

Abstract
Mutations of MYO15A are generally known to cause severe to profound hearing loss throughout all frequencies. Here, we found two novel MYO15A mutations, c.3871C>T (p.L1291F) and c.5835T>G (p.Y1945X) in an affected individual carrying congenital profound sensorineural hearing loss (SNHL) through targeted resequencing of 134 known deafness genes. The variant, p.L1291F and p.Y1945X, resided in the myosin motor and IQ2 domains, respectively. The p.L1291F variant was predicted to affect the structure of the actin-binding site from three-dimensional protein modeling, thereby interfering with the correct interaction between actin and myosin. From the literature analysis, mutations in the N-terminal domain were more frequently associated with residual hearing at low frequencies than mutations in the other regions of this gene. Therefore we suggest a hypothetical genotype-phenotype correlation whereby MYO15A mutations that affect domains other than the N-terminal domain, lead to profound SNHL throughout all frequencies and mutations that affect the N-terminal domain, result in residual hearing at low frequencies. This genotype-phenotype correlation suggests that preservation of residual hearing during auditory rehabilitation like cochlear implantation should be intended for those who carry mutations in the N-terminal domain and that individuals with mutations elsewhere in MYO15A require early cochlear implantation to timely initiate speech development.
Keywords: cochlear implantation, mutation, MYO15A, nonsyndromic sensorineural hearing loss (NSHL)
INTRODUCTION
Nonsyndromic hearing loss (NSHL) accounts for 70% of hereditary hearing loss, and most NSHL (∼80%) is inherited in an autosomal recessive fashion (Hilgert et al., 2009). The DFNB3 locus, which is implicated in autosomal recessive congenital deafness (OMIM 600316), was first identified in families from an Indonesian village (Friedman et al., 1995). This locus was mapped to the 17p11.2 chromosome region in this population (Liang et al., 1998). A subsequent study revealed MYO15A (MIM# 600316, NM_016239) to be the causative gene of DFNB3 (Wang et al., 1998).
MYO15A has 66 coding exons and encodes a 3,530-aminoacid protein. Depending on the presence of exon 2, which encodes an N-terminal extension, myosin XVA has two isoforms; class 1 and class 2 (Liang et al., 1999). The class 1 isoform is composed of an N-terminal domain, a motor head domain, a neck region and a tail region. The neck region contains three light chain-binding motifs (IQ). The long tail region contains two myosin tail homology 4 (MyTH4) domains, two F, ezrin, radixin, and moesin (FERM) domains, an SH3 domain and a C-terminal PDZ binding motif (Berg et al., 2001; Garcia-Alvarez et al., 2003; Krendel and Mooseker, 2005; Liang et al., 1999). The class 2 isoform, which has no N-terminal domain, is also present in the human inner ear (Liang et al., 1999).
Myosin XVa plays a crucial role in the graded elongation of stereocilia (Abecasis et al., 2001; Belyantseva et al., 2003; Krendel and Mooseker, 2005) and actin-organization in inner ear hair cells (Berg et al., 2001; Wang et al., 1998), which are vital for sound transduction. Myosin XVa is therefore an essential component of normal auditory function. The effects of mutations in MYO15A on hearing have been investigated extensively. MYO15A mutations have been regarded as leading to congenital severe to profound hearing loss (Belguith et al., 2009; Brownstein et al., 2011; Gao et al., 2013; Kalay et al., 2007; Liburd et al., 2001; Shearer et al., 2009; Wang et al., 1998). In contrast, it was also reported that a mutation in exon 2 of MYO15A resulted in some residual hearing (Bashir et al., 2012; Cengiz et al., 2010; Nal et al., 2007), especially at low frequencies, suggesting that there may be a genotype-phenotype correlation.
Here, we report two novel MYO15A mutations identified by targeted exome sequencing from a non-consanguineous Korean family, showing congenital profound sensorineural hearing loss (SNHL). Based on our observations, which are compatible with previous suggested genotype-phenotype correlations for MYO15A, we propose how this finding can be utilized clinically as regards early bilateral cochlear implantation and residual hearing preservation during cochlear implantation.
MATERIALS AND METHODS
Human subjects
All procedures in this study were approved by the Institutional Review Boards at Seoul National University Hospital (IRBY-H-0905-041-281) and Seoul National University Bundang Hospital (IRB-B-1007-105-402). Written informed consent was obtained from all individuals (or guardians, in the case of children). One family (SB156), which showed profound SNHL in an autosomal recessive fashion, was included in this study. The family (SB156) comprised 12 individuals, 3 of whom participated in the study. The family members (SB156-272, 327 and 328) covered two generations and were evaluated at Seoul National University Bundang Hospital (Fig. 1A). Phenotypic evaluations included medical and developmental history interviews, physical examinations, and audiometric evaluation.
Fig. 1.
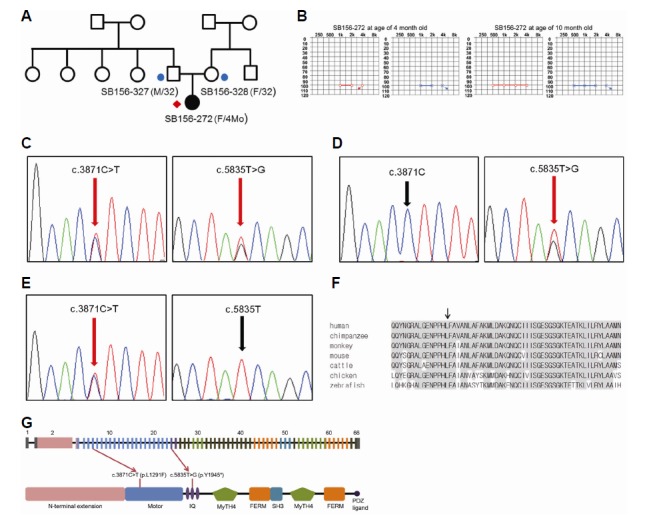
Pedigree, auditory steady state response (ASSR) and segregation of the c.3871C>T and c.5835T>G variants of MYO15A in family SB156. (A) Targeted sequencing was performed for one affected individual (red diamond). An additional two affected individuals (circles) were recruited for Sanger validation and further analyses. (B) The ASSR test revealed that the average hearing threshold of SB156-272 was 100 dB at 4 and 10 months of age. (C) Sanger sequencing traces for the c.3871C>T (p.L1291F) + c.5835T>G (p.Y1945*) compound heterozygote (SB156-272). (D) Sanger sequencing traces for the c.5835T>G carrier (SB156-327). (E) Sanger sequencing traces for the c.3871C>T carrier (SB156-328). (F) Conservation of mutant residues among orthologs from several species; p.L1291 is conserved among all species, ranging from humans to zebrafish. (G) The sequence variants c.3871C>T and c.5835T>G reside in exon 6 (motor domain) and exon 24 (IQ2 domain) of MYO15A, respectively (adapted from Nal et al., 2007).
