

Abstract
The amphibian fungal disease chytridiomycosis, which affects species across all continents, recently emerged as one of the greatest threats to biodiversity. Yet, many aspects of the basic biology and epidemiology of the pathogen, Batrachochytrium dendrobatidis (Bd), are still unknown, such as when and from where did Bd emerge and what is its true ecological niche? Here, we review the ecology and evolution of Bd in the Americas and highlight controversies that make this disease so enigmatic. We explore factors associated with variance in severity of epizootics focusing on the disease triangle of host susceptibility, pathogen virulence, and environment. Reevaluating the causes of the panzootic is timely given the wealth of data on Bd prevalence across hosts and communities and the recent discoveries suggesting co‐evolutionary potential of hosts and Bd. We generate a new species distribution model for Bd in the Americas based on over 30,000 records and suggest a novel future research agenda. Instead of focusing on pathogen “hot spots,” we need to identify pathogen “cold spots” so that we can better understand what limits the pathogen's distribution. Finally, we introduce the concept of “the Ghost of Epizootics Past” to discuss expected patterns in postepizootic host communities.
Keywords: Amphibian, chytrid, Emerging infectious disease, fungi, immunogenetics, species distribution model, virulence
Introduction
Infectious diseases emerge because of changes in host–pathogen–environment interactions and, increasingly, anthropogenic habitat alterations are directly affecting these interactions (Jones et al. 2008). The number of emerging diseases caused by fungi relative to other types of pathogens has risen steeply during the last two decades, although the causes for this bias are unclear (Fisher et al. 2012). Among these emerging fungal diseases is the amphibian‐killing chytrid fungus Batrachochytrium dendrobatidis [hereafter Bd (Longcore et al. 1999)], which has, in the last 15 years, captured the attention of scientists and conservationists. Although chytridiomycosis (Box 1) is one of a long list of fungal diseases of animals and plants that have emerged in the last century (Fisher et al. 2012), over a thousand studies (1088) have already been published on Bd and its effects on amphibians (Web of Science search using term “chytridiomycosis” on 3 June 2015). Most of these studies have been descriptive in scope: establishing baseline patterns of Bd distribution [summarized in (Olson et al. 2013)], correlating disease prevalence with abiotic and biotic factors (Kriger and Hero 2007; Liu et al. 2013), and documenting pathogen genetic variation to identify its mode of spread (James et al. 2009; Rosenblum et al. 2013).
Box 1. Glossary.
aneuploidy: Having an atypical number of chromosomal homologs, not multiples of the baseline haploid number. May be represented by additional or fewer copies of a homologous chromosome(s).
chytridiomycosis: The disease of amphibians caused by the fungus Batrachochytrium. Animals that test positive for the presence of Bd may show no symptoms of the disease.
dilution effect: The concept that disease risk of a generalist pathogen is ameliorated with increased biodiversity through mechanisms that reduce the probability of transmission.
endemic pathogen hypothesis (EPH): Posits that Bd co‐existed with its host in equilibrium before the panzootic was triggered by some other factor, such as environmental change.
enzootic: Describes host–pathogen dynamics that support coexistence over time.
epizootic: Describes pathogens that are increasing in frequency, that is, have not reached a stable equilibrium.
global panzootic lineage (gpl): The most frequently encountered lineage of Bd, which is highly virulent in the laboratory, genetically depauperate with only two alleles per locus, and the only genotype that has been associated with amphibian die‐offs in the field. GPL contains two sublineages, GPL‐1 (which predominates in North America and Europe) and GPL‐2 (which predominates in the Neotropics, Australia, and Africa).
loss of heterozygosity: In diploid or polyploid organisms, genotypes may lose heterozygosity during mitosis through the action of nondisjunction of chromosomes, crossing over, or gene conversion.
novel pathogen hypothesis (NPH): Posits that pathogens emerge by the translocation of a virulent strain into a new geographic location or into a host species that has no evolved resistance.
pathogenicity: Describes the ability of an organism to cause disease.
resistance: Refers to the natural ability of an organism to resist microorganisms or toxins produced in disease.
ribosomal internal transcribed spacer: The DNA region used for diagnostic Bd PCR detection that lies between the large and small subunits of ribosomal DNA. Multiple variants per strain make it problematical for use in population genetics.
saprobic: Describes microbes and fungi that feed on dead or decaying organic matter.
tolerance: Refers to the development of the host capacity to endure and become less responsive to a substance or a physiological insult especially with repeated exposure.
virulence: Describes the degree to which an organism can cause damage to a host.
zoospore: Flagellated motile spore. In Bd, the zoospore possesses a single flagellum and lacks a rigid cell wall.
A major reason for this focus on Bd is that it is a generalist amphibian pathogen and close to 41% of amphibians are threatened, making them one of the most threatened vertebrate lineages (Monastersky 2014). Bd emergence demonstrates that host–pathogen interactions can play a major role in species declines and even extinctions (Crawford et al. 2010). Bd has now been reported from over 500 amphibian host species, has a cosmopolitan distribution, and has been detected at 48% of localities that have been surveyed (Olson et al. 2013). This wide distribution has been documented just since chytridiomycosis was first described (Longcore et al. 1999), when it was the first known vertebrate pathogen from an obscure phylum of fungi whose mechanism of pathogenesis and life cycle (Fig. 1) were incompletely known. This obscurity has meant studies on the biology of the pathogen have lagged behind those of hosts. Indeed, most of the questions that the research community set out to answer when the disease was first described, including “Where did it come from?”, “How does it spread?”, “Why are some species resistant or tolerant?”, “Does it have an alternate host or environmental stage?”, and “Why now?”, have yet to be definitively answered (Collins and Crump 2009; Kilpatrick et al. 2010).
Figure 1.

Batrachochytrium dendrobatidis develops by either of two pathways, depending on whether growth is on nutrient agar (A–E; Longcore et al. 1999) or inside of amphibian cells (A, F, I; Greenspan et al. 2012). On agar, the zoospore (A) encysts and forms anucleate rhizoids. Over the course of 4 days (A–D), the zoospore cyst matures into a zoosporangium that releases zoospores through discharge papillae (E). Colonial thalli divided by septae (arrow) occur occasionally (B′), and their presence has been used to confirm the identity of B. dendrobatidis. On skin, the zoospore encysts on the surface of a cell (F), and forms a germ tube (arrow), which grows through one or more host cell layers (G). The zoosporangium with sparse rhizoids forms from a swelling of the germ tube (G, H). By the time zoospores are released, the outer skin layer (arrow) has sloughed (I).
The chytridiomycosis research community has struggled to reconcile geographic and host patterns of epizootics that result from complex interactions among host, pathogen, and the environment. Overlain upon a pan‐global distribution of Bd is a set of more restricted geographic regions where Bd has caused massive loss of amphibian biodiversity, such as eastern Australia, Central America, northwestern South America, and western North America (Berger et al. 1998; Lips et al. 2006; Vredenburg et al. 2010). Surprisingly, all continents, whether they have declining or persisting amphibian populations, harbor highly similar pathogen genotypes (the global panzootic lineage [GPL]) that are highly virulent in animal models (Fisher et al. 2009b). However, numerous advances in the study of Bd in recent years have radically altered our perspective of a homogeneous pathogen and a homogeneous host response across the globe. For example, the discovery of multiple enzootic Bd lineages (Farrer et al. 2011; Schloegel et al. 2012; Bataille et al. 2013), a new, related species of Batrachochytrium specific to salamanders (Martel et al. 2013), and the suggestion of alternative pathogen niches, such as in the GI tracts of crayfish (McMahon et al. 2012), challenge the notion of pathogen homogeneity. Variation in host responses to infection is also now better understood. The idea that amphibian species are equivalent targets for Bd has been challenged by results that indicate frogs may be capable of acquiring immunity (McMahon et al. 2014) and that frog immune genotype matters for population persistence (Savage and Zamudio 2011). Recent research has also focused on the identification of mechanisms leading to variation in effects of Bd across hosts or communities, such as understanding how behavior (Venesky et al. 2011), environmental contaminants (Hanlon and Parris 2014), microbial skin communities (Bletz et al. 2013), seasonality (Longo et al. 2010), and community structure (Becker et al. 2014) influence a species' susceptibility.
Here we review how recent research has helped explain the patterns of chytridiomycosis across space, time, and host. We highlight advances in the field that accompany a shift from the panzootic phase, where emphasis was placed on surveys of the disease, to the postpanzootic phase, in which mechanistic questions are being addressed. We provide a comprehensive species distribution model for Bd in the Americas to summarize what we know about the geographic and environmental factors that control Bd distribution. We chose to emphasize the distribution of Bd in the Americas for several reasons. First, the Americas have good geographic coverage with respect to disease surveillance, genetic data, and contemporary amphibian surveys. Second, the response of host populations to Bd in the Americas is highly variable. Finally, host diversity and pathogen diversity are both high in the Americas, and the region contains many endangered species, making the study of Bd important from a conservation perspective. Our review is framed using the concept of the disease triangle (environment, host, and pathogen interactions), which highlights interactions among hosts and pathogens under different environmental conditions and is therefore an excellent framework for explaining complex infection outcomes. We outline potential research areas that will help explain the mosaic geographic patterns of morbidity left in the wake of an apparent global panzootic, and we discuss lessons learned that could be useful when considering other emerging infectious diseases.
Environmental factors predicting chytridiomycosis epidemics
The disease triangle model is commonly used to explain how variation in environmental factors, host susceptibility, and pathogen virulence lead to varying disease outcomes, yet only a small parameter space in the model results in an epidemic (Scholthof 2007; Gurr et al. 2011). We know that some aspects of chytridiomycosis epizootics show environmental correlates (Olson et al. 2013), and these are expected because temperature and precipitation affect Bd‐amphibian dynamics by physiologically limiting vital processes such as pathogen growth and host immune responses. All amphibians need moist skin for water uptake and to maintain electrolyte balance, and many species also require standing water for reproduction. Because Bd reproduces by zoospores, it requires at least water films to disperse. Bd has a surprisingly narrow optimal growth range of 17–25°C, but can tolerate temperatures between 4 and 28°C (Piotrowski et al. 2004), matching most amphibian temperature tolerance ranges.
