

Abstract
Tripartite motif protein 25 (TRIM25), mediates K63-linked polyubiquitination of Retinoic acid inducible gene I (RIG-I) that is crucial for downstream antiviral interferon signaling. Here, we demonstrate that TRIM25 is required for melanoma differentiation-associated gene 5 (MDA5) and MAVS mediated activation of NF-κB and interferon production. TRIM25 is required for the full activation of NF-κB at the downstream of MAVS, while it is not involved in IRF3 nuclear translocation. Mechanical studies showed that TRIM25 is involved in TRAF6-mediated NF-κB activation. These collectively indicate that TRIM25 plays an additional role in RIG-I/MDA5 signaling other than RIG-I ubiquitination via activation of NF-κB.
Keywords: MAVS, MDA5, NF-κB, TRAF6, TRIM25
INTRODUCTION
Retinoic acid inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) are members of the DExD/H box RNA helicase family, and act as intracellular RNA sensors detecting viral RNA to elicit antiviral immune responses including production of type-I interferons (Kato et al., 2006; Yoneyama et al., 2004). RIG-I has been implicated in recognizing various clinically important viruses including influenza virus and hepatitis C virus. MDA5 plays a crucial role in antiviral immune responses against picornaviruses and paramyxoviruses (Kato et al., 2006; Loo et al., 2008; Nakhaei et al., 2009; Sumpter et al., 2005). RIG-I and MDA5 share similar domain structures: two N-terminal caspase activation and recruitment domain (CARD) domains, a central helicase domain and a C-terminal domain. Once RIG-I or MDA5 recognize viral RNAs, they undergo a conformational change to enable interaction with MAVS/IPS-1/VISA/Cardif, a critical downstream adaptor protein located at the mitochondrial membrane (Kawai et al., 2005; Meylan et al., 2005; Seth et al., 2005; Xu et al., 2005). Activation of MAVS leads to the recruitment of signaling complexes such as the TANK-binding kinase 1 (TBK1) complex and I kappa B kinase (IKK) complex to activate transcription factors including IRF3/7 and NF-κB (Goubau et al., 2013; Ramos and Gale, 2011).
Upon recognition of viral RNA, RIG-I undergoes K63-linked polyubiquitination catalyzed by tripartite motif protein 25 (TRIM25), an E3-ubiquitin ligase (Gack et al., 2007). Since the delivery of K63-linked polyubiquitin to RIG-I is required for interaction between RIG-I and MAVS (IPS-1, VISA, Cardiff), a crucial downstream adaptor protein, TRIM25 is critical for RIG-I-mediated antiviral signaling and interferon production (Gack et al., 2007). Various viruses, including influenza A virus and Kaposi’s sarcoma-associated human herpesvirus (KSHV), have evolved strategies to target this pivotal ubiquitination of RIG-I (Gack et al., 2009; Inn et al., 2011b). An alternative mechanism of RIG-I activation suggested by Zeng et al. (2010) involves RIG-I activation by interaction of the its CARD domain with unanchored K63-linked polyubiquitin chains generated by TRIM25 catalyzation instead of covalent conjugation to RIG-I. Although the suggested mechanism of RIG-I activation is different, their study confirms the critical roles of TRIM25 and K63-linked polyubiquitin chains in RIG-I activation.
While recognition of RNA and activation of RIG-I have been extensively studied, little is known about RNA recognition and activation of MDA5. It has been shown that MDA5 preferentially recognizes long stable double-stranded RNA and higher order RNA complexes, but the exact endogenous ligand for MDA5 remains elusive (Kato et al., 2008; Pichlmair et al., 2009). Although it is known that RIG-I and MDA5 share the same MAVS-mediated downstream signaling pathway, the details of the activation process of MDA5 have not been extensively studied. In this study, we investigated the potential role of TRIM25 in the MDA5-mediated antiviral signaling pathway and demonstrate the positive regulatory role of TRIM25.
MATERIALS AND METHODS
Plasmids and reagents
GST-RIG-2CARD (RIG-IN), MDA5-2CARD (MDA5N; aa1-295), MAVS, eGFP-IRF3 and TRIM25 plasmids were previously described (Gack et al., 2007; Wies et al., 2013). Influenza A NS1 wild-type and E96A/E97A mutant were described elsewhere (Gack et al., 2009). TBK1, IKKα, IKKβ, and IKKγ constructs were kindly provided by Dr. Jae U. Jung. Poly I:C was purchased from Invivogen.