Audiometric evaluation
Auditory steady state response (ASSR), auditory brain stem response threshold (ABRT) and distortion product otoacoustic emission (DPOAE) tests were carried out on SB156-272 at 4 and 10 months of age. The ASSR results are shown in Fig. 1B. Temporal bone computed tomography (CT) and internal auditory canal magnetic resonance image (MRI) of SB156-272 were used to identify any inner ear anomalies related to the hearing loss.
Molecular genetic testing
Targeted resequencing and bioinformatics analyses were performed as described previously (Choi et al., 2013). DNA samples from SB156-272 were subjected to targeted resequencing of 134 known deafness genes (TRS-134) by Otogenetics (USA) (Supplementary Table S1). The acquired reads were mapped onto the UCSC hg19 reference genome. Further bioinformatics analyses were performed to identify variants. As a basic filtering step, non-synonymous single nucleotide polymorphisms (SNPs) with read depths >40 were chosen. These non-synonymous SNPs were compared against an in-house database, an independent cohort consisting of 54 normal Korean individuals and the Single Nucleotide Polymorphism Database (dbSNP build 138). Only novel SNPs or known disease-causing SNPs remained. Inheritance patterns were checked and SNPs that did not coincide with an autosomal recessive pattern were excluded. The remaining SNPs were validated in other family members (SB156-327 and SB156-328) by Sanger sequencing. The SNPs were also checked against an additional 426 unrelated Korean control chromosomes (Fig. 2).
Fig. 2.
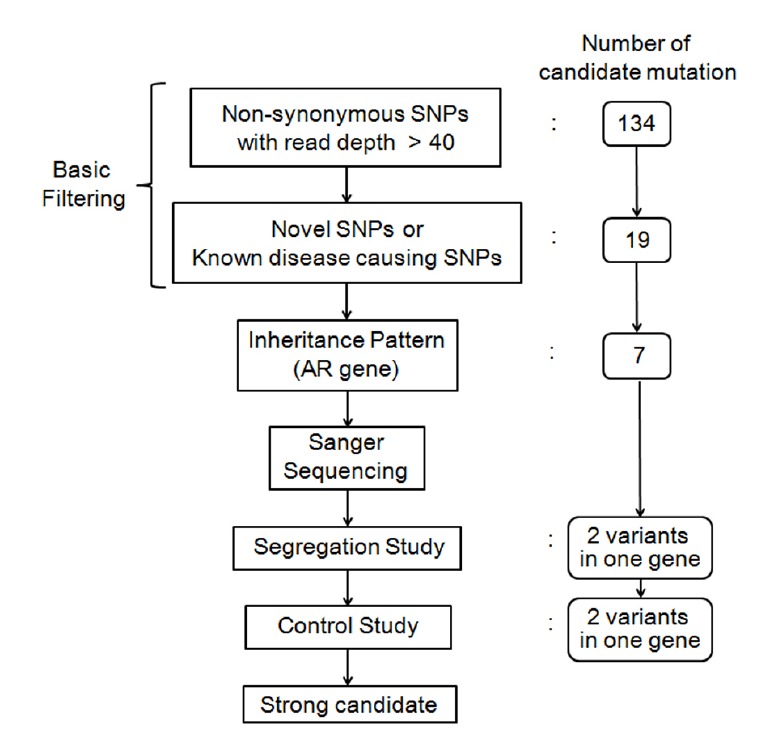
Schematic flow chart of the filtering of causative variants in this study
To predict the pathogenicity of the missense variants, SIFT and PolyPhen-2 analyses were carried out. The evolutionary conservation of the amino acid sequence was estimated using the GERP++ score in the UCSC Genome Browser (http://genome.ucsc.edu/).
Three-dimensional protein modeling of the motor domain of myosin XVA
To assess the pathogenicity of a candidate variant in the motor domain of myosin XVA, three-dimensional protein modeling of the motor domain was performed. Since there is no known 3D structure of motor domain of MYO15A in the Protein Data Bank, the structure for motor domain of MYO15A was built using the homolog modeling method (BLAST, http://blast.ncbi.nlm.nih.gov/Blast.cgi/; HHpred, http://toolkit.tuebingen.mpg.de/hhpred/; PDB, http://pdb.org/pdb/home/home.do/; UNIPROT. http://www.uniprot.org/). The template structure that was sequentially most similar to the motor domain of myosin XVA using BLAST and HHPred was sought, and the myosin II heavy chain from Dictyostelium discoideum (pdb ID: 1W9J A) was selected. The sequence identity between this chain and human myosin XVA was 44%. The structure model of human myosin was built using MODELLER, a homology-modeling software package (Eswar et al., 2006). A mutant model was also constructed from the wild type structure model using FoldX (http://foldx.crg.es/). To ensure the quality of the models, the wild type and mutant models were examined using the Pymol visualization tool (http://www.pymol.org/).
Analysis of audiologic features of MYO15A mutations
We tried to delineate any correlation between the locations of MYO15A mutations and the audiotory phenotype, relying on the audiograms available in the literature. In case audiogram was unavailable, description about auditory phenotype was used. Subjects without audiogram or description were excluded from the analysis. A literature search was made in Pubmed covering the period 2001 to 2014. Subjects with MYO15A mutations were divided into two groups, with mutations in the N-terminal domain or in the other regions. The frequency of subjects with residual hearing, of which thresholds were better than 70dB HL at any frequency, was calculated in each group.