These physiological limits likely influence Bd distribution and disease outcome, but the data reveal complex and even conflicting patterns. At the individual level, preferences for higher temperatures among hosts correlate with reduced probability of Bd infection (Rowley and Alford 2013) and warmer or drier areas serve as refugia from Bd (Puschendorf et al. 2009). At the population level, epidemics in Central America were found in the middle or end of the rainy season (Lips 1998), but the drier months of the year were associated with higher Bd prevalence in Puerto Rico (Longo et al. 2010). At the landscape level, temperature likely is responsible for a positive correlation between prevalence and both elevation (Gründler et al. 2012) and latitude (Kriger et al. 2007). On the other hand, a study in the Sierra Nevada demonstrated no relationship between elevation and temperature on Bd prevalence (Knapp et al. 2011).
Since its discovery, much effort has been spent on mapping the spatial distribution of Bd to provide a landscape view of areas with high environmental suitability. Olson et al. (2013) provided the most recent overview of the global distribution of Bd, analyzing the host and geographic patterns of 4281 individually swabbed frogs from 56 countries, of which 1814 (48%) were Bd positive by molecular detection methods. Using logistic regression, Olson et al. examined associations between Bd occurrence at a site and latitude, elevation, biome, amphibian species richness, and global temperature and precipitation metrics. While these methods are powerful to detect environmental correlations, they cannot determine whether an unsampled site may be environmentally suitable for Bd. Species distribution models (SDMs), on the other hand, predict the geographic extent of a species and identify the contribution of habitat parameters in explaining that distribution. Rödder et al. (2009) and Liu et al. (2013) developed the first Bd SDMs that were global in scope, based on 365 and 1829 Bd records, respectively. Here we present a comprehensive SDM for Bd in the Americas (Fig. 2), based on 6071 Bd positives from 30,382 analyzed swabs from fieldwork of the authors plus an intensive literature review (Box 2). The most intensively surveyed areas include western and eastern United States, Costa Rica, Panama, Puerto Rico, the Andes, and the Brazilian Atlantic Forest. Knowledge gaps include the central United States, northern Mexico, the Amazon Basin, the Brazilian Cerrado and Pantanal, and large regions in Argentina and Bolivia (Fig. 2).
Figure 2.
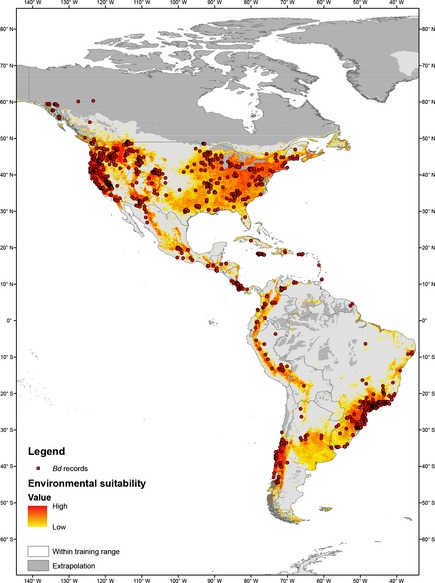
Positive records of Batrachochytrium dendrobatidis and potential distribution of the fungus according to an ensemble species distribution model. Warmer colors indicate higher probability of environmental suitability. Areas exceeding the environmental training range of the SDM are indicated in gray.
Box 2. Species Distribution Model for Bd in the Americas.
Based on the most up‐to‐date information on the realized distribution of Bd, we developed an updated SDM based on the predictions of an ensemble of eight different algorithms and both environmental factors (temperature, humidity) and land cover information (normalized difference vegetation index, NDVI), thus capturing the pathogen's Grinnellian niche as well as anthropogenic factors such as human footprint (Liu et al. 2013). The ensemble techniques within the biomod2 framework (Thuiller et al. 2014), which were employed here represent recent advances in SDM methodology, acknowledging that no single algorithm provides the best solution and that uncertainties in different steps of SDM development are best acknowledged in a comparative framework.
Environmental data: As a first step, we obtained a comprehensive set of 19 bioclimatic variables with a spatial resolution of 2.5 arc min from www.wordclim.org as well as variables characterizing seasonal changes in the normalized difference vegetation index (NDVI; derived from the data set GIMMS [global inventory modeling and mapping studies] NDVI: 1981–2006; available through www.edc.uri.edu/ATMT-DSS/data_gateway/modis/gimms.zip; NDVI scores are coded as 8 bit integer ranging from 0:255), potential evapotranspiration (Trabucco and Zomer 2009), and “human footprint” (Sanderson et al. 2002). The human footprint ranges from 0 (no human influence) to 100 (strongest human influence) and characterizes the human influence on land surface based on accessibility, anthropogenic land transformation, human population density, and electrical power infrastructure. As multicollinearity of environmental predictors may violate statistical assumptions of SDM algorithms, we computed pairwise Spearman rank correlations among all variables and selected among those pairs with R 2 < 0.75, the putatively most relevant for Bd. The final set of predictors included “Mean Diurnal Temperature Range” (Bio2; Mean of monthly (max temperature – min temperature)), “Temperature Annual Range” (Bio7), “Mean Temperature of Warmest Quarter” (Bio10), “Annual Precipitation” (Bio12), “Precipitation Seasonality” (Bio15; Coefficient of Variation of monthly Precipitation), “Precipitation of Warmest Quarter” (Bio18), “Annual Mean NDVI” (NDVI_Bio1), “NDVI Annual Range” (NDVI_Bio7), “minimum monthly potential evapotranspiration” (PET_HE_Bio6), “annual range of potential evapotranspiration” (PET_HE_Bio7), modified from monthly raw data in (Trabucco and Zomer 2009), and the “human footprint”.
Based on our review, we compiled a set of 6071 georeferenced Bd positives from 30,382 swabs from adult amphibians of 749 named species. However, for successful SDM development the spatial structure of Bd records needs to be taken into account as spatial autocorrelation may hamper inference of Bd environmental niche from distribution data. Therefore, the spatial autocorrelation structure of Bd records was assessed via a semivariogram based on Moran's I and Bd records were subsequently spatially subsampled to a minimum distance between two records of 12.11 km leaving 765 records for model development. Bd prevalence in amphibian populations was visualized by aggregating Bd‐positive and Bd‐negative records within a distance of four km and computing the percentage of positives from the total sample size.
For full details regarding biomod2 algorithms and model parameters, see Appendix S1. To account for inherent uncertainties arising from the modeling and evaluation procedures, we (1) created ten different random subsets of Bd records which were used for model calibration (70%) and evaluation (30%) and (2) created three different sets of pseudo‐absences which were randomly sampled from the environmental space available within the Americas, but outside of the realized environmental space for Bd records (SRE option in biomod2). From the 240 single models (8 algorithms * 3 pseudo‐absence data sets * 10 evaluation runs), 180 SDMs had TSS scores >0.6 (TSSaverage = 0.67; Kappaaverage = 0.43, ROCaverage = 0.90). On average, “minimum monthly potential evapotranspiration” had the highest contribution to the Bd SDMs (20.3%), followed by “human footprint” (14.4%), “Annual Precipitation” (11.5%), “Mean Temperature of Warmest Quarter” (11.7%), “Precipitation of Warmest Quarter” (8.7%), “annual range of potential evapotranspiration” (7.7%), “Temperature Annual Range” 7.2%), “Mean Diurnal Temperature Range” (7.2%), and “Annual Mean NDVI” (7.1%). The remaining variables contributed less than 5%. Response curves of the final ensemble SDM are shown in Supplementary Fig. 1.
In comparison with previous SDMs for Bd, our new model employs an ensemble model approach, which has superior performance when compared to single algorithms (Meller et al. 2014). This enables the prediction of both hot spots and cold spots of environmental suitability for Bd. The new SDM predicts that most parts of eastern and western USA, mountainous areas in Central America, the Northern Andes, lowlands of Chile, the Brazilian Atlantic Forest, and adjacent areas in Uruguay and Argentina provide suitable environmental conditions for Bd (Fig. 2). Most parts of the Amazon basin are not predicted to be suitable for Bd, most likely because their comparatively high annual mean temperatures exceed the critical thermal maximum of the pathogen. Our results highlight similar hot spots for Bd as those predicted by Rödder et al. (2009) and Liu et al. (2013), but allow more differentiation between suitable and unsuitable sites because of increased sampling efforts and the use of analytical methods that are an ensemble of different algorithms (Box 2). A major difference between this new SDM and the previous projections is the lower suitability for Bd in the Amazon basin and higher suitability for Bd in western North America relative to the earlier model by Rödder et al. (2009). Our model also differs from that of Liu et al. (2013) in showing less suitability for eastern North America. We compared the results of the SDM with the prevalence of Bd at given localities for which we had population‐level data (Fig. 3). Overall, Bd prevalence correlates well with the SDM, but neither picture is able to completely explain the epidemiological patterns observed. For example, eastern US amphibian populations have among the lowest mean prevalence (mean = 12.4%), whereas eastern Brazilian populations display among the highest prevalence (mean = 28.5%) (Fig. 3). Yet, both regions show limited evidence for Bd‐related declines.
Figure 3.

Prevalence of Batrachochytrium dendrobatidis and potential distribution of the fungus according to an ensemble species distribution model. Warmer colors indicate higher probability of environmental suitability. Prevalence was computed only for those grid cells with more than 10 samples, wherein the size of the circles represent sample size. Areas exceeding the environmental training range of the SDM are indicated in gray.
The first 15 years of chytridiomycosis surveys have limited the space of the disease triangle to a set of environmental parameters that are coincident with hot spots on the SDM. Altogether, our SDM provides a strong prediction of where Bd will occur on the basis of climate and land cover (Box 2). Yet, currently, environmental factors alone fail to provide a clear explanation for why Bd is such a problem right now or what species will be affected. Beyond basic climatic variables, we clearly need more data on how other physical, chemical, and biotic characteristics of environments predict Bd presence and disease outcome. For example, the SDM suggests significant impact of “human footprint” on Bd prevalence (Box 2), yet the mechanisms underlying this footprint need greater investigation.
