

Abstract
The global regulatory veA gene governs development and secondary metabolism in numerous fungal species, including Aspergillus flavus. This is especially relevant since A. flavus infects crops of agricultural importance worldwide, contaminating them with potent mycotoxins. The most well-known are aflatoxins, which are cytotoxic and carcinogenic polyketide compounds. The production of aflatoxins and the expression of genes implicated in the production of these mycotoxins are veA dependent. The genes responsible for the synthesis of aflatoxins are clustered, a signature common for genes involved in fungal secondary metabolism. Studies of the A. flavus genome revealed many gene clusters possibly connected to the synthesis of secondary metabolites. Many of these metabolites are still unknown, or the association between a known metabolite and a particular gene cluster has not yet been established. In the present transcriptome study, we show that veA is necessary for the expression of a large number of genes. Twenty-eight out of the predicted 56 secondary metabolite gene clusters include at least one gene that is differentially expressed depending on presence or absence of veA. One of the clusters under the influence of veA is cluster 39. The absence of veA results in a downregulation of the five genes found within this cluster. Interestingly, our results indicate that the cluster is expressed mainly in sclerotia. Chemical analysis of sclerotial extracts revealed that cluster 39 is responsible for the production of aflavarin.
INTRODUCTION
Aspergillus flavus is a saprophytic filamentous fungus that is also able to colonize economically important crops such as peanuts, cotton, maize, and other oil seed crops during preharvest or storage. Its most efficient mode of dissemination is the production of airborne conidia. In addition, A. flavus produces resistant structures called sclerotia, which allow this fungus to survive adverse environmental conditions for long periods of time (1–3). This opportunistic pathogen produces a wide range of secondary metabolites, including aflatoxins (AFs). Among them, AFB1 is the most mutagenic and carcinogenic natural compound known (4–8). Ingestion of food products contaminated with AFs has been associated with hepatotoxicity, teratogenicity, immunosuppression, and liver cancer (6, 9). AF contamination also results in a negative impact on the economy in developed countries. In the United States alone A. flavus causes more than a billion dollar in losses per year due to contaminated crops (10). In addition to AFs, A. flavus is known to produce other mycotoxins, including cyclopiazonic acid (CPA), a suppressor of the calcium-dependent ATPase in the sarcoplasmic reticulum, and aflatrem, a tremogenic mycotoxin causative of neurological disorders (11, 12).
Studies of the A. flavus genome have revealed many gene clusters possibly connected to the synthesis of other secondary metabolites. Specifically, 55 different clusters were predicted based on the presence of genes encoding polyketide synthases (PKSs), nonribosomal peptide synthetases (NRPSs), hybrid PKS-NRPSs, and prenyltransferases (PTRs) within the clusters (13, 14). An additional gene cluster, responsible for the synthesis of kojic acid, has also been recently identified (15). In spite of these new findings, only a few metabolites have been associated with these clusters (16), among them the clusters associated with the synthesis of kojic acid, as well as the synthesis of AFs, CPA, aflatrem, and asparasone. Expression of these last four gene clusters was shown to be dependent on the global regulatory gene veA, which is also involved in developmental regulation in A. flavus (17–20) and in other fungi (21–26). Previous studies, conducted mainly with the model fungus Aspergillus nidulans, revealed that the VeA protein forms a complex with LaeA, a chromatin-modifying protein, and the regulator VelB, another protein of the velvet family. It also interacts with light-sensing proteins such as the red phytochrome FphA, which interacts with the blue-light-sensing proteins LreA-LreB (19, 27, 28). It seemed likely that additional secondary metabolite gene clusters will also be veA dependent or influenced by light in this agriculturally important fungus. In our study, we used whole-genome microarray transcript profiling to examine whether the expression of a number of genes, particularly those associated with predicted secondary metabolite gene clusters, was influenced by veA or by light in A. flavus. We found that among the genes regulated by veA are all the genes in cluster 39 (as designated by Georgianna et al. [13]). We show that the genes in cluster 39 are expressed mainly in sclerotia, resulting in the formation of a compound know to have anti-insectan activity.
MATERIALS AND METHODS
Strains used in this study and culture conditions.
The strains used in this study are listed in Table 1. A. flavus CA14 (ku70 niaD pyrG ptrA) served as the host for transformation experiments. A. flavus CA14 pyrG-1 served as a control for cluster 39 afvA to -E deletion strains, while CA14 pPTRI was used as the control for the overexpression afvB (OEafvB) (Table 1). Cultures were point inoculated onto 2× V8 agar (50 ml V8 juice per liter, pH 5.2) supplemented when required with 3 mg/ml (NH4)2SO4 and/or 1 mg/ml uracil and were incubated at 30°C in the light. Conidia were collected from plates by addition of 10 ml 0.01% Triton X-100 and gentle scraping of the colony surface with a cell scraper (BD Biosciences, San Jose, CA). Conidia were stored at 4°C. The strains were cultured on YGT medium, GMM–2% sorbitol medium (GMM-S), or Wickerham medium as indicated in each case. Cultures were incubated at 30°C and stored at −80°C as glycerol stocks.
TABLE 1.
Fungal strains used in this study
| Strain | Pertinent genotype | Reference |
|---|---|---|
| A. flavus 70s | Wild type | 17 |
| A. flavus ΔveA | ΔveA | 17 |
| A. flavus CA14 | pyrG ptrA niaD ku70 | This study |
| A. flavus CA14 pyrG-1 | ptrA niaD ku70 | This study |
| A. flavus ΔafvA | ΔafvA::pyrG ptrA niaD ku70 | This study |
| A. flavus ΔafvB | ΔafvB::pyrG ptrA niaD ku70 | This study |
| A. flavus ΔafvC | ΔafvC::pyrG ptrA niaD ku70 | This study |
| A. flavus ΔafvD | ΔafvD::pyrG ptrA niaD ku70 | This study |
| A. flavus ΔafvE | ΔafvE::pyrG ptrA niaD ku70 | This study |
| A. flavus ΔafvB-com | ΔafvB::pyrG gpdA(p)::afvB::ptrA+ niaD ku70 | This study |
| A. flavus CA14 pPTRI | ptrA+ pyrG niaD ku70 | This study |
| A. flavus OEafvB | gpdA(p)::afvB::ptrA+ pyrG niaD ku70 | This study |
Transcriptome analysis.
An Aspergillus flavus whole-genome 70-mer oligonucleotide microarray was fabricated at the J. Craig Venter Institute. The array chips included a total of 27,648 spots, containing 11,820 protein-coding genes. Among them, 11,139 (94%) match 10,477 gene models (i.e., with AFL2G gene numbers) of the Broad Institute's Aspergillus flavus genome release based on a BLAST search, corresponding to 83% of the 12,604 A. flavus genes annotated by the Broad Institute.
Conidia (106/ml) from A. flavus 70S and ΔveA strains were inoculated into 100 ml liquid YGT and incubated at 30°C with agitation for 14 h. Mycelia were vacuum filtered onto nylon membranes that were transferred onto the surfaces of YGT plates, and cultures were incubated at 30°C in the light or dark. Mycelia were collected from the surface of the nylon membrane following 6, 24, 48, and 72 h of incubation, immediately frozen in liquid nitrogen, and stored at −80°C. The experiment was performed with three replicates. RNA was isolated from about 100 mg of ground tissue using the Aurum total RNA minikit (Bio-Rad, Hercules, CA) and DNase I treatment. RNA quality and quantity were determined using the Experion automated electrophoresis station (Bio-Rad). RNA aliquots were preserved at −80°C. cDNA was synthesized from RNA using the iScript cDNA Synthesis kit (Bio-Rad). The microarray experimental design is shown in Table 2.
TABLE 2.
Microarray experimental design
| Expt | Pair at time pointa: |
|||
|---|---|---|---|---|
| 6 h | 24 h | 48 h | 72 h | |
| KO/WT, light | koL vs wtL | koL vs wtL | koL vs wtL | koL vs wtL |
| KO/WT, dark | koD vs wtD | koD vs wtD | koD vs wtD | koD vs wtD |
| WT, dark/light | wtD vs wtL | wtD vs wtL | wtD vs wtL | wtD vs wtL |
wtL, wild type in light; wtD, wild type in dark; koL, ΔveA in light; koD, ΔveA in dark.
To avoid labeling bias of the Cy5 and Cy3 dyes, each treatment was labeled with each dye (dye flip). Hybridized slides were scanned using either a ScanArray5000XL (GSI Lumonics, Packard Biochip, Packard BioScience, Billerica, MA, USA) or an Axon GenePix 4000B (Axon Instruments, Molecular Devices, Sunnyvale, CA, USA). The independent TIFF images generated from each channel were analyzed using the J. Craig Venter Institute Spotfinder software to acquire image intensity for relative transcript levels (29). To remove the nonspecific background signals or the artifacts introduced by the Cy3 or Cy5 dye bias, the raw data were normalized using a local regression technique, LOWESS (locally weighted scatterplot smoothing), implemented in the MIDAS software tool (29). The resulting data were averaged over two duplicate genes on each array and over two duplicate arrays (dye flip) for each experiment. Some genes are represented by two separate nonoverlapping fragments; therefore, because a gene may be detected by each fragment, they are defined as features so that the gene count will not be overreported.