RESULTS
Auditory phenotype and cochlear implantation
SB156-272 showed no response to 90 dB click sounds at 4 and 10 months of age in ABRT testing. ASSR testing using louder stimuli revealed an average threshold of 100 dB for SB156-272 at 4 and 10 months. There was no change in the hearing ability of SB156-272 over the 6-month follow-up period. The guardians of SB156-272 denied any exposure to risk factors such as drugs or loud noises. No syndromic features were detected in the physical examination. Temporal bone CT and internal auditory canal MRI revealed no abnormal findings. SB156-272 had started wearing hearing aids in both ears at 5 months and underwent simultaneous bilateral cochlear implantation at 11 months.
Targeted resequencing data analysis
Targeted resequencing was performed in SB156-272. The reads were aligned to a human reference genome. Bioinformatics analyses were carried out as mentioned above (Fig. 2). Following the basic filtering step, 19 SNPs were selected as candidate mutations (Supplementary Table S2). The inheritance pattern of the family was regarded as autosomal recessive (Fig. 1A) and SNPs that did not fit with an autosomal recessive inheritance pattern were excluded. Candidate variants were validated by Sanger sequencing in the parents of SB156-272 (SB156-327 and SB156-328). Two candidate variants from a single gene, MYO15A, remained (Table 1). These were c.3871C>T (p.L 1291F) in exon 6 and c.5835T>G (p.Y1945*) in exon 24 (Fig. 1C). The mother of SB156-272 (SB156-328) was heterozygous for c.3871C>T (p.L1291F) and the father (SB156-327) was heterozygous for c.5835T>G (p.Y1945*) (Figs. 1D and 1E). These two variants were not found in 426 control chromosomes from unrelated Koreans with normal hearing. In addition, the SIFT and Polyphen-2 analyses consistently identified the MYO15A c.3871C>T (p.L1291F) SNP as “damaging.” Furthermore, p.L1291 was well-conserved in several species, as indicated by the high GERP++ score of 5.01 (Fig. 1F). These results strongly supported the pathogenicity of the conversion of leucine to phenylalanine at residue 1291 of myosin XVA. The c.3871C>T and c.5835T>G mutations resided in the myosin motor and IQ2 domains, respectively. The p.Y1945* was the second mutation detected in the IQ2 domain (Fig. 1G).
Table 1.
Causal Variant MYO15A
Three-dimensional protein modeling of myosin XVA
The p.L1291F model was constructed from the wild-type structure model using FoldX by replacing p.L1291 with phenylalanine. The p.L1291F mutation was evaluated by comparing that of the wild type to the mutant model. Visual inspection of the three-dimensional structure revealed that the mutation site, although it was distant from the actin binding region (from 1972 to 1799) in terms of protein sequence, was located near the actin binding region which is crucial to the proper function of myosin (Fig. 3). In particular, mutations at this site would alter the position of the actin-binding region. As phenylalanine is larger than leucine, p.L1291F would push away the actin-binding region as a result of direct contact between p.F1291 and p.M1793 (Figs. 4A and 4B). This structural change in the actin-binding region would decrease the binding stability between actin and the myosin motor domain, which would have a detrimental effect on the interaction between actin and myosin.
Fig. 3.
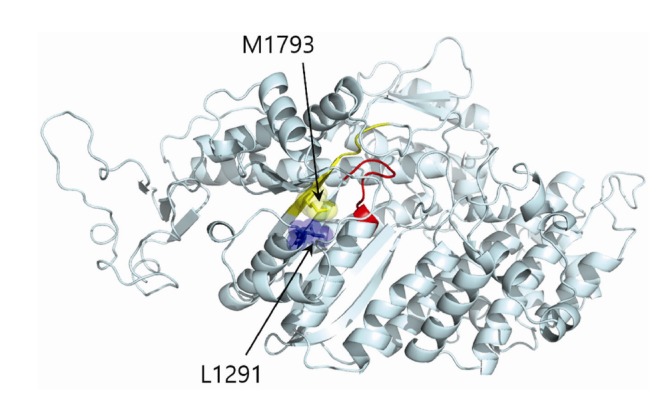
The location of the p.L1291F mutation (blue) is close to two functionally important binding sites; the ATP-binding site (red) and the actin-binding site (yellow).
Fig. 4.
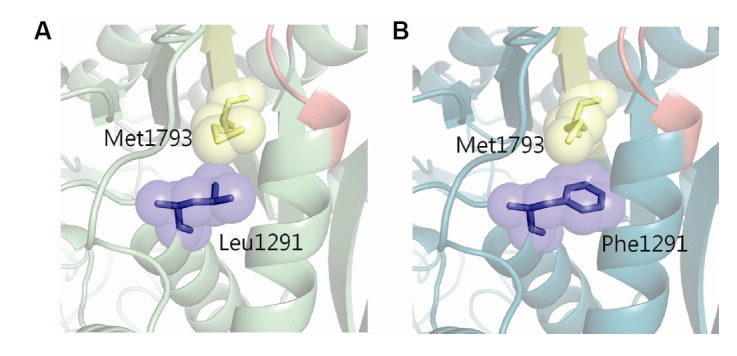
Pathogenicity of the p.L1291F mutation. The mutation p.L1291F would alter the structure of the actin-binding site. (A) Wild type and (B) mutation show different positions of Met1793. Yellow area is the actin-binding site and red area is ATP binding site.
Analysis of audiologic features of MYO15A mutations
From the analysis of literature, 9 (64.2%) of 14 subjects with mutations in the N-terminal domain showed significant residual hearing of which thresholds were better than 70dB HL especially at low frequencies. In contrast, among 93 subjects harboring mutations in the other regions of this gene, only one (1.0%) subjects retained a significant residual hearing, while the others were associated with severe to profound hearing loss throughout all frequencies. Therefore, mutations in the N-terminal domain of MYO15 were more frequently associated with less severe hearing loss than were mutations in the other regions of this gene (Fisher’s exact test, P < 0.001) (Table 2).
Table 2.
