

Abstract
Gintonin is a novel ginseng-derived lysophosphatidic acid (LPA) receptor ligand. Oral administration of gintonin ameliorates learning and memory dysfunctions in Alzheimer’s disease (AD) animal models. The brain cholinergic system plays a key role in cognitive functions. The brains of AD patients show a reduction in acetylcholine concentration caused by cholinergic system impairments. However, little is known about the role of LPA in the cholinergic system. In this study, we used gintonin to investigate the effect of LPA receptor activation on the cholinergic system in vitro and in vivo using wild-type and AD animal models. Gintonin induced [Ca2+]i transient in cultured mouse hippocampal neural progenitor cells (NPCs). Gintonin-mediated [Ca2+]i transients were linked to stimulation of acetylcholine release through LPA receptor activation. Oral administration of gintonin-enriched fraction (25, 50, or 100 mg/kg, 3 weeks) significantly attenuated scopolamine-induced memory impairment. Oral administration of gintonin (25 or 50 mg/kg, 2 weeks) also significantly attenuated amyloid-β protein (Aβ)-induced cholinergic dysfunctions, such as decreased acetylcholine concentration, decreased choline acetyltransferase (ChAT) activity and immunoreactivity, and increased acetylcholine esterase (AChE) activity. In a transgenic AD mouse model, long-term oral administration of gintonin (25 or 50 mg/kg, 3 months) also attenuated AD-related cholinergic impairments. In this study, we showed that activation of G protein-coupled LPA receptors by gintonin is coupled to the regulation of cholinergic functions. Furthermore, this study showed that gintonin could be a novel agent for the restoration of cholinergic system damages due to Aβ and could be utilized for AD prevention or therapy.
Keywords: acetylcholine, cholinergic systems, ginseng, gintonin, LPA receptors
INTRODUCTION
Alzheimer’s disease (AD) is the most common age-associated neurodegenerative disease (Alzheimer, 1907). It is characterized clinically by progressive memory impairment and deterioration of other cognitive functions (Van Der Flier et al., 2002). Neuropathological hallmarks of AD include senile plaques and neurofibrillary tangles in neocortical and limbic brain lesions, which are coupled with neuronal loss or dysfunction (Braak et al., 1986). The formation of senile plaques and neurofibrillary tangles are well characterized in AD neuropathy. Senile plaques contain amyloid-β protein (Aβ) (Yankner, 1996). Aβ is produced from the amyloid-β protein precursor (AβPP) via amyloidogenic proteolytic cleavage pathways (Vassar et al., 1999). Another important phenomenon in AD is the dysfunction of the cholinergic system by amyloid plaques in several brain areas such as the basal forebrain, cortical regions, and the hippocampus. The loss of cholinergic neurons in the brain is closely associated with the reduction of acetylcholine synthesis; this is known as the cholinergic hypothesis of AD (Bartus et al., 1982).
Ginseng and ginkgo or ginseng extracts increased acetylcholine release (Liu et al., 2004; Su et al., 2007) and enhanced cognitive performance in human and AD patients (Heo et al., 2008; Kennedy and Scholey, 2003; Lee et al., 2008). However, relatively little is known about the active ingredient of ginseng and its signaling mechanisms. In our previous work, we showed that ginseng contains a novel G protein-coupled lysophosphatidic acid (LPA) receptor ligand, gintonin (Hwang et al., 2012a; Pyo et al., 2011). Gintonin enhanced synaptic transmission in hippocampal slices through LPA receptor signaling pathways (Park et al., 2015). We showed that gintonin is the active component of ginseng to attenuate AD-related neuropathies via activation of non-amyloidogenic pathways; gintonin significantly improved Aβ-induced cognitive dysfunctions in mice. In addition, long-term oral administration of gintonin attenuated amyloid plaque deposition in the hippocampus as well as short- and long-term memory impairment in a transgenic AD mouse model (Hwang et al., 2012b).
LPA receptors play an important role in learning and memory functions in aged animals. For example, LPAR1-null mice exhibited impaired performances in hippocampus-mediated spatial memory and cognitive tests (Castilla-Ortega et al., 2010; 2011; 2012; Dash et al., 2004). LPAR1-null mice also showed impaired fear extinction (Pedraza et al., 2014). Thus, although the previous reports showed that ginseng extract could induce acetylcholine release and improve cognitive functions in human and AD patients (Heo et al., 2008; Kennedy and Scholey, 2003; Lee et al., 2008; Liu et al., 2004; Su et al., 2007) and that LPA treatment to embryonic neural stem cells cultured stimulates the differentiation to cholinergic neurons (Cui and Qiao, 2006), it was not shown whether gintonin affects the cholinergic system through LPA receptors or whether activation of the LPA receptor by gintonin exhibits ameliorating effects on the cholinergic system impaired by scopolamine, by Aβ-infusion into the brain, and in a transgenic AD mouse model (Pákáski and Kálmán, 2008).
