012) and are strongly expressed in Müller glial end feet (Fig. 1). Analysis of Aqp4−/− retinas (n = 8) showed a substantial (69.48 ± 8.27%; p < 0.0001) reduction in the abundance of Kir4.1 transcripts and a nonsignificant 38.71 ± 15.43% decrease in Trpv4 mRNA (Fig. 2). Likewise, Aqp4 and Kir4.1 transcript levels were markedly reduced in Trpv4−/− retinas [76.72 ± 2.55% (Aqp4) and 65.85 ± 8.04% (Kir4.1); n = 8; p < 0.0001; Fig. 2]. Analysis of genes presumably unrelated to TRPV4 and AQP4 function, i.e., GS (Glul), glyceraldehyde-3-phosphate dehydrogenase (Gapdh), β-actin (Actb), and the 18S ribosomal subunit genes, showed no significant changes in transcription upon deletion of Aqp4 or Trpv4, arguing against global downregulation of transcription in KO retinas. These findings suggest that TRPV4 and AQP4 activation not only modulate fluxes of cations and water but also influence the transcriptional regulation of osmoregulation-relevant genes in the retina.
AQP4 is not required for TRPV4 activation
Given TRPV4–AQP4 colocalization and the proposed obligatory interaction in cortical astrocytes (Benfenati et al., 2011), we investigated whether AQP4 is required for agonist-induced glial TRPV4 activation in mouse Müller glia. Müller cells were voltage clamped and stimulated with GSK101, a selective TRPV4 agonist at an approximate EC50 for Müller glia (∼25 nm; Ryskamp et al., 2014). Voltage ramps over time revealed that GSK101 stimulated cation currents with slight outward rectification typical of TRPV4 (Fig. 3A). A representative time course of the whole-cell currents in an Aqp4−/− Müller cell is shown in Figure 3B. The cell exhibited the typical delayed activation of the TRPV4-mediated current, with a gradual increase-to-peak over several minutes of both inward and outward components (Fig. 3B) that reversed close to 0 mV (Fig. 3A). GSK101-evoked responses were completely blocked by the selective antagonist HC-06 (Ryskamp et al., 2014). The response amplitudes and I-V characteristics of GSK101-evoked currents in WT (n = 6) and Aqp4−/− Müller (n = 5) glia revealed no significant differences (−402 ± 195 vs −458 ± 220 pA at −100 mV and 652 ± 226 vs 636 ± 189 pA at 100 mV, respectively). AQP4 channels have been shown previously to influence TRPV4-dependent Ca2+ signals after swelling-induced stretch of the glial plasma membrane (Benfenati et al., 2011; Ryskamp et al., 2014). Therefore, we tested whether AQP4 was required for TRPV4-influenced Ca2+ homeostasis. The average spontaneous activity in Aqp4−/− Müller glia at baseline, measured by the 340/380 ratio of fura-5F fluorescence, was 0.26 ± 0.03 (n = 44), not significantly different from WT cells (n = 12; p = 0.8705, Holm–Sidak test), suggesting that AQP4 channels do not contribute to the resting Müller cell [Ca2+]i. To assess the activation properties of TRPV4 channels, we stimulated the cells with GSK101. [Ca2+]i levels in Aqp4−/− cells maintain the sensitivity to GSK101 stimulation, as indicated by the 88.9 ± 10.3% (n = 44) increase in the fluorescence ratio over the basal levels in the presence of the agonist (Fig. 3C). Nonetheless, comparison with signals in WT cells (124.4 ± 13.4% over baseline) revealed a modest (∼28.6%) but significant (p = 0.0453, Holm–Sidak test) decrease in GSK101-induced Aqp4−/− [Ca2+]i compared with WT. These data suggest the following: (1) AQP4 is not required for basal regulation of Müller cell [Ca2+]i levels; (2) AQP4 modulates, but is not obligatory for, agonist-induced TRPV4 activation in Müller cells; and (3) decreased Trpv4 transcription in Aqp4−/− cells might be associated with functional decreases in the cellular response to swelling-evoked membrane stretch. The unaltered I-V relationship and amplitude of the GSK-induced TRPV4-mediated transmembrane current (Fig. 3B) suggest that loss of Aqp4 might predominantly affect secondary components of TRPV4-dependent Ca2+ homeostasis, such as ATP release, release from internal stores, and/or activation of store-operated channels.
Figure 3.

AQP4 is not required for agonist-induced TRPV4 activation. A, Averaged I–V curves of GSK101-induced currents in WT (black trace) and Aqp4−/− (blue trace) Müller cells. B, Representative time courses of the GSK101-evoked whole-cell current in Aqp4−/− and WT Müller cells. Shown are current amplitudes for WT cells (black trace) and Aqp4−/− cells (blue trace) obtained at 100 mV (circles) and −100 mV (triangles). C, The TRPV4 agonist GSK101 evokes a 5.3-fold increase in the fura-5F ratio in WT Müller cells over baseline activity (n = 12; gray bars). GSK101 responses were 3.4-fold larger than baseline activity in Aqp4−/− Müller cells (n = 44; blue bars) but significantly diminished compared with responses in WT cells. Shown are mean ± SEM values.
TRPV4 and AQP4 regulate hypotonic Müller glial swelling and RVD
To test for functional interactions between TRPV4 and AQP4, we compared the magnitude and dynamics of volume changes during HTS in WT, Trpv4−/−, and Aqp4−/− Müller cells. Water influx across the plasma membrane was measured indirectly by tracking the changes in cell area and/or volume. Cell area was determined by outlining the perikaryal perimeter, whereas changes in glial volume were determined by the algebraic summation 340 and 380 nm emission fluorescence of fura-5F, yielding a Ca2+-independent signal inversely proportional to the cell volume (see Materials and Methods; Fig. 4A). HTS evoked sustained, dose-dependent and reversible increases in cross-sectional area and volume in cells from all three genotypes, whereas hypertonic stimuli resulted in cell shrinkage (Fig. 4B). The initial response of Müller cells to osmotic challenge tracked the stressor magnitude, suggestive of the “osmometer-like” properties reported for cortical astrocytes (Risher et al., 2009). Anisotonicity-induced responses were reversible, with cell volume typically returning to baseline levels after 1–5 min in control saline (Fig. 4A,C). Trpv4−/− cells showed significant decreases in peak swelling during HTS, whereas the maximal swelling in Aqp4−/− cells was similar to controls (Fig. 4B). A 160 mOsm tonicity decrease (46.7% tonicity) enlarged the mean Müller cell area in Trpv4−/− cells by 19.84 ± 1.23% (n = 28), significantly less compared with 39.47 ± 0.73% (n = 33) in control cells (p < 0.0001, Dunnett's test), whereas the mean area of Aqp4−/− cells increased by 37.59 ± 0.52% (n = 69; p = 0.104; Fig. 4B). The reduction in the extent of Müller cell swelling by genetic elimination of the cation channel is consistent with the previously reported effects induced by pharmacological blockade of the channel (Ryskamp et al., 2014).
Figure 4.