Log odds ratios using base 2 were calculated for each gene to measure the fold changes in expression between each pair of experiments (see Tables S1 to S3 in the supplemental material), e.g., wild type (WT) dark 6 h versus ΔveA dark 6 h. This generated three data matrix files corresponding to the three experiments in Table 2, each containing log2 values for genes of four time points. Using a log2 of ratio >1 or <−1 as the cutoff, meaning at least 2-fold upregulation or downregulation, respectively, we selected differentially expressed genes. The accession numbers were then submitted to the DAVID (Database for Annotation, Visualization and Integrated Discovery) v6.7 web server (30), utilizing annotations from various databases, including Gene Ontology, InterPro, COG, KEGG, and UniProt. In addition, searches against the A. nidulans genome in order to find the best hit were also useful to predict putative functions for the differentially expressed genes.
Characterization of A. flavus gene cluster 39. (i) Phylogenetic analysis.
Based on the study by Georgianna et al. (13), the cluster includes five genes, encoding a putative NADH oxidase (AFLA_108540, afvA), a PKS (AFLA_108550, afvB), an O-methyltransferase (AFLA_108560, afvC), a methyltransferase (AFLA_108570, afvD), and a cytochrome P450 monooxygenase (AFLA_108580, afvE). The possible conservation of this gene cluster in other species of the genus Aspergillus was examined. Cluster 39 genes were used as a query against other genome databases of phylogenetically related species using BLAST (blastp) analysis. Sequence information was obtained from the Aspergillus comparative database (http://www.broadinstitute.org/annotation/genome/aspergillus_group) and from the National Center for Biotechnology Information [NCBI] (http://blast.ncbi.nlm.nih.gov). Gene entries with the highest bit score and the lowest E value for each one of the species, including A. oryzae, A. nidulans, A. niger, A. terreus, A. fumigatus, A. fischerium, and A. clavatus, were selected, and their locations on the chromosome were mapped.
(ii) Domain architecture analysis of AfvB.
The PKS gene constitutes the backbone gene in cluster 39. Fungal PKSs consist of a minimal domain set of ketosynthase (KS), acyl transferase (AT), and acyl carrier protein (ACP). Additionally, other domains, such as ketoreductase (KR), enoyl reductase (ER) (70), and methyltransferase (MT) (71), may also be present in the fungal PKSs. The information about the domain structure of the predicted PKS encoded by afvB was obtained from the Pfam database (http://pfam.sanger.ac.uk/). Conservation of the domains in AfvB was compared to those homologs in other species of the genus Aspergillus. Multiple comparisons of the deduced amino acid sequences of PKS genes were performed using CLUSTALW-Omega (http://www.ebi.ac.uk), provided by EMBL-EBI.
(iii) Gene expression analysis.
The transcriptome results indicating the veA dependence of cluster 39 were validated by quantitative reverse transcription-PCR (qRT-PCR). RNA was obtained from AF70 and ΔveA strains as described above following culture under conditions identical to those used for the transcriptome analysis. Quantitative RT-PCR was performed using SYBR green I chemistry and the iCycler iQ5 Multicolor real-time PCR detection system (Bio-Rad) as described previously (31). The sequences of the oligonucleotide primers used for qRT-PCR amplification are listed in Table S4 in the supplemental material. Gene expression levels at each time point were normalized (by ΔΔCT analysis [32]) to A. flavus 18S rRNA gene expression levels utilizing the gene expression analysis software package for the Bio-Rad iQ5.
Additionally, expression of afvB was determined from isolated mycelium, conidia, and sclerotia of the AF70 pyrG-1 control. AF70 pyrG-1 was grown on GMM–2% sorbitol agar for 5 days, and sclerotia and conidia were separated from mycelium by gentle scraping of the colony surface followed by repeated washing with sterile water to remove all traces of sclerotia and conidia. Sclerotia were collected by adding 10 ml sterile, deionized water to the agar surface and gently scraping colonies with a cell scraper. Sclerotia were washed 5 times by addition of 40 ml water to remove residual conidia and mycelia, resuspended in a final volume of 25 ml water, and stored at 4°C. RNA was collected from ground fungal tissues as described above. qRT-PCR analysis was carried out as described above using primers afvB-F and afvB-R (see Table S4 in the supplemental material).
(iv) Generation of the deletion, complementation, and overexpression afvB strains.
A ΔafvB::pyrG knockout (KO) plasmid in which a 1.1-kb region within the beta-ketoacyl synthase N-terminal domain of the afvB coding region is replaced by the A. parasiticus pyrG selectable marker gene was generated essentially as described by Cary et al. (20). PCR primers used in construction of pAfvB-pyrG are listed in Table S4 in the supplemental material. Transformation was performed as previously described (33) using A. flavus CA14 as the host. Conidia were inoculated in potato dextrose broth supplemented with 1 mg/ml uracil (PDB-U), and transformants were regenerated on Czapek solution agar (Difco, BD, Sparks, MD) supplemented with 10 mM ammonium sulfate (CZ-AS). A plasmid vector for genetic complementation and overexpression of afvB was generated by placing the approximately 5.44-kb afvB coding region under the control of the A. nidulans glyceraldehyde-3-phosphate (gpdA) promoter and trpC transcriptional terminator in plasmid pPTRI (TaKaRa Bio, Inc.) harboring the A. oryzae pyrithiamine selectable marker gene. Plasmid pPTRI-afvB-trpC was constructed by amplifying the approximately 5.44-kb afvB coding region of CA14 genomic DNA. PCR amplification was performed with PrimeSTAR Max DNA polymerase according to the manufacturer's instructions (TaKaRa Bio, Inc.) using primers 5′ afvB NotI and 3′ afvB RsrII (see Table S4 in the supplemental material). The PCR product was ligated to NotI-RsrII-digested pPTRI-gpd::trpC plasmid using the In-Fusion HD cloning kit (Clontech, Mountain View, CA). Plasmid pPTRI-afvB-trpC was used to transform CA14 or CA14 ΔafvB mutant 4 protoplasts, and transformants were regenerated on CZ-AS supplemented with 1 mg/ml uracil and 0.1 μg/ml pyrithiamine.
Additional knockout mutants of putative cluster 39 biosynthetic genes were generated using the fusion PCR technique as described in reference 34. Cluster 39 gene fragments were amplified using Platinum Pfx DNA polymerase (Invitrogen) and A. flavus CA14 genomic DNA and pPG2.8, containing the A. parasiticus pyrG selectable marker, as templates. Primers used in these PCRs are shown in Table S4 in the supplemental material.
(v) Assessment of sclerotial production.
The A. flavus CA14 pyrG-1 control, ΔafvB, and ΔafvB-com strains, along with OEafvB and its isogenic control (CA14 pPTRI), were point inoculated on GMM-S. The cultures were incubated for 8 days at 30°C in the dark. Sixteen-millimeter cores were taken 1 cm from the center of each culture and sprayed with 70% ethanol to improve the visualization of sclerotia. These structures were quantified under a Leica MZ75 stereomicroscope.
(vi) Identification of the compound associated with A. flavus gene cluster 39. (a) Sample preparation.
The A. flavus CA14 pyrG-1 control, ΔafvB, and ΔafvB-com strains, along with the OEafvB and its isogenic control (CA14 pPTRI), were point inoculated on Wickerham medium. The cultures were incubated for 8 days at 30°C in the dark. Sclerotia were collected from these cultures and cleaned by rolling them on 3% agar plates. Sclerotia were lyophilized, placed in a mortar, and ground with a pestle in 5 ml ethyl acetate-acetone (1:1)–formic acid (0.1%), with 0.5 g of sterile fine sand added to assist with breakage of sclerotia. The ground sclerotium-solvent mixture was kept at room temperature for 15 min and then briefly ground again and kept at room temperature for another 15 min. The solvent was collected in a 100-ml glass beaker and allowed to dry overnight at room temperature. The dried samples were dissolved in 0.5 ml of methanol-acetonitrile-water (30:30:40, vol/vol/vol) and passed through a 0.22-mm filter before analysis.
(b) LC-Orbitrap MS analysis.
Liquid chromatography-mass spectrometry (LC-MS) analyses were performed on an Orbitrap Exactive mass analyzer (Thermo Fisher Scientific) coupled to an Accela ultra-high-performance liquid chromatography (UHPLC) system (Thermo Fisher Scientific). Chromatographic separation was achieved using a Zorbax RRHD Eclipse Plus reverse-phase C18 column (1.8 μm, 100 mm by 2.1 mm [inner diameter]) (Agilent) maintained at 30°C. A gradient elution of solvent A (H2O-methanol; 95:5, vol/vol) and solvent B (H2O-methanol; 5:95, vol/vol), both containing 0.1% formic acid and 10 mM ammonium formate, was applied as follows: 0% solvent B from 0 to 0.5 min, 0 to 99% solvent B from 0.5 to 20 min, 99% solvent B from 20 to 21 min, 99 to 0% solvent B from 21 to 24 min, and reequilibration with 0% solvent B from 24 to 28 min. Five microliters of sample was injected into the column. For a comprehensive metabolite profiling, both the electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI) ion sources were investigated in positive as well as in negative mode. ESI positive-ion mass spectrometry (ESI+/MS) allowed differential detection of peaks in the control compared to the mutant. The ESI+/MS parameters were as follows: spray voltage, 4.5 kV; capillary temperature, 250°C; heater temperature, 250°C; sheath gas flow rate, 45 arbitrary units (a.u.); and auxiliary gas flow rate, 10 a.u. The data were processed using the Xcalibur 2.1 and Exactive Tune software (Thermo Fisher Scientific). The instrument was operated in full-scan mode with a resolution of 100,000 full width at half maximum (FWHM). The maximum injection time was 250 ms, and the number of microscans per scan was 1.