Here, we report that gintonin stimulates acetylcholine release in cells expressing endogenous LPA receptor. In addition, oral administration of gintonin restored scopolamine-induced memory dysfunctions, blocked Aβ-induced reductions of acetylcholine concentration and choline acetyltransferase (ChAT) activity, and also reduced acetylcholinesterase (AChE) activity in the mouse hippocampus. Furthermore, in a transgenic AD mouse model, long-term treatment of gintonin blocked amyloid plaque-induced reductions of acetylcholine concentration and ChAT activity and reduced AChE activity in the mouse hippocampus. We discuss how gintonin-mediated LPA receptor activation is coupled to the reinforcement of the cholinergic system that was damaged by a cholinergic blocker, by Aβ-infusion into the brain, and in the transgenic AD mouse model. We propose that gintonin could be utilized as an agent for restoration of the cholinergic system damaged by Aβ.
MATERIALS AND METHODS
Materials
Gintonin was prepared from Panax ginseng according to a previous method (Pyo et al. 2011). Because of gintonin’s scarcity in animal experiments, we used a gintonin-enriched fraction. The gintonin-enriched fraction was prepared as follows. One kilogram of 4-year-old ginseng was ground into small pieces (> 3 mm) and refluxed with 70% fermented ethanol eight times for 8 h at 80°C each. The extracts (150 g) were concentrated, dissolved in distilled, cold water at a ratio of 1 to 10, and stored at 4°C for 24–96 h. The supernatant and precipitate of water fractionation after ethanol extraction of ginseng were separated by centrifugation (3000 rpm, 20 min). The precipitate after centrifugation was lyophilized. This fraction was designated the gintonin-enriched fraction, since this fraction contains most of the gintonin (Choi et al., 2015a). For in vitro study, gintonin was dissolved in dimethyl sulfoxide (DMSO) and then diluted with bath medium before use. The final DMSO concentration was less than 0.01%. For in vivo studies, the gintonin-enriched fraction was dissolved in saline. The Amplex® Red Acetylcholine/Acetylcholinesterase Assay Kit (A-12217) was purchased from Invitrogen (USA). The primary antibody against choline acetyltransferase was purchased from Millipore (Bedford, MA, USA). All other reagents were purchased from Sigma-Aldrich (USA).
Animals
Male ICR or C57BL/6 mice (4- or 8-weeks-old) were purchased from Koatech Co., Ltd. (Pyongtaek, Korea). Breeding pairs of double Tg mice expressing the mutant swe-AβPP (AβPPswe) gene and the mutant presenilin-1 (PSEN-1) gene (deletion of exon 9) [AβPPswe/PSEN-1 double Tg mice; B6C3-Tg (AβPPswe/PSEN1dE9) 85Dbo/J, The Jackson Laboratory, Bar Harbor, ME, USA] were bred and housed in an approved animal facility at Kangwon National University (Korea). Six-month-old AβPPswe/PSEN1dE9 double Tg mice were treated with gintonin (25 or 50 mg/kg, p.o. administration) or donepezil (DPZ) (1 mg/kg, i.p. injection) thrice a week (Monday, Wednesday, and Friday) for 3 months. The immunohistochemistry for choline acetyltransferase commenced when the mice were 9-months-old. The animals were housed five per cage, allowed access to water and food ad libitum, and maintained under a constant temperature (23 ± 1°C) and humidity (55 ± 5%) under a 12 h light/dark cycle (lights on 07:00 to 19:00 h). All experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committees of the Kangwon (Permit Number: 13–156) and Konkuk (Permit Number: 14–956) Universities. All surgeries were performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering.
Drug treatment
β-Amyloid (Aβ)40-1 and Aβ1–40 (American Peptide Co., USA) were dissolved in 0.1 M phosphate-buffered saline (PBS) (pH 7.4), and aliquots were stored at −20°C. Each aliquot was aggregated by incubation in sterile distilled water at 37°C for 4 days. Two month-old C57BL/6 mice were administered Aβ40-1 or Aβ1–40 [400 pmol, i.c.v. injection] according to the procedure established by Hwang et al. (2012b). Each mouse was injected consciously at the bregma with a 10-μl microsyringe (Hamilton, USA) fitted with a 26-gauge needle that was inserted to a depth of 2.4 mm. The injection volume was 5 μl. The injection placement or needle track was visible and was verified at the time of dissection.