Pathogen evolution, life history, and predictions of chytridiomycosis epidemiology
Another side of the disease triangle is the virulence or propagule pressure of the pathogen (Scholthof 2007). Here, we explore how both pathogen genotype and environmental niche influence the virulence and transmission of the pathogen. Early Bd studies speculated that a recent change in pathogen virulence occurred, leading to rapid spread of the pathogen across the landscape. If true, this would support the novel pathogen hypothesis (NPH) over the alternate endemic pathogen hypothesis (EPH), which posits that Bd was endemic and emerged because of environmental change (Rachowicz et al. 2005; Fisher et al. 2009b). Here, we review evidence supporting both NPH and ancient endemism and progress toward identifying a source population.
The earliest genetic studies of Bd isolates found low global variation at both microsatellite and sequenced loci, with both types of markers having only two alleles (Morgan et al. 2007; James et al. 2009). However, sampling in these initial studies was biased to localities with documented die‐offs, such as the Sierra Nevada of California, USA, Coclé, Panama, and Queensland, Australia. The absence of allelic diversity, however, supported the NPH model in which the GPL arose from a single diploid genotype, and its descendants were shaped by loss of heterozygosity (LOH). This tight bottleneck also suggested a recent emergence of chytridiomycosis from a single source population, but the lack of any geographic pattern left ample room for speculation about the source population.
Because of this potential recent emergence, researchers turned to museum specimens to connect Bd occurrence to a place and time, initially with histology (Weldon et al. 2004), and subsequently with molecular methods (Cheng et al. 2011). Based on museum specimens from southern Africa, where massive capture and export of Xenopus laevis occurred in the 20th century, Weldon et al. (2004) suggested a possible African origin of Bd, correlating the earliest occurrence of Bd in the museum record (1938) with the onset of frog exportation. This hypothesis, however, rested solely on the specimens from Africa being the oldest infected specimens known at that time. Subsequent studies have refuted the African origin by demonstrating infected museum amphibians collected considerably earlier: 1894 in the Atlantic Forest of Brazil (Rodriguez et al. 2014) and 1888 in central USA (Talley et al. 2015).
The picture of global pathogen genetic homogeneity stood until Goka et al. (2009) identified a phylogenetically novel lineage of Bd from the giant salamander in Japan. This study was the first to indicate that additional genetic diversity might exist and led to the hypothesis of an Asian origin of panzootic Bd. Soon thereafter, screening of nondeclining populations was intensified and combined with thorough molecular analyses (Box 3). Within a span of 3 years, novel genotypes putatively endemic to the Cape of South Africa (Bd‐Cape), Switzerland (Bd‐CH), Brazil (Bd‐Brazil), and Korea (Bd‐Korea) (Farrer et al. 2011; Schloegel et al. 2012; Bataille et al. 2013) were described. By increasing the sampling of Bd strains from regions where populations were not declining, it became apparent that the earlier perspective on genetic diversity was biased toward epizootic strains. Now, in addition to the globally prevalent Bd‐GPL, we know of several divergent lineages of Bd, distributed on each continent that has been surveyed. Another major breakthrough was the recent discovery of a congeneric species B. salamandrivorans (Bsal). The new species is morphologically, genetically, and functionally distinct from Bd. It was discovered as a pathogen of fire salamanders (Salamandra salamandra) in northwestern Europe (Martel et al. 2013), but was probably introduced from eastern Asia (Martel et al. 2014). The discovery of this new form pushes the association of the Batrachochytrium genus as an amphibian parasite to an age of at least 25 million years, showing that the emergence of Bd is not associated with a recent host jump to amphibians.
Box 3. Advanced molecular methods for Bd population genetics.
Our understanding of Bd population genetics in the last five years has improved through deeper sampling of isolates and an increased sampling of genetic loci. Although PCR amplification of the ribosomal internal transcribed spacer (ITS) region has been the workhorse for diagnosing Bd infections, each strain contains multiple and variable copies of the locus (Schloegel et al. 2012; Longo et al. 2013), limiting its utility as a population genetic marker. On the other hand, multilocus sequence typing or microsatellite markers provide low resolution among strains for the level of effort/expense (Morgan et al. 2007; James et al. 2009). With the reduced cost of next‐generation sequencing, population genetics by genome resequencing is starting to replace marker‐based studies (Farrer et al. 2011; Rosenblum et al. 2013). These genome resequencing studies reveal rampant loss of heterozygosity and aneuploidy or polyploidy (Rosenblum et al. 2013); however, with respect to geographic conclusions, critical patterns within the GPL are unclear because of low sample sizes and geographically disparate isolates grouping together. Currently, sequencing a single locus (e.g., BdC24 (James et al. 2009)) can typically distinguish each of the major groups of strains and increasing this to a handful of marker loci may be useful for identifying clones within populations. Extensive clonal reproduction, however, suggests that genome sequencing is unnecessary at the local level (e.g., within a stream). Nonetheless, given the limitations to obtaining cultures, it seems probable that future studies will work toward genome resequencing of many isolates. These studies must be carried out with improved methods, however, and with high sequence coverage to deal with the variable and high ploidy common in Bd. Insufficient coverage can lead to low‐quality genotyping, which increases the noise:signal ratio.
A major need is to develop marker approaches for population genetics using skin swabs. When infection levels are high, MLST markers can be genotyped from swabs (Garner et al. 2006; Velo‐Anton et al. 2012) with modest success rates. Enrichment techniques to increase the recovery of Bd DNA by hybridization to Bd‐specific probes attached to magnetic beads show promise to increase the success of genotyping from swabs when coupled with whole genome amplification (Rodriguez et al. 2012). A danger with low DNA input methods is the inability to distinguish allele drop out from true loss of heterozygosity (LOH), and LOH is exactly the information being targeted. As single‐cell genomic methods continue to develop, it is likely that future epidemiological studies can be carried out from swabs. However, first, by comparing genotypes of cultures to relevant phenotypes (e.g., morphometrics and virulence testing), we as a community must determine what genotypic information we really are after.
The finding of enzootic lineages of Bd that are more restricted in their distribution contrasts with the broad distribution and spread of the virulent genotype (Bd‐GPL). Enzootic genotypes appear to be rarer and may represent a pattern of historical genetic diversity that is in the process of being erased or outcompeted by the current panzootic. Only in Korea has Bd‐GPL been shown to have low prevalence, which might indicate that the enzootic genotype can outcompete other Bd lineages in this environment or that GPL has only recently been introduced (Bataille et al. 2013). Because >90% of strains isolated from nature are Bd‐GPL (Schloegel et al. 2012), a leading hypothesis is that Bd‐GPL is a hypervirulent genotype that is replacing the rarer, enzootic lineages. The replacement of enzootic with panzootic genotypes in nature is also consistent with infection experiments showing that strains of Bd‐Cape have lower virulence than the Bd‐GPL (Farrer et al. 2011), but several studies demonstrate that even Bd‐GPL genotypes vary in virulence when tested on common hosts (Berger et al. 2005; Fisher et al. 2009a). Unfortunately, at this point we cannot generalize that certain lineages or genotypes are less virulent because so few host species and Bd genotypes have been tested. More standardized studies are needed to quantify the pathogenicity of Bd genotypes across various hosts and continents, focusing on standardized housing, doses, and using strains with low passaging history (Kilpatrick et al. 2010; Langhammer et al. 2013). Other traits such as temperature optima, growth rates, and morphology are worth investigating across genotypes. For example, the optimum growth temperature of the newly described salamander parasite Bsal (Martel et al. 2013) is markedly lower (15 C) than that of Bd (17–25 C). Unfortunately, growth rates and temperature optima for other lineages or other isolates of Bd‐GPL have not yet been determined.
The basis for these phenotypic distinctions among lineages may be a product of genomic differences among them. A prominent feature of the GPL lineages is the presence of particular LOH events that must have occurred before the global dispersal of the GPL because they occur in every strain (Rosenblum et al. 2013); other LOH events occurred after GPL began to diversify and led to the formation of two clades within GPL, GPL‐1 and GPL‐2 (Schloegel et al. 2012). Based on the LOH model, we infer that GPL‐1 is the more ancestral variant because it differs from GPL‐2 by the absence of particular LOH events. GPL‐2 is the most common lineage in the tropics (Fig. 4) and is the genotype isolated from massive die‐offs in Central America and Australia (Berger et al. 1998). In contrast, GPL‐1 is most common in North America and is the lineage associated with epizootics of Rana muscosa in the Sierra Nevada (Schloegel et al. 2012). GPL‐1 also predominates in Europe, but has not been found in Australia or Africa. If we are correct in our inference that GPL‐1 is the ancestral panzootic lineage, this indicates that GPL first emerged in the northern temperate zone and later dispersed into the tropics. Interestingly, only rarely is the dominant tropical form (GPL‐2) found in temperate regions, and vice versa for GPL‐1. Importantly, the spatial distribution of GPL‐2 points in North America suggests a role for anthropogenic movement of frogs or pathogen (Fig. 4), because most of the GPL‐2 points represent isolates from animals in captivity, including one from Dendrobates azureus at the National Zoo in Washington, D.C. (the type strain JEL197), and another isolate from a Xenopus laevis strain imported to U.C. Berkeley from Africa in the 1980s (Morgan et al. 2007).
Figure 4.
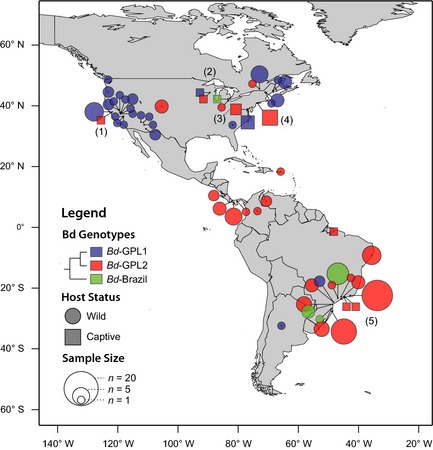
Distribution of Batrachochytrium dendrobatidis genotypes determined from multilocus sequence typing of cultured isolates. Bd clades are identified by color, and the captive status of the host amphibians is indicated by shape. The area of each shape represents the sample size of genotypes from each locality. Notable samples include the following: a captive an isolate of Bd‐GPL‐2 from Xenopus laevis imported to U.C. Berkeley, California (1), an isolate of a novel Bd‐Brazil strain from Lithobates catesbeianus in a Michigan market (2), captive isolates of GPL‐2 from the National Zoo, Washington, D.C. (3), and the Bronx Zoo and markets in New York City (4), and a region of high genetic heterogeneity in the Atlantic Forest of southeastern Brazil (5). Isolates are considered GPL‐1 if they are heterozygous or homozygous for diagnostic alleles at loci BdC24, R6046, or both. Data are compiled from published sources (Morgan et al. 2007; James et al. 2009; Schloegel et al. 2012; Velo‐Anton et al. 2012) and unpublished data.