Cell lines
Human embryonic kidney (HEK293T) cells were maintained in Dulbecco’s modified Eagle’s (DMEM) media containing 10% fetal bovine serum (FBS) and penicillin/streptomycin (100 U/ml). Wild-type (WT) and TRIM25-knockout mouse embryonic fibroblasts (MEFs) were previously described (Gack et al., 2007).
Luciferase assay
Luciferase assays were conducted using Interferon-β promoter reporter, NF-κB reporter, and interferon-sensitive responsive element (ISRE) reporter plasmids to analyze activation of Interferon-β signaling pathway. All the luciferase assays were perfomed using a Dual-luciferase assay kit according to the er’s protocol (Promega). HEK293T cells and Mouse embryonic fibroblasts (MEFs) were transfected with the indicated plasmids and reporter plasmids. To deplete TRIM25 from HEK293T cells, pSUPER.retro.puro plasmids encoding shRNAs for TRIM25 were transfected. Details of the shRNA sequence and tion method have been described previously (Gack et al., 2007; Wies et al., 2013). All reporter plasmids were described previously (Inn et al., 2011a). Data were normalized by Tymidine-kinase (TK)-Renilla lucilferase activity to compensate for transfection efficiency. For polyI:C stimulation, polyI:C (2 μg/well, 6-well plates) was transfected 24 h after reporter plasmids transfection using Lipofectamine2000 (Invitrogen) according to the manufacturer’s instructions.
RNA extraction and quantitative RT-PCR (RT-qPCR)
Synthesis of interferon-β was analyzed by RT-qPCR using CFX-9000 (Bio-Rad) real-time PCR. Total RNA was extracted and cDNA was reverse transcribed from 1 μg RNA by Superscript III reverse transcriptase (Invitrogen) and oligo20(dT) primers. Quantitative PCR was performed using 2 μl of synthesized cDNA as a template. IFN-β was amplified using the primer pair 5′-GAACTTTGACATCCCTGAGGAGATT-3′ (forward) and 5′-TGCGGCGTCCTCCTTCT-3′ (reverse). Human IFN-β mRNA levels were normalized to those of human β-actin, amplified with the primer pair 5′-TGCCGCATCCTCTTCCTC-3′ (forward) and 5′-CGCCTTCACCGTTCCAGT-3′ (reverse). TRIM25 knock-down efficiency was analyzed using the primer pair 5′-AGCAGCTACAACAAGAATACACG-3′ (forward) and 5-GGCTCTGTTCAATCTCCTCCT-3′.
Analysis of IRF3 nucleus translocation
HEK293T cells were transfected with eGFP-IRF3 and the indicated plasmids or shRNAs. Twenty four hours after transfection, cells were transfected with polyI:C (2 μg/ml) using Lipofectamin2000 (Invitrogen) according to the manufacturer’s instructions. Cells were further incubated for 12 h and then fixed with 4% paraformaldehyde for 20 min. Nuclei were stained with DAPI. Nuclear localization of eGFP-IRF3 was examined under fluorescent microscope. Six fields were randomly chosen from two different slides for each group. More than three hundreds cells were examined.
co-IP (co-immunoprecipitation) and immunoblotting assay
HEK293T cells were cotransfected with the indicated plasmids and then lysed with Triton X-100 lysis buffer (25 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5% Triton X-100) containing protease inhibitor cocktail (Pierce). After centrifugation, supernatants were incubated at 4°C with the indicated antibodies overnignt. Mixtures were further incubated with protein A/G beads for 2–4 h. After washing with lysis buffer, bound proteins were eluted with 1X sample buffer and subjected to SDS-PAGE and immunoblotting.
Statistical analysis
Statistical comparisons between the control and treated groups were performed using the Student’s t-test. A value of p ≤ 0.05 was considered to be significant.