Hippocampal Neural Progenitor Cell (NPC) Culture
Hippocampal neural progenitor cultures were prepared according to the method described by Kim et al. (2011). Briefly, on embryonic day 14.5 (E14.5), embryos were dissected from adult, female, pregnant C57BL/6 mice. The hippocampal regions of embryonic brains were isolated in calcium/magnesium-free Hank’s balanced salt solution (HBSS). The cells were plated at 2.5 × 104 cells/cm2 on 10-cm-diameter dishes coated with 15 μg/ml poly-l-ornithine and 1 μg/ml fibronectin (Invitrogen). The cells were placed in N2 media supplemented with B27 (Invitrogen) at 37°C in a 95% air/5% CO2 gas incubator. Basic fibroblast growth factor (bFGF, 20 ng/ml, R&D Systems) and epidermal growth factor (EGF, 20 ng/ml, R&D Systems) were required daily in order to expand the population of neural precursors, and the medium was changed every other day. Cells at 80% confluency were passaged and maintained at 6 × 104 cells/cm2 in B27-supplemented N2 media containing bFGF and EGF. These sub-cultured progenitors were induced to differentiate by withdrawing bFGF and EGF and kept in differentiation media (Neurobasal medium supplemented with B27) for 3 to 5 days.
Measurement of acetylcholine (ACh) level
Mice were sacrificed one day after the behavioral study. The hippocampal tissues were homogenated in ice-cold 20 mM sodium phosphate buffer (pH 7.4), and tissue homogenates were centrifuged at 12,000 × g for 30 min at 4°C. The supernatant was assayed for ACh levels using an Amplex® Red Acetylcholine/Acetylcholinesterase Assay Kit (A-12217; Invitrogen, USA). According to the manufacturer’s instructions, reactions were initiated by adding 100 μl of the working solution, containing 400 μM Amplex Red reagent, 2 U/ml horseradish peroxidase (HRP), 0.2 U/ml choline oxidase, and 1 U/ml acetylcholinesterase, to each microplate well containing 100 μl of the standard or test sample. Each reaction was incubated for 1 h at room temperature with plate agitation and protection from light. Absorbance was then measured using a microplate reader (Molecular Devices, USA) at a wavelength of 563 nm. Hippocampal acetylcholine levels were calculated from a standard curve and expressed as nmol/mg protein (Jin et al., 2009). Protein concentrations were determined using a Quant-iT™ Protein assay kit (Invitrogen).
Acetylcholinesterase (AChE) and choline acetyltransferase (ChAT) activity
AChE activity was also measured using an Amplex® Red Acetylcholine/Acetylcholinesterase Assay Kit (A-12217; Invitrogen). A working solution, containing 400 μM Amplex Red reagent, 2 U/ml HRP, 0.2 U/ml choline oxidase, and 100 μM acetylcholine, was used for AChE activity measurement. For ChAT activity, 5% tissue homogenates were prepared in ice-cold 20 mM sodium phosphate buffer (pH 7.4) and kept frozen overnight at −20°C. They were thawed on the following day and centrifuged at 12,000 × g for 1 h at 4°C. The supernatant was assayed for ChAT activity according to the method of Chao and Wolfgram (Chao and Wolfgram, 1972). Each reaction mixture (0.4 ml), containing 25 mM sodium phosphate buffer (pH 7.2), 0.31 mM acetyl-CoA, 50 mM choline chloride, 38 μM neostigmine sulfate, 0.15 M NaCl, and 55 μM EDTA, was preincubated in centrifuge tubes in a water bath at 37°C for 5 min. One-hundred microliters of the homogenate was then added to the reaction mixture, and the tubes were kept at 37°C for an additional 20 min. The reaction was stopped by boiling for 2 min, and 1 ml of oxygen-free distilled water was added to the tubes. The denatured protein was removed by centrifugation. One milliliter of supernatant was added to a tube containing 30 μl of 1 mM 4,4′-dithiopyridine. After a 15-min incubation, the absorbance was measured at 324 nm with a spectrophotometer. One unit of enzyme activity was defined as 1 μmol of reduced coenzyme formed/min·mg protein. Protein was assayed as mentioned above.