Auditory Phenotype According to The Affected Domain
| Nucleotide change | Amino acid change | Affected domain | Inheritance | Hearing loss | Reference |
|---|---|---|---|---|---|
| c.867C>G | p.Tyr289* | N-terminal | Homozygous | *Residual hearing at low frequency | Cengiz et al., 2010 |
| c.867C>G | p.Tyr289* | N-terminal | Homozygous | Profound | Cengiz et al., 2010 |
| c.1185dup | p.Glu396Argfs*36 | N-terminal | Homozygous | *Residual hearing at low frequency | Bashir et al., 2012 |
| c.3313G>T | p.Glu1105* | N-terminal | Homozygous | Severe to profound | Nal et al., 2007 |
| c.3336del | p.Arg1113Valfs*12 | N-terminal | Homozygous | *Residual hearing at low frequency | Nal et al., 2007 |
| c.3685C>T | p.Gln1229* | Motor | Homozygous | Profound | Liburd et al., 2001 |
| c.3756+1G>T | Homozygous | Profound | Liburd et al., 2001 | ||
| c.3758C>T | p.Thr1253Ile | Motor | Homozygous | Severe to profound | Nal et al., 2007 |
| c.3866+1G>A | Homozygous | Severe to profound | Nal et al., 2007 | ||
| c.3871C>T | p.Leu1291Phe | Motor | Heterozygous | Profound | This study |
| c.4176C>A | p.Tyr1392* | Motor | Homozygous | Severe to profound | Nal et al., 2007 |
| c.4198G>A | p.Val1400Met | Motor | Homozygous | Severe to profound | Cengiz et al., 2010 |
| c.4240G>A | p.Glu1414Lys | Homozygous | Profound | Brownstein et al., 2011 | |
| c.4320+1G | Motor | Heterozygous | Severe to profound | Woo et al,.2013 | |
| c.4351G>A | p.Asp1451Asn | Motor | Homozygous | Severe to profound | Nal et al., 2007 |
| c.4441T>C | p.Ser1481Pro | Motor | Homozygous | Severe to profound | Cengiz et al., 2010 |
| c.4669A>G | p.Lys1557Glu | Motor | Homozygous | Severe to profound | Nal et al., 2007 |
| c.4998C>A | p.Cys1666* | Motor | Homozygous | Severe to profound | Belguith et al., 2009 |
| c.5117_5118del | p.Gly1706Glufs*102 | Motor | Homozygous | Severe to profound | Nal et al., 2007 |
| c.5189T>C | p.Leu1730Pro | Motor | Homozygous | Severe to profound | Nal et al., 2007 |
| c.5492G>T | p.Gly1831Val | Motor | Homozygous | Profound | Kalay et al., 2007 |
| c.5808_5814del | p.Arg1937Thrfs*10 | IQ 2 | Homozygous | Severe to profound | Cengiz et al., 2010 |
| c.5835T>G | p.Tyr1945* | IQ 2 | Heterozygous | Profound | This study |
| c.5913G>A | Heterozygous | Severe to profound | Gao et al,.2013 | ||
| c.6061C>T | p.Gln2021* | ? | Severe to profound | Nal et al., 2007 | |
| c.6217C>T | p.Pro2073Ser | MyTH4 1 | Homozygous | Severe to profound | Shearer et al., 2009 |
| c.6308dup | p.Ala2104Cysfs*19 | MyTH4 1 | Heterozygous | Severe to profound | Yang et al., 2013 |
| c.6340G>A | p.Val2114Met | MyTH4 1 | Heterozygous | Severe to profound | Yang et al., 2013 |
| c.6371G>A | p.Arg2124Gln | MyTH4 1 | Homozygous | Severe to profound | Shearer et al., 2009 |
| c.6437G>A | p.Arg2146Gln | MyTH4 1 | Heterozygous | Severe to profound | Woo et al,.2013 |
| c.6614C>T | p.Thr2205Ile | MyTH4 1 | Hemizygous | *Residual hearing at low frequency | Liburd et al., 2001 |
| c.6731G>A | p.Gly2244Glu | Homozygous | Severe to profound | Nal et al., 2007 | |
| c.6796G>A | p.Val2266Met | Homozygous | Severe to profound | Nal et al., 2007 | |
| c.6956+9C>G | Heterozygous | Severe to profound | Yang et al., 2013 | ||
| c.6956+9C>G | Heterozygous | Severe to profound | Yang et al., 2013 | ||
| c.7395+3G>C | Homozygous | Severe to profound | Belguith et al., 2009 | ||
| c.8148G>T | p.Gln2716His | Homozygous | Profound | Liburd et al., 2001 | |
| c.8158G>C | p.Asp2720His | Homozygous | Severe to profound | Nal et al., 2007 | |
| c.8324G>A | p.Arg2775His | Heterozygous | Severe to profound | Yang et al., 2013 | |
| c.8375T>C | p.Val2792Ala | Heterozygous | Severe to profound | Gao et al,.2013 | |
| c.8767C>T | p.Arg2923* | SH3 | Heterozygous | Severe to profound | Yang et al., 2013 |
| c.8821_8822insTG | p.Gly2941Valfs*94 | SH3 | Homozygous | Severe to profound | Nal et al., 2007 |
| c.8968−1G>C | Homozygous | Profound | Kalay et al., 2007 | ||
| c.9229+1G>T | Homozygous | Severe to profound | Belguith et al., 2009 | ||
| c.9478C>T | p.Leu3160Phe | MyTH4 2 | Homozygous | Severe to profound | Nal et al., 2007 |
| c.9958_9961del | p.Asp3320Thrfs*2 | FERM | Heterozygous | Severe to profound | Lezirovitz et al., 2008 |
| c.9995_10002dup | p.Ser3335Alafs*121 | FERM | Homozygous | Severe to profound | Cengiz et al., 2010 |
| c.10258_10260del | p.Phe3420del | FERM | Heterozygous | Severe to profound | Yang et al., 2013 |
| c.10474C>T | p.Gln3492* | FERM | Homozygous | Severe to profound | Nal et al., 2007 |
| c.10573del | p.Ser3525Alafs*29 | FERM | Heterozygous | Severe to profound | Lezirovitz et al., 2008 |
| c.10573del | p.Ser3525Alafs*29 | FERM | Homozygous | Severe to profound | Lezirovitz et al., 2008 |
DISCUSSION
Myosin belongs to the superfamily of actin-dependent molecular motors, and facilitates the movement of actin filaments via the force generated by hydrolysis of adenosine triphosphate (ATP) (Kim et al., 2008; Krendel and Mooseker, 2005; Mooseker and Cheney, 1995). Myosin consists of three evolutionarily conserved domains: the motor domain, the neck region, and the tail region. The major structural components of the motor domain are highly conserved. The neck region includes one or more light-chain-binding IQ motifs. The tail region is diverse in length and sequence (Friedman et al., 1999). The myosin superfamily is subdivided into 1 conventional and 20 unconventional classes, based on the amino acid sequence variation of the motor domain (Berg et al., 2001; Krendel and Mooseker, 2005). Two conventional myosins; MYH9 and MYH14, and five unconventional myosins; Ia, IIIa, VI, VIIa and XVa are important in normal auditory function. Mutations in the genes encoding these myosins are responsible for hearing loss (Donaudy et al., 2003; 2004; Friedman et al., 1999; Lalwani et al., 2000; Walsh et al., 2002).