(c) LC-ion trap MS analysis.
The HPLC-ion trap MS system (Thermo Fisher Scientific) was used for the fragmental analysis of the targeted analytes. The column used was a Symmetry C18 column (5 μm, 2.1 by 150 mm) supplied by Waters (Milford, MA, USA). The mobile phase consisted of (A) water-methanol (95:5, vol/vol) containing 0.1% formic acid and 10 mM ammonium formate and (B) water-methanol (5:95, vol/vol) containing 0.1% formic acid and 10 mM ammonium formate. The linear gradient elution program for LC-ion trap analysis was as follows: 0 to 10 min with B = 0% to 100%, 10 to 11 min with B = 100%, 11 to 12 min with B = 100% to 0%, and hold on for a further 4 min for reequilibration, giving a total run time of 16 min. The mass spectrometer was operated both in HESI+ and in HESI− with the following settings: source voltage of 5 kV, capillary temperature of 250°C heater temperature of 175°C sheath gas flow of 45 a.u., and auxiliary gas of 10 a.u. The Xcalibur 2.0.7 software (Thermo Scientific) was used for instrument control and data acquisition and processing.
RESULTS
Aspergillus flavus veA-dependent transcriptome.
Functional genomics studies revealed which genes are regulated by veA in A. flavus. The numbers of differentially expressed genes in the three experiments at different time points are shown in Fig. 1. Overall, the comparison between the ΔveA sample and the wild type in the dark (wtD) or the ΔveA sample and the wild type in the light (wtL) showed that, as expected, more genes were downregulated than upregulated by the absence of veA. In addition, the comparison of genes expressed by the wild type in the dark compared to those expressed in light clearly indicated that a higher number of genes were upregulated than were downregulated in the absence of light. In total, 468 genes were differentially expressed in wtD versus ΔveAD experiments, 391 in wtL versus ΔveA L experiments, and 477 in wtL versus wtD experiments. Eighty-seven of those genes were differentially expressed in the three described comparisons. In the wild type, the number of differentially expressed genes sharply increased at 48 h depending on presence or absence of light, while when comparing ΔveAD versus wtD, the increase in the number of genes occurred at 24 h. In the light, when comparing ΔveA and wild type, numerous differentially expressed genes appeared as early as 6 h.
FIG 1.
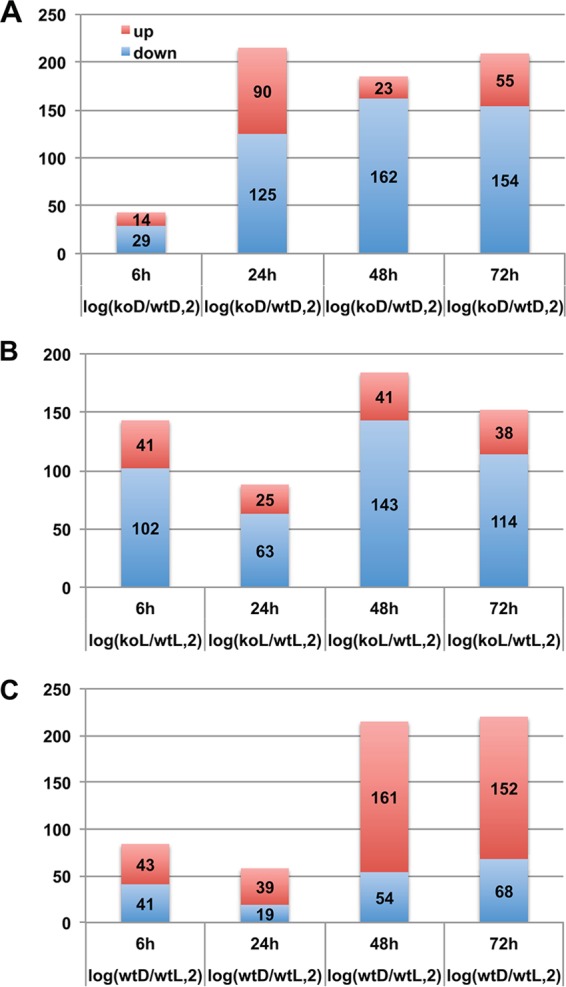
Number of differentially expressed genes in the following experiments: WT versus ΔveA (KO) cultures in the dark (A), WT versus ΔveA cultures in the light (B), and WT cultures in the dark versus WT cultures in the light (C). x axis, four time points; y axis, number of differentially expressed genes (red, upregulated; blue, downregulated).
In addition, we examined how many genes were differentially expressed at more than one time point in an experiment (see Fig. S1 in the supplemental material). Very few genes were differentially expressed in all four time points. In the dark, 2 genes, AFLA_117430 encoding a putative histidinol dehydrogenase and AFLA_131310 encoding a hypothetical protein, were consistently downregulated in the ΔveA mutant with respect to the wild type grown under the same conditions. Two genes, AFLA_014260 encoding a putative hydrophobin and AFLA_035260 encoding a protein with unknown function, were consistently upregulated at all time points. In cultures exposed to light, 3 genes, AFLA_013230 encoding a flavin-binding monooxygenase-like family protein, AFLA_020690 encoding a predicted glycosylphosphatidylinositol (GPI)-anchored protein, and AFLA_099790 encoding a conserved hypothetical protein, were downregulated in the mutant compared to the wild type, and 1 was upregulated (AFLA_035260 encoding a conserved hypothetical protein). In the wild type, no genes were differentially expressed at all time points analyzed when comparing light versus dark conditions.
The complete lists of differentially expressed genes together with the corresponding annotation information (such as IDs from different databases and best hit in A. nidulans and gene names) are provided in Tables S5 to S28 in the supplemental material, and their detailed annotations are provided in Tables S29 to S34 in the supplemental material. Each experiment had 55 to 75% differentially expressed genes with unknown functions (not annotated by any of the searched databases). For the differentially expressed genes that could be functionally annotated (Fig. 2; see Fig. S2 in the supplemental material), GO:0043167∼ion binding (including secondary metabolism), GO:0000166∼nucleotide binding, and GO:0016021∼integral to membrane functions are enriched not only in genes in all experiments but also in both veA-dependent upregulated and downregulated genes. In addition, GO:0030528∼transcription regulator activity, GO:0016052∼carbohydrate catabolic process, GO:0008233∼peptidase activity, and GO:0019842∼vitamin binding and methyltransferase functions are enriched in all experiments, while GO:0009165∼nucleotide biosynthetic process, GO:0048037∼cofactor binding, PIRSF006060:AA_transporter, sugar transport, IPR006163:phosphopantetheine binding, and IPR016181:acyl-CoAN-acyltransferase functions are unique to the comparison of wild type versus ΔveA in dark cultures, while GO:0008610∼lipid biosynthetic process, GO:0044271∼nitrogen compound biosynthetic process, and GO:0055114∼oxidation reduction are unique to the same comparison in the presence of light, and GO:0004180∼carboxypeptidase activity, GO:0006811∼ion transport, GO:0015031∼protein transport, GO:0030529∼ribonucleoprotein complex, GO:0046914∼transition metal ion binding, and afv00010:glycolysis/gluconeogenesis are unique to the comparison of the wild type growing in the light versus dark. Additional details are shown in Tables S29 to S35 in the supplemental material.
FIG 2.

Functional annotation of differentially expressed genes in WT versus ΔveA (KO) cultures growing in the dark and in the light. Genes from different time points are combined for the analysis using the DAVID server. Categories are from the GO database (GO), InterPro database (IPR), PIR database (PIRSF), SMART database (SM), KEGG database (afv), or UniProt (no accession numbers). The x axis shows the number of genes in each category. (A) Dark cultures. (B) Light cultures.
(i) Comparison of expression of developmental genes.
It is known that veA also controls morphological development in filamentous fungi (17, 35, 36). This is also reflected in our results. Using information obtained for 70 developmental genes from four different Aspergillus genomes (A. nidulans, A. fumigatus, A. niger, and A. oryzae) (37), blastx searches of the NCBI database revealed best hits to 61 A. flavus homologs. We found that, besides veA, 7 of these 61 A. flavus genes were differentially expressed in our experiments (see Table S38 in the supplemental material), including ganB (AFLA_018540, G protein complex alpha subunit GpaB), abaA (AFLA_029620, transcription factor), chiB (AFLA_031380, class V chitinase), tpsA (AFLA_034030, trehalose-phosphate synthase/phosphatase complex subunit Tps1), velB (AFLA_081490, VeA-interacting protein), brlA (AFLA_082850, C2H2 type conidiation transcription factor BrlA), and pkaA (AFLA_135040, cyclic AMP [cAMP]-dependent protein kinase catalytic subunit PkaC1). Of these, brlA, abaA, and velB are upregulated in the ΔveAD-versus-wtD comparison, chiB, tpsA, and pkaA are downregulated in the ΔveAL-versus-wtL comparison, and veA, velB, ganB, abaA, and tpsA are upregulated, while pkaA is downregulated, in the wtD-versus-wtL comparison. Lastly, chiB is differentially expressed in all three experiments.
(ii) Comparison of expression of 55 secondary metabolism gene clusters.