Immunocytochemistry
Mice were first perfused transcardially with a 50-mL syringe containing ice-cold PBS (10 ml/10 g body weight) and then followed by 4% paraformaldehyde (20 ml/10 g body weight) for immunocytochemical analysis (Hooijmans et al., 2007; Jung et al., 2010). The brains were collected and stored in 4% paraformaldehyde overnight. To quench endogenous peroxidase activity, sections were pre-incubated with 0.3% hydrogen peroxide in PBS for 30 min and then incubated in PBS containing 0.4% Triton X-100 for 20 min and 1% normal serum for 20 min. Sections were incubated for 48 h with the primary antibody against AChE (1:100, Millipore, Millipore, USA) or ChAT (1:100, Millipore, Millipore, USA), and were further incubated with the secondary biotinylated antisera (1:1000, Vector Laboratories, USA) for 1 h. Then, sections were immersed in a solution containing an avidin-biotin-peroxidase complex (Vector Laboratories) for 1 h, and 3,3′-diaminobenzidine was used as the chromogen. Digital images were acquired on an Olympus microscope (BX51, Olympus®, Tokyo, Japan) using an attached digital microscope camera (DP72, Olympus®) and IBM PC. Region of interest (ROI) was created by Optimas® version 6.51 (Media Cybernetics, Inc. USA). Subsequent quantification was performed using ImageJ version 1.47 software (National Institutes of Health, USA) as described previously (Wang et al., 2012). Briefly, background was subtracted using the rolling ball “Subtract Background” command to correct uneven background. ChAT-immunopositive neurites were selected by adjusting threshold values for hue (0–255), saturation (0–255), and brightness (0–240) in the “Adjust Color Threshold” dialog box, and then the integrated density was measured. The results are expressed as the percentage of control mice.
Passive avoidance test
The passive avoidance test was performed as previously described using a Gemini Avoidance System (San Diego Instruments, USA) (Kim et al., 2013). Briefly, during the acquisition trial, each mouse was first placed into a dark compartment as the start chamber. After 20 s, this chamber was illuminated, and the door was opened to allow the mouse to freely move into the second dark chamber. After the mouse had entered the dark chamber, the door was immediately closed. One unavoidable and scrambled electric shock (0.8 mA, 2 s) was given through the floor grid. The mouse was then returned to its home cage. Each mouse was placed in the start chamber again 24 h later. The interval between the placement in the illuminated chamber and entry into the dark chamber was measured as the step-through latency in both the acquisition and the retention trials up to 300 s. Mice were daily treated with gintonin-enriched fraction (control vehicle, 25, 50, or 100 mg/kg) for three weeks by oral administration. Acquisition trial was performed one hour after the last treatment with gintonin-enriched fraction. Thirty minutes before the acquisition trial (i.e., 30 min after the last treatment with gintonin-enriched fraction), memory impairment was induced by administering scopolamine (0.5 mg/kg, i.p.). The control group has been received saline rather than gintonin-enriched fraction.
Morris water-maze test
A rounded pool (97 cm in diameter and 60 cm in height) was used for Morris water maze test according to the previous procedure with slight modifications (Kim et al., 2013). Briefly, the circular pool was filled to a depth height of 30 cm with clouded water (20 ± 1°C) with powdered milk. The top of the platform (6 cm in diameter and 29 cm in height) was 1 cm below the water surface in the center of one quadrant of the maze. The first day of the experiment was performed for swimming training for 60 s in the absence of the platform. From the next day, the mice were received trial sessions for four days. In each daily training session, the mice were subjected to four successive training trials. During each trial, the escape latency of each mouse was measured using a stopwatch. This parameter was averaged for each session of trials and for each mouse. Once the mouse located the platform, the mouse was allowed to stay on it for 10 s. If the mouse did not locate the platform within 60 s, the mouse was placed on the platform for 10 s. The time interval between each trial was 30 s. On the last day of training, mice were given a probe trial session, in which the platform from the pool was removed and mice were allowed to swim in search of it for 60 s. The swimming time was recorded in the pool quadrant, where the platform had previously been placed was maintained. Mice were also daily treated with gintonin-enriched fraction (control vehicle, 25, 50, or 100 mg/kg) for three weeks by oral administration including the last four days of the training sessions. During the training sessions, gintonin-enriched fraction was administered one hour before each training session. Thirty minutes later, mice were treated with scopolamine (0.5 mg/kg, i.p.) to induce memory impairment.
Statistical analysis
Statistical comparisons between controls and treated experimental groups were made using Student’s t-test unless otherwise stated. Statistical analyses were performed using one-way analysis of variance (ANOVA) or repeated measures one-way ANOVA. A post-hoc Fisher’s protected least significant difference (PLSD) test followed. Data are expressed as the mean ± SEM. A P value < 0.05 was accepted as statistically significant.