Among the myosin superfamily, changes in the unconventional myosin XVA, which is encoded by MYO15A, are known to be responsible for DFNB3. The pathophysiology of deafness due to MYO15A mutations has been studied in shaker-2 mice, which contain homozygous recessive mouse mutations (sh2) in exon 20 (comprising the motor domain), and are regarded as a mouse homolog of human DFNB3 deafness (Cope et al., 1996; Probst et al., 1998). In a wild-type mouse, during auditory and vestibular hair cell development, myosin XVa is present at the tips of the stereocilia when the stereocilia bundle begins to develop its staircase architecture. Myosin XVa contributes to the differential elongation of each stereocilium (Tilney and Tilney, 1986; Tilney et al., 1992). In shaker-2 mice, myosin XVa is absent and the stereocilia are short and nearly equal in length. This leads to a loss of the staircase architecture of the stereocilia bundle, which is fundamental for sound transduction (Belyantseva et al., 2003; 2005). The loss of the staircase architecture of the stereocilia bundles is regarded as the main defect of the MYO15A mutation.
Studies of the MYO15A mutation in humans have usually been performed in the Middle East, where consanguineous families are more common (Bashir et al., 2012; Belguith et al., 2009; Brownstein et al., 2011; Cengiz et al., 2010; Diaz-Horta et al., 2012; Duman et al., 2011; Fattahi et al., 2012; Kalay et al., 2007; Liburd et al., 2001; Nal et al., 2007; Shearer et al., 2009). However, as next-generation sequencing methods have become more popular, MYO15A mutations have been reported from various parts of the world, and the importance of the MYO15A mutation has emerged (Besnard et al., 2014; Gao et al., 2013; Woo et al., 2013; Yang et al., 2013). The frequency of the MYO15A mutation was first reported by Friedman et al. (1995), who reported that 2% of the residents of Bengkala, Bali, had nonsyndromic autosomal recessive deafness due to the MYO15A mutation (Friedman et al., 1995). In 2002, Friedman et al. reported the frequency of the MYO15A mutation as 5% in Pakistan (Friedman et al., 2002). Recently, Fattahi et al. (2012) reported that in Iran, 5.71% of patients affected by nonsyndromic autosomal recessive deafness were associated with MYO15A. In Turkey, the frequency of the MYO15A mutation related with nonsyndromic autosomal recessive deafness has been reported as 9.9%, which represents the second most common deafness gene (Duman et al., 2011). In our previous study, the frequency of the MYO15A mutation in nonsyndromic autosomal recessive deafness in patients requesting cochlear implants was 2.1%, which represents the fourth most common common deafness gene in Korea after SLC26A4, GJB2 and CDH23 (Park et al., 2014). The frequency of the MYO15A mutation in Korea is not high as in the Middle East and this could be attributed to the scarcity of consanguineous families in Korea. However, MYO15A remains an important deafness gene in the Korean population.
Several MYO15A mutations have been reported according to the domains they affect. MYO15A mutations occur primarily in the motor domain, such as the p. L1291F (c.3871C>T) mutation identified in this study. The motor domain contains the ATP- and actin-binding sites, the switch I and II helices, relay helix, SH3 helix, SH1/SH2 helix and a converter domain, and acts as a molecular motor. Therefore, it is conceivable that mutations in the motor domain would be more detrimental to the functioning of the protein and would be more prevalent than mutations in other myosin XVA domains. In this study, the 3D model created of the motor domain demonstrated how the p. L1291F (c.3871C>T) mutation altered the structure of the actin binding site and decreased the stability of actin binding. This could induce a disorder of the binding process between myosin and the actin filament and result in hearing loss. Recent successful purification of the motor domain of this protein by Bird et al. (2014) would contribute significantly to evaluation of pathogenic potential and mechanisms of variants located in this domain in the near future.
The second mutation found was a nonsense mutation: p.Y1945* (c.5835T>G). It resides in the IQ2 domain, which is highly conserved throughout myosin and is known to contribute to the reinforcement of the motor function of myosin by increasing conformational changes (Bahler and Rhoads, 2002). The p.Y1945* (c.5835T>G) mutation is predicted to truncate the translation of mRNA and result in myosin XVA that lacks half of its usual length. Several other nonsense mutations; p.Q2021*, p.S2661*, p.R2923* and p.Q3492*, located downstream to p.Y1945*, have already been reported to cause severe to profound hearing loss (Duman et al., 2011; Nal et al., 2007; Yang et al., 2013). Therefore it seems likely that the p.Y1945* (c.5835T>G) mutation that results in a shorter protein product would definitely be pathogenic.
MYO15A mutations have been regarded as leading to congenital severe to profound hearing loss (Belguith et al., 2009; Brownstein et al., 2011; Gao et al., 2013; Kalay et al., 2007; Liburd et al., 2001; Shearer et al., 2009; Wang et al., 1998;). Recently, conflicting descriptions of auditory phenotypes of the MYO15A mutation have been reported; Fattahi et al. (2012) reported seven mutations throughout the N-terminal extension, motor domain, IQ domain and FERM domain of MYO15A and suggested that a down-sloping audioprofile was the defining audiologic feature of MYO15A-associated deafness. However, the audiograms included in the study by Fattahi et al. (2012) were not present in association with the affected domain, which precluded any genotype-phenotype correlations. Later, Bashir et al. (2012) proposed that the audiologic features of MYO15A mutations could be related to the affected domain. Specifically, they reported that almost all patients with mutations in exon 2 of MYO15A (p.E396Rfs*36 homozygote) had residual hearing at low frequencies but profound hearing loss at high frequencies. This feature has been observed in other studies in which mutations in exon 2 (p.E1105*, p.R1113Vfs*12 and p.Y289* homozygote), which encodes the N-terminal extension of MYO15A, resulted in better residual hearing at low frequencies, or even moderate to severe hearing loss compared with mutations in other regions of MYO15A (Cengiz et al., 2010; Nal et al., 2007). This observation has been confirmed by our analysis of audiologic features of MYO15A mutations (Table 2).