RESULTS
TRIM25 is involved in MDA5-mediated antiviral signaling
It has been shown that commercial polyI:C is mainly recognized by MDA5 rather than RIG-I (Kato et al., 2006; 2008). Thus, we initially tested the effect of depletion of TRIM25 on polyI:C-induced interferon-β production. Knock-down of TRIM25 using shRNAs resulted in a decrease of interferon-β mRNA synthesis in polyI:C transfected cells (Fig. 1A). To confirm the activation of MDA5 signaling activity by TRIM25, TRIM25 was co-expressed with MDA5-2Card (MDA5N), a constitutively active form of MDA5. As shown in Figs. 1B and 1C, TRIM25 enhanced both RIG-IN- and MDA5N-mediated interferon-β promoter activity and NF-κB transcriptional activity to a similar extent. In addition, MDA5N-mediated interferon-β promoter activity in TRIM25 deficient cells (TRIM25−/− MEF) was much lower compared to wild-type MEF, suggesting that TRIM25 is required for efficient activation of the MDA5 signaling pathway (Fig. 1D).
Fig. 1.
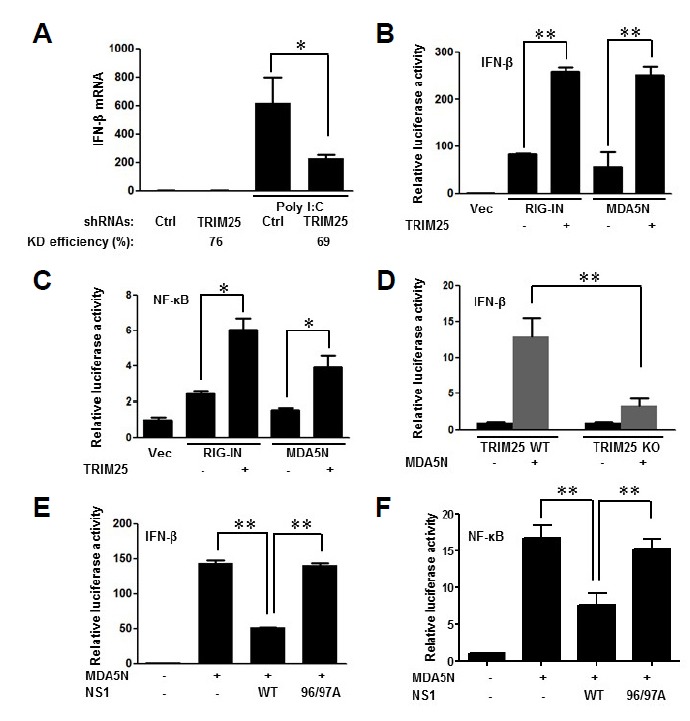
TRIM25 is involved in MDA5-mediated antiviral signaling. (A) Effect of TRIM25 depletion on polyI:C induced interferon-β synthesis. HEK293T cells were transfected with control shRNA or shRNAs against TRIM25. Cells were transfected with polyI:C 24 h later. Cells were lysed at 6 h after transfection and analyzed for interferon (IFN)-β mRNA levels by RT-qPCR, as described in “Materials and Methods”. KD efficiency (%): Knock-down efficiency compared to control (Ctrl). (B) Enhanced RIG-I and MDA5-mediated IFN-β promoter activity by ectopic expression of TRIM25. HEK293T cells were transfected with a RIG-IN, MDA5N expression plasmids or control vector together with vector or TRIM25 expression plasmids as indicated. Cells were co-transfected with IFN-β promoter-reporter and TK-Renilla reporter plasmids. Promoter activities were determined by Dual-Luciferase assay 16 h after transfection. (C) Enhanced RIG-I and MDA5-mediated NF-κB promoter activity by ectopic expression of TRIM25. HEK293T cells were transfected with expression plasmids and NF-κB reporter plasmids as indicated. Promoter activities were determined using procedures similar to those in (B). (D) Requirement of TRIM25 for efficient activation IFN-β promoter by MDA5. Wild-type (WT) and TRIM25 knock-out (TRIM25 KO) MEFs were transfected with vector or MDA5N plasmids together with reporter plasmids. IFN-β promoter activity was determined by luciferase assay as describe above. (E) Inhibition of MDA5 signaling by influenza A NS1. HEK293T cells were transfected with vector or MDA5N plasmids together with wild-type influenza A NS1 (WT) or mutant NS1 (E96A/E97A) as indicated. IFN-β promoter activity was determined by luciferase assay as describe above. Results of experiments, performed in triplicate, are presented as means ± standard deviation. *p < 0.05, **p < 0.01
Previously, it was reported that Non-structural protein 1 (NS1) of influenza A virus suppresses RIG-I-mediated antiviral signaling through disruption of TRIM25 (Gack et al., 2009). To further confirm the activity of TRIM25 on MDA5 signaling, we tested whether influenza A NS1 can suppress MDA5 signaling. As shown in Figs. 1E and 1F, ectopic expression of NS1 suppressed interferon-β promoter and NF-κB promoter activation induced by MDA5N. In addition, the NS1 E96/97A mutant (Gack et al., 2009), which does not interact with TRIM25 and therefore cannot inhibit TRIM25, did not exert inhibitory activity on MDA5N-mediated signaling (Figs. 1E and 1F).