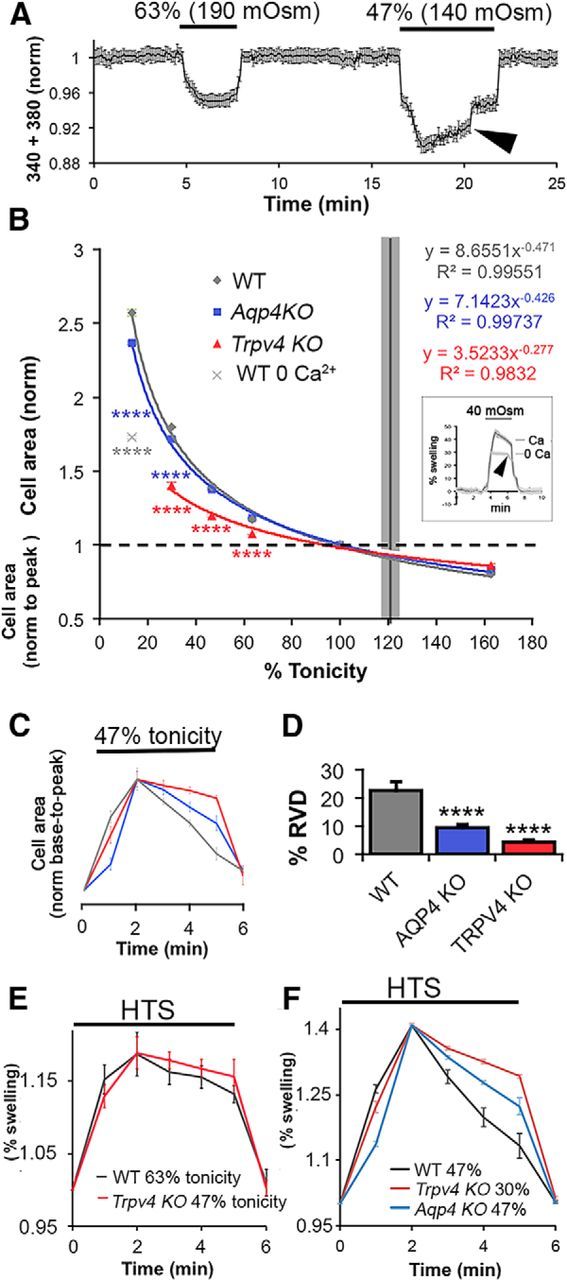
TRPV4-induced Ca2+ entry regulates Müller glial swelling and RVD. A, Cell volume changes in a fura-5F-loaded Müller cell. The sum of 340/380 emissions, adjusted to yield calcium-insensitive intensity values, shows dose-dependent decreases as cell volume increases in the presence of HTS. RVD is observed during the 140 mOsm (46.67% tonicity) but not 190 mOsm (63.33% tonicity) stimulus (arrowhead). B, Dose–response curves for changes in peak perikaryal cross-sectional area in the presence of osmotic challenge show reduced glial swelling in Trpv4−/− (n = 25–31 cells per stimulus) but not substantially in Aqp4−/− cells (n = 31–71) compared with WT cells (n = 27–61). Removal of extracellular Ca2+ during swelling of WT cells (13.3% tonicity) shifted swelling to the extrapolated Trpv4−/− curve (light gray data point; n = 27). Inset, Volume measurement (340 + 380 signals, normalized to baseline) in WT cells (n = 8) exposed to extreme HTS (40 mOsm) and shown with inverted traces. Note the lower fluorescence decrease in Ca2+-free saline and the absence of RVD under these conditions (arrow). C, The time course of swelling in WT, Trpv4−/−, and Aqp4−/− cells during a 5 min exposure to 140 mOsm HTS, normalized to base and peak cross-sectional areas to compare swelling kinetics. D, RVD during exposure to 140 mOsm HTS is impaired in Trpv4−/− (n = 38) and Aqp4−/− (n = 57) cells compared with WT cells (n = 52). E, WT and Trpv4−/− cell area over time normalized to baseline (20% peak swelling; 190 mOsm for WT, n = 9; 140 mOsm for Trpv4−/− cells, n = 8). F, WT, Trpv4−/−, and Aqp4−/− cell area over time normalized to baseline (40% peak swelling; 140 mOsm for WT, n = 10; 90 mOsm for Trpv4−/− cells, n = 10; 140 mOsm for Aqp4−/− cells, n = 5).
We next evaluated the time dependence of RVD in the response to sustained hypotonic stress. As illustrated in Figure 4, in WT cells, sustained exposure to large decreases in extracellular tonicity evokes slow but consistent decreases in cross-sectional area or volume relative to peak swelling, consistent with RVD. After reaching peak swelling, cell volume in HTS (140 mOsm)-stimulated cells gradually declined toward the original volume (to 22.6 ± 3.1% within a 5 min timeframe in WT Müller cells; n = 52). Traces of cell area over time were overlaid by normalization to baseline and peak volume to compare the time course of swelling and RVD at 47% tonicity (Fig. 4C). This analysis indicated that, relative to WT Müller cells, swelling is delayed in Aqp4−/− Müller cells and that RVD is impaired in Aqp4−/− cells (n = 57) and Trpv4−/− Müller cells (n = 38 cells; p < 0.0001 for both, Dunnett's test; Fig. 4C,D,F). Because the peak HTS-induced swelling was reduced for the Trpv4−/− cells (Fig. 4B), a reduction in RVD in these cells compared with WT may be observed as a consequence of the different level of membrane stretch from which RVD is initiated. To examine the extent of RVD evoked from a similar amount of swelling, WT and Trpv4−/− Müller cells were exposed to sustained HTS of different magnitudes to obtain a similar level of swelling ∼15–20% (Fig. 4E) and ∼40% (Fig. 4F). Under these comparable conditions, these genotypes responded similarly to the milder HTS stimulus (Fig. 4E). However, at 40% swelling, RVD was impaired in Trpv4−/− and Aqp4−/− Müller cells, suggesting that suppression of TRPV4 activation in these conditions (see below) may reduce RVD by inhibiting ion fluxes associated with triggering and enacting RVD (Fig. 4F). These data suggest the following: (1) TRPV4 activation contributes to HTS-induced glial swelling; (2) AQP4 accelerates swelling; (3) the degree of RVD depends on swelling amplitude; and (4) TRPV4 and AQP4 contribute to RVD for adaptation to extreme, nonphysiological hypotonic challenges.
TRPV4 activation increases swelling in WT and Aqp4−/− Müller glia
Given that the extent of HTS-induced swelling was reduced in Trpv4−/− Müller cells (Fig. 4B) and that GSK101- and HTS-induced Ca2+ elevations are abolished by the removal of external Ca2+ (Ryskamp et al., 2014), we tested whether TRPV4-dependent changes in Müller cell area/volume require Ca2+ influx. Removal of extracellular Ca2+ reduced the HTS-induced cell swelling as determined by cell area (Fig. 4B) or volume (Fig. 4B, inset), suggesting that HTS-dependent swelling is promoted by TRPV4-mediated Ca2+ entry.