Twenty-eight of the 55 gene clusters described by Georgianna et al. (13) have at least one gene within the predicted cluster that is differentially expressed at at least one time point (highlighted in Table S35 in the supplemental material), corresponding to a total 64 genes. Some of these genes are differentially expressed at several time points in different experiments (Fig. 3; see Tables S36 and S37 in the supplemental material). Among the 64 genes, 37 genes (18 clusters) are differentially expressed at at least one time point in the ΔveAD-versus-wtD experiments, 40 genes (21 clusters) are differentially expressed at at least one time point in ΔveAL-versus-wtL experiments, and 32 genes (15 clusters) are differentially expressed at at least one time point in wtD-versus-wtL experiments. Furthermore, 25 genes (13 clusters) are differentially expressed at at least one time point in both ΔveAD-versus-wtD and ΔveAL-versus-wtL experiments, and 12 genes (6 clusters: 1 gene of cluster 14, 2 genes of cluster 18, 5 genes of cluster 23, 1 gene of cluster 24, 1 gene of cluster 27, and 2 genes of cluster 39) are differentially expressed at at least one time point in all three experiments (see Table S37 in the supplemental material). For example, AFLA_066970 (conserved hypothetical protein) of cluster 23 and AFLA_082150 (polyketide synthase, putative) of cluster 27 are differentially expressed at six time points in the three established comparisons.
FIG 3.

Gene expression heat map of 64 differentially expressed secondary metabolism genes. Genes have log2 ratios of >1 or <−1 at at least one time point. A gene with the corresponding gene cluster number, GenBank ID, chromosome number, and functional description is shown in each row.
In cluster 39, an example of a veA-dependent uncharacterized cluster (Fig. 4), all the genes except afvA (AFLA_108540) were differentially expressed in at least one of the comparisons of wild type to the veA mutant; afvC (AFLA_108560) and afvE (AFLA_108580) were differentially expressed in all the three comparisons. Both afvC and afvE are downregulated in the ΔveA mutant compared to the wild type under both light and dark conditions (koD versus wtD and koL versus wtL). Expression of the PKS gene, afvB, was downregulated in the ΔveA mutant, particularly in the dark. In terms of the extent, afvC is the most differentially expressed gene in this cluster, in the presence or absence of veA, as well as in the illumination regimen comparison. Validation of the effect of veA on the expression of the genes in cluster 39 by qRT-PCR showed that all the genes in this cluster are under veA regulation, including afvA at 96 h, as well as adjacent genes AFLA_108520 and AFLA_108590 (Fig. 5).
FIG 4.

Genes, their locations, and fold changes in expression (log2) in cluster 39. Fold changes in expression for four time points are shown as blue (6 h), red (24 h), green (48 h), and yellow (72 h) circles. Circles with log2 values of >1 or <−1 represent time points for differentially expressed genes. Plots were made using the genoPlotR package.
FIG 5.

Expression analysis of veA-dependent cluster 39 biosynthetic genes. Expression of SMURF-identified cluster 39 genes (AFLA_108540 to _108580) and flanking genes (AFLA_108520, AFLA_108590, and AFLA_108600) is shown. Our transcriptome analysis indicated that this secondary metabolite gene cluster is veA dependent. These results were validated by qRT-PCR (shown above). RNA was isolated from the Af70 pSL82 control and ΔveA strains at 24, 48, 72, and 96 h. Gene expression levels at each time point were normalized (ΔΔCT analysis) to those for the A. flavus 18S rRNA gene, utilizing the gene expression analysis software package for the Bio-Rad iQ5. Bars: black, Af70 pSL82 control; gray, A. flavus ΔveA mutant.
Phylogenetic analysis of cluster 39.
The predicted backbone gene, afvB, encoding a PKS includes the following domains from N-terminal to C-terminal orientation: beta-ketoacyl synthase (KS), acyl transferase (AT), and malonyl coenzyme A (malonyl-CoA)-acyl carrier protein transacylase (ACP). This domain structure was highly conserved in A. oryzae, A. nidulans, A. niger, A. terreus, A. fumigatus, A. fischerianus, and A. clavatus. The domain structure comparison of the cluster 39 PKS specific to four selected homologs in A. flavus, A. oryzae, A. niger, and A. clavatus is represented in Fig. S3 in the supplemental material.
Other genes present in this cluster encode an O-methyltransferase (OMT), a methyltransferase (MT), a cytochrome P450 (CP450), and an NADH oxidase. In this work, A. flavus gene cluster 39 was used as a query against other Aspergillus genome databases, specifically those for A. oryzae, A. niger, A. clavatus, A. terreus, A. fischerianus, and A. nidulans, to identify fully or partially conserved clusters (Fig. 6; see Table S39 in the supplemental material). In silico analysis indicated that the genes constituting cluster 39 in A. flavus were highly conserved in A. oryzae, A. niger, and A. clavatus and partially conserved in A. terreus (Fig. 6). However, the GenBank and Aspergillus comparative database BLAST searches with the gene encoding the putative NADH oxidase as a query did not yield any significant matches in A. clavatus and A. terreus. In A. niger, the gene with the Aspergillus comparative database locus number fge1_pg_C_5000666 (Entrez gene locus tag, ANI_1_3002024) was found to have nearly 80% amino acid identity with the gene encoding the putative NADH oxidase (AFLA_108540). However, it was not found in proximity of the genes forming cluster 39 in A. niger. Also, the CP450 gene was present near the 3′ end of the cluster PKS gene in both A. clavatus and A. niger and in opposite orientation compared to the CP450 gene of A. flavus that is adjacent to the methyltransferase gene. Intergenic distances were similar when comparing this gene cluster in A. flavus and A. oryzae, while it varied in A. niger, A. clavatus, and A. terreus (Fig. 6). Comparison of the genes forming cluster 39 in A. niger and A. clavatus with those in other Aspergillus species also yielded significant matches in A. flavus and A. oryzae. NCBI GenBank blast searches using A. flavus cluster 39 gene sequences as queries yielded significant matches in other fungal genera only with respect to the PKS gene (data not shown).
FIG 6.

Comparison of gene cluster 39 in selected species of the genus Aspergillus. In A. flavus (strain NRRL3357), cluster 39 includes the following genes: afvA, encoding NADH oxidase, putative (NADH) (AFLA_108540); afvB, encoding a polyketide synthase (PKS) (AFLA_108550); afvC, encoding an O-methyltransferase (OMT) (AFLA_108560); afvD, encoding a methyltransferase (MT) (AFLA_108570); and afvE, encoding a cytochrome P450 (CP450) (AFLA_108580). The gene lengths (base pairs) and the intergenic distances are drawn to scale.
Interestingly, several conserved putative transcription factor binding sites were identified in the intergenic regions of these genes (data not shown; see Fig. S4 in the supplemental material). For example, putative binding sites for the transcription factors AflR, PacC, CreA, FlbB, AreA, SltA, and Crz1P were found between afvB and afvC (see Fig. S4 in the supplemental material).
Gene cluster 39 is expressed predominantly in sclerotia.
In order to elucidate whether the production of the compound corresponding to gene cluster 39 is associated with a particular fungal structure, qRT-PCR was used to examine the expression of afvB, encoding the PKS, in mycelium, conidia, and sclerotia of wild-type A. flavus (Fig. 7). The results of this analysis revealed that afvB is expressed mainly in sclerotia, with expression levels 20-fold and 10-fold greater than the expression levels of this gene found in mycelium and conidia, respectively.
FIG 7.
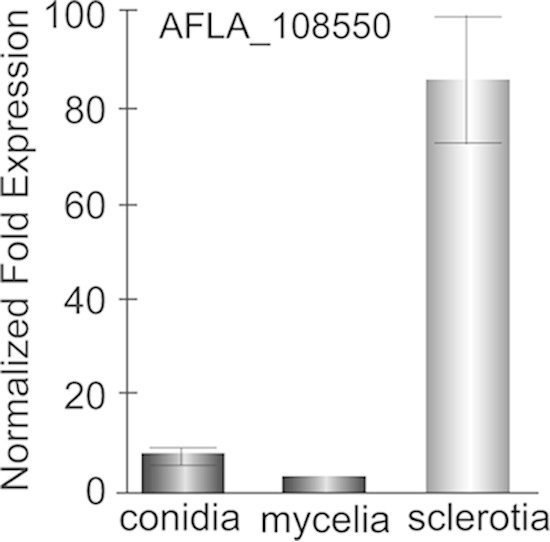
Analysis of afvB expression in mycelium, conidia, and sclerotia. Expression of afvB (AFLA_108550) in mycelium, conidia, and sclerotia was determined following growth of Af70 pyrG-1 on GMM–2% sorbitol plates at 30°C for 5 days in the dark. Sclerotia and conidia were separated from mycelium by gentle scraping of the colony surface followed by repeated washing with sterile water to remove all traces of sclerotia and conidia. Sclerotia were collected by adding 10 ml sterile, deionized water to the agar surface and gently scraping colonies with a cell scraper. Sclerotia were washed 5 times by addition of 40 ml water to remove residual conidia and mycelia, resuspended in a final volume of 25 ml water, and stored at 4°C. Gene expression levels at each time point were normalized (ΔΔCT analysis) to those for the A. flavus 18S rRNA gene, utilizing the gene expression analysis software package for the Bio-Rad iQ5.
Cluster 39 affects sclerotial production.