Role of TRIM25 in MAVS-mediated signaling
Upon activation of RIG-I and MDA5, both are recruited to MAVS, a critical downstream molecule. The effect of TRIM25 on MAVS-induced interferon signaling was investigated to determine whether TRIM25 acts upstream or downstream of MAVS. MAVS-induced interferon-β promoter activity and NF-κB transcriptional activity in TRIM25 deficient MEFs were lower than those in wild-type MEFs, indicating that TRIM25 may act downstream of MAVS (Figs. 2A and 2B). In addition, ectopic expression of TRIM25 augmented MAVS-induced activation of interferon-β promoter activity and NF-κB transcriptional activities (Figs. 2C and 2D).
Fig. 2.
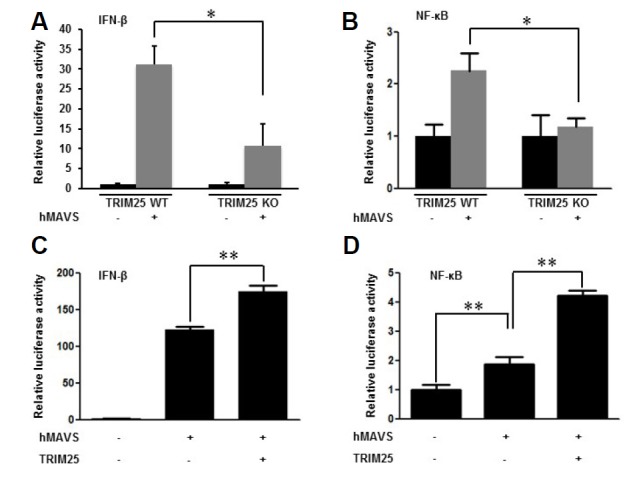
Role of TRIM25 in MAVS-mediated signaling. (A) TRIM25 is required for efficient activation of IFN-β promoter by MAVS. Wild-type (WT) and TRIM25 knock-out (TRIM25 KO) MEFs were transfected with vector or MAVS plasmids (hMAVS). Cells were co-transfected with IFN-β promoter-reporter and TK-Renilla reporter plasmids. Promoter activities were determined by Dual-Luciferase assay 16 h after transfection. (B) TRIM25 is required for efficient activation of NF-κB promoter by MAVS. WT and TRIM25 KO MEFs were transfected with vector or MAVS plasmids together with NF-κB reporter and TK-Renilla reporter plasmids. Promoter activities were determined using procedures similar to those in (A). (C) Enhanced MAVS-induced IFN-β promoter activity by TRIM25 overexpression. HEK293T cells were transfected with vector, MAVS and TRIM25 plasmids as indicated, together with IFN-β reporter and TK-Renilla reporter plasmids. Promoter activities were determined using procedures similar to those in (A). (D) Enhanced MAVS-induced NF-κB promoter activity by TRIM25 overexpression. HEK293T cells were transfected with expression plasmids and NF-κB reporter plasmids as indicated. Promoter activities were determined using procedures similar to those in (C). Results of experiments, performed in triplicate, are presented as means ± standard deviation.