RESULTS
Effect of gintonin on [Ca2+]i Transients in hippocampal neural progenitor cells (NPCs) and its signal transduction pathway
In previous reports, we showed that gintonin derived from ginseng is a novel ligand for LPA receptors (Hwang et al., 2012a). Since LPA receptors are well expressed during the developmental stages of the brain (Hecht et al., 1996) and LPA promotes differentiation of rat embryonic neural stem cells to cholinergic neurons (Cui and Qiao, 2006), we examined the effects of gintonin on the coupling of [Ca2+]i transients to acetylcholine release using cultured hippocampal NPCs, which mainly express the LPA1 receptor (Sun et al., 2010). We first examined the effects of gintonin on [Ca2+]i transients in hippocampal NPCs. As shown in Figs. 1A and 1B, gintonin treatment induced a transient rise of [Ca2+]i in NPCs in a reversible and concentration-dependent manner. The EC50 was 0.21 ± 0.03 μg/ml. Gintonin-induced [Ca2+]i transients were initiated without a detectable lag and reached peak values within a few seconds, and [Ca2+]i gradually decreased and returned to basal level. We observed that treatment of NPC12 cells with LPA C18:1 also induced a [Ca2+]i transient, similar to gintonin (data not shown). We examined the effects of gintonin on [Ca2+]i transients in the absence or presence of the LPA1/3 receptor antagonist Ki16425. As shown in Figs. 1C and 1E, the presence of Ki16425 significantly attenuated the gintonin-mediated [Ca2+]i transient. The active phospholipase C inhibitor U73122, the inositol 1,4,5-triphosphate receptor antagonist 2-APB, and the intracellular Ca2+ chelator BAPTA-AM all blocked gintonin-mediated [Ca2+]i transients in NPCs (Figs. 1D and 1E). These results show that gintonin, via activation of the LPA receptor-phospholipase C-intracellular IP3 receptor signaling transduction pathway, elicits the release of Ca2+ from intracellular stores to increase [Ca2+]i.
Fig. 1.
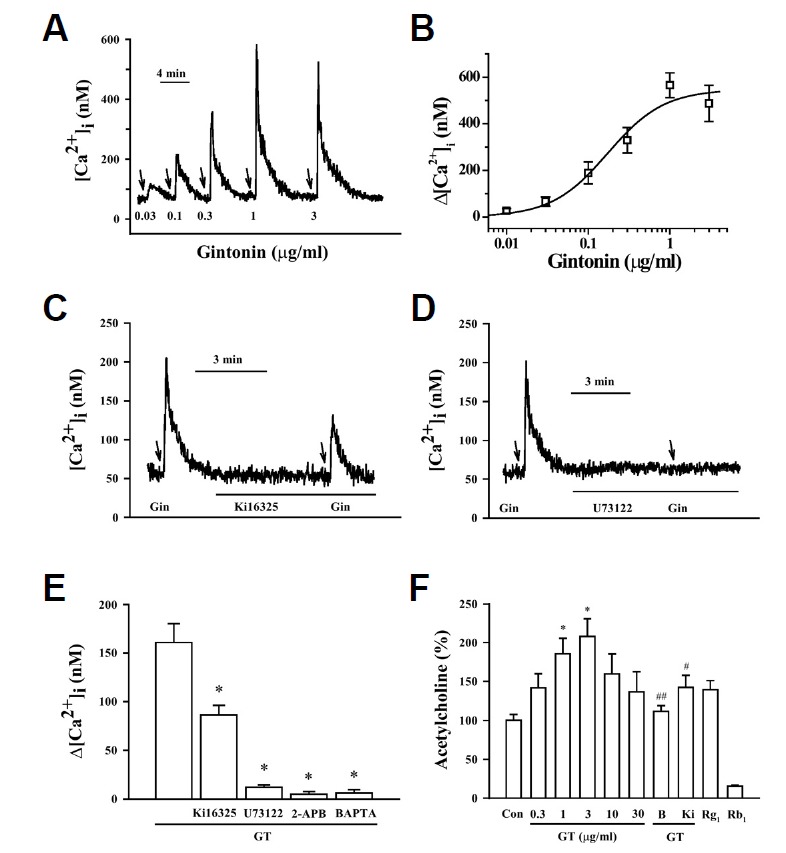
Effects of gintonin (GT) on [Ca2+]i transients in neural progenitor cells (NPCs) and its signal transduction. (A) A representative trace obtained after gintonin treatment in NPCs. Gintonin treatment (0.03–3 μg/ml) induces a [Ca2+]i transient. (B) Concentration-response relationship curve for gintonin-induced [Ca2+]i transients in NPCs. Each point represents the mean ± S.E.M. (n = 3–4). (C–E) Representative traces of gintonin-mediated [Ca2+]i transients in the absence or presence of various antagonists. The arrows indicate the application of gintonin (1 μg/ml). An LPA1/3 receptor antagonist (Ki16425, 10 μM), PLC inhibitor (U73122, 5 μM), or Ca2+ chelator (BAPTA-AM, 50 μM) was added before gintonin application. (F) Histograms representing net increases of gintonin-mediated [Ca2+]i transients calculated from traces obtained in the absence or presence of various pharmacological agents. Effect of gintonin (0, 0.3, 1, 3, 10, and 30 μg/ml) and BAPTA-AM (B), a calcium chelator, (50 μM) on acetylcholine release in the NPCs, determined using an Amplex Red Acetylcholine/Acetylcholinesterase Assay Kit (Invitrogen, #A12217). Ki16425 (Ki) also significantly attenuated gintonin-mediated acetylcholine release. NPCs were pretreated with BAPTA or Ki16425 for 1 h before gintonin treatment and then treated with gintonin or each ginsenoside for 16 h. Each value is the mean ± S.E.M. (n = 5). *P < 0.05 vs. control; #P < 0.05 or ##P < 0.01 vs. GT (3 μg/ml).