We know surprisingly little about the requirements of various Bd life‐cycle stages; Might free‐living stages, environmental resting stages, or alternate hosts exist? This information is crucial when considering any mitigation or reintroduction program. Bd DNA was recently reported from the GI tract and the surface of crayfish [Procamberus alleni, P. clarkia, and Oronectes virilis (McMahon et al. 2012)]. Further, the authors inoculated crayfish with Bd and found higher mortality and gill recession than for controls. They also documented that Bd could be transmitted from crayfish to larval amphibians. The possibility of a saprobic reservoir for Bd has been discussed since the description of the species, when it was noted that because Bd was able to grow in pure culture on nutrient media and limitedly on snake skin, it might also be able to live saprobically in nature (Longcore et al. 1999). Bd has survived in autoclaved lake water for 6 weeks, and for 3 weeks in autoclaved tap water (Johnson and Speare 2003). The lack of living microbes, protists, and small invertebrates in these experiments, however, makes inferences about Bd survival in nature difficult. Although tested in sterile conditions, the ability of Bd to remain viable in natural sources of water and on keratinaceous substrates supports the view that the environment can, at least temporarily, support the viability of Bd outside of amphibian hosts. Moreover, the detection of Bd DNA throughout the entire year from filtered North American water samples suggests the persistence of the fungus during a time when amphibians are dormant (Chestnut et al. 2014). In pure culture, Bd sporangia develop from a zoospore without a germ tube first being formed (Fig. 1); this differs from development in vivo with a germ tube, as shown by transmission electron microscopy (Greenspan et al. 2012). Development without forming a germ tube is a feature of many chytrid species that form a sporangium on the top of their substrate, whereas formation of a germ tube is characteristic of chytrid species that develop a sporangium within their substrate. The presence of these two developmental pathways, the ability to grow in pure culture, and the presence of Bd DNA in environmental sources all predict saprobic reproduction, yet a nonliving reservoir for Bd has yet to be identified. Identifying such reservoirs will be important to understanding the effects of Bd on amphibian populations, as demonstrated by model predictions of the long‐term dynamics of Bd with and without a hypothesized saprobic phase (Mitchell et al. 2008).
The past decade of studies on the pathogen has uncovered deeper complexity in pathogen genotype, phenotype, and novel biotic interactions. Combined, the data indicate a hypervirulent lineage that is primarily responsible for epizootics; however, we still lack a clear indication of why and from where the Bd‐GPL lineage emerged, and we lack good studies on the phenotypes and virulence of newly discovered enzootic lineages. Genetic studies of Bd and other pathogens suggest the origin of Bd‐GPL will not be found by looking at sites of die‐offs, and therefore, the hunt for genetically diverse source populations from nondeclining populations is a high research priority. Ideally, these genetic diversity surveys would use markers ascertainable from DNA extracted from swabbed animals, but such a marker system has proven difficult to implement (Velo‐Anton et al. 2012) (Box 3). Other priorities involve investigating pathogen genotype dynamics (such as hybridization and competition) in regions where multiple genotypes coexist, such as Mallorca and the Brazilian Atlantic Forest (Walker et al. 2008; Farrer et al. 2011; Schloegel et al. 2012). Studies are also needed to confirm whether nonamphibian hosts are part of the life cycle or just a dead end, and what substrates might support saprobic life styles.
Variation in host disease susceptibility and disease dynamics
The final side of the disease triangle is the host, and here we review evidence that variance in host traits can explain susceptibility to Bd. Despite our greater knowledge of amphibian hosts, with over 7400 species described, the immunology, distribution, and ecology of many species are still poorly characterized. Field and laboratory studies document large variation in susceptibility and resistance to Bd across species (Lips et al. 2006; Crawford et al. 2010; Searle et al. 2011b; Gahl et al. 2012). Multiple factors most likely lead to this variation, including host differences in innate and acquired immunological response, host associated microbes, and behavioral and life‐history traits. Behavioral, life history, and habitat traits have taken center stage because these characteristics are easy to measure. The emerging consensus is that life‐history traits matter, such as lower susceptibility in direct developers that lack an aquatic larval stage (Kriger and Hero 2007; Bielby et al. 2008). However, these generalizations do not sufficiently capture differences in host range that could be predictive of epizootics. Here we focus on recent findings showing that community composition and individual species genetic variation may influence not only the outcome of infection but also the potential for evolution of resistance.
The role of amphibian community composition in regulating Bd dynamics has been addressed from the perspectives of host diversity and identity. One potential consequence of high species richness is a “dilution effect” resulting in reduced risk of disease (Keesing et al. 2006). Dilution effects occur because more diverse communities should be buffered from epizootics of generalist pathogens because encounters and potential transmission will often occur between susceptible and resistant hosts. Tests for dilution effects in the amphibian‐Bd system have been conducted by several authors with results showing host diversity decreases (Searle et al. 2011a; Becker et al. 2014; Venesky et al. 2014), increases (Becker and Zamudio 2011), or has no impact (Liu et al. 2013) on the risk of infection. All laboratory studies have shown a dilution effect, whereas these effects are more difficult to detect in the field, perhaps due to the differences in both host diversity and habitat complexity. One laboratory study (Becker et al. 2014) found that host diversity decreased Bd infection due to changes in species interactions, specifically by reducing shared habitat use and transmission among hosts. Additionally, one particular terrestrial species showed reduced infection loads in diverse assemblages at the expense of neighboring aquatic hosts becoming heavily infected. Therefore, despite the fact that Bd is a highly generalist pathogen, these findings show the importance of understanding community‐wide transmission dynamics and species‐specific interactions for predicting disease outcome.
The idiosyncratic results from testing the dilution effect suggest that species diversity may be less important than the presence of particular species in a community. Amphibians that are highly susceptible to Bd, like Atelopus zeteki, can function as “acute supershedders,” thus amplifying disease transmission (DiRenzo et al. 2014). Non‐native, Bd‐tolerant species, such as the American bullfrog (Lithobates catesbeianus) and the African clawed frog (Xenopus laevis), may function as reservoir species, those carrier species that are highly tolerant of infections. Importantly, these two invasive species have been implicated in the global spread of the disease (Daszak et al. 2001; Vredenburg et al. 2013). In Colorado, where L. catesbeianus is invasive, the density of L. catesbeianus was positively correlated with Bd infection prevalence and load in co‐occurring native fauna (Peterson and McKenzie 2014). On the other hand, a recent study failed to find evidence of increased Bd infection on native UK fauna due to the presence of invasive and Bd‐infected Xenopus (Tinsley et al. 2015). These different results across regions highlight the important interactions between amphibian communities and environmental factors in determining infection outcomes.
Moreover, variable outcomes of infection are observed in the field, with some populations persisting after the arrival of Bd, while others go extinct (Briggs et al. 2010). These variable outcomes suggest potential differences in host genotype and prompt the question of the potential for host evolution of increased resistance or tolerance to Bd. Amphibians can rely on innate and adaptive immune responses to manage Bd infections, and at least some of these immune responses have a genetic basis (Savage and Zamudio 2011; Ellison et al. 2014), suggesting host genotypic variation may be an important factor explaining persistence or mortality. For instance, alleles of the major histocompatibility complex (MHC), an important family of genes in the adaptive immune response, were significantly associated with resistance and survival in Lithobates yavapaiensis (Savage and Zamudio 2011) and Litoria verreauxii (Bataille et al. 2015). Various immunogenetic studies have reported either a strong or weak adaptive immune response post‐Bd infection (Rosenblum et al. 2012; Ellison et al. 2014), underscoring variation among species in their potential for the evolution of resistance or tolerance. Recently, a study suggested both adaptive behavioral avoidance and partial immunity could be acquired following Bd exposure (McMahon et al. 2014), although vaccination by prior infection has not proven effective in at least one species (Cashins et al. 2013). Altogether, the data suggest natural variation in both pathogen virulence and host immunity, but the interactions between these two components have not been adequately addressed to allow predictions of which species or communities have the potential to recover after exposure to Bd.
The epizootic space of the disease triangle largely excludes direct developing species lacking a larval stage, aggressive invasive species, and those with a large clutch size (Bielby et al. 2008). However, communities diverse and species‐poor alike have suffered declines, and predicting the outcome of Bd infection for any given species remains elusive. Rare species may have lower genetic variation for parasite resistance, and the role of genetic variation in buffering disease through genetic fitness correlations needs to be better explored (Allentoft and O'Brien 2010). What happens in a resistant response is largely unknown, but now that we know that there is meaningful variation in immunological response, further research can also address variation in immunogenetic diversity across species with a range of susceptibilities (Ellison et al. 2015).
Future research prognosis: cold spots, rather than hot spots, may be the key to understanding enigmatic disease
Improved modeling techniques and additional survey data have refined our understanding of Bd distribution and prevalence across the globe. Bd has a broad distribution that is correlated with colder temperatures and more moist environments (Fig. 2), yet distribution maps and SDMs highlight important, yet often enigmatic details about the distribution of Bd. First, the SDM identifies a number of hot spots including high‐elevation forests in Central America, the Sierra Nevada, and the Brazilian Atlantic Forest (Fig. 2). These are regions of the world where we know Bd is already present at high prevalence, and the disease largely seems to have become enzootic, although not all of these regions have suffered declines. Second, the Bd distribution model contains several cold spots, such as the Amazon Basin and the Great Plains region of North America. Although the Amazon Basin is an apparent cold spot for Bd based on climatic variables, Bd has been detected there (McCracken et al. 2009). Based on the results of our SDM, we suggest the time is right to rephrase questions regarding distribution of Bd to: “Where are the cold spots in Bd distribution?” and “Why are they cold?” Now, studies are needed to identify regions and populations where Bd is absent to learn about the biotic and abiotic mechanisms underlying this distribution. More surveys in tropical regions are clearly warranted given the diversity of amphibians in these regions and the relative paucity of studies. Moreover, studies at smaller spatial and temporal scales are needed to understand environmental regulation and transmission patterns that lead to variation in community‐level prevalence.