In order to study the potential biological roles of cluster 39 in A. flavus, we generated an afvB deletion strain. Successful replacement of a region within the beta-ketoacyl synthase domain of afvB with the pyrG selectable marker gene in fungal transformants was verified by PCR (see Fig. S5 in the supplemental material). Loss of afvB expression was also confirmed in ΔafvΒ mutant 4, which was selected for further studies. Complementation of ΔafvΒ 4 (afvB-com) and generation of a CA14 afvB overexpression strain (OEafvB) was confirmed following transformation of CA14 ΔafvΒ 4 and CA14, respectively, with plasmid pPTRI-gpd-afvB-trpC (see Fig. S6 in the supplemental material). Successful integration of the overexpression plasmid was confirmed by PCR. Overexpression of afvB was also confirmed by qRT-PCR of RNA generated from afvB-com 1 and OEafvB 2.
No difference in colony growth was observed between ΔafvΒ and the control strains (data not shown). Similarly, overexpression of afvΒ did not affect A. flavus fungal growth. However, sclerotial production was approximately 40% reduced in the afvB mutant compared to the wild type and complementation controls on GMM-S medium (Fig. 8). No significant difference was observed when the afvB was overexpressed.
FIG 8.

afvB influences sclerotial production. (A and B) The A. flavus CA14 PyrG-1 (WT), ΔafvB, and complementation strain (A), as well as CA14 pPTRI (WT) and the afvB overexpression strain (B), were point inoculated on GMM–1% sorbitol medium and incubated at 30°C in the dark for 8 days. (C and D) Quantification of sclerotia in the cultures shown in panels A and B, respectively. Cores (16-mm diameter) were collected approximately 1 cm from the center of each plate. The cores were then sprayed with 70% ethanol to improve visualization, and sclerotia were quantified under a Leica dissecting microscope. The experiment was carried out with three replicates. Standard errors are shown. Bars with different letters represent values that are statistically significantly different (P < 0.05).
Gene cluster 39 is responsible for the synthesis of aflavarin in A. flavus.
Comparison of the metabolome of wild type with that of the CA14 ΔafvB mutant using HR-Orbitrap MS and multiple-stage MS indicated that cluster 39 is responsible for producing a series of bicoumarins (compounds 1 to 6) (Fig. 9; see Fig. S7 to S9 in the supplemental material), which are putatively synthesized by dimerization of the monomeric coumarins. Deletion of afvA and afvC to -E was verified by DNA analysis (see Fig. S10 in the supplemental material). The major compound (compound 1) of this series of differentially expressed metabolites (more abundant in the wild type than in the ΔafvB strain) was shown to possess a molecular formula of C24H23O9 ([M + H+]/z = 455.1328; error = −1.843 ppm). The mass was identical to the mass of aflavarin, a bicoumarin metabolite previously reported in sclerotial extracts of A. flavus (38). This assignment was further confirmed by comparison of retention time and mass in LC-MS to those of an authentic sample of aflavarin. Mass spectral analysis demonstrated a perfect match between metabolite 1 and the standard solution of aflavarin in terms of exact mass, retention time, and fragmentation data (see Fig. S11 in the supplemental material). As depicted in Fig. S7 in the supplemental material, aflavarin exists in at least 4 isomers, which elute at 12.19, 12.47, 12.74, and 14.14 min. The isomeric diversity is a result of the C-C bridge (biaryl axis), as is the case for bicoumarins (39). Compound 2, with a [M + H+]/z of 453.1171 (C24H21O9; error = −2.005 ppm), has two hydrogens fewer than aflavarin, implying an extra double bond in its structure. Considering the structure of aflavarin, the only possibility for an additional double bond is by oxidation of the hydroxy-methyl group (—CH2OH) to an aldehyde (—CHO). The molecular formula of compound 3 was established as C23H21O9 ([M + H+]/z = 441.1173; error = −1.516 ppm). The 14-Da difference compared to aflavarin is consistent with the lack of a methyl group. The structure of this metabolite was also supported by analysis of two other metabolomes of deletion mutants of the tailoring genes, afvD and afvE. Compound 4 has a molecular formula of C24H23O8 ([M + H+]/z = 439.1378; error = −2.059 ppm), indicating a mass 16 Da (an oxygen atom) less than that of aflavarin. Therefore, based on the comparison with the known structure of aflavarin, compound 4 must be a dehydroxylated analog of it. Compound 5, with a [M + H+]/z of 425.1221 (C23H21O8; error = −2.503 ppm), was shown to be a demethylated analog of compound 4, whereas metabolite 6 ([M + H+]/z = 411.1066 [C22H19O8, error = −2.053 ppm]) proved to be a demethylated analog of compound 5.
FIG 9.

Structures of different identified metabolites associated with gene cluster 39 in CA14 A. flavus.
In the wild-type extracts we were able to detect trace amounts of coumarin monomers (compounds 7 to 10), which represent the biosynthetic precursors of the above-mentioned bicoumarins. A more clear occurrence and accumulation (around 100 times more abundant than in the WT) of these monomers can be seen in the afvE cytochrome P450 monooxygenase knockout mutant (see Fig. S9 in the supplemental material). Metabolite 7, with its chemical formula C12H13O4 ([M + H+]/z = 221.0807; error = −0.431 ppm) is consistent with a monomeric coumarin siderin, whereas compound 8 is suggested to be a hydroxylated analog of siderin with its corresponding molecular formula of C12H13O5 ([M + H+]/z = 237.0757; error = −0.090 ppm). Monomer 9, possessing a molecular formula of C11H11O4 ([M + H+]/z = 207.0649; error = −0.305 ppm) is consistent with a 7-O-demethyl siderin, whereas metabolite 10 appears to be the hydroxylated analog of 9 with a chemical formula of C11H11O5 ([M + H+]/z = 223.0600; error = −0.070 ppm). Monomers 7 and 9 are already described in the literature as being important precursors in the biosynthesis of several classes of bicoumarins (40, 41).
DISCUSSION
Functional genomics and metabolomics contribute to accelerate the pace in the elucidation of the genetic regulatory networks governing activation and modulation of different cellular processes in response to environmental stimuli. The velvet regulatory system has been shown to play a central role in the fibers of these networks. Our knowledge of velvet has greatly expanded through studies of the model fungus A. nidulans (19, 36, 42, 43), providing insight into its mechanism of action in response to environmental cues such as light (28, 42). The master regulator veA is the keystone of the velvet system, and it is conserved in numerous fungal species, including A. flavus (17, 18, 44). The present transcriptome analysis of genes regulated by VeA showed that in A. flavus, as in the case of A. nidulans and A. fumigatus homologs (45), it affects the transcription of numerous genes. The number of genes differentially expressed increases over time in culture. While for some genes veA acts as a negative regulator, most of the genes governed by veA are positively regulated, particularly in the dark, a condition that was shown to favor VeA accumulation in nuclei (42). Additionally, the observation that very few genes are differentially expressed at all four time points analyzed in this study suggests dynamic genetic changes over time in response to changes in environmental conditions as the culture ages.
Similar to that observed for A. nidulans and A. fumigatus (45), the veA-dependent transcriptome in A. flavus includes genes involved in morphological differentiation and the production of secondary metabolites, also called natural products. Fungi have the capacity to produce a wide range of these bioactive compounds. Whereas some of these compounds are clearly beneficial, such as those utilized as therapeutic drugs, other secondary metabolites are detrimental (e.g., mycotoxins). Among them, aflatoxins, commonly produced by A. flavus, are the most well known and studied. The aflatoxin gene cluster, as well as the A. flavus aflatrem and asparasone A gene clusters (17, 20), has been previously demonstrated to be positively regulated by veA. The present study revealed numerous additional veA-dependent secondary metabolic gene clusters, including cluster 39.
In A. flavus, some of these secondary metabolites are associated with developmental structures as pigments or protective factors. Sclerotia are resistance structures that represent a source of primary inoculum for some phytopathogenic fungi. A. flavus, as well as other fungi, accumulates a variety of secondary metabolites in sclerotia (44, 46, 47). Some of these compounds protect the fungus against fungivorous predators (48). Our study revealed for the first time that the genes in cluster 39 are expressed mainly in sclerotia and are necessary for the formation of aflavarin. Interestingly, like many other sclerotial metabolites (for instance, asparasone A [49]), aflavarin has been shown to exhibit anti-insectan activity (38), thus serving an important role in A. flavus ecology and contributing to its survival. Furthermore, our data indicated that a functional cluster 39 is necessary for normal levels of sclerotial production, suggesting that aflavarin probably has an additional role in sclerotial development besides its detrimental effect on fungivorous insects. It is likely that other A. flavus secondary metabolites, which have not yet been assigned to a predicted gene cluster, could also be accumulating in the sclerotium, functioning to protect these resistance structures from predators, competitors, and other environmental biotic as well as abiotic stressors. Although no putative pathway-specific transcriptional activator gene was present within cluster 39, in silico analysis of the shared afvB-afvC promoter identified numerous putative binding sites for DNA-binding proteins responsive to internal and external signals. This suggests that expression of genes in cluster 39 is governed by a plurality of factors, including stress conditions. In addition, consensus binding sites for AflR, the transcription factor gene responsible for the activation of aflatoxin cluster genes (50, 51), were also found in this promoter as well as in other promoters of genes in cluster 39, suggesting a regulatory cross talk between these clusters. A regulatory connection between gene clusters could contribute to the fungus' fitness, particularly if both biosynthetic pathways share the same metabolic precursors.