An alternative method for determining changes in cell volume is by using calcein, a fluorescein derivative that is independent of changes in intracellular ion concentrations (Crowe et al., 1995) and signals proportional changes in the volume of retinal neurons and glia (Ryskamp et al., 2011, 2014). Previously, we used calcein to show that HC-06 suppressed swelling in HTS-stimulated WT Müller glia (Ryskamp et al., 2014). Analysis of Aqp4−/− Müller glia revealed that the antagonist similarly suppresses the increase in cell volume in KO cells (Fig. 5A,B). Measurements of cell area likewise revealed a similar extent of peak glial swelling induced by 190 mOsm HTS (measured at 5 min after onset) between WT cells (n = 24) and Aqp4−/− cells (n = 23; Figs. 4B, 5C). The HTS-evoked increase in the cell area of Aqp4−/− Müller cells (n = 12) continued to be suppressed by HC-06 (p < 0.05; Fig. 5C), indicating that TRPV4 activation contributes to Müller cell swelling even in the absence of AQP4. WT, Trpv4−/−, and Aqp4−/− cells showed similar responses to the hypertonic stimulus (data not shown), suggesting that TRPV4 is not required for regulating the extent of osmotically induced volume decreases in Müller glia.
Figure 5.

TRPV4 blockers suppress the swelling of Aqp4−/− Müller glia loaded with calcein AM. A, The TRPV4 antagonist HC-06 (1 μm) suppresses HTS-induced increases in Aqp4−/− glial volume (190 mOsm; n = 9 cells from 5 slides). B, Cumulative data for cell volume measurements in Aqp4−/− cells stimulated with 190 mOsm in the presence/absence of HC-06 (n = 13 and 20, respectively). C, Cell area measurements revealed a similar extent of swelling after 5 min of HTS (190 mOsm) perfusion in WT (n = 24) and Aqp4−/− (n = 23) Müller cells. HC-06 (1 μm) significantly decreased the extent of HTS-induced glial swelling in WT (n = 10) and Aqp4−/− (n = 12) Müller cells.
Together, these experiments suggest that TRPV4 channels regulate peak glial swelling by increasing [Ca2+]i, in a manner independent of the presence of AQP4. AQP4 functions as a passive conduit for osmotic water fluxes and increases, i.e., the rate of HTS-induced cell swelling while not being implicated in setting the end volume after an osmotic challenge. Thus, TRPV4-dependent Ca2+ signals might reorganize the mechanical properties of Müller glia, wherein the final volume is determined by a shift in the balance of tensile forces (e.g., from cytoskeletal elements) that counteract swelling and osmotic pressure forces that drive cell swelling.
AQP4 channels amplify HTS-induced TRPV4-mediated currents and Ca2+ responses in Müller cells
Exposure of HTS (190 mOsm) induced inward and outward currents in Müller end feet, which reached −3982 ± 751 and 3703 ± 635 pA at holding potentials of −100 and +100 mV, respectively (n = 8; Fig. 6A,D). Consistent with the strong immunoreactivity for TRPV4 (Fig. 1), the current amplitude was ∼10-fold larger compared with signals measured previously in cell somata (Ryskamp et al., 2014). The full block of the HTS-evoked end foot current by HC-06 (Fig. 6C,D) indicates that it was primarily mediated by TRPV4. In addition to the larger response amplitude compared with the agonist-evoked response (Fig. 3), the I–V relationship of the HTS-evoked current was more linear at positive potentials (Fig. 6C), and the response was characterized by faster time-to-peak and desensitization of inward and outward components (Fig. 6A). Consistent with the slower rate of swelling-induced membrane stretch resulting from decreased water permeability, together with lower Trpv4 expression, Aqp4−/− cells showed a reduction in HTS-induced current amplitude to −917 ± 628 and 1001 ± 515 pA at −100 and 100 mV, respectively (n = 8 cells, p < 0.01; Fig. 6B–D). Aqp4−/− cells also displayed slower activation; the swelling-evoked current reached its maximum at 202.5 ± 32.2 s in Aqp4−/− compared with 61.3 ± 22.4 s in WT cells (p < 0.01; Fig. 6A,B). The magnitude and inactivation features of the HTS-induced current in Aqp4−/− cells appeared to be qualitatively similar to agonist-induced currents (Fig. 3). As opposed to WT cells, inactivation was notably absent in Aqp4−/− glia, at least for the duration of our stimulus (300 s; Fig. 6B). The antagonist HC-06 inhibited the HTS-induced current in WT and KO cells (Fig. 6C,D); however, the transmembrane current in HC-06-treated Aqp4−/− cells was not significantly different from the response of cells that were not exposed to the TRPV4 antagonist (Fig. 6C,D). These electrophysiological findings indicate that the presence of AQP4 dramatically influences the time course and magnitude of HTS-induced TRPV4-dependent currents in Müller glia.
Figure 6.

AQP4 regulates the amplitude and kinetics of the swelling-induced TRPV4-mediated transmembrane current. A, B, Representative time course of the HTS (235 mOsm)-evoked whole-cell current in WT (black traces) and Aqp4−/− (blue traces) Müller cells at positive (100 mV; open circles) and negative (−100 mV; filled circles) holding potentials. C, Averaged I-V relationship of HTS-induced currents in WT (white; n = 14) and Aqp4−/− (blue; n = 13) Müller cells, WT cells treated with HC-06 (gray; n = 5), and Aqp4−/− cells treated with HC-06 (red; n = 8). Individual I–V curves of HTS-induced currents were subtracted from pre-HTS baselines. HC-06 was applied 4 min before the application of HTS and was present throughout the experiment. D, Summary for HTS-induced currents at 100 mV (open bars) and −100 mV (shadowed bars) for WT (white) and Aqp4−/− (blue) Müller cells, WT cells treated with HC-06 (gray), and Aqp4−/− cells treated with HC-06.
HTS-induced [Ca2+]i signals were likewise strongly suppressed in Trpv4−/− Müller glia (Fig. 7A,C). The amplitude of HTS (140 mOsm)-evoked Ca2+ responses was reduced by ∼54% in Aqp4−/− cells (ΔR/R of 0.34 ± 0.03, n = 13) compared with WT Müller cells (ΔR/R of 0.74 ± 0.05, n = 20; p < 0.0001, two-way ANOVA, Holm–Sidak test) and completely abolished in the presence of HC-06 (Fig. 7B). Interestingly, in cells challenged with exceedingly severe HTS (70% reduction in tonicity), Ca2+ elevations in Aqp4−/− Müller glia were substantial (p < 0.0001, Dunnett's test) yet moderately (if significantly) lower than in WT cells (p < 0.001; Fig. 7C). The reliance of swelling-induced, but not agonist-induced, TRPV4 signals on AQP4 at modest-to strong levels of hypotonic stress suggests that the water channel primarily regulates TRPV4 through the swelling response, whereas the disappearance of this effect with extreme hypotonic stimuli indicates that the increased driving force for swelling compensated for the loss of AQP4 or alternative swelling mechanisms contributed to TRPV4 activation independently of AQP4.
Figure 7.