We have defined cold spots as regions where the pathogen is absent or predicted to be absent, or where it occurs at low prevalence; these spots may exist for a number of reasons. First, they could be artifacts due to limited sampling of habitats within that specific niche parameter space. However, as global Bd survey has progressed in the last decade, these potential sampling artifacts are becoming less likely. Second, Bd may never have dispersed there. Third, environmental conditions may be outside of Bd's tolerance window. Lastly, Bd might historically have been present but the frogs have evolved defenses (or only the resistant species remain through pathogen‐driven selection), and Bd later disappeared or persists at low population prevalence.
The existence of Bd cold spots raises a number of questions. If they are the result of environmental restriction, why is the fungus unable to adapt to higher temperatures? More experimental work is needed to understand physiological plasticity and propensity for local adaptation in Bd. In the laboratory, changes in virulence and other phenotypes have been noted over time in culture, indicating that the fungus can adapt rapidly (Langhammer et al. 2013; Voyles et al. 2014a). The distribution of Bd, which includes warm tropical lowland forests, is inconsistent with the high level of growth inhibition seen in the laboratory at 28°C. A major research need is characterizing differences in the fungus by the collection, genotyping, and temperature profiling of strains from these habitats seemingly outside of the Bd physiological envelope.
Cold spots could also represent areas within the distribution of Bd that appear to be hot in terms of prevalence, but cold in terms of negative effects on the fitness of the amphibian hosts. These regions, including eastern Brazil, Chile, and eastern North America, apparently have not experienced species declines despite widespread Bd occurrence and high prevalence of infection. Because Bd has only been known for 15 years, however, missing baseline data may be obscuring proper inferences, and the absence of mass mortalities is certainly not evidence that disease‐related declines did not occur. Indeed, species in Brazil, Chile, and Wisconsin of the United States underwent enigmatic declines near the time that Bd was implicated in declines in nearby areas (Hine et al. 1981; Eterovick et al. 2005; Soto‐Azat et al. 2013). Gradual declines of amphibian species are especially hard to detect because populations naturally fluctuate and quantitative population data are lacking (Adams et al. 2013). Some extant amphibian communities might be remnants, that is, amphibian communities affected by epizootics before the last three decades of high vigilance. If so, could these surviving communities show signs of “The Ghost of Epizootics Past” and how could we distinguish them?
The theory of disease ecology predicts that when a disease enters a naïve population, lack of host immunity often results in epizootics, characterized by high intensity and high prevalence of disease; in contrast, in enzootic scenarios the pathogen is predicted to be present at lower levels of infection once hosts and pathogens have reached an equilibrium state in susceptibility and infection (Ewald 1994). Over time, enzootic pathogens are expected to coevolve with their hosts and adapt to their shared environment. Given the long‐term co‐existence of Bd and amphibians (Rodriguez et al. 2014; Talley et al. 2015) and the presence of putative enzootic Bd lineages, the amphibians we see today may be postepizootic relics that have adapted to coexist with their now enzootic Bd lineage.
If the Ghost of Epizootics Past exists, we should be able to detect it using community and population genetic data. These methods can be tuned to identify signatures expected in communities that suffered epizootics by comparison with control communities for which we have clear evidence against disease‐related declines. Possible regions for defining control expectations are Far East Asia and the Amazon basin, which have well‐studied amphibian fauna with no evidence of declines. Korea is one compelling control population because it essentially only has enzootic Bd genotypes present, and exhibits high prevalence with low loads, suggesting that it has not suffered epizootics (Bataille et al. 2013).
We expect that communities that have undergone declines may have lost lineages or species with increased susceptibility to the disease, which can be tracked by comparative analyses across communities. One expected signature is the loss of specific susceptible hosts from amphibian communities. For example, postenzootic communities may be enriched for terrestrial breeders, large clutch sizes, and small body size, given the relationship of these variables and Bd infection observed across communities (Kriger and Hero 2007; Bielby et al. 2008). Analyses of community diversity using traits as variables could allow identification of outliers with less diversity than expected in particular traits (e.g., breeding behaviors) relative to control communities. Other less typical phenotypes could also be analyzed, such as correlates of innate immunity (e.g., antimicrobial peptide production), skin microbial communities, or average genetic diversity (heterozygosity) of populations or species. As an example, phenotype frequencies from killing assays where immune cells of animals are challenged with bacteria have been shown to change following an epizootic in Florida scrub jays caused by an unknown pathogen (Wilcoxen et al. 2010).
Phylogenetic methods analyzing the distribution of species in regional amphibian fauna may also help identify communities that are phylogenetically overdispersed or underdispersed relative to expectations (Cavender‐Bares et al. 2009), as if many of the leaves had been pruned by disease. Similar phylogenetic methods could be applied to candidate genes, such as the MHC genes (Savage and Zamudio 2011), where tree‐based methods could detect patterns that deviate from the default signature of balancing selection (Schierup et al. 2001), which is expected to occur if epizootics were selected for particular alleles by directional selection as evidenced in field studies (Savage and Zamudio 2011; Bataille et al. 2015). A particularly exciting prospect is that community‐level approaches may have the advantage of leveraging museum collections, which extend deep into the early days of exploration of the New World, to characterize control populations. Shifts in taxonomic richness over time can test for selective loss of clades, as Bd prevalence appears to be nonrandomly phylogenetically distributed (Baláž et al. 2014). Testing the predictors we outline could utilize Panamanian and Peruvian communities where declines and extirpations are well documented (Crawford et al. 2010; Catenazzi et al. 2011).
These approaches are not easy to implement, so what other signatures might allow us to detect an earlier, community changing epizootic? While we cannot resurrect extinct populations that have experienced historical declines, we can utilize animals in extant communities with this signature of enzootic disease and test their current susceptibility to the pathogen. Under predictions of the Ghost of Epizootics Past model, the surviving animals should be adapted to resist endemic pathogen genotypes, but testing them against foreign genotypes should reveal higher susceptibility. This calls for inoculation studies, for example, of Brazilian endemic herpetofauna, using enzootic strains of Bd from all groups: Bd‐GPL, Bd‐Brazil, Bd‐Cape, and Bd‐Korea. If coevolution following epizootics has occurred, the surviving amphibians in Brazil will be highly susceptible only to the latter two strain types, which are absent from the native range. In contrast, if the populations have not experienced adaptation, the lineages present will be those with the highest virulence. Determining the traits responsible for this adaptation may be a key to understanding the signatures of the Ghost of Epizootics Past.
Lessons learned
Despite the many unanswered questions remaining regarding Bd and its interaction with amphibian hosts, major advances have occurred in the last 15 years. These advances have improved our preparedness to document and prevent future emergence of infectious diseases of wildlife (Voyles et al. 2014b). We end this review with four unanticipated lessons learned.
Generalist pathogens can be a cause of extinction
The most important lesson is that an infectious disease can be a major cause of biodiversity loss. Before chytridiomycosis emerged, we had only a handful of examples where infectious disease was linked to severe declines or extinctions, including the American elm, the American chestnut, and six animal cases (Collins and Crump 2009). As a result, conservation biologists generally ignored pathogens as a cause of extinction. During the amphibian chytridiomycosis crisis, we may have lost dozens of species and witnessed severe population declines of hundreds more. That a pathogen can decimate vertebrate populations of multiple species has highlighted the importance of studies in taxonomy and systematics, not only in terms of urgency, but because knowledge on all aspects of biodiversity is critical in preparing us for future outbreaks.
Most emerging infectious diseases (EIDs) of wildlife are host generalists: Chytridiomycosis, rabies, white nose syndrome (WNS), West Nile virus, avian cholera, and snake fungal disease all have a wide number of hosts, making them a significant threat to both biodiversity and ecosystem functions. However, impacts across host species are highly variable due to biotic and abiotic determinants. For example, just as with chytridiomycosis, WNS occurs in seven bat species but only populations of the four most gregarious species are endangered by the disease (Langwig et al. 2012). However, as we have highlighted here, EIDs caused by host generalists require consideration of the whole host community because of the presence of amplifier species and the possibility of dilution effects.
Fungal diseases are on the rise
Although the underlying causes are unclear, fungal EIDs of wildlife (including WNS, snake fungal disease, sea fan disease, and chytridiomycosis) appear to be emerging faster than those caused by bacteria, viruses, and protozoa (Fisher et al. 2012). Common themes of fungal EIDs include being host generalists, invaders of soft tissues rather than blood, and typically on ectotherms or the colder extremities of endotherms (such as the wings and noses of hibernating bats). As is the case for Bd, environmental filtering also plays a role in other wildlife fungal diseases, such WNS, where the pathogen grows optimally at the same temperatures which are typically found in bat hibernacula (Blehert et al. 2009). The presence of facultative saprobic life cycles or environmental reservoirs may also be a common thread that ties together fungal EIDs, allowing pathogens to continue transmission even after causing host mortality (Fisher et al. 2012). These saprobic phases would also facilitate dispersal because they eliminate the need for hosts. As many of the fungal EIDs (e.g., WNS, chytridiomycosis, and snake fungal disease) appear to have evolved from saprobic ancestors, this may perhaps explain their necrotrophic pathogenicity with gross tissue destruction.
The emergences of a number of fungal EIDs, such as chytridiomycosis, WNS, and sudden oak death, are hypothesized to follow a recent introduction and movement of pathogens. Therefore, it may be that a sudden increase in transmissibility or movement of fungi is what has led to this increase in fungal EIDs. Though not readily obvious, these diseases can spread through international movement of infected hosts, such as when infected chestnut trees were brought to the New York area and led to the epidemic that nearly led to their extinction (Anagnostakis and Hillman 1992). Evidence to support these point introductions comes from characterization of low pathogen genetic diversity. As observed for Bd, the rapid emergence of WNS in eastern North America is due to a single clonal genotype of Pseudogymnoascus destructans, presumably introduced from Europe (Ren et al. 2012). These examples of recent spread indicate that we should consider the ever‐increasing potential for anthropogenic movement of pathogen propagules as an explanation for the global rise in fungal EIDs.