The possibility of less severe hearing loss associated with mutations residing in the N-terminal extension domain had already been suggested; Belyantseva et al. (2005) transfected a class 2 myosin XVa isoform that had no N-terminal extension, into shaker-2 hair cells, which had abnormally short stereocilia. In this explant culture experiment, elongation of the stereocilia occurred, and a wild-type-like staircase architecture of the hair bundle was obtained (Belyantseva et al., 2005). In other words, the staircase architecture of the hair bundle, the loss of which had been considered a crucial defect of DFNB3, was restored without N-terminal extension. Therefore, it is possible that the defect caused by mutations in exon 2 of MYO15A might be compensated for in part by the presence of the class 2 isoform without the N-terminal extension. Alternatively, alterations in the N-terminal extension domain, or even the absence of the class 1 myosin XVA, might be related to functional problems or minor structural defects in the staircase architecture of the hair bundle (Nal et al., 2007), thereby leading to a milder auditory phenotype. Conversely, it can be hypothesized that if MYO15A mutations affect a common region shared by both isoforms, other than exon 2, then the phenotype would be profound throughout all frequencies and have no, or minimal, residual hearing to trigger linguistic development, thereby requiring early cochlear implantation for speech development. The mutation identified in SB156-272 resided in exons 24 and 6. Based on the locations of the mutations, we predicted that SB156-272 would retain no residual hearing, which was later confirmed by behavioral audiometry and electrically evoked auditory testing. Since minimal residual hearing would not allow language development even with a conventional hearing aid, simultaneous bilateral cochlear implantation was performed using a full-length electrode in SB156-272 at 11 months.
Based on our findings and analysis of audiologic features of MYO15A mutations, we suggest a hypothetical genotype-phenotype correlation whereby MYO15A mutations that affect domains other than the N-terminal domain, lead to profound SNHL throughout all frequencies, while mutations that affect the N-terminal domain, result in residual hearing at low frequencies, which could make contribution to language development and speech perception. Nowadays cochlear implantation, which converts sound into electrical pulse and stimulates the spiral ganglion or auditory nerve directly bypassing auditory hair cells, is the only auditory rehabilitation for severe to profound SNHL patients and gives a great benefit to them. As the technique of cochlear implantation has progressed and the indication of cochlear implantation has been expanded, severe to profound SNHL patients with residual hearing could also get a benefit from cochlear implantation. However cochlear implantation itself could damage auditory hair cells and basilar membrane and result in loss of residual hearing. Consequently the presence of residual hearing has been an important issue for cochlear implantation and the techniques of preserving residual hearing have been getting increased attention. Therefore our hypothetical genotype-phenotype correlation suggests a customized treatment that preservation of residual hearing during cochlear implantation should be intended for those who carry mutations in the N-terminal domain and that the individuals with mutations elsewhere in MYO15A require early cochlear implantation to timely initiate speech development.
Here we identified two novel MYO15A mutations and suggested hypothetical genotype-phenotype correlation. Basing decisions of appropriate timings and modality of auditory rehabilitation of DFNB3 deafness on the location of the mutations in MYO15A would provide a good model for customized auditory rehabilitation in this field.
Acknowledgments
No potential conflict of interest relevant to this article was reported. This study was supported by a grant of the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI12C0014). And no potential conflict of interest relevant to this article was reported.
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. GRR: graphical representation of relationship errors. Bioinformatics. 2001;17:742–743. doi: 10.1093/bioinformatics/17.8.742. [DOI] [PubMed] [Google Scholar]
- Bahler M., Rhoads A. Calmodulin signaling via the IQ motif. FEBS Lett. 2002;513:107–113. doi: 10.1016/s0014-5793(01)03239-2. [DOI] [PubMed] [Google Scholar]
- Bashir R., Fatima A., Naz S. Prioritized sequencing of the second exon of MYO15A reveals a new mutation segregating in a Pakistani family with moderate to severe hearing loss. Eur. J. Med. Genet. 2012;55:99–102. doi: 10.1016/j.ejmg.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belguith H., Aifa-Hmani M., Dhouib H., Said M.B., Mosrati M.A., Lahmar I., Moalla J., Charfeddine I., Driss N., Arab S.B., et al. Screening of the DFNB3 locus: identification of three novel mutations of MYO15A associated with hearing loss and further suggestion for two distinctive genes on this locus. Genet. Test. Mol. Biomarkers. 2009;13:147–151. doi: 10.1089/gtmb.2008.0077. [DOI] [PubMed] [Google Scholar]
- Belyantseva I.A., Boger E.T., Friedman T.B. Myosin XVa localizes to the tips of inner ear sensory cell stereocilia and is essential for staircase formation of the hair bundle. Proc. Natl. Acad. Sci. USA. 2003;100:13958–13963. doi: 10.1073/pnas.2334417100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyantseva I.A., Boger E.T., Naz S., Frolenkov G.I., Sellers J.R., Ahmed Z.M., Griffith A.J., Friedman T.B. Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat. Cell. Biol. 2005;7:148–156. doi: 10.1038/ncb1219. [DOI] [PubMed] [Google Scholar]