Activation of TRAF6-mediated NF-κB by TRIM25
Upon activation of RIG-I or MDA5 signaling pathways, IKK and TBK complexes are recruited to MAVS to activate NF-κB and IRF3/7 transcriptional factors, respectively. To further elucidate the action of TRIM25 in the MDA5-MAVS signaling pathway, we examined the effect of TRIM25 on IRF3 nuclear localization and NF-κB activation. As depicted in Fig. 3A, ectopic expression or depletion of TRIM25 did not significantly change GFP-IRF3 nuclear translocation upon polyI:C transfection. Furthermore, ectopic expression of TRIM25 or TRIM25 RINGCS mutant which lose its E3-ligase activity did not affect the MAVS-induced IRF3 phosphorylation (Fig. 3B). Also, TBK1-induced activation of ISRE was not affected by ectopic expression of TRIM25 (Fig. 3C). Since we have shown that MDA5-MAVS induced NF-κB activation was enhanced by the ectopic expression of TRIM25 (Figs. 1D and 2D) and TRIM25 is required for efficient MDA5-MAVS-mediated NF-κB activation (Fig. 2B), it is likely that TRIM25 is involved in the activation of NF-κB via the MDA5-MAVS signaling pathway. Furthermore, a physical interaction between TRIM25 and IKK complex component proteins (IKKα, IKKβ, and IKKγ) was detected by coimmunoprecipitation assay in an overexpression system (Fig. 3D). Interestingly, phosphorylation of NF-κB (p65) induced by ectopic expression of IKKα or IKKβ was not decreased by depletion of TRIM25, suggesting that TRIM25 working at the level between MAVS and IKK complex to regulate NF-κB signaling (Fig. 3E).
Fig. 3.
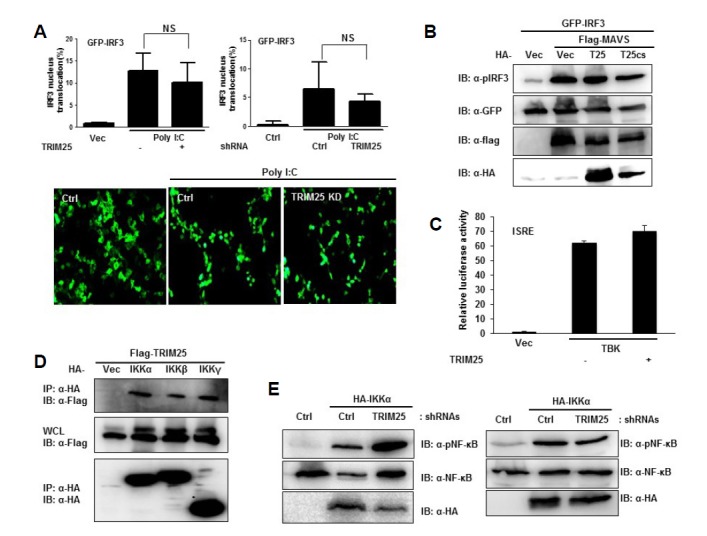
TRIM25 is dispensible for MAVS-mediated IRF3 activation. (A) Role of TRIM25 in polyI:C-induced IRF3 nuclear translocation. HEK293T cells were transfected with eGFP-IRF3 together with vector or TRIM25 expression plasmids. Cells were re-transfected with polyI:C 24 h after the first transfection. Localization of eGFP-IRF3 was examined under fluorescent microscopy (upper left panel). To see the effect of TRIM25 depletion, HEK293T cells were transfected with eGFP-IRF3 together with control shRNA (Ctrl) or shRNAs against TRIM25 (Right panel; TRIM25 KD). Three randomly chosen fields were examined for each group. Lower panel shows representative images from the shRNA experiment. NS. Not significant. (B) TRIM25 ectopic expression does not affect MAVS-induced IRF3 phosphorylation. HEK293T cells were transfected with indicated plasmids. Cell lysates were subjected to immunoblotting using indicated antibodies. (C) TRIM25 does not enhance TBK1-induced ISRE promoter activation. HEK293T cells were transfected with indicated plamids and ISRE reporter plasmid. ISRE promoter activity was analyzed by dual-luciferase assay. (D) Interaction between TRIM25 and IKK complex proteins. TRIM25 physically interacts with the IKK complex. HEK293T cells were transfected with Flag-TRIM25 and vector, HA-IKKα, HA-IKKβ, or HA-IKKγ. The 24 h after transfection, cell lysates were subjected to immunoprecipitation using an anti-HA antibody and analyzed by immunoblotting using the indicated antibodies. (E) TRIM25 does not affect the NF-κB (p65) phosphorylation upon IKKα or IKKβ expression. HEK293T cells were transfected with indicated plasmids and shRNAs. Knock-down efficiency was confirmed by measuring TRIM25 mRNA using RT-qPCR. Cell lysates were subjected to immunoblotting using indicated antibodies.