AQP4 amplifies glial HTS-induced TRPV4 activation with moderate (140 mOsm) but not extreme (90 mOsm) HTS. A, B, Ca2+ signals in response to 140 mOsm HTS were blocked in Trpv4−/− (n = 50) and Aqp4−/− (n = 64) Müller cells compared with WT cells (n = 20). HC-06 inhibited HTS-induced [Ca2+]i elevations in WT cells (n = 14) but had no additional effect in Aqp4−/− cells (n = 7), indicating that TRPV4 activation by HTS requires Aqp4. C, At excessively large hypotonic gradients (90 mOsm), Aqp4−/− cells (blue bar) displayed robust HTS-induced [Ca2+]i responses approaching those of the WT cells. Trpv4−/− cells (red bar) showed no [Ca2+]i response when challenged with HTS.
AQP channels facilitate TRPV4 activation in the oocyte model system
To further test whether AQP4 gates TRPV4 function through steric interactions or indirectly via swelling-induced membrane strain, the two channels were expressed, either alone or in combination, in the Xenopus oocyte expression system (Fig. 8). The cell volume and membrane currents were determined simultaneously after 60 s stimulation with HTS (Δ125 mOsm) or GSK101. As expected, exposure to GSK101 induced inward currents in TRPV4-expressing oocytes (at Vm = 60 mV; 2.4 ± 0.6 μA, n = 7 for TRPV4-expressing oocytes and 3.6 ± 1.1 μA, n = 8 for TRPV4/AQP4-expressing oocytes; Fig. 8A-C). The GSK101-induced currents were of similar magnitude whether AQP4 was coexpressed or not (p = 0.9, Student's t test), whereas no current was observed in AQP4-expressing controls (0.08 ± 0.01 μA, n = 5; Fig. 8A–C). Hypotonic cell swelling was more prominent in AQP4 expressors (5.76 ± 0.37%, n = 5 for AQP4-expressing oocytes, 4.25 ± 0.24%, n = 9 for TRPV4/AQP4-expressing oocytes vs 0.33 ± 0.04%, n = 8 in TRPV4-expressing oocytes, p < 0.001, one-way ANOVA, with Dunnett's post hoc test; for representative volume traces, see Fig. 8D). The I-V relationship measured 60 s after imposition of HTS yielded markedly larger currents in TRPV4/AQP4-expressing oocytes than oocytes expressing only TRPV4 (at Vm = 60 mV; 6.3 ± 1.5 μA, n = 9 vs 1.7 ± 0.4 μA, n = 8, p < 0.05, Student's t test; Fig. 8D,E). The increased HTS-induced current in oocytes coexpressing TRPV4 and AQP4 could be attributable to a required presence of AQP4 for full TRPV4 activity or to the increased AQP4-dependent oocyte swelling. Thus, we determined HTS-induced TRPV4-mediated current in TRPV4-expressing oocytes and TRPV4/AQP4-expressing oocytes exposed to different osmotic gradients to obtain a similar degree of cell swelling (Δ125 mOsm for TRPV4-expressing oocytes, 0.33 ± 0.04% cell swelling after 60 s, n = 8 and Δ15 mOsm for TRPV4 + AQP4-expressing oocytes, 0.27 ± 0.03% cell swelling after 60 s, n = 9; p = 0.2, Student's t test). The I-V curves showed distinct overlap (p = 0.4, Student's t test; Fig. 8F). Thus, AQP4 channels mediate activation of TRPV4 via their ability to facilitate cell swelling.
Figure 8.

AQP4 channels amplify stretch-induced TRPV4 activation. A, Oocytes expressing TRPV4 and AQP4 alone or in combination were voltage clamped at Vm = −30 mV and exposed to 100 nm GSK101 (indicated by black bar). After 60 s GSK exposure, an I–V curve was obtained by jumping the clamp potential to potentials ranging from 60 to −140 mV at 20 mV intervals (seen as the vertical lines in the current trace). B, Representative I–V relations from oocytes expressing TRPV4 + AQP4, TRPV4, or AQP4 in control solution (left), in GSK101 solutions (right), or when exposed to Δ125 mOsm hypotonic challenge (middle). C, Summarized I-V relations obtained with GSK101 (as in B; n = 5–8). D, Representative current (black lines) and volume (gray lines) traces recorded simultaneously from oocytes expressing TRPV4 and AQP4 alone or in combination during exposure to a Δ125 mOsm hypotonic stimulus (indicated by the black bar; n = 5–9). E, Summarized I-V relations obtained during Δ125 mOsm. F, Oocytes expressing TRPV4 alone or in combination with AQP4 were exposed to hypotonic challenges of different magnitudes to observe similar degree of cell swelling (Δ125 mOsm for TRPV4-expressing oocytes and Δ15 mOsm for TRPV4/AQP4-expressing oocytes), and I–V relations were obtained after 60 s of cell swelling (n = 8–9).
Discussion
Our results provide new insight into the molecular mechanisms that mediate the perception and transduction of osmotic stress in retinal Müller cells. The prominent colocalization of TRPV4 and AQP4 in perivascular Müller end feet suggests that the two channels are likely to play important functions in the exchange of water and ions across the blood–retina barrier. Consistent with this, functional analysis revealed a profound regulatory and functional interdependence between osmotically induced water transport and calcium signaling. Although data from KO mice indicate that TRPV4 and AQP4 are trafficked independently and are capable of autonomous activation, they also show that AQP4 augments swelling-induced activation of TRPV4 and that TRPV4 channels modulate water influx during a range of hypotonic challenges by increasing the membrane permeability to Ca2+. Additional evidence for the significance of TRPV4–AQP4 interactions in Müller glia was obtained in Trpv4−/− retinas, which exhibited marked reduction in the transcription of genes that include Aqp4 and Kir4.1, and in the oocyte expression system in which AQP4 expression facilitated the swelling-evoked activation of TRPV4 channels.
Ca2+ elevations in response to osmotic swelling have been observed across all biotic kingdoms, from plants (Johansson et al., 2000) and hagfish (Herr et al., 2014) to mammalian cells that include primary, cultured, and intact astrocytes (Kimelberg et al., 1992; Hoffmann et al., 2009). In vivo imaging studies showed that HTS-induced Ca2+ signals in astrocytes require Aqp4 (Thrane et al., 2011), whereas we found that TRPV4 is a main mediator of HTS-induced Ca2+ entry in Müller cells (Ryskamp et al., 2014). Here, we combine the insights from those studies to show that hypotonic challenges affect Ca2+ homeostasis and modulate glial cell volume via symbiotic interactions between the two channels. Functional AQP4 appears to be required for the maximal HTS-induced activation of TRPV4-mediated currents and [Ca2+]i responses, whereas the vanilloid channel facilitates Müller cell volume increases and regulation in a Ca2+-dependent manner. Under our experimental conditions, Müller cells behaved in osmotic disequilibria as cellular osmometers, showing proportional swelling in hypotonic, and shrinking in hypertonic, solutions. These results are consistent with in vivo and in vitro data from the majority of cell types studied so far, including brain astrocytes (Pasantes-Morales et al., 2006; Risher et al., 2009), but differ from studies on rodent Müller glia that concluded that these cells resist hypotonic stress (Pannicke et al., 2004, Hirrlinger et al., 2008). The reasons for discrepant results are unclear but might involve different preparations, experimental means of detection of volume changes, and/or types of hypotonic stimuli (i.e., adding distilled water vis à vis mannitol supplementation; retinal slices vs dissociated cells). It is noteworthy that arachidonic acid, a proinflammatory ω6 polyunsaturated lipid, augments Müller cell sensitivity to hypotonic swelling (Reichenbach and Bringmann, 2010; Ryskamp et al., 2014), is required for activation of glial TRPV4 (Dunn et al., 2013), and also inhibits volume-regulated anion channels (VRACs) that mediate osmolyte release (Hoffmann et al., 2009). Therefore, as illustrated in Figure 9, loss of TRPV4-mediated signals in Aqp4−/− Müller glia might have been influenced by reduced stimulation of Ca2+-activated isoforms of PLA2 (Lambert et al., 2006).