- Berg J.S., Powell B.C., Cheney R.E. A millennial myosin census. Mol. Biol. Cell. 2001;12:780–794. doi: 10.1091/mbc.12.4.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard T., Garcia-Garcia G., Baux D., Vache C., Faugere V., Larrieu L., Leonard S., Millan J.M., Malcolm S., Claustres M., et al. Experience of targeted Usher exome sequencing as a clinical test. Mol. Genet. Genomic. Med. 2014;2:30–43. doi: 10.1002/mgg3.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird J.E., Takagi Y., Billington N., Strub M.P., Sellers J.R., Friedman T.B. Chaperone-enhanced purification of unconventional myosin 15, a molecular motor specialized for stereocilia protein trafficking. Proc. Natl. Acad. Sci. USA. 2014;111:12390–12395. doi: 10.1073/pnas.1409459111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownstein Z., Friedman L.M., Shahin H., Oron-Karni V., Kol N., Abu Rayyan A., Parzefall T., Lev D., Shalev S., Frydman M., et al. Targeted genomic capture and massively parallel sequencing to identify genes for hereditary hearing loss in Middle Eastern families. Genome Biol. 2011;12:R89. doi: 10.1186/gb-2011-12-9-r89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cengiz F.B., Duman D., Sirmaci A., Tokgoz-Yilmaz S., Erbek S., Ozturkmen-Akay H., Incesulu A., Edwards Y.J., Ozdag H., Liu X.Z., et al. Recurrent and private MYO15A mutations are associated with deafness in the Turkish population. Genet. Test. Mol. Biomarkers. 2010;14:543–550. doi: 10.1089/gtmb.2010.0039. [DOI] [PubMed] [Google Scholar]
- Choi B.Y., Park G., Gim J., Kim A.R., Kim B.J., Kim H.S., Park J.H., Park T., Oh S.H., Han K.H., et al. Diagnostic application of targeted resequencing for familial nonsyndromic hearing loss. PLoS One. 2013;8:e68692. doi: 10.1371/journal.pone.0068692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope M.J., Whisstock J., Rayment I., Kendrick-Jones J. Conservation within the myosin motor domain: implications for structure and function. Structure. 1996;4:969–987. doi: 10.1016/s0969-2126(96)00103-7. [DOI] [PubMed] [Google Scholar]
- Diaz-Horta O., Duman D., Foster J., 2nd, Sirmaci A., Gonzalez M., Mahdieh N., Fotouhi N., Bonyadi M., Cengiz F.B., Menendez I., et al. Whole-exome sequencing efficiently detects rare mutations in autosomal recessive nonsyndromic hearing loss. PLoS One. 2012;7:e50628. doi: 10.1371/journal.pone.0050628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaudy F., Ferrara A., Esposito L., Hertzano R., Ben-David O., Bell R.E., Melchionda S., Zelante L., Avraham K.B., Gasparini P. Multiple mutations of MYO1A, a cochlear-expressed gene, in sensorineural hearing loss. Am. J. Hum. Genet. 2003;72:1571–1577. doi: 10.1086/375654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaudy F., Snoeckx R., Pfister M., Zenner H.P., Blin N., Di Stazio M., Ferrara A., Lanzara C., Ficarella R., Declau F., et al. Nonmuscle myosin heavy-chain gene MYH14 is expressed in cochlea and mutated in patients affected by autosomal dominant hearing impairment (DFNA4) Am. J. Hum. Genet. 2004;74:770–776. doi: 10.1086/383285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman D., Sirmaci A., Cengiz F.B., Ozdag H., Tekin M. Screening of 38 genes identifies mutations in 62% of families with nonsyndromic deafness in Turkey. Genet. Test. Mol. Biomarkers. 2011;15:29–33. doi: 10.1089/gtmb.2010.0120. [DOI] [PubMed] [Google Scholar]
- Eswar N., Webb B., Marti-Renom M.A., Madhusudhan M.S., Eramian D., Shen M.Y., Pieper U., Sali A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics Chapter. 2006;5 doi: 10.1002/0471250953.bi0506s15. Unit 5.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fattahi Z., Shearer A.E., Babanejad M., Bazazzadegan N., Almadani S.N., Nikzat N., Jalalvand K., Arzhangi S., Esteghamat F., Abtahi R., et al. Screening for MYO15A gene mutations in autosomal recessive nonsyndromic, GJB2 negative Iranian deaf population. Am. J. Med. Genet. A. 2012;158a:1857–1864. doi: 10.1002/ajmg.a.34411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman T.B., Liang Y., Weber J.L., Hinnant J.T., Barber T.D., Winata S., Arhya I.N., Asher J.H., Jr. A gene for congenital, recessive deafness DFNB3 maps to the pericentromeric region of chromosome 17. Nat. Genet. 1995;9:86–91. doi: 10.1038/ng0195-86. [DOI] [PubMed] [Google Scholar]
- Friedman T.B., Sellers J.R., Avraham K.B. Unconventional myosins and the genetics of hearing loss. Am. J. Med. Genet. 1999;89:147–157. doi: 10.1002/(sici)1096-8628(19990924)89:3<147::aid-ajmg5>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Friedman T.B., Hinnant J.T., Ghosh M., Boger E.T., Riazuddin S., Lupski J.R., Potocki L., Wilcox E.R. DFNB3, spectrum of MYO15A recessive mutant alleles and an emerging genotype-phenotype correlation. Adv. Otorhinolaryngol. 2002;61:124–130. doi: 10.1159/000066824. [DOI] [PubMed] [Google Scholar]
- Gao X., Zhu Q.Y., Song Y.S., Wang G.J., Yuan Y.Y., Xin F., Huang S.S., Kang D.Y., Han M.Y., Guan L.P., et al. Novel compound heterozygous mutations in the MYO15A gene in autosomal recessive hearing loss identified by whole-exome sequencing. J. Transl. Med. 2013;11:284. doi: 10.1186/1479-5876-11-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Alvarez B., de Pereda J.M., Calderwood D.A., Ulmer T.S., Critchley D., Campbell I.D., Ginsberg M.H., Liddington R.C. Structural determinants of integrin recognition by talin. Mol. Cell. 2003;11:49–58. doi: 10.1016/s1097-2765(02)00823-7. [DOI] [PubMed] [Google Scholar]