Effects of gintonin on acetylcholine release in hippocampal NPCs
Since the induction of depolarization by elevation of extracellular K+ or receptor ligands that induce a [Ca2+]i transient is coupled to neurotransmitter release, we next examined the effects of gintonin on acetylcholine release in hippocampal NPCs. As shown in Fig. 1F, gintonin stimulated acetylcholine release in a concentration-dependent manner. The maximal gintonin-induced acetylcholine release increase was 2-fold with a concentration of 3 μg/ml; higher concentrations of gintonin had no additional effect (Fig. 1F). LPA1/3 receptor antagonist Ki16425 blocked gintonin-induced acetylcholine release. Treatment with an intracellular calcium chelator, BAPTA-AM, abolished the gintonin action, indicating that the gintonin effect on acetylcholine release is LPA receptor- and calcium-dependent (Fig. 1F). The representative ginsenoside such as ginsenoside Rb1 had no effect, but ginsenoside Rg1 inhibited acetylcholine release (Fig. 1F). These results indicate that gintonin, but not ginsenosides, stimulates acetylcholine release through LPA receptor activation and its signal transduction pathway.
Effects of gintonin on choline acetyltransferase (ChAT) expression in hippocampal NPCs and in the adult mouse hippocampus
Since ChAT is a key enzyme for acetylcholine synthesis, we further examined whether gintonin treatment to hippocampal NPCs affects on the expression level of ChAT. We found that gintonin increased ChAT expression through immunostaining and immunoblotting (Supplementary Figs. S1A and S1B). To explore if oral administration of gintonin could also increase ChAT expression in the brain, we examined the effect of gintonin on in vivo ChAT immunoreactivity in wild-type adult mouse hippocampal neurons. As shown in Supplementary Fig. 2, saline administration did not show much ChAT immunoreactivity in hippocampal regions but oral administration of gintonin for 3 weeks significantly increased ChAT immunoreactivity of the hippocampal regions. Interestingly, in ChAT immunoreactivity of the hippocampal regions, gintonin treatment (50 and 100 mg/kg) showed stronger ChAT immunoreactivity at the CA1 and dentate gyrus (DG) compared to the CA3 area (Supplementary Fig. S2). These results indicate that gintonin might have cholinergic stimulating or upregulating effects mediated by the increase of expression of ChAT in neurons during the developmental stage of the hippocampus as well as in the adult mouse hippocampus.
Gintonin-enriched fraction ameliorates memory impairment induced by scopolamine, a cholinergic blocker
Next, we examined whether oral administration of gintonin-enriched fraction could ameliorate scopolamine-induced memory dysfunction. As shown in Fig. 2A, the effect of gintonin on memory function was first examined in a passive avoidance test. Treatment with scopolamine alone decreased the step-through latency time (Fig. 2A, *P < 0.01, compared with control vehicle), whereas the gintonin-enriched fraction treatment groups (25, 50, and 100 mg/kg, p.o.) demonstrated significantly improved latency times in a dose-dependent manner compared with the control group (Fig. 2A, #P < 0.05, compared with control). In addition, we also measured the effects of gintonin-enriched fraction using the Morris water maze task (Figs. 2B and 2C). The scopolamine-treated group exhibited longer escape latencies throughout the training days than the control vehicle group (*P < 0.01). The gintonin-enriched fraction significantly shortened the escape latencies prolonged by treatment with scopolamine on the last day of the training trial sessions in a dose-dependent manner (#P < 0.05, compared with scopolamine treatment group) (Fig. 2B). On the day following the final day of the training trial sessions, swimming times within the platform quadrant for the scopolamine-treated group were significantly lower than those of the control vehicle group (*P < 0.01). However, treatment with gintonin-enriched fraction significantly increased the swimming time shortened by scopolamine (#P < 0.05) (Fig. 2C). These results indicate that oral administration of gintonin-enriched fraction ameliorates scopolamine-induced memory dysfunctions.