Figure 9.

Proposed schema of TRPV4–AQP4 interactions in the Müller cell end foot. Hypotonic stress stimulates fluxes of water after the osmotic gradient. The resulting increase in cell volume stretches the plasma membrane, activating TRPV4 and a Ca2+- and stretch-sensitive PLA2. The product, arachidonic acid (AA), is a precursor for the cytochrome P450-mediated synthesis of eicosanoid metabolites (EETs) that serve as final activators of TRPV4 channels (Ryskamp et al., 2015) but may also suppress Kir4.1 (Bringmann et al., 1998). The expression of Kir4.1 (Reichenbach and Bringmann, 2010) and reactive gliosis (Ryskamp et al., 2014) are modulated by HTS-induced influx of Ca2+. It remains to be determined whether stretch activates PLA2 simultaneously with TRPV4 or whether its activation, which is Ca2+ dependent, amplifies the initial TRPV4 signal (blue arrows). Ca2+ induces both cell swelling (short term) and RVD (long term) and may be required for Ca2+-dependent gene expression (Aqp4, Kir4.1, Trpv4, Gfap). High levels of Ca2+ may also stimulate the activity of BK and VRAC channels and facilitate RVD.
TRPV4–AQP4 interactions are particularly important for responses to hypotonic challenges, whereas WT, Trpv4−/−, and Aqp4−/− cells showed similar decreases in cross-sectional area during exposure to hypertonic saline. Thus, the two channels together may contribute to regulation of glial swelling but not shrinkage. Removal of Ca2+ reproduced the effect of TRPV4 ablation by reducing the overall extent of HTS-induced swelling, suggesting that Ca2+ influx itself plays a key role in the initial volume increase. Decreased swelling in Trpv4−/− cells might, in part, arise as a result of reduced expression of Aqp4 and Kir4.1 genes. We propose that the water channel modulates TRPV4 indirectly by facilitating swelling-induced stretch of the plasma membrane (Fig. 9). TRPV4 current amplitudes elicited at the EC50 for the agonist were an order of magnitude higher compared with currents elicited with a 40% reduction in tonicity, whereas the capacity of Aqp4−/− cells to respond to moderate swelling with increases in [Ca2+]i was compromised even as the cells maintained responsiveness to GSK101. Notably, the facilitatory effect of AQP4 on the rate of HTS-induced glial swelling and the magnitude of cation influx was reproduced in TRPV4/AQP4-expressing oocytes. Of particular interest is the observation that coexpression with AQP4 confers on TRPV4-expressing cells the capability to respond to considerably smaller osmotic gradients.
Our data expand the context of calcium involvement in volume regulation by showing an intriguing dual effect of TRPV4 activation consisting of positive (increased swelling) and negative (increased RVD) feedback loops. In contrast to cultured astrocytes, which show strong (∼100%) RVD even in mild HTS (250 mOsm; Kimelberg et al., 1992; Benfenati et al., 2011), RVD in Müller glia and intact astrocytes is limited even in the presence of substantial hypotonic stress (Andrew and MacVicar, 1994; Risher et al., 2009; present study). Compared with WT cells, RVD was compromised in Trpv4−/− Müller glia during prolonged and substantial HTS. Thus, TRPV4 might contribute to adaptive volume regulation in retinal glia, as observed in airway epithelial, salivary gland, keratinocyte, and cortical glia and heterologously expressing CHO cells (Arniges et al., 2004; Becker et al., 2005; Liu et al., 2006; Benfenati et al., 2011). Under physiological conditions in which Trpv4−/− cells swelled to a level comparable with that of the WT, the extent of volume regulation was comparable between the two genotypes. Given that RVD was observed mainly at hypotonic challenges that were out of the physiological range, we suggest that it may have limited physiological significance for Müller cell osmoregulation in vivo. The molecular mechanism underlying the net export of ions during RVD is unclear but could include activation of Ca2+-dependent channels, release of organic osmolytes, such as glutamate, taurine, and myo-inositol (Fischer et al., 1997; Hoffmann et al., 2009; Fernández et al., 2013), stimulation of VRACs that are sensitized by swelling-evoked Ca2+ microdomains (Akita and Okada, 2011), and/or activation of water-translocating cotransporters (MacAulay and Zeuthen 2010).
Benfenati et al. (2011) proposed that TRPV4-dependent regulation of osmotic fluxes within the end foot requires an obligatory physical interaction with AQP4, whereas HTS-induced activation of TRPV4 in salivary gland epithelial cells appears to depend primarily on the interaction with AQP5 (Liu et al., 2006). Although we cannot exclude the possibility of direct association between the two proteins or their embedment within macromolecular entities involving Kir4.1, α-syntrophin and/or dystrophins (Puwarawuttipanit et al., 2006; Lanciotti et al., 2012), our data suggest that the communication between the volume sensor and water influx can be explained adequately by indirect interaction mediated through transcription and cell swelling (Fig. 9). Thus, the amplitude of GSK101-mediated Ca2+ entry in Aqp4−/− cells mirrored signals in WT cells, whereas the cognate water channel AQP1 could substitute for AQP4 by facilitating swelling-induced activation of TRPV4 (N.M. and D.K., unpublished observations). This conjecture is supported by the functional independence of Kir4.1 from AQP4 (Ruiz-Ederra et al., 2007; Zhang and Verkman, 2008), despite the ability of AQP4 to facilitate hypotonic-induced activation of Kir4.1 (Soe et al., 2009). The precise mechanism responsible for the modest reduction in GSK101-induced [Ca2+]i signals observed in Aqp4−/− cells remains to be determined but could involve reduced expression of Trpv4 (Fig. 2), release of ATP from cytosol or Ca2+ from internal stores (Lipp et al., 2009; Thrane et al., 2011).
Swelling of glial cells is a major cause of brain and retinal edema caused by liver disease, water intoxication, epilepsy, diabetes, stroke, and traumatic brain injury and can result in excitotoxicity, metabolic injury, and mechanical damage of blood vessels and neurons (Häussinger et al., 2000; Hirrlinger et al., 2008; Risher et al., 2009). Cysts, formed by swollen and dying Muller cells, are associated with macular edema that may drive ischemia and neuronal loss in diabetic retinopathy (Reichenbach and Bringmann, 2010). Edema formation, resulting from osmotic imbalances between retinal and extraretinal sites, is potentiated by fatty acids known to activate TRPV4 (Fig. 9), whereas deletion of AQP4 decreases the amplitude and frequency of Ca2+ signals during a hypotonic challenge and was associated with protection against the pathological effects of swelling, edema, and infarct (Manley et al., 2000; Da and Verkman, 2004; Thrane et al., 2011). Currently accepted interventions to treat brain swelling include cranial decompression and application of hyperosmolar hydrogels. We propose that cytotoxic Ca2+ signaling during glial swelling might instead be regulated by targeting TRPV4. The advantages of such interventions are the availability of specific antagonists (Vincent and Duncton, 2011) and the general absence of observable phenotypes in Trpv4−/− mice (Everaerts et al., 2010). In the retina, additional benefit might be derived from neuroprotection conferred by inhibition of TRPV4 channels in RGCs (Križaj et al., 2014).