Baseline data are vital for defining epidemics
Generally, we suffer from a lack of long‐term data on wildlife populations, which severely impedes the detection of epidemics. How often do declines occur? A meta‐analysis of frog declines conducted early in the days of Bd research (Houlahan et al. 2000) was only able to draw from data from 1950 to 2000. These data suggested a downward slide of amphibian populations since 1960, but in the absence of copious data from before 1960, it is difficult to know how early this trend began. Wildlife and plant monitoring programs need to be supported so that when declines and epizootics occur, baseline data are in place for comparison. The relatively new North American Amphibian Monitoring Program aims at cataloging trends in amphibian populations across the continent using citizen scientists. However, this program is solely based on frog calls, and we need more efforts in monitoring of disease and its effects using molecular diagnostics and mark–recapture studies. Such efforts would expedite our response to epizootics and form the basis for proactive rather than reactive science.
Baseline population and community data can also be drawn from museum collections, and examples exist in which new bird species of the Brazilian Atlantic Forest were described using museum specimens, yet the same species had already become locally extinct. (Lees and Pimm 2015). Museum specimens not only yield locality records, but also allow detection of time periods in which Bd increased in prevalence (Cheng et al. 2011). Recently, museum specimens have been used to time introductions of particular genotypes of Aphanomyces astaci causing epizootics of crayfish plague in Norway beginning in 1971 (Vrålstad et al. 2014). Studies of crayfish plague and of Bd in the Brazilian Atlantic Forest (Rodriguez et al. 2014) that utilize genetic markers provide information regarding gene flow and a more precise migration history. To increase the utility of material for microbial work, collectors should make sure to preserve the integrity of samples for pathogen DNA analysis and histology, and given the importance of amphibian skin and its microbial communities, future amphibian collectors should consider archiving swabs or skin samples with accessioned specimens.
Never stop sampling
Major breakthroughs in understanding the genetic diversity of Bd occurred only after many years of sampling. Understanding biases in sampling and geographic coverage is essential for identifying source populations and rare, but informative, genotypes. Likewise, important information on the history and virulence of Bd will require sampling additional genes (Box 3). Meaningful genetic variation goes deeper than sequence polymorphism. Both chromosome number variation and LOH are highly variable in Bd at the same time that sequence variation is extremely low. We recently characterized GPL‐1 and GPL‐2 sublineages, and these, as well as the enzootic lineages, have geographic and genomic patterns in great need of further exploration. The first 10 years of sampling only revealed GPL (James et al. 2009); the last five years uncovered five additional lineages of Bd, including a clearly sexually produced isolate (Schloegel et al. 2012), and a new species with a different host range (Martel et al. 2013). Sampling biases should also serve as a cautionary tale. For example, failure to detect Bsal in North America does not necessarily mean that the species, or a close relative, is not present. In fact, it seems unlikely that Bsal or a closely related species is not in North America given that it diverged from Bd so long ago and thus has existed for at least 25 million years. An alternative is that we just are unable to detect it and that the Old World Bsal is analogous to Bd‐GPL, only restricted to salamanders. If anything, our previous experience indicates that we need to keep sampling to find additional lineages of Bsal that we may likely be missing.
Conflict of Interest
None declared.
Supporting information
Figure S1. Response curves of the ensemble species distribution model for Bd.
Appendix S1. Algorithms and model options for biomod2.
Acknowledgments
This paper stemmed from a joint US National Science Foundation (OISE‐1159513) and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP 2011/51694‐7) project to catalyze international collaboration between Brazil and the United States. LFT thanks the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for grants (FAPESP 2011/51694‐7; CNPq 405285/2013‐2) and fellowships (CNPq 302589/2013‐9). TYJ and LFT acknowledge support from a grant through the USFWS Amphibians Without Borders Program (F12AP00997) and a fellowship from CNPq (300980/2014‐0). We thank Danelle Larson (Russell) and Mike Lannoo for contributing Bd field prevalence data and Guilherme Becker for helpful discussions on the dilution effect.
Ecology and Evolution 2015; 5(18): 4079–4097
References
- Adams, M. J. , Miller D. A. W., Muths E., Corn P. S., Grant E. H. C., Bailey L. L., et al. 2013. Trends in amphibian occupancy in the United States. PLoS ONE 8:e64347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allentoft, M. E. , and O'Brien J.. 2010. Global amphibian declines, loss of genetic diversity and fitness: a review. Diversity 2:47–71. [Google Scholar]
- Anagnostakis, S. L. , and Hillman B.. 1992. Evolution of the chestnut tree and its blight. Arnoldia 52:3–10. [Google Scholar]
- Baláž, V. , Vörös J., Civiš P., Vojar J., Hettyey A., Sós E., et al. 2014. Assessing risk and guidance on monitoring of Batrachochytrium dendrobatidis in Europe through identification of taxonomic selectivity of infection. Conserv. Biol. 28:213–223. [DOI] [PubMed] [Google Scholar]
- Bataille, A. , Fong J. J., Cha M., Wogan G. O. U., Baek H. J., Lee H., et al. 2013. Genetic evidence for a high diversity and wide distribution of endemic strains of the pathogenic chytrid fungus Batrachochytrium dendrobatidis in wild Asian amphibians. Mol. Ecol. 22:4196–4209. [DOI] [PubMed] [Google Scholar]
- Bataille, A. , Cashins S. D., Grogan L., Skerratt L. F., Hunter D., McFadden M., et al. 2015. Susceptibility of amphibians to chytridiomycosis is associated with MHC class II conformation. Proc. Biol. Sci. 282:20143127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, C. G. , and Zamudio K. R.. 2011. Tropical amphibian populations experience higher disease risk in natural habitats. Proc. Natl. Acad. Sci. USA 108:9893–9898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, C. G. , Rodriguez D., Toledo L. F., Longo A. V., Lambertini C., Correa D. T., et al. 2014. Partitioning the net effect of host diversity on an emerging amphibian pathogen. Proc. Biol. Sci. 281:20141796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger, L. , Speare R., Daszak P., Green D. E., Cunningham A. A., Goggin C. L., et al. 1998. Chytridiomycosis causes amphibian mortality associated with population declines in the rain forests of Australia and Central America. Proc. Natl. Acad. Sci. USA 95:9031–9036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger, L. , Marantelli G., Skerratt L. L., and Speare R.. 2005. Virulence of the amphibian chytrid fungus Batrachochytrium dendrobatidis varies with the strain. Dis. Aquat. Organ. 68:47–50. [DOI] [PubMed] [Google Scholar]
- Bielby, J. , Cooper N., Cunningham A. A., Garner T. W. J., and Purvis A.. 2008. Predicting susceptibility to future declines in the world's frogs. Conserv. Lett. 1:82–90. [Google Scholar]
- Blehert, D. S. , Hicks A. C., Behr M., Meteyer C. U., Berlowski‐Zier B. M., Buckles E. L., et al. 2009. Bat white‐nose syndrome: An emerging fungal pathogen? Science 323:227–227. [DOI] [PubMed] [Google Scholar]
- Bletz, M. C. , Loudon A. H., Becker M. H., Bell S. C., Woodhams D. C., Minbiole K. P. C., et al. 2013. Mitigating amphibian chytridiomycosis with bioaugmentation: characteristics of effective probiotics and strategies for their selection and use. Ecol. Lett. 16:807–820. [DOI] [PubMed] [Google Scholar]
- Briggs, C. J. , Knapp R. A., and Vredenburg V. T.. 2010. Enzootic and epizootic dynamics of the chytrid fungal pathogen of amphibians. Proc. Natl. Acad. Sci. USA 107:9695–9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashins, S. D. , Grogan L. F., McFadden M., Hunter D., Harlow P. S., Berger L., et al. 2013. Prior infection does not improve survival against the amphibian disease chytridiomycosis. PLoS ONE 8:e56747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catenazzi, A. , Lehr E., Rodriguez L. O., and Vredenburg V. T.. 2011. Batrachochytrium dendrobatidis and the collapse of anuran species richness and abundance in the Upper Manu National Park, southeastern Peru. Conserv. Biol. 25:382–391. [DOI] [PubMed] [Google Scholar]
- Cavender‐Bares, J. , Kozak K. H., Fine P. V. A., and Kembel S. W.. 2009. The merging of community ecology and phylogenetic biology. Ecol. Lett. 12:693–715. [DOI] [PubMed] [Google Scholar]
- Cheng, T. L. , Rovito S. M., Wake D. B., and Vredenburg V. T.. 2011. Coincident mass extirpation of neotropical amphibians with the emergence of the infectious fungal pathogen Batrachochytrium dendrobatidis . Proc. Natl. Acad. Sci. USA 108:9502–9507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chestnut, T. , Anderson C., Popa R., Blaustein A. R., Voytek M., Olson D. H., et al. 2014. Heterogeneous occupancy and density estimates of the pathogenic fungus Batrachochytrium dendrobatidis in waters of North America. PLoS ONE 9:e106790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, J. P. , and Crump M. L.. 2009. Extinction in our times. Global amphibian decline. Oxford University Press, Oxford. [Google Scholar]