Previously, it has been shown that TRAF6 is crucial for RIG-I-mediated NF-κB activation (Yoboua et al., 2010; Yoshida et al., 2008). To further delineate the role of TRIM25 in MAVS-mediated NF-κB activation, we analyzed the role of TRIM25 in TRAF6-mediated NF-κB activation. As depicted in Figs. 4A and 4B, ectopic expression of TRIM25 enhanced TRAF6-mediated NF-κB activation, whereas depletion of TRIM25 resulted in suppression of TRAF6-mediated NF-κB activation. Furthermore, ubiquitination of TRAF6, which is critical for its activity, was reduced by depletion of TRIM25, further supporting the role of TRIM25 in TRAF6-mediated downstream NF-κB activation (Fig. 4C). To examine whether the E3-ubiqutin ligase enzyme activity of TRIM25 is required for its role in TRAF6-mediated NF-κB signaling, we compared the activities of wild-type and TRIM25CS (C50S/C53S), a mutant TRIM25 that does not carry E3-ubiquitin ligase activity. TRIM25CS was unable to enhance TRAF6-mediated NF-κB activation, suggesting that E3-ubiquitin ligase activity is required for its downstream NF-κB activating function (Fig. 4D).
Fig. 4.
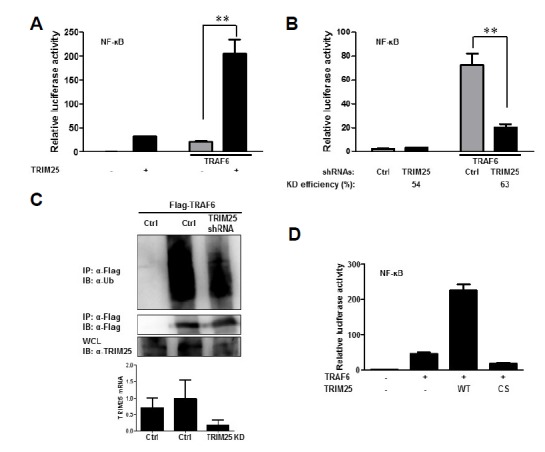
TRIM25 is involved in TRAF6-mediated NF-κB signaling. (A) Enhanced TRAF6-induced NF-κB activation by ectopic expression of TRIM25. HEK293T cells were transfected with plasmids as indicated. Cells were cotransfected with NF-κB luciferase reporter and TK-Renilla reporter plasmids. Promoter activities were determined by Dual-Luciferase assay 16 h after transfection. (B) Suppression of TRAF6-induced NF-κB activation by depletion of TRIM25. HEK293T cells were transfected with control shRNA (Ctrl) or shRNAs targeting TRIM25 as indicated together with reporter plasmids. Promoter activities were determined using procedures similar to those in (A). Knock-down efficiency was confirmed by measuring TRIM25 mRNA using RT-qPCR. (C) Role of TRIM25 in TRAF6 ubiquitination. HEK293T cells were transfected with flag-TRAF6 together with control shRNA (Ctrl) or shRNAs targeting TRIM25. Cell lysates were subjected to immunoprecipitation using an anti-flag antibody and analyzed by immunoblotting using the indicated antibodies to examine the level of TRAF6 ubiquitination. Suppression of TRIM25 mRNA synthesis was confirmed by RT-qPCR. (D) E3-ligase activity of TRIM25 is required for TRAF6-induced NF-κB activation. HEK293T cells were transfected with wild-type (WT) and E3-ligase activity dead C50S/C53S mutant (CS) TRIM25 together with vector or TRAF6 plasmid. NF-κB luciferase reporter and TK-Renilla reporter plasmids were co-transfected. Promoter activities were determined by Dual-Luciferase assay 16 h after transfection.
DISCUSSION
TRIM25 has been demonstrated to play a critical role in RIG-I-mediated antiviral signaling (Gack et al., 2007; Zeng et al., 2010). Previously, it was shown that TRIM25-mediated K63 linked polyubiquitination of RIG-I Lys172 is critical for its activation, linked polyubiquitination of RIG-I Lys172 is critical for its activation, although another study showed that non-covalently bound K63-linked polyubiquitin chain was sufficient to activate RIG-I signaling (Gack et al., 2007; Zeng et al., 2010). Whichever the case, the critical role of TRIM25 and K63-linked polyubiquitination has been proven by these studies. Ubiquitination of RIG-I results in oligomerization and interaction with MAVS through CARD-CARD homotypic interaction (Jiang et al., 2012). However, it is not clear if TRIM25 and/or K63-linked polyubiquitina tion is involved in MDA5 activation and downstream signaling pathway. So far, there is no evidence of direct conjugation of ubiquitin chain on the CARD domains of MDA5, and Lysine172, which is critical for RIG-I ubiquitination, is not conserved in MDA5 (Gack et al., 2007). Although an interaction between MDA5 and K63-linked ubiquitin has been reported, another group showed that MDA5 can be directly activated by forming a filamentous structure around ds-RNA in a ubiquitin-independent manner (Jiang et al., 2012; Wu et al., 2013).