Cluster 39 is not only present in A. flavus; it is also semiconserved in A. clavatus and also in A. niger, where it is involved in the synthesis of kotanin (41). In the present study, we also elucidated the biosynthetic pathway that leads to the production of aflavarin in A. flavus by the generation of mutants with deletions of other genes in cluster 39. A large number of natural biaryl compounds have been extensively described in plants, fungi, and bacteria (52–54). In particular, fungi of the genus Aspergillus have been shown to be capable of producing a diverse class of bicoumarins, including C8-C8′-linked bicoumarins (e.g., kotanin and orlandin [39, 55]), C6-C8′-linked bicoumarins (e.g., desertorin A-C [56]), C6-C6′-linked bicoumarins (e.g., isokotanin A-C [57]), and C3-C3′-linked bicoumarins (e.g., bicoumanigrin [58]). All these metabolites share the C-C biaryl axis as a characteristic structural feature. Aflavarin and its structural derivatives identified in this study, as products of A. flavus CA14 gene cluster 39, belong to the C3-C8′-linked regioisomeric subclass of bicoumarins. Up to the present, there is no evidence for the existence of any other natural bicoumarin harboring the C3-C8′ regioisomeric linkage.
Previous reports on the biosynthesis of bicoumarins have shown that the main step in the biosynthesis of these dimeric metabolites is the dimerization of the polyketide monomers via an intermolecular oxidative phenol coupling reaction. All examples of previously described C-C-cross-linked bicoumarins have clearly demonstrated that the stereo- and regioselectivity during oxidative phenol coupling reactions is fundamentally controlled by cytochrome P450 enzymes (41, 53, 54).
Iterative type I PKSs, in general represent a hallmark of fungal polyketide biosynthesis compared to bacterial PKSs (59). According to the presence or absence of β-keto processing domains, fungal PKSs are subdivided as nonreducing (NR), partially reducing (PR), and highly reducing (HR) PKSs (60). Based on the fact that aflavarin has an unsaturated cyclic system, with no reduction step required in its formation, the backbone gene, afvB, can be considered a nonreducing polyketide synthase (NR PKS) gene. Disruption of afvB led to complete loss in coumarin biosynthesis (see ΔafvB in Fig. S7 to S9 in the supplemental material). LC-MS of the cluster 39 deletion mutants allowed a model of the aflavarin biosynthetic pathway to be proposed (Fig. 10). Deletion of the afvC (O-methyltransferase) gene totally abolished the biosynthesis of coumarin (see ΔafvC, [ΔAFLA_108560] in Fig. S7 to S9 in the supplemental material). In previous studies (41, 53), it was clearly demonstrated that the methoxy group at position C-4 of the bicoumarin skeleton is a prerequisite for the oxidative phenol coupling reaction. This suggests that in the case of aflavarin, the O-methyltransferase is involved in the initial steps of O-methylation of the 4,7-didesmethyl-siderin (I), which results in the subsequent formation of the monomers (compounds 9 and 7).
FIG 10.

Proposed A. flavus biosynthetic pathway of aflavarin and its structural analogs. AFLA_108540 (putative NADH oxidase), afvA; AFLA_108550 (polyketide synthase), afvB (pks39); AFLA_108560 (ortho-methyltransferase), afvC; AFLA_108570 (methyltransferase), afvD; AFLA_108580 (cytochrome P450 monooxygenase), afvE.
The disruption of the afvE (cytochrome P450 monooxygenase) also led to the breakdown of bicoumarin biosynthesis (compounds 1 to 6) (see ΔafvE [ΔAFLA_108580] in Fig. S7 and S8 in the supplemental material). However, as expected, in this intermediate mutant gene the monomeric coumarins (compounds 7 to 10) were accumulated (see ΔafvE in Fig. S9 in the supplemental material), proving the concept that cytochrome P450 monooxygenase is responsible for phenolic coupling reactions. On the other hand, the deletion of the putative methyltransferase gene, afvD, is associated with the accumulation of compounds 3, 5, and 6, which suggests that this gene is responsible for converting these metabolites (compounds 3, 5, and 6) to their respective products 1, 4 and 5 (see ΔafvD [ΔAFLA_108570] in Fig. S7 and S8 in the supplemental material). These data suggest that ΔafvD is mainly responsible for the final O-methylation steps after the reactions of oxidative phenolic coupling. Deletion of the putative NADH oxidase gene, afvA, in general did not affect the total biosynthesis of bicoumarins, although the peak intensity of these metabolites is decreased (see ΔafvA [ΔAFLA_108540] in Fig. S7 and S8 in the supplemental material). Interestingly, ΔafvA mutants showed accumulation of the monomeric coumarins 7 and 9; however, we observed only trace amount of the hydroxylated monomers (compounds 8 and 10) (see ΔafvA [ΔAFLA_108540] in Fig. S9 in the supplemental material). This suggests that the putative NADH oxidase is possibly catalyzing the hydroxylation of siderin (compound 7) and 7-O-demethyl siderin (compound 9) to their respective hydroxylated analogs, compounds 8 and 10. Regarding metabolite 2, we speculate that it is synthesized by further oxidation of aflavarin which results in production of this aldehyde derivative. Since none of the tailoring genes of cluster 39 seem to be involved in this process, this biosynthetic step is possibly carried out by an oxidase present elsewhere in the genome, outside cluster 39.
Based on the results we obtained from deletion mutants, a model for the biosynthetic pathway of aflavarin is established, as follows. The PKS AfvB catalyzes four repetitive Claisen condensations to synthesize the pentaketidic precursor I (4,7-didesmethyl-siderin) which is then O-methylated to give compounds 7 and 9 by the O-methyltransferase, AfvC. Afterwards, these monomers are hydroxylated to produce compounds 8 and 10, possibly by the activity of the putative NADH oxidase AfvA. From this point, aflavarin can be synthesized through two different paths, either by a direct oxidative phenol coupling of the monomers 7 and 8 or indirectly through formation of metabolite 3, which is then O-methylated to give compound 1. The oxidative phenol coupling reaction part of this biosynthetic network are catalyzed by the cytochrome P450 monooxygenase AfvE. In the same fashion as for aflavarin, other identified bicoumarins are also produced by cluster 39 (Fig. 10).
It is known that fungal secondary metabolism and morphological development are genetically linked (19, 44, 61). As expected, our study showed that in A. flavus, some developmental genes are regulated by veA in a light-dependent manner, including the asexual development-associated transcription factor genes brlA and abaA (62), velB (encoding a well-known VeA-interacting protein) (63), and ganB, encoding a G protein complex alpha subunit that negatively affects conidiation (64, 65). Additionally, we found that although an illumination regimen is sufficient to change the expression of pkaA, encoding cAMP-dependent kinase A (66), and chiB, encoding a chitinase (67), both genes are also veA dependent. The fact that chiB expression is altered in the veA deletion mutant also suggests possible alterations in cell wall composition in this A. flavus strain. This, together with the observed alterations in the expression of tpsA, encoding a phosphate synthase important in trehalose biosynthesis (68), could in part contribute the greater sensitivity to environmental stresses, such as oxidative stress, in the ΔveA strain (69).
In conclusion, the current study revealed that hundreds of genes are regulated by veA and are also influenced by the presence or absence of light, including developmental genes and secondary metabolite gene clusters, in A. flavus. Some of the veA-dependent secondary metabolites gene clusters identified in this study are orphans, including the uncharacterized cluster 39. Our metabolomic analysis demonstrated that the product associated with this cluster is the polyketide-derived compound aflavarin, which is known for its anti-insectan activity associated with A. flavus sclerotia. It is possible that veA-dependent accumulation of protective secondary metabolites could also occur in sclerotia or fruiting bodies in other fungal species. In addition to contributing to the elucidation of the regulatory pathways controlling secondary metabolism and its role in fungal biology, these findings could be applied to develop genetic strategies to reduce the detrimental effects of fungal secondary metabolites on humans and animals.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the Department of Biological Sciences at NIU and by USDA grant 58-6435-4-015.
A.M.C. thanks Barbara Ball and Sourabh Dhingra for their technical support.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00092-15.