In conclusion, the synergistic mechanisms described here could represent a general cellular mechanism linking swelling-dependent gene expression, Ca2+ homeostasis, and volume regulation in the brain. It is noteworthy that TRPV4/AQP4 channels are ideally positioned to sense and modulate ion/water fluxes across the blood–brain and blood–retina barrier, which is compromised during the formation of cytotoxic edema. TRPV4–AQP4 interactions might also contribute to light-evoked ion fluxes and/or neuronal hyperexcitation within the retina (Huang and Karwoski, 1992; Dmitriev et al., 1999). Finally, our results point toward alternative strategies for mitigating the debilitating consequences of acute and chronic swelling in the retina and the brain.
Footnotes
This work was supported by National Institutes of Health Grants T32DC008553 and F32NS093786 (D.A.R.), EY013574 (A.S.V.), and EY022076 and P30EY014800 (D.K.), Howard Hughes Medical Institute Med into Grad Initiative (D.A.R.), Department of Defense Grant W81XWH-12-1-0244, the Glaucoma Research Foundation, the State of Utah Technology Commercialization and Innovation Program, the University of Utah Neuroscience Initiative (D.K.), the University of Utah Undergraduate Research Opportunity Program (A.O.J.), Willard L. and Ruth P. Eccles Foundation, and unrestricted support from Research to Prevent Blindness (to Moran Eye Institute). We thank Dr. Wolfgang Liedtke (Duke University, Durham, NC) for Trpv4−/− mice and Alomone Labs for the anti-Kir4.1 antibody sample.
References
- Akita T, Okada Y. Regulation of bradykinin-induced activation of volume-sensitive outwardly rectifying anion channels by Ca2+ nanodomains in mouse astrocytes. J Physiol. 2011;589:3909–3927. doi: 10.1113/jphysiol.2011.208173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew RD, MacVicar BA. Imaging cell volume changes and neuronal excitation in the hippocampal slice. Neuroscience. 1994;62:371–383. doi: 10.1016/0306-4522(94)90372-7. [DOI] [PubMed] [Google Scholar]
- Arniges M, Vázquez E, Fernández-Fernández JM, Valverde MA. Swelling-activated Ca2+ entry via TRPV4 channel is defective in cystic fibrosis airway epithelia. J Biol Chem. 2004;279:54062–54068. doi: 10.1074/jbc.M409708200. [DOI] [PubMed] [Google Scholar]
- Becker D, Blase C, Bereiter-Hahn J, Jendrach M. TRPV4 exhibits a functional role in cell-volume regulation. J Cell Sci. 2005;118:2435–2440. doi: 10.1242/jcs.02372. [DOI] [PubMed] [Google Scholar]
- Benfenati V, Caprini M, Dovizio M, Mylonakou MN, Ferroni S, Ottersen OP, Amiry-Moghaddam M. An aquaporin-4/transient receptor potential vanilloid 4 (AQP4/TRPV4) complex is essential for cell-volume control in astrocytes. Proc Natl Acad Sci U S A. 2011;108:2563–2568. doi: 10.1073/pnas.1012867108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringmann A, Skatchkov SN, Biedermann B, Faude F, Reichenbach A. Alterations of potassium channel activity in retinal Müller glial cells induced by arachidonic acid. Neuroscience. 1998;86:1291–1306. doi: 10.1016/S0306-4522(98)00079-7. [DOI] [PubMed] [Google Scholar]
- Chiavaroli C, Bird G, Putney JW., Jr Delayed “all-or-none” activation of inositol 1, 4, 5-trisphosphate-dependent calcium signaling in single rat hepatocytes. J Biol Chem. 1994;269:25570–25575. [PubMed] [Google Scholar]
- Crowe WE, Altamirano J, Huerto L, Alvarez-Leefmans FJ. Volume changes in single N1E-115 neuroblastoma cells measured with a fluorescent probe. Neuroscience. 1995;69:283–296. doi: 10.1016/0306-4522(95)00219-9. [DOI] [PubMed] [Google Scholar]
- Da T, Verkman AS. Aquaporin-4 gene disruption in mice protects against impaired retinal function and cell death after ischemia. Invest Ophthalmol Vis Sci. 2004;45:4477–4483. doi: 10.1167/iovs.04-0940. [DOI] [PubMed] [Google Scholar]
- Dmitriev AV, Govardovskii VI, Schwahn HN, Steinberg RH. Light-induced changes of extracellular ions and volume in the isolated chick retina–pigment epithelium preparation. Vis Neurosci. 1999;16:1157–1167. doi: 10.1017/S095252389916615X. [DOI] [PubMed] [Google Scholar]
- Dunn KM, Hill-Eubanks DC, Liedtke WB, Nelson MT. TRPV4 channels stimulate Ca2+-induced Ca2+ release in astrocytic endfeet and amplify neurovascular coupling responses. Proc Natl Acad Sci U S A. 2013;110:6157–6162. doi: 10.1073/pnas.1216514110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everaerts W, Zhen X, Ghosh D, Vriens J, Gevaert T, Gilbert JP, Hayward NJ, McNamara CR, Xue F, Moran MM, Strassmaier T, Uykal E, Owsianik G, Vennekens R, De Ridder D, Nilius B, Fanger CM, Voets T. Inhibition of the cation channel TRPV4 improves bladder function in mice and rats with cyclophosphamide-induced cystitis. Proc Natl Acad Sci U S A. 2010;107:19084–19089. doi: 10.1073/pnas.1005333107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton RA, Moeller HB, Zelenina M, Snaebjornsson MT, Holen T, MacAulay N. Differential water permeability and regulation of three aquaporin 4 isoforms. Cell Mol Life Sci. 2010;67:829–840. doi: 10.1007/s00018-009-0218-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández JM, Di Giusto G, Kalstein M, Melamud L, Rivarola V, Ford P, Capurro C. Cell volume regulation in cultured human retinal Müller cells is associated with changes in transmembrane potential. PLoS One. 2013;8:e57268. doi: 10.1371/journal.pone.0057268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer R, Schliess F, Häussinger D. Characterization of the hypo-osmolarity-induced Ca2+ response in cultured rat astrocytes. Glia. 1997;20:51–58. doi: 10.1002/(SICI)1098-1136(199705)20:1<51::AID-GLIA5>3.0.CO%3B2-8. [DOI] [PubMed] [Google Scholar]
- Florence CM, Baillie LD, Mulligan SJ. Dynamic volume changes in astrocytes are an intrinsic phenomenon mediated by bicarbonate ion flux. PLoS One. 2012;7:e51124. doi: 10.1371/journal.pone.0051124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galizia L, Pizzoni A, Fernandez J, Rivarola V, Capurro C, Ford P. Functional interaction between AQP2 and TRPV4 in renal cells. J Cell Biochem. 2012;113:580–589. doi: 10.1002/jcb.23382. [DOI] [PubMed] [Google Scholar]
- Goodyear MJ, Crewther SG, Junghans BM. A role for aquaporin-4 in fluid regulation in the inner retina. Vis Neurosci. 2009;26:159–165. doi: 10.1017/S0952523809090038. [DOI] [PubMed] [Google Scholar]