- Crawford, A. J. , Lips K. R., and Bermingham E.. 2010. Epidemic disease decimates amphibian abundance, species diversity, and evolutionary history in the highlands of central Panama. Proc. Natl. Acad. Sci. USA 107:13777–13782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daszak, P. , Cunningham A. A., and Hyatt A. D.. 2001. Anthropogenic environmental change and the emergence of infectious diseases in wildlife. Acta Trop. 78:103–116. [DOI] [PubMed] [Google Scholar]
- DiRenzo, G. V. , Langhammer P. F., Zamudio K. R., and Lips K. R.. 2014. Fungal infection intensity and zoospore output of Atelopus zeteki, a potential acute chytrid supershedder. PLoS ONE 9:e93356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison, A. R. , Savage A. E., DiRenzo G. V., Langhammer P., Lips K. R., and Zamudio K. R.. 2014. Fighting a losing battle: vigorous immune response countered by pathogen suppression of host defenses in the chytridiomycosis‐susceptible frog Atelopus zeteki . G3 (Bethesda), 4:1275–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison, A. R. , Tunstall T., DiRenzo G. V., Hughey M. C., Rebollar E. A., Belden L. K., et al. 2015. More than skin deep: Functional genomic basis for resistance to amphibian chytridiomycosis. Genome Biol. Evol. 7:286–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eterovick, P. C. , Carnaval A., Borges‐Nojosa D. M., Silvano D. L., Segalla M. V., and Sazima I.. 2005. Amphibian declines in Brazil: an overview. Biotropica 37:166–179. [Google Scholar]
- Ewald, P. W. 1994. Evolution of infectious disease. Oxford University Press, Oxford. [Google Scholar]
- Farrer, R. A. , Weinert L. A., Bielby J., Garner T. W., Balloux F., Clare F., et al. 2011. Multiple emergences of genetically diverse amphibian‐infecting chytrids include a globalized hypervirulent recombinant lineage. Proc. Natl. Acad. Sci. USA 108:18732–18736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, M. C. , Bosch J., Yin Z., Stead D. A., Walker J., Selway L., et al. 2009a. Proteomic and phenotypic profiling of the amphibian pathogen Batrachochytrium dendrobatidis shows that genotype is linked to virulence. Mol. Ecol. 18:415–429. [DOI] [PubMed] [Google Scholar]
- Fisher, M. C. , Walker S. F., and Garner T. W. J.. 2009b. The global emergence of Batrachochytrium dendrobatidis in space, time, and host. Annu. Rev. Microbiol. 63:291–310. [DOI] [PubMed] [Google Scholar]
- Fisher, M. C. , Henk D. A., Briggs C. J., Brownstein J. S., Madoff L. C., McCraw S. L., et al. 2012. Emerging fungal threats to animal, plant and ecosystem health. Nature 484:186–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahl, M. K. , Longcore J. E., and Houlahan J. E.. 2012. Varying responses of northeastern North American amphibians to the chytrid pathogen Batrachochytrium dendrobatidis . Conserv. Biol. 26:135–141. [DOI] [PubMed] [Google Scholar]
- Garner, T. W. J. , Perkins M. W., Govindarajulu P., Seglie D., Walker S., Cunningham A. A., et al. 2006. The emerging amphibian pathogen Batrachochytrium dendrobatidis globally infects introduced populations of the North American bullfrog, Rana catesbeiana . Biol. Lett. 2:455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenspan, S. E. , Longcore J. E., and Calhoun A. J. K.. 2012. Host invasion by Batrachochytrium dendrobatidis: fungal and epidermal ultrastructure in model anurans. Dis. Aquat. Organ. 100:201–210. [DOI] [PubMed] [Google Scholar]
- Gründler, M. C. , Toledo L. F., Parra‐Olea G., Haddad C. F. B., Giasson L. O. M., Sawaya R. J., et al. 2012. Interaction between breeding habitat and elevation affects prevalence but not infection intensity of Batrachochytrium dendrobatidis in Brazilian anuran assemblages. Dis. Aquat. Organ. 97:173–184. [DOI] [PubMed] [Google Scholar]
- Goka, K. , Yokoyama J., Une Y., Kuroki T., Suzuki K., Nakahara M., Kobayashi A., Inaba S., Mizutani T., and Hyatt A. D.. 2009. Amphibian chytridiomycosis in Japan: distribution, haplotypes and possible route of entry into Japan. Mol. Ecol. 18:4757‐4774. [DOI] [PubMed] [Google Scholar]
- Gurr, S. , Samalova M., and Fisher M.. 2011. The rise and rise of emerging infectious fungi challenges food security and ecosystem health. Fungal Biol. Rev. 25:181–188. [Google Scholar]
- Hanlon, S. M. , and Parris M. J.. 2014. The interactive effects of chytrid fungus, pesticides, and exposure timing on gray treefrog (Hyla versicolor) larvae. Environ. Toxicol. Chem. 33:216–222. [DOI] [PubMed] [Google Scholar]
- Hine, R. L. , Les B. L., and Hellmich B. F.. 1981. Leopard frog populations and mortality in Wisconsin, 1974–1976. Technical Bulletin No. 122, Department of Natural Resources, Madison, Wisconsin. [Google Scholar]
- Houlahan, J. E. , Findlay C. S., Schmidt B. R., Meyer A. H., and Kuzmin S. L.. 2000. Quantitative evidence for global amphibian population declines. Nature 404:752–755. [DOI] [PubMed] [Google Scholar]
- James, T. Y. , Litvintseva A. P., Vilgalys R., Morgan J. A. T., Taylor J. W., Fisher M. C., et al. 2009. Rapid expansion of an emerging fungal disease into declining and healthy amphibian populations. PLoS Pathogens 5:e1000458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, M. L. , and Speare R.. 2003. Survival of Batrachochytrium dendrobatidis in water: Quarantine and disease control implications. Emerg. Infect. Dis. 9:922–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, K. E. , Patel N. G., Levy M. A., Storeygard A., Balk D., Gittleman J. L., et al. 2008. Global trends in emerging infectious diseases. Nature 451:990–U4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keesing, F. , Holt R. D., and Ostfeld R. S.. 2006. Effects of species diversity on disease risk. Ecol. Lett. 9:485–498. [DOI] [PubMed] [Google Scholar]
- Kilpatrick, A. M. , Briggs C. J., and Daszak P.. 2010. The ecology and impact of chytridiomycosis: an emerging disease of amphibians. Trends Ecol. Evol. 25:109–118. [DOI] [PubMed] [Google Scholar]
- Knapp, R. A. , Briggs C. J., Smith T. C., and Maurer J. R.. 2011. Nowhere to hide: impact of a temperature‐sensitive amphibian pathogen along an elevation gradient in the temperate zone. Ecosphere, 2(8): doi:10.1890/ES11‐00028.1 [Google Scholar]
- Kriger, K. M. , and Hero J. M.. 2007. The chytrid fungus Batrachochytrium dendrobatidis is non‐randomly distributed across amphibian breeding habitats. Divers. Distrib. 13:781–788. [Google Scholar]
- Kriger, K. M. , Pereoglou F., and Hero J. M.. 2007. Latitudinal variation in the prevalence and intensity of chytrid (Batrachochytrium dendrobatidis) infection in Eastern Australia. Conserv. Biol. 21:1280–1290. [DOI] [PubMed] [Google Scholar]
- Langhammer, P. F. , Lips K. R., Burrowes P. A., Tunstall T., Palmer C. M., and Collins J. P.. 2013. A fungal pathogen of amphibians, Batrachochytrium dendrobatidis, attenuates in pathogenicity with in vitro passages. PLoS ONE 8:e77630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langwig, K. E. , Frick W. F., Bried J. T., Hicks A. C., Kunz T. H., and Kilpatrick A. M.. 2012. Sociality, density‐dependence and microclimates determine the persistence of populations suffering from a novel fungal disease, white‐nose syndrome. Ecol. Lett. 15:1050–1057. [DOI] [PubMed] [Google Scholar]
- Lees, A. C. , and Pimm S. L.. 2015. Species, extinct before we know them? Curr. Biol. 25:R177–R180. [DOI] [PubMed] [Google Scholar]
- Lips, K. R. 1998. Decline of a tropical amphibian fauna. Conserv. Biol. 12:106–117. [Google Scholar]
- Lips, K. R. , Brem F., Brenes R., Reeve J. D., Alford R. A., Voyles J., et al. 2006. Emerging infectious disease and the loss of biodiversity in a Neotropical amphibian community. Proc. Natl. Acad. Sci. USA 103:3165–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Rohr J. R., and Li Y. M.. 2013. Climate, vegetation, introduced hosts and trade shape a global wildlife pandemic. Proc. Biol. Sci., 280, doi: 10.1098/rspb.2012.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longcore, J. E. , Pessier A. P., and Nichols D. K.. 1999. Batrachochytrium dendrobatidis gen et sp nov, a chytrid pathogenic to amphibians. Mycologia 91:219–227. [Google Scholar]
- Longo, A. V. , Burrowes P. A., and Joglar R. L.. 2010. Seasonality of Batrachochytrium dendrobatidis infection in direct‐developing frogs suggests a mechanism for persistence. Dis. Aquat. Organ. 92:253–260. [DOI] [PubMed] [Google Scholar]
- Longo, A. V. , Rodriguez D., Leite D. D., Toledo L. F., Almeralla C. M., Burrowes P. A., et al. 2013. ITS1 copy number varies among Batrachochytrium dendrobatidis strains: Implications for qPCR estimates of infection intensity from field‐collected amphibian skin swabs. PLoS ONE 8:e59499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel, A. , Spitzen‐van der Sluijs A., Blooi M., Bert W., Ducatelle R., Fisher M. C., et al. 2013. Batrachochytrium salamandrivorans sp nov causes lethal chytridiomycosis in amphibians. Proc. Natl. Acad. Sci. USA 110:15325–15329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel, A. , Blooi M., Adriaensen C., Van Rooij P., Beukema W., Fisher M. C., et al. 2014. Recent introduction of a chytrid fungus endangers Western Palearctic salamanders. Science 346:630–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken, S. , Gaertner J. P., Forstner M. R. J., and Hahn D.. 2009. Detection of Batrachochytrium dendrobatidis in amphibians from the forest floor to the upper canopy of an Ecuadorian Amazon lowland rainforest. Herpetol. Rev. 40:190. [Google Scholar]