In this study, we examined the role of TRIM25 in the MDA5 signaling pathway. Studies using ectopic expression and TRIM25 depleted cells clearly showed that TRIM25 is involved in not only RIG-I but also MDA5 signaling pathway. In addition, influenza A NS1 protein, which has been shown to inhibit RIG-I signaling by disrupting TRIM25 oligomerization, also suppressed MDA5 signaling. Since both RIG-I and MDA5 shares the MAVS-mediated signaling to activate IRF3 and NF-κB, it is conceivable that TRIM25 may have additional role at the downstream of MAVS in addition to RIG-I ubiquitination. Indeed, MAVS-induced interferon signaling was augmented by TRIM25 overexpression and suppressed by the depletion of TRIM25, indicating that TRIM25 may function at the level of or downstream of MAVS. MAVS has been shown to turn on TBK1-IRF3 signaling axis and TRAF6-IKK signaling pathways to activate IRF3 and NF-κB, respectively. In experiments to examine which signaling axis is affected, TRIM25 did not significantly affect the activation of TBK1-IRF3 signaling axis as determined by IRF3 phosphorylation, IRF3 nuclear translocation and TBK1-mediated activation of interferon promoter activity. Given that MDA5-induced NF-κB activation was affected by TRIM25, it is likely that TRIM25 is not involved in direct activation of MDA5, but rather regulates NF-κB activation via the MDA5-MAVS signaling axis. Upon activation, MAVS recruits TRAF6 to induce TAK1-mediated IKK complex activation (Yoshida et al., 2008). We showed NF-κB activation by ectopic expression of MAVS or TRAF6 was enhanced by TRIM25 overexpression and suppressed by TRIM25 depletion, indicating that TRIM25 act at the level of TRAF6 or downstream of TRAF6. We also showed the suppression of TRAF6 ubiquitination in the TRIM25-depleted cells. Given that TRAF6 K63-ubiquitination is a key event in NF-κB activation, suppression of TRAF6 ubiquitination by depletion of TRIM25 suggests a possibility that TRIM25 may regulate NF-κB signaling through modulating the ubiquitination of TRAF6. Despite several attempts, we could not detect a physical interaction between TRIM25 and TRAF6. However, we could show an interaction between overexpressed TRIM25 and IKK complex proteins, further supporting the possible role of TRIM25 in regulating NF-κB activation by MDA5-MAVS signaling. Depletion of TRIM25 has failed to suppress the NF-κB (p65) phosphorylation by ectopic expression of IKKα or IKKβ. We also observed that activation of NF-κB transcriptional activity by ectopic expession of IKK complex could not be suppressed by depletion of TRIM25 using luciferase assays (Data not shown). These results support that TRIM25 modulates MDA5-MAVS mediated NF-κB signaling at the level of TRAF6. Although E3-ligase activity of TRIM25 seems to be involved in this additional function, uncovering the underlying molecular mechanism by which TRIM25 modulates MDA5-mediated NF-κB activation requires further investigation.
In conclusion, we showed that TRIM25 acts as a key modulator in antiviral signaling by positively regulating not only RIG-I-mediated signaling, but also MDA5-mediated signaling. Mechanistic studies suggest that TRIM25 is involved in the activation of NF-κB and interaction with the IKK complex.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) Funded by the Ministry of Science, ICT & Future Planning (NRF-2012R1A1A1015130).