- Haj-Yasein NN, Bugge CE, Jensen V, Østby I, Ottersen OP, Hvalby Ø, Nagelhus EA. Deletion of aquaporin-4 increases extracellular K+ concentration during synaptic stimulation in mouse hippocampus. Brain Struct Funct. 2015;220:2469–2474. doi: 10.1007/s00429-014-0767-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häussinger D, Kircheis G, Fischer R, Schliess F, vom Dahl S. Hepatic encephalopathy in chronic liver disease: a clinical manifestation of astrocyte swelling and low-grade cerebral edema? J Hepatol. 2000;32:1035–1038. doi: 10.1016/S0168-8278(00)80110-5. [DOI] [PubMed] [Google Scholar]
- Herr JE, Clifford AM, Goss GG, Fudge DS. Defensive slime formation in Pacific hagfish requires Ca2+- and aquaporin-mediated swelling of released mucin vesicles. J Exp Biol. 2014;217:2288–2296. doi: 10.1242/jeb.101584. [DOI] [PubMed] [Google Scholar]
- Hirrlinger PG, Wurm A, Hirrlinger J, Bringmann A, Reichenbach A. Osmotic swelling characteristics of glial cells in the murine hippocampus, cerebellum, and retina in situ. J Neurochem. 2008;105:1405–1417. doi: 10.1111/j.1471-4159.2008.05243.x. [DOI] [PubMed] [Google Scholar]
- Hoffmann EK, Lambert IH, Pedersen SF. Physiology of cell volume regulation in vertebrates. Physiol Rev. 2009;89:193–277. doi: 10.1152/physrev.00037.2007. [DOI] [PubMed] [Google Scholar]
- Huang B, Karwoski CJ. Light-evoked expansion of subretinal space volume in the retina of the frog. J Neurosci. 1992;12:4243–4252. doi: 10.1523/JNEUROSCI.12-11-04243.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Xing W, Ryskamp DA, Punzo C, Križaj D. Localization and phenotype-specific expression of ryanodine calcium release channels in C57BL6 and DBA/2J mouse strains. Exp Eye Res. 2011;93:700–709. doi: 10.1016/j.exer.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson I, Karlsson M, Johanson U, Larsson C, Kjellbom P. The role of aquaporins in cellular and whole plant water balance. Biochim Biophys Acta. 2000;1465:324–342. doi: 10.1016/S0005-2736(00)00147-4. [DOI] [PubMed] [Google Scholar]
- Kanemaru K, Kubota J, Sekiya H, Hirose K, Okubo Y, Iino M. Calcium-dependent N-cadherin up-regulation mediates reactive astrogliosis and neuroprotection after brain injury. Proc Natl Acad Sci U S A. 2013;110:11612–11617. doi: 10.1073/pnas.1300378110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelberg HK, Sankar P, O'Connor ER, Jalonen T, Goderie SK. Functional consequences of astrocytic swelling. Prog Brain Res. 1992;94:57–68. doi: 10.1016/S0079-6123(08)61739-2. [DOI] [PubMed] [Google Scholar]
- Križaj D, Copenhagen DR. Compartmentalization of calcium extrusion mechanisms in the outer and inner segments of photoreceptors. Neuron. 1998;21:249–256. doi: 10.1016/S0896-6273(00)80531-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Križaj D, Ryskamp DA, Jo AO, Verkman A, MacAulay N. Molecular coupling between TRPV4 and aquaporin 4 mediates osmosensation in Müller glial cells. Invest Ophthalmol Vis Sci. 2013;54:2673–2673. [Google Scholar]
- Križaj D, Ryskamp DA, Tian N, Tezel G, Mitchell CH, Slepak VZ, Shestopalov VI. From mechanosensitivity to inflammatory responses: new players in the pathology of glaucoma. Curr Eye Res. 2014;39:105–119. doi: 10.3109/02713683.2013.836541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert IH, Pedersen SF, Poulsen KA. Activation of PLA2 isoforms by cell swelling and ischaemia/hypoxia. Acta Physiologica. 2006;187:75–85. doi: 10.1111/j.1748-1716.2006.01557.x. [DOI] [PubMed] [Google Scholar]
- Lanciotti A, Brignone MS, Molinari P, Visentin S, De Nuccio C, Macchia G, Aiello C, Bertini E, Aloisi F, Petrucci TC, Ambrosini E. Megalencephalic leukoencephalopathy with subcortical cysts protein 1 functionally cooperates with the TRPV4 cation channel to activate the response of astrocytes to osmotic stress: dysregulation by pathological mutations. Hum Mol Genet. 2012;21:2166–2180. doi: 10.1093/hmg/dds032. [DOI] [PubMed] [Google Scholar]
- Li J, Patil RV, Verkman AS. Mildly abnormal retinal function in transgenic mice without Müller cell aquaporin-4 water channels. Invest Ophthalmol Vis Sci. 2002;43:573–579. [PubMed] [Google Scholar]
- Liedtke W, Friedman JM. Abnormal osmotic regulation in trpv4−/− mice. Proc Natl Acad Sci U S A. 2003;100:13698–13703. doi: 10.1073/pnas.1735416100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipp S, Wurm A, Pannicke T, Wiedemann P, Reichenbach A, Chen J, Bringmann A. Calcium responses mediated by type 2 IP 3-receptors are required for osmotic volume regulation of retinal glial cells in mice. Neurosci Lett. 2009;457:85–88. doi: 10.1016/j.neulet.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Bandyopadhyay B, Nakamoto T, Singh B, Liedtke W, Melvin JE, Ambudkar I. A role for AQP5 in activation of TRPV4 by hypotonicity: concerted involvement of AQP5 and TRPV4 in regulation of cell volume. J Biol Chem. 2006;281:15485–15495. doi: 10.1074/jbc.M600549200. [DOI] [PubMed] [Google Scholar]
- Loukin S, Zhou X, Su Z, Saimi Y, Kung C. Wild-type and brachyolmia-causing mutant TRPV4 channels respond directly to stretch force. J Biol Chem. 2010;285:27176–27181. doi: 10.1074/jbc.M110.143370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Yang B, Gillespie A, Carlson EJ, Epstein CJ, Verkman AS. Generation and phenotype of a transgenic knockout mouse lacking the mercurial-insensitive water channel aquaporin-4. J Clin Invest. 1997;100:957–962. doi: 10.1172/JCI231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAulay N, Zeuthen T. Water transport between CNS compartments: contributions of aquaporins and cotransporters. Neuroscience. 2010;168:941–956. doi: 10.1016/j.neuroscience.2009.09.016. [DOI] [PubMed] [Google Scholar]
- Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159–163. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- Minieri L, Pivonkova H, Harantova L, Anderova M, Ferroni S. Intracellular Na+ inhibits volume-regulated anion channel in rat cortical astrocytes. J Neurochem. 2015;132:286–300. doi: 10.1111/jnc.12962. [DOI] [PubMed] [Google Scholar]