- Hilgert N., Smith R.J., Van Camp G. Function and expression pattern of nonsyndromic deafness genes. Curr. Mol. Med. 2009;9:546–564. doi: 10.2174/156652409788488775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalay E., Uzumcu A., Krieger E., Caylan R., Uyguner O., Ulubil-Emiroglu M., Erdol H., Kayserili H., Hafiz G., Baserer N., et al. MYO15A (DFNB3) mutations in Turkish hearing loss families and functional modeling of a novel motor domain mutation. Am. J. Med. Genet. A. 2007;143a:2382–2389. doi: 10.1002/ajmg.a.31937. [DOI] [PubMed] [Google Scholar]
- Kim H.Y., Rhim T., Ahnm M.H., Yoon P.O., Kim S.H., Lee S.H., Park C.S. The fast skeletal muscle myosin light chain is differentially expressed in smooth muscle cells of OVA-challenged mouse trachea. Mol. Cells. 2008;25:78–85. [PubMed] [Google Scholar]
- Krendel M., Mooseker M.S. Myosins: tails (and heads) of functional diversity. Physiology (Bethesda) 2005;20:239–251. doi: 10.1152/physiol.00014.2005. [DOI] [PubMed] [Google Scholar]
- Lalwani A.K., Goldstein J.A., Kelley M.J., Luxford W., Castelein C.M., Mhatre A.N. Human nonsyndromic hereditary deafness DFNA17 is due to a mutation in nonmuscle myosin MYH9. Am. J. Hum. Genet. 2000;67:1121–1128. doi: 10.1016/s0002-9297(07)62942-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y., Wang A., Probst F.J., Arhya I.N., Barber T.D., Chen K.S., Deshmukh D., Dolan D.F., Hinnant J.T., Carter L.E., et al. Genetic mapping refines DFNB3 to 17p11.2, suggests multiple alleles of DFNB3, and supports homology to the mouse model shaker-2. Am. J. Hum. Genet. 1998;62:904–915. doi: 10.1086/301786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y., Wang A., Belyantseva I.A., Anderson D.W., Probst F.J., Barber T.D., Miller W., Touchman J.W., Jin L., Sullivan S.L., et al. Characterization of the human and mouse unconventional myosin XV genes responsible for hereditary deafness DFNB3 and shaker 2. Genomics. 1999;61:243–258. doi: 10.1006/geno.1999.5976. [DOI] [PubMed] [Google Scholar]
- Liburd N., Ghosh M., Riazuddin S., Naz S., Khan S., Ahmed Z., Riazuddin S., Liang Y., Menon P.S., Smith T., et al. Novel mutations of MYO15A associated with profound deafness in consanguineous families and moderately severe hearing loss in a patient with Smith-Magenis syndrome. Hum. Genet. 2001;109:535–541. doi: 10.1007/s004390100604. [DOI] [PubMed] [Google Scholar]
- Mooseker M.S., Cheney R.E. Unconventional myosins. Annu. Rev. Cell. Dev. Biol. 1995;11:633–675. doi: 10.1146/annurev.cb.11.110195.003221. [DOI] [PubMed] [Google Scholar]
- Nal N., Ahmed Z.M., Erkal E., Alper O.M., Luleci G., Dinc O., Waryah A.M., Ain Q., Tasneem S., Husnain T., et al. Mutational spectrum of MYO15A: the large N-terminal extension of myosin XVA is required for hearing. Hum. Mutat. 2007;28:1014–1019. doi: 10.1002/humu.20556. [DOI] [PubMed] [Google Scholar]
- Park J.H., Kim NK.D., Kim A.R., Rhee J., Oh S.H., Koo J.W., Nam J.Y., Park W.Y., Choi B.Y. Comprehensive exploration of molecular genetic etiology for cochlear implantees with severe to profound hearing loss and its implication. orphanet. J. Rare Dis. 2014;9:167. doi: 10.1186/s13023-014-0167-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst F.J., Fridell R.A., Raphael Y., Saunders T.L., Wang A., Liang Y., Morell R.J., Touchman J.W., Lyons R.H., Noben-Trauth K., et al. Correction of deafness in shaker-2 mice by an unconventional myosin in a BAC transgene. Science. 1998;280:1444–1447. doi: 10.1126/science.280.5368.1444. [DOI] [PubMed] [Google Scholar]
- Shearer A.E., Hildebrand M.S., Webster J.A., Kahrizi K., Meyer N.C., Jalalvand K., Arzhanginy S., Kimberling W.J., Stephan D., Bahlo M., et al. Mutations in the first MyTH4 domain of MYO15A are a common cause of DFNB3 hearing loss. Laryngoscope. 2009;119:727–733. doi: 10.1002/lary.20116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney L.G., Tilney M.S. Functional organization of the cytoskeleton. Hear. Res. 1986;22:55–77. doi: 10.1016/0378-5955(86)90077-8. [DOI] [PubMed] [Google Scholar]
- Tilney L.G., Tilney M.S., DeRosier D.J. Actin filaments, stereocilia, and hair cells: how cells count and measure. Annu. Rev. Cell. Biol. 1992;8:257–274. doi: 10.1146/annurev.cb.08.110192.001353. [DOI] [PubMed] [Google Scholar]
- Walsh T., Walsh V., Vreugde S., Hertzano R., Shahin H., Haika S., Lee M.K., Kanaan M., King M.C., Avraham K.B. From flies’ eyes to our ears: mutations in a human class III myosin cause progressive nonsyndromic hearing loss DFNB30. Proc. Natl. Acad. Sci. USA. 2002;99:7518–7523. doi: 10.1073/pnas.102091699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A., Liang Y., Fridell R.A., Probst F.J., Wilcox E.R., Touchman J.W., Morton C.C., Morell R.J., Noben-Trauth K., Camper S.A., et al. Association of unconventional myosin MYO15 mutations with human nonsyndromic deafness DFNB3. Science. 1998;280:1447–1451. doi: 10.1126/science.280.5368.1447. [DOI] [PubMed] [Google Scholar]
- Woo H.M., Park H.J., Baek J.I., Park M.H., Kim U.K., Sagong B., Koo S.K. Whole-exome sequencing identifies MYO15A mutations as a cause of autosomal recessive nonsyndromic hearing loss in Korean families. BMC Med. Genet. 2013;14:72. doi: 10.1186/1471-2350-14-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T., Wei X., Chai Y., Li L., Wu H. Genetic etiology study of the non-syndromic deafness in Chinese Hans by targeted next-generation sequencing. Orphanet. J. Rare Dis. 2013;8:85. doi: 10.1186/1750-1172-8-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.