Fig. 2.
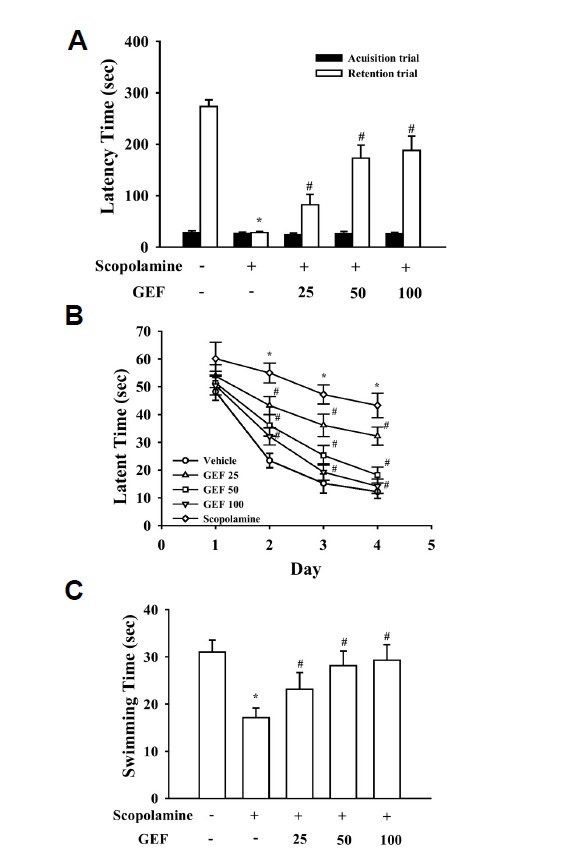
Effects of gintonin-enriched fraction (GEF) on scopolamine-induced memory deficit in the passive avoidance (A) and Morris water maze tests (B, C). (A) GEF (25, 50, and 100 mg/kg, p.o.) was daily administered through oral administration for three weeks. Memory impairment was induced by treatment with scopolamine (0.5 mg/kg, i.p.). Values are expressed as means ± SEM (n = 10). *P < 0.01 vs. control vehicle group). #P < 0.05 vs. scopolamine-treated control group. (B, C) The first trial session was performed 30 min after treatment with scopolamine. The training trial and the probe trial sessions were performed for 4 days as described in the Materials and Methods. Values are expressed as means ± SEM (n = 10). Mice were treated with GEF for 3 weeks by oral administration. *P < 0.01 vs. control vehicle group. #P < 0.05 vs. scopolamine-treated group.
Gintonin attenuates Aβ (1–40)-induced changes in acetylcholine level and AChE and ChAT activities in the hippocampus
Aβ-induced cholinergic dysfunctions are considered the main causes of memory impairments in patients with AD or in AD animal models (Bales et al., 2006; Maurice et al., 1996). In our previous study, we showed that oral administration of gintonin ameliorated Aβ (1–40)-induced memory impairment (Hwang et al., 2012a). However, it remains unknown whether the ameliorating effects of gintonin against memory impairment caused by Aβ (1–40) are achieved via improvement of the cholinergic system. In this study, since we showed that gintonin stimulates acetylcholine release and attenuates scopolamine-induced memory impairment (Figs. 1 and 2), we further investigated whether gintonin also protects against Aβ-induced cholinergic system disturbances including acetylcholine concentration and AChE or ChAT activity. We first examined the effects of gintonin on Aβ-induced cholinergic system dysfunction according to the procedure described in Fig. 3A. We observed that the hippo-campal acetylcholine levels significantly decreased in the Aβ (1–40)-infused mice [*P < 0.01 vs. Sal + Aβ (40-1)] (Fig. 3B), and treatment with gintonin significantly attenuated this effect [25 or 50 mg/kg gintonin + Aβ (1–40) vs. Sal + Aβ (1–40), #P < 0.05 or ##P < 0.01] (Fig. 3B). In addition, Aβ (1–40) significantly increased AChE activity [*P < 0.01 vs. Sal + Aβ (40-1)], but significantly decreased ChAT activity [*P < 0.01 vs. Sal + Aβ (40-1)] in the mouse hippocampus; these results are consistent with previous reports of Aβ-induced cholinergic dysfunction (Bales et al., 2006; Maurice et al., 1996). However, oral administration of gintonin 3 days before Aβ (1–40) infusion significantly attenuated the changes in AChE and ChAT activities [both AChE and ChAT activity: 25 or 50 mg/kg gintonin + Aβ (1–40) vs. Sal + Aβ (1–40), #P < 0.05 or ##P < 0.01] (Fig. 3C). The effect of gintonin on Aβ (1–40)-induced cholinergic dysfunction was comparable to that of donepezil (1 mg/kg) (Figs. 3B and 3C). These results indicate that gintonin significantly protects the cholinergic system from Aβ-induced cholinergic dysfunction in the mouse hippocampus.