- McMahon, T. A. , Brannelly L. A., Chatfield M. W. H., Johnson P. T. J., Joseph M. B., McKenzie V. J., et al. 2012. Chytrid fungus Batrachochytrium dendrobatidis has nonamphibian hosts and releases chemicals that cause pathology in the absence of infection. Proc. Natl. Acad. Sci. USA 110:210–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon, T. A. , Sears B. F., Venesky M. D., Bessler S. M., Brown J. M., Deutsch K., et al. 2014. Amphibians acquire resistance to live and dead fungus overcoming fungal immunosuppression. Nature 511:224–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller, L. , Cabeza M., Pironon S., Barbet‐Massin M., Maiorano L., Georges D., et al. 2014. Ensemble distribution models in conservation prioritization: from consensus predictions to consensus reserve networks. Divers. Distrib. 20:309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, K. M. , Churcher T. S., Garner T. W. J., and Fisher M. C.. 2008. Persistence of the emerging pathogen Batrachochytrium dendrobatidis outside the amphibian host greatly increases the probability of host extinction. Proc. Biol. Sci. 275:329–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monastersky, R. 2014. Biodiversity: Life–a status report. Nature, 516:158–161. [DOI] [PubMed] [Google Scholar]
- Morgan, J. A. T. , Vredenburg V. T., Rachowicz L. J., Knapp R. A., Stice M. J., Tunstall T., et al. 2007. Population genetics of the frog‐killing fungus Batrachochytrium dendrobatidis . Proc. Natl. Acad. Sci. USA 104:13845–13850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson, D. H. , Aanensen D. M., Ronnenberg K. L., Powell C. I., Walker S. F., Bielby J., et al. 2013. Mapping the global emergence of Batrachochytrium dendrobatidis, the amphibian chytrid fungus. PLoS ONE 8:e56802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson, A. C. , and McKenzie V. J.. 2014. Investigating differences across host species and scales to explain the distribution of the amphibian pathogen Batrachochytrium dendrobatidis . PLoS ONE 9:e107441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotrowski, J. S. , Annis S. L., and Longcore J. E.. 2004. Physiology of Batrachochytrium dendrobatidis, a chytrid pathogen of amphibians. Mycologia 96:9–15. [PubMed] [Google Scholar]
- Puschendorf, R. , Carnaval A. C., VanDerWal J., Zumbado‐Ulate H., Chaves G., Bolanos F., et al. 2009. Distribution models for the amphibian chytrid Batrachochytrium dendrobatidis in Costa Rica: proposing climatic refuges as a conservation tool. Divers. Distrib. 15:401–408. [Google Scholar]
- Rachowicz, L. J. , Hero J. M., Alford R. A., Taylor J. W., Morgan J. A. T., Vredenburg V. T., et al. 2005. The novel and endemic pathogen hypotheses: Competing explanations for the origin of emerging infectious diseases of wildlife. Conserv. Biol. 19:1441–1448. [Google Scholar]
- Ren, P. , Haman K. H., Last L. A., Rajkumar S. S., Keel M. K., and Chaturvedi V.. 2012. Clonal spread of Geomyces destructans among bats, midwestern and southern United States. Emerg. Infect. Dis. 18:883–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rödder, D. , Kielgast J., Bielby J., Schmidtlein S., Bosch J., Garner T. W. J., et al. 2009. Global amphibian extinction risk assessment for the panzootic chytrid fungus. Diversity 1:52–66. [Google Scholar]
- Rodriguez, D. , Longo A. V., and Zamudio K. R.. 2012. Magnetic capture hybridization of Batrachochytrium dendrobatidis genomic DNA. J. Microbiol. Methods 90:156–159. [DOI] [PubMed] [Google Scholar]
- Rodriguez, D. , Becker C. G., Pupin N. C., Haddad C. F. B., and Zamudio K. R.. 2014. Long‐term endemism of two highly divergent lineages of the amphibian‐killing fungus in the Atlantic Forest of Brazil. Mol. Ecol. 23:774–787. [DOI] [PubMed] [Google Scholar]
- Rosenblum, E. B. , Poorten T. J., Settles M., and Murdoch G. K.. 2012. Only skin deep: shared genetic response to the deadly chytrid fungus in susceptible frog species. Mol. Ecol. 21:3110–3120. [DOI] [PubMed] [Google Scholar]
- Rosenblum, E. B. , James T. Y., Zamudio K. R., Poorten T. J., Ilut D., Rodriguez D., et al. 2013. Complex history of the amphibian‐killing chytrid fungus revealed with genome resequencing data. Proc. Natl. Acad. Sci. USA 110:9385–9390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley, J. J. L. , and Alford R. A.. 2013. Hot bodies protect amphibians against chytrid infection in nature. Sci. Rep. 3:1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson, E. W. , Jaiteh M., Levy M. A., Redford K. H., Wannebo A. V., and Woolmer G.. 2002. The human footprint and the last of the wild. Bioscience 52:891–904. [Google Scholar]
- Savage, A. E. , and Zamudio K. R.. 2011. MHC genotypes associate with resistance to a frog‐killing fungus. Proc. Natl. Acad. Sci. USA 108:16705–16710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierup, M. H. , Mikkelsen A. M., and Hein J.. 2001. Recombination, balancing selection and phylogenies in MHC and self‐incompatibility genes. Genetics 159:1833–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloegel, L. M. , Toledo L. F., Longcore J. E., Greenspan S. E., Viera C. A., Lee M., et al. 2012. Novel, panzootic, and hybrid genotypes of amphibian chytridiomycosis associated with the bullfrog trade. Mol. Ecol. 21:5162–5177. [DOI] [PubMed] [Google Scholar]
- Scholthof, K. B. G. 2007. The disease triangle: pathogens, the environment and society. Nat. Rev. Microbiol. 5:152–156. [DOI] [PubMed] [Google Scholar]
- Searle, C. L. , Biga L. M., Spatafora J. W., and Blaustein A. R.. 2011a. A dilution effect in the emerging amphibian pathogen Batrachochytrium dendrobatidis . Proc. Natl. Acad. Sci. USA 108:16322–16326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searle, C. L. , Gervasi S. S., Hua J., Hammond J. I., Relyea R. A., Olson D. H., et al. 2011b. Differential host susceptibility to Batrachochytrium dendrobatidis, an emerging amphibian pathogen. Conserv. Biol. 25:965–974. [DOI] [PubMed] [Google Scholar]
- Soto‐Azat, C. , Valenzuela‐Sanchez A., Collen B., Rowcliffe J. M., Veloso A., and Cunningham A. A.. 2013. The population decline and extinction of Darwin's frogs. PLoS ONE 8:e66957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talley, B. L. , Muletz C. R., Vredenburg V. T., Fleischer R. C., and Lips K. R.. 2015. A century of Batrachochytrium dendrobatidis in Illinois amphibians (1888‐1989). Biol. Conserv. 182:254–261. [Google Scholar]
- Thuiller, W. , Georges D., and Engler R.. 2014. biomod2: Ensemble platform for species distribution modeling. R package version 3.1‐64. http://CRAN.R-project.org/package=biomod2.
- Tinsley, R. C. , Coxhead P. G., Stott L. C., Tinsley M. C., Piccinni M. Z., and Guille M. J.. 2015. Chytrid fungus infections in laboratory and introduced Xenopus laevis populations: assessing the risks for U.K. native amphibians. Biol. Conserv. 184:380–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabucco, A. , and Zomer R. J.. 2009. Global Aridity Index (Global‐Aridity) and Global Potential Evapo‐Transpiration (Global‐PET) Geospatial Database. In. Published online, available from the CGIAR‐CSI GeoPortal at: http://www.csi.cgiar.org/ CGIAR Consortium for Spatial Information.
- Velo‐Anton, G. , Rodriguez D., Savage A. E., Parra‐Olea G., Lips K. R., and Zamudio K. R.. 2012. Amphibian‐killing fungus loses genetic diversity as it spreads across the New World. Biol. Conserv. 146:213–218. [Google Scholar]
- Venesky, M. D. , Kerby J. L., Storfer A., and Parris M. J.. 2011. Can differences in host behavior drive patterns of disease prevalence in tadpoles? PLoS ONE 6:e24991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venesky, M. D. , Liu X., Sauer E. L., and Rohr J. R.. 2014. Linking manipulative experiments to field data to test the dilution effect. J. Anim. Ecol. 83:557–565. [DOI] [PubMed] [Google Scholar]
- Voyles, J. , Johnson L. R., Briggs C. J., Cashins S. D., Alford R. A., Berger L., et al. 2014a. Experimental evolution alters the rate and temporal pattern of population growth in Batrachochytrium dendrobatidis, a lethal fungal pathogen of amphibians. Ecol. Evol. 4:3633–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voyles, J. , Kilpatrick A. M., Collins J. P., Fisher M. C., Frick W. F., McCallum H., et al. 2014b. Moving beyond too little, too late: Managing emerging infectious diseases in wild populations requires international policy and partnerships. EcoHealth, 2014:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrålstad, T. , Strand D. A., Grandjean F., Kvellestad A., Håstein T., Knutsen A. K., et al. 2014. Molecular detection and genotyping of Aphanomyces astaci directly from preserved crayfish samples uncovers the Norwegian crayfish plague disease history. Vet. Microbiol. 173:66–75. [DOI] [PubMed] [Google Scholar]
- Vredenburg, V. T. , Knapp R. A., Tunstall T. S., and Briggs C. J.. 2010. Dynamics of an emerging disease drive large‐scale amphibian population extinctions. Proc. Natl. Acad. Sci. USA 107:9689–9694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vredenburg, V. T. , Felt S. A., Morgan E. C., McNally S. V. G., Wilson S., and Green S. L.. 2013. Prevalence of Batrachochytrium dendrobatidis in Xenopus collected in Africa (1871–2000) and in California (2001–2010). PLoS ONE 8:e63791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, S. F. , Bosch J., James T. Y., Litvintseva A. P., Valls J. A. O., Piña S., et al. 2008. Introduced pathogens threaten species recovery programs. Curr. Biol. 18:R853–R854. [DOI] [PubMed] [Google Scholar]
- Weldon, C. , du Preez L. H., Hyatt A. D., Muller R., and Speare R.. 2004. Origin of the amphibian chytrid fungus. Emerg. Infect. Dis. 10:2100–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcoxen, T. E. , Boughton R. K., and Schoech S. J.. 2010. Selection on innate immunity and body condition in Florida scrub‐jays throughout an epidemic. Biol. Lett. 6:552–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Response curves of the ensemble species distribution model for Bd.
Appendix S1. Algorithms and model options for biomod2.