- Molnar T, Barabas P, Birnbaumer L, Punzo C, Kefalov V, Križaj D. Store-operated channels regulate intracellular calcium in mammalian rods. J Physiol. 2012;590:3465–3481. doi: 10.1113/jphysiol.2012.234641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagelhus EA, Ottersen OP. Physiological roles of aquaporin-4 in brain. Physiol Rev. 2013;93:1543–1562. doi: 10.1152/physrev.00011.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagelhus EA, Veruki ML, Torp R, Haug FM, Laake JH, Nielsen S, Agre P, Ottersen OP. Aquaporin-4 water channel protein in the rat retina and optic nerve: polarized expression in Müller cells and fibrous astrocytes. J Neurosci. 1998;18:2506–2519. doi: 10.1523/JNEUROSCI.18-07-02506.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Membrane physiology of retinal glial (Muller) cells. J Neurosci. 1985;5:2225–2239. doi: 10.1523/JNEUROSCI.05-08-02225.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannicke T, Iandiev I, Uckermann O, Biedermann B, Kutzera F, Wiedemann P, Wolburg H, Reichenbach A, Bringmann A. A potassium channel-linked mechanism of glial cell swelling in the postischemic retina. Mol Cell Neurosci. 2004;26:493–502. doi: 10.1016/j.mcn.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Pannicke T, Wurm A, Iandiev I, Hollborn M, Linnertz R, Binder DK, Kohen L, Wiedemann P, Steinhäuser C, Reichenbach A, Bringmann A. Deletion of aquaporin-4 renders retinal glial cells more susceptible to osmotic stress. J Neurosci Res. 2010;88:2877–2888. doi: 10.1002/jnr.22437. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Verkman AS. Aquaporin water channels in the nervous system. Nat Rev Neurosci. 2013;14:265–277. doi: 10.1038/nrn3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasantes-Morales H, Lezama RA, Ramos-Mandujano G, Tuz KL. Mechanisms of cell volume regulation in hypo-osmolality. Am J Med. 2006;119:S4–S11. doi: 10.1016/j.amjmed.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Puwarawuttipanit W, Bragg AD, Frydenlund DS, Mylonakou MN, Nagelhus EA, Peters MF, Kotchabhakdi N, Adams ME, Froehner SC, Haug FM, Ottersen OP, Amiry-Moghaddam M. Differential effect of α-syntrophin knockout on aquaporin-4 and Kir4.1 expression in retinal macroglial cells in mice. Neuroscience. 2006;137:165–175. doi: 10.1016/j.neuroscience.2005.08.051. [DOI] [PubMed] [Google Scholar]
- Reichenbach A, Bringmann A. Müller cells in the healthy and diseased retina. New York: Springer Science and Business Media; 2010. [DOI] [PubMed] [Google Scholar]
- Risher WC, Andrew RD, Kirov SA. Real-time passive volume responses of astrocytes to acute osmotic and ischemic stress in cortical slices and in vivo revealed by two-photon microscopy. Glia. 2009;57:207–221. doi: 10.1002/glia.20747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Ederra J, Zhang H, Verkman AS. Evidence against functional interaction between aquaporin-4 water channels and Kir4. 1 potassium channels in retinal Müller cells. J Biol Chem. 2007;282:21866–21872. doi: 10.1074/jbc.M703236200. [DOI] [PubMed] [Google Scholar]
- Ryskamp DA, Witkovsky P, Barabas P, Huang W, Koehler C, Akimov NP, Lee SH, Chauhan S, Xing W, Rentería RC, Liedtke W, Križaj D. The polymodal ion channel transient receptor potential vanilloid 4 modulates calcium flux, spiking rate, and apoptosis of mouse retinal ganglion cells. J Neurosci. 2011;31:7089–7101. doi: 10.1523/JNEUROSCI.0359-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryskamp DA, Jo AO, Frye AM, Vazquez-Chona F, MacAulay N, Thoreson WB, Križaj D. Swelling and eicosanoid metabolites differentially gate TRPV4 channels in retinal neurons and glia. J Neurosci. 2014;34:15689–15700. doi: 10.1523/JNEUROSCI.2540-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryskamp DA, Iuso A, Križaj D. TRPV4 links inflammatory signaling and neuroglial swelling. Channels. 2015;9:70–72. doi: 10.1080/19336950.2015.1017998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliess F, Sinning R, Fischer R, Schmalenbach C, Häussinger D. Calcium-dependent activation of Erk-1 and Erk-2 after hypo-osmotic astrocyte swelling. Biochem J. 1996;320:167–171. doi: 10.1042/bj3200167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki K, Ikenaka K, Tamalu F, Tominaga M, Ishizaki Y. A novel subtype of astrocytes expressing TRPV4 (transient receptor potential vanilloid 4) regulates neuronal excitability via release of gliotransmitters. J Biol Chem. 2014;289:14470–14480. doi: 10.1074/jbc.M114.557132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soe R, Macaulay N, Klaerke DA. Modulation of Kir4.1 and Kir4.1-Kir5.1 channels by small changes in cell volume. Neurosci Lett. 2009;457:80–84. doi: 10.1016/j.neulet.2009.04.010. [DOI] [PubMed] [Google Scholar]
- Szikra T, Barabas P, Bartoletti TM, Huang W, Akopian A, Thoreson WB, Križaj D. Calcium homeostasis and cone signaling are regulated by interactions between calcium stores and plasma membrane ion channels. PLoS One. 2009;4:e6723. doi: 10.1371/journal.pone.0006723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrane AS, Rappold PM, Fujita T, Torres A, Bekar LK, Takano T, Peng W, Wang F, Rangroo Thrane V, Enger R, Skare Ø, Holen T, Klungland A, Ottersen OP, Nedergaard M, Nagelhus EA. Critical role of aquaporin-4 (AQP4) in astrocytic Ca2+ signaling events elicited by cerebral edema. Proc Natl Acad Sci U S A. 2011;108:846–851. doi: 10.1073/pnas.1015217108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent F, Duncton MA. TRPV4 agonists and antagonists. Curr Top Med Chem. 2011;11:2216–2226. doi: 10.2174/156802611796904861. [DOI] [PubMed] [Google Scholar]
- Witkovsky P, Gábriel R, Križaj D. Anatomical and neurochemical characterization of dopaminergic interplexiform processes in mouse and rat retinas. J Comp Neurol. 2008;510:158–174. doi: 10.1002/cne.21784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeuthen T, Belhage B, Zeuthen E. Water transport by Na+-coupled cotransporters of glucose (SGLT1) and of iodide (NIS). The dependence of substrate size studied at high resolution. J Physiol. 2006;570:485–499. doi: 10.1113/jphysiol.2005.100933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Verkman AS. Aquaporin-4 independent Kir4. 1 K+ channel function in brain glial cells. Mol Cell Neurosci. 2008;37:1–10. doi: 10.1016/j.mcn.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]