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Gack M.U., Shin Y.C., Joo C.H., Urano T., Liang C., Sun L., Takeuchi O., Akira S., Chen Z., Inoue S., et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- Gack M.U., Albrecht R.A., Urano T., Inn K.S., Huang I.C., Carnero E., Farzan M., Inoue S., Jung J.U., Garcia-Sastre A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe. 2009;5:439–449. doi: 10.1016/j.chom.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goubau D., Deddouche S., Reis E.S.C. Cytosolic sensing of viruses. Immunity. 2013;38:855–869. doi: 10.1016/j.immuni.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inn K.S., Gack M.U., Tokunaga F., Shi M., Wong L.Y., Iwai K., Jung J.U. Linear ubiquitin assembly complex negatively regulates RIG-I- and TRIM25-mediated type I interferon induction. Mol. Cell. 2011a;41:354–365. doi: 10.1016/j.molcel.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inn K.S., Lee S.H., Rathbun J.Y., Wong L.Y., Toth Z., Machida K., Ou J.H., Jung J.U. Inhibition of RIG-I-mediated signaling by Kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J. Virol. 2011b;85:10899–10904. doi: 10.1128/JVI.00690-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X., Kinch L.N., Brautigam C.A., Chen X., Du F., Grishin N.V., Chen Z.J. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity. 2012;36:959–973. doi: 10.1016/j.immuni.2012.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H., Takeuchi O., Sato S., Yoneyama M., Yamamoto M., Matsui K., Uematsu S., Jung A., Kawai T., Ishii K.J., et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- Kato H., Takeuchi O., Mikamo-Satoh E., Hirai R., Kawai T., Matsushita K., Hiiragi A., Dermody T.S., Fujita T., Akira S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008;205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T., Takahashi K., Sato S., Coban C., Kumar H., Kato H., Ishii K.J., Takeuchi O., Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- Loo Y.M., Fornek J., Crochet N., Bajwa G., Perwitasari O., Martinez-Sobrido L., Akira S., Gill M.A., Garcia-Sastre A., Katze M.G., et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 2008;82:335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan E., Curran J., Hofmann K., Moradpour D., Binder M., Bartenschlager R., Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- Nakhaei P., Genin P., Civas A., Hiscott J. RIG-I-like receptors: sensing and responding to RNA virus infection. Semin. Immunol. 2009;21:215–222. doi: 10.1016/j.smim.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Pichlmair A., Schulz O., Tan C.P., Rehwinkel J., Kato H., Takeuchi O., Akira S., Way M., Schiavo G., Reis e Sousa C. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J. Virol. 2009;83:10761–10769. doi: 10.1128/JVI.00770-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos H.J., Gale M., Jr. RIG-I like receptors and their signaling crosstalk in the regulation of antiviral immunity. Curr. Opin. Virol. 2011;1:167–176. doi: 10.1016/j.coviro.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth R.B., Sun L., Ea C.K., Chen Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Sumpter R., Jr., Loo Y.M., Foy E., Li K., Yoneyama M., Fujita T., Lemon S.M., Gale M., Jr. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 2005;79:2689–2699. doi: 10.1128/JVI.79.5.2689-2699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wies E., Wang M.K., Maharaj N.P., Chen K., Zhou S., Finberg R.W., Gack M.U. Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity. 2013;38:437–449. doi: 10.1016/j.immuni.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B., Peisley A., Richards C., Yao H., Zeng X., Lin C., Chu F., Walz T., Hur S. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell. 2013;152:276–289. doi: 10.1016/j.cell.2012.11.048. [DOI] [PubMed] [Google Scholar]
- Xu L.G., Wang Y.Y., Han K.J., Li L.Y., Zhai Z., Shu H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Yoboua F., Martel A., Duval A., Mukawera E., Grandvaux N. Respiratory syncytial virus-mediated NF-kappa B p65 phosphorylation at serine 536 is dependent on RIG-I, TRAF6, and IKK beta. J. Virol. 2010;84:7267–7277. doi: 10.1128/JVI.00142-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M., Kikuchi M., Natsukawa T., Shinobu N., Imaizumi T., Miyagishi M., Taira K., Akira S., Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- Yoshida R., Takaesu G., Yoshida H., Okamoto F., Yoshioka T., Choi Y., Akira S., Kawai T., Yoshimura A., Kobayashi T. TRAF6 and MEKK1 play a pivotal role in the RIG-I-like helicase antiviral pathway. J. Biol. Chem. 2008;283:36211–36220. doi: 10.1074/jbc.M806576200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W., Sun L., Jiang X., Chen X., Hou F., Adhikari A., Xu M., Chen Z.J. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. 2010;141:315–330. doi: 10.1016/j.cell.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.