REFERENCES
- 1.Coley-Smith JR, Cooke RC. 1971. Survival and germination of fungal sclerotia. Annu Rev Phytopathol 9:65–92. doi: 10.1146/annurev.py.09.090171.000433. [DOI] [Google Scholar]
- 2.Malloch D, Cain RF. 1972. The Trichocomataceae: ascomycetes with Aspergillus, Paeciloyces and Penicillium imperfect states. Can J Bot 50:2613–2628. doi: 10.1139/b72-335. [DOI] [Google Scholar]
- 3.Wicklow DT. 1987. Survival of Aspergillus flavus sclerotia in soil. Trans Br Mycol Soc 89:131–134. doi: 10.1016/S0007-1536(87)80073-6. [DOI] [Google Scholar]
- 4.Payne GP, Brown MP. 1998. Genetics and physiology of aflatoxin biosynthesis. Annu Rev Phytopathol 36:329–362. doi: 10.1146/annurev.phyto.36.1.329. [DOI] [PubMed] [Google Scholar]
- 5.Sweeney MJ, Dobson AD. 1999. Molecular biology of mycotoxin biosynthesis. FEMS Microbiol Lett 175:149–163. doi: 10.1111/j.1574-6968.1999.tb13614.x. [DOI] [PubMed] [Google Scholar]
- 6.Trail F, Mahanti N, Linz J. 1995. Molecular biology of aflatoxin biosynthesis. Microbiology 141:755–765. doi: 10.1099/13500872-141-4-755. [DOI] [PubMed] [Google Scholar]
- 7.Probst C, Schulthess F, Cotty PJ. 2010. Impact of Aspergillus section Flavi community structure on the development of lethal levels of aflatoxins in Kenyan maize (Zea mays). J Appl Microbiol 108:600–610. doi: 10.1111/j.1365-2672.2009.04458.x. [DOI] [PubMed] [Google Scholar]
- 8.Turner PC, Moore SE, Hall AJ, Prentice AM, Wild CP. 2003. Modification of immune function through exposure to dietary aflatoxin in Gambian children. Environ Health Perspect 111:217–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dvorackova I, Kusak V. 1990. Hepatocellular carcinoma (a 28-year necropsy review). J Environ Pathol Toxicol Oncol 10:220–224. [PubMed] [Google Scholar]
- 10.Vardon PJ. 2003. Mycotoxins: risks in plant, animal and human systems, p 136–142. In Potential economic costs of mycotoxins in the United States. CAST task force report, no. 139 CAST, Ames, IA. [Google Scholar]
- 11.Tokuoka M, Seshime Y, Fujii I, Kitamoto K, Takahashi T, Koyama Y. 2008. Identification of a novel polyketide synthase-nonribosomal peptide synthetase (PKS-NRPS) gene required for the biosynthesis of cyclopiazonic acid in Aspergillus oryzae. Fungal Genet Biol 45:1608–1615. doi: 10.1016/j.fgb.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 12.Zhang S, Monahan BJ, Tkacz JS, Scott B. 2004. Indole-diterpene gene cluster from Aspergillus flavus. Appl Environ Microbiol 70:6875–6883. doi: 10.1128/AEM.70.11.6875-6883.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Georgianna DR, Fedorova ND, Burroughs JL, Dolezal AL, Bok JW, Horowitz-Brown S, Woloshuk CP, Yu J, Keller NP, Payne GA. 2010. Beyond aflatoxin: four distinct expression patterns and functional roles associated with Aspergillus flavus secondary metabolism gene clusters. Mol Plant Pathol 11:213–226. doi: 10.1111/j.1364-3703.2009.00594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khaldi N, Seifuddin FT, Turner G, Haft D, Nierman WC, Wolfe KH, Fedorova ND. 2010. SMURF: genomic mapping of fungal secondary metabolite clusters. Fungal Genet Biol 47:736–741. doi: 10.1016/j.fgb.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Terabayashi Y, Sano M, Yamane N, Marui J, Tamano K, Sagara J, Dohmoto M, Oda K, Ohshima E, Tachibana K, Higa Y, Ohashi S, Koike H, Machida M. 2010. Identification and characterization of genes responsible for biosynthesis of kojic acid, an industrially important compound from Aspergillus oryzae. Fungal Genet Biol 47:953–961. doi: 10.1016/j.fgb.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 16.Forseth RR, Amaike S, Schwenk D, Affeldt KJ, Hoffmeister D, Schroeder FC, Keller NP. 2012. Homologous NRPS-like gene clusters mediate redundant small-molecule biosynthesis in Aspergillus flavus. Angew Chem Int Ed Engl 51:1–7. doi: 10.1002/anie.201106864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duran RM, Cary JW, Calvo AM. 2007. Production of cyclopiazonic acid, aflatrem, and aflatoxin by Aspergillus flavus is regulated by veA, a gene necessary for sclerotial formation. Appl Microbiol Biotechnol 73:1158–1168. [DOI] [PubMed] [Google Scholar]
- 18.Duran RM, Cary JW, Calvo AM. 2009. The role of veA in Aspergillus flavus infection of peanut, corn and cotton. Open Mycol J 3:27–36. doi: 10.2174/1874437000903010027. [DOI] [Google Scholar]
- 19.Calvo AM. 2008. The VeA regulatory system and its role in morphological and chemical development in fungi. Fungal Genet Biol 45:1053–1061. doi: 10.1016/j.fgb.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 20.Cary JW, Harris-Coward PY, Ehrlich KC, Di Mavungu JD, Malysheva SV, De Saeger S, Dowd PF, Shantappa S, Martens SL, Calvo AM. 2014. Functional characterization of a veA-dependent polyketide synthase gene in Aspergillus flavus necessary for the synthesis of asparasone, a sclerotium-specific pigment. Fungal Genet Biol 64:25–35. doi: 10.1016/j.fgb.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Dhingra S, Andes D, Calvo AM. 2012. VeA regulates conidiation, gliotoxin production, and protease activity in the opportunistic human pathogen Aspergillus fumigatus. Eukaryot Cell 11:1531–1543. doi: 10.1128/EC.00222-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Myung K, Zitomer NC, Duvall M, Glenn AE, Riley RT, Calvo AM. 2012. The conserved global regulator VeA is necessary for symptom production and mycotoxin synthesis in maize seedlings by Fusarium verticillioides. 61:152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li S, Myung K, Guse D, Donkin B, Proctor RH, Grayburn WS, Calvo AM. 2006. FvVE1 regulates filamentous growth, the ratio of microconidia to macroconidia and cell wall formation in Fusarium verticillioides. Mol Microbiol 62:1418–1432. doi: 10.1111/j.1365-2958.2006.05447.x. [DOI] [PubMed] [Google Scholar]
- 24.Laskowski-Peak MC, Calvo AM, Rohrssen J, Smulian AG. 2012. VEA1 is required for cleistothecial formation and virulence in Histoplasma capsulatum. Fungal Genet Biol 49:838–846. doi: 10.1016/j.fgb.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 25.Dreyer J, Eichhorn H, Friedlin E, Kurnsteiner H, Kuck U. 2007. A homologue of the Aspergillus velvet gene regulates both cephalosporin C biosynthesis and hyphal fragmentation in Acremonium chrysogenum. Appl Environ Microbiol 73:3412–3422. doi: 10.1128/AEM.00129-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Calvo AM, Bok J-W, Brooks W, Keller NP. 2004. VeA is required for toxin and sclerotial production in Aspergillus parasiticus. Appl Environ Microbiol 70:4733–4739. doi: 10.1128/AEM.70.8.4733-4739.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bayram O, Krappmann S, Ni M, Bok JW, Helmstaedt K, Valerius O, Braus-Stromeyer S, Kwon NJ, Keller NP, Yu JH, Braus GH. 2008. VelB/VeA/LaeA complex coordinates light signal with fungal development and secondary metabolism. Science 320:1504–1506. doi: 10.1126/science.1155888. [DOI] [PubMed] [Google Scholar]
- 28.Purschwitz J, Muller S, Kastner C, Schoser M, Haas H, Espeso EA, Atoui A, Calvo AM, Fischer R. 2008. Functional and physical interaction of blue- and red-light sensors in Aspergillus nidulans. Curr Biol 18:255–259. doi: 10.1016/j.cub.2008.01.061. [DOI] [PubMed] [Google Scholar]
- 29.Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. 2003. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34:374–378. [DOI] [PubMed] [Google Scholar]
- 30.Huang da W, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 31.Cary JW, GR OB, Nielsen DM, Nierman W, Harris-Coward P, Yu J, Bhatnagar D, Cleveland TE, Payne GA, Calvo AM. 2007. Elucidation of veA-dependent genes associated with aflatoxin and sclerotial production in Aspergillus flavus by functional genomics. Appl Microbiol Biotechnol 76:1107–1118. doi: 10.1007/s00253-007-1081-y. [DOI] [PubMed] [Google Scholar]
- 32.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 33.Cary JW, Ehrlich KC, Bland JM, Montalbano BG. 2006. The aflatoxin biosynthesis cluster gene, aflX, encodes an oxidoreductase involved in conversion of versicolorin A to demethylsterigmatocystin. Appl Environ Microbiol 72:1096–2101. doi: 10.1128/AEM.72.2.1096-1101.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szewczyk E, Chiang YM, Oakley CE, Davidson AD, Wang CC, Oakley BR. 2008. Identification and characterization of the asperthecin gene cluster of Aspergillus nidulans. Appl Environ Microbiol 74:7607–7612. doi: 10.1128/AEM.01743-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim H, Han K, Kim K, Han D, Jahng K, Chae K. 2002. The veA gene activates sexual development in Aspergillus nidulans. Fungal Genet Biol 37:72–80. doi: 10.1016/S1087-1845(02)00029-4. [DOI] [PubMed] [Google Scholar]
- 36.Kato N, Brooks W, Calvo AM. 2003. The expression of sterigmatocystin and penicillin genes in Aspergillus nidulans is controlled by veA, a gene required for sexual development. Eukaryot Cell 2:1178–1186. doi: 10.1128/EC.2.6.1178-1186.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krijgsheld P, Bleichrodt R, van Veluw GJ, Wang F, Muller WH, Dijksterhuis J, Wosten HA. 2013. Development in Aspergillus. Stud Mycol 74:1–29. doi: 10.3114/sim0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.TePaske MR, Gloer JB, Wicklow DT, Dowd PF. 1992. Aflavarin and beta-aflatrem: new anti-insectan metabolites from the sclerotia of Aspergillus flavus. J Nat Prod 55:1080–1086. doi: 10.1021/np50086a008. [DOI] [Google Scholar]
- 39.Hussain H, Hussain J, Al-Harrasi A, Krohn K. 2012. The chemistry and biology of bicoumarins. Tetrahedron 68:2553–2578. doi: 10.1016/j.tet.2012.01.035. [DOI] [Google Scholar]
- 40.Stothers JB, Stoessl A. 1988. Confirmation of the polyketide origin of kotanin by incorporation of [1,2-13C2]acetate. Can J Chem 66:2816–2818. doi: 10.1139/v88-435. [DOI] [Google Scholar]
- 41.Gil Girol C, Fisch KM, Heinekamp T, Günther S, Hüttel W, Piel J, Brakhage AA, Müller M. 2012. Regio- and stereoselective oxidative phenol coupling in Aspergillus niger. Angew Chem Int Ed 51:9788–9791. doi: 10.1002/anie.201203603. [DOI] [PubMed] [Google Scholar]
- 42.Stinnett SM, Espeso EA, Cobeno L, Araujo-Bazan L, Calvo AM. 2007. Aspergillus nidulans VeA subcellular localization is dependent on the importin alpha carrier and on light. Mol Microbiol 63:242–255. doi: 10.1111/j.1365-2958.2006.05506.x. [DOI] [PubMed] [Google Scholar]
- 43.Bayram O, Braus GH. 2012. Coordination of secondary metabolism and development in fungi: the velvet family of regulatory proteins. FEMS Microbiol Rev 36:1–24. doi: 10.1111/j.1574-6976.2011.00285.x. [DOI] [PubMed] [Google Scholar]
- 44.Calvo AM, Cary JW. 2015. Association of fungal secondary metabolism and sclerotial biology. Front Microbiol 6:62. doi: 10.3389/fmicb.2015.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lind AL, Wisecaver JH, Smith TD, Feng X, Calvo AM, Rokas A. 2015. Examining the evolution of the regulatory circuit controlling secondary metabolism and development in the fungal genus Aspergillus. PLoS Genet 11:e1005096. doi: 10.1371/journal.pgen.1005096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gloer JB, TePaske MR, Sima JS. 1988. Antiinsectan aflavinine derivatives from the sclerotia of Aspergillus flavus. J Org Chem 53:5457–5460. doi: 10.1021/jo00258a011. [DOI] [Google Scholar]
- 47.Gloer JB. 2007. Applications of fungal ecology in the search for new bioactive natural products, 2nd ed, p257–283. Springer-Verlag, New York, NY. [Google Scholar]
- 48.Rohlfs M, Albert M, Keller NP, Kempken F. 2007. Secondary chemicals protect mould from fungivory. Biol Lett 3:523–525. doi: 10.1098/rsbl.2007.0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sobolev VS, Cole RJ, Dorner JW, Horn BW, Harrigan GG, Gloer JB. 1997. Isolation and structure elucidation of a new metabolite produced by Aspergillus parasiticus. J Nat Prod 60:847–850. doi: 10.1021/np970131m. [DOI] [Google Scholar]
- 50.Chang P-K, Cary JW, Bhatnagar D, Cleveland TE, Bennett JW, Linz JE, Woloshuk CP, Payne GA. 1993. Cloning of the Aspergillus parasiticus apa-2 gene associated with the regulation of aflatoxin biosynthesis. Appl Environ Microbiol 59:3273–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ehrlich KC, Montalbano BG, Cary JW. 1999. Binding of the C6-zinc cluster protein, AFLR, to the promoters of aflatoxin pathway biosynthesis genes in Aspergillus parasiticus. Gene 230:249–257. doi: 10.1016/S0378-1119(99)00075-X. [DOI] [PubMed] [Google Scholar]
- 52.Davin LB, Wang HB, Crowell AL, Bedgar DL, Martin DM, Sarkanen S, Lewis NG. 1997. Stereoselective bimolecular phenoxy radical coupling by an auxiliary (dirigent) protein without an active center. Science 275:362–366. doi: 10.1126/science.275.5298.362. [DOI] [PubMed] [Google Scholar]
- 53.Huttel W, Muller M. 2007. Regio- and stereoselective intermolecular oxidative phenol coupling in kotanin biosynthesis by Aspergillus niger. Chembiochem 8:521–529. doi: 10.1002/cbic.200600434. [DOI] [PubMed] [Google Scholar]
- 54.Prag A, Gruning BA, Hackh M, Ludeke S, Wilde M, Luzhetskyy A, Richter M, Luzhetska M, Gunther S, Muller M. 2014. Regio- and stereoselective intermolecular oxidative phenol coupling in Streptomyces. J Am Chem Soc 136:6195–6198. doi: 10.1021/ja501630w. [DOI] [PubMed] [Google Scholar]
- 55.Cutler HG, Crumley FG, Cox RH, Hernandez O, Cole RJ, Dorner JW. 1979. Orlandin: a nontoxic fungal metabolite with plant growth inhibiting properties. J Agric Food Chem 27:592–595. doi: 10.1021/jf60223a043. [DOI] [PubMed] [Google Scholar]
- 56.Nowara D, Gay A, Lacomme C, Shaw J, Ridout C, Douchkov D, Hensel G, Kumlehn J, Schweizer P. 2010. HIGS: host-induced gene silencing in the obligate biotrophic fungal pathogen Blumeria graminis. Plant Cell 22:3130–3141. doi: 10.1105/tpc.110.077040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Laakso JA, Narske ED, Gloer JB, Wicklow DT, Dowd PF. 1994. Isokotanins A-C: new bicoumarins from the sclerotia of Aspergillus alliaceus. J Nat Prod 57:128–133. doi: 10.1021/np50103a018. [DOI] [PubMed] [Google Scholar]
- 58.Hiort J, Maksimenka K, Reichert M, Perovic-Ottstadt S, Lin WH, Wray V, Steube K, Schaumann K, Weber H, Proksch P, Ebel R, Muller WE, Bringmann G. 2004. New natural products from the sponge-derived fungus Aspergillus niger. J Nat Prod 67:1532–1543. doi: 10.1021/np030551d. [DOI] [PubMed] [Google Scholar]
- 59.Cox RJ, Simpson TJ. 2009. Fungal type I polyketide synthases. Methods Enzymol 459:49–78. doi: 10.1016/S0076-6879(09)04603-5. [DOI] [PubMed] [Google Scholar]
- 60.Hertweck C. 2009. The biosynthetic logic of polyketide diversity. Angew Chem Int Ed Engl 48:4688–4716. doi: 10.1002/anie.200806121. [DOI] [PubMed] [Google Scholar]
- 61.Calvo AM, Wilson RA, Bok JW, Keller NP. 2002. Relationship between secondary metabolism and fungal development. Microbiol Mol Biol Rev 66:447–459. doi: 10.1128/MMBR.66.3.447-459.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Adams TH, Wieser JK, Yu JH. 1998. Asexual sporulation in Aspergillus nidulans. Microbiol Mol Biol Rev 62:35–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chang PK, Scharfenstein LL, Li P, Ehrlich KC. 2013. Aspergillus flavus VelB acts distinctly from VeA in conidiation and may coordinate with FluG to modulate sclerotial production. Fungal Genet Biol 58–59:71–79. doi: 10.1016/j.fgb.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 64.Chang MH, Chae KS, Han DM, Jahng KY. 2004. The GanB Galpha-protein negatively regulates asexual sporulation and plays a positive role in conidial germination in Aspergillus nidulans. Genetics 167:1305–1315. doi: 10.1534/genetics.103.025379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Emri T, Molnar Z, Szilagyi M, Pocsi I. 2008. Regulation of autolysis in Aspergillus nidulans. Appl Biochem Biotechnol 151:211–220. doi: 10.1007/s12010-008-8174-7. [DOI] [PubMed] [Google Scholar]
- 66.Shimizu K, Keller NP. 2001. Genetic involvement of a cAMP-dependent protein kinase in a G protein signaling pathway regulating morphological and chemical transitions in Aspergillus nidulans. Genetics 157:591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yamazaki H, Yamazaki D, Takaya N, Takagi M, Ohta A, Horiuchi H. 2007. A chitinase gene, chiB, involved in the autolytic process of Aspergillus nidulans. Curr Genet 51:89–98. doi: 10.1007/s00294-006-0109-7. [DOI] [PubMed] [Google Scholar]
- 68.Fillinger S, Chaveroche MK, van Dijck P, de Vries R, Ruijter G, Thevelein J, d'Enfert C. 2001. Trehalose is required for the acquisition of tolerance to a variety of stresses in the filamentous fungus Aspergillus nidulans. Microbiology 147:1851–1862. [DOI] [PubMed] [Google Scholar]
- 69.Baidya S, Duran RM, Lohmar JM, Harris-Coward PY, Cary JW, Hong SY, Roze LV, Linz JE, Calvo AM. 2014. VeA is associated with the response to oxidative stress in the aflatoxin producer Aspergillus flavus. Eukaryot Cell 13:1095–1103. doi: 10.1128/EC.00099-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schumann J, Hertweck C. 2006. Advances in cloning, functional analysis and heterologous expression of fungal polyketide synthase genes. J Biotechnol 124:690–703. doi: 10.1016/j.jbiotec.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 71.Hutchinson CR, Kennedy J, Park C, Kendrew S, Auclair K, Vederas J. 2000. Aspects of the biosynthesis of non-aromatic fungal polyketides by iterative polyketide synthases. Antonie van Leeuwenhoek 78:287–295. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.