Fig. 3.
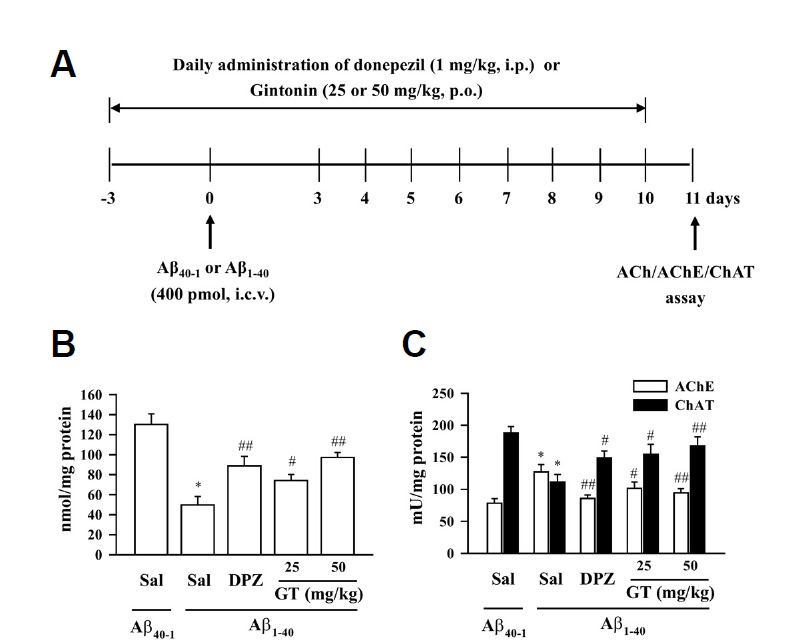
Effect of gintonin (GT; 25 or 50 mg/kg, p.o.) on the Aβ (1–40)-induced changes in cholinergic system. (A) Experimental schedule to evaluate acute gintonin effect in Aβ (1–40)-induced cholinergic system impairment. Donepezil (DPZ; 1 mg/kg, i.p.) was used as a reference drug. Gintonin or DPZ administration started 3 days before the Aβ i.c.v. injection, and the drug administration was continued once a day for consecutive 14 days. Mice were sacrificed at 1 day after the final treatment with gintonin or DPZ. (B) Acetylcholine level. (C) Acetylcholinesterase activity and choline acetyltransferase activity in the hippocampus of the mouse. Sal = saline. Each value is the mean ± S.E.M of eight mice. *P < 0.01 vs. Sal + Aβ (40-1), #P < 0.05, ##P < 0.01 vs. Sal + Aβ (1–40) (one-way ANOVA followed by Fisher’s PLSD test).
Gintonin attenuates Aβ (1–40)-induced decrease in ChAT immunoreactivity in the hippocampus
Since it was reported that central injection of Aβ (1–40) could induce direct axonal toxicity of septohippocampal cholinergic neurons (Colom et al., 2011), the effects of Aβ on ChAT-immunoreactivity (ChAT-IR) in the mouse hippocampus were examined next. Our results were in line with previous study, showing that injection of Aβ (1–40) resulted in a significant decrease in ChAT-IR in the hippocampus [CA1: *P < 0.05 vs. Saline + Aβ (40-1); CA3: **P < 0.01 vs. Saline + Aβ (40-1)] (Fig. 4, upper panel). Oral administration of gintonin significantly attenuated the Aβ (1–40)-induced decrease in ChAT-IR [CA1: 25 or 50 mg/kg gintonin + Aβ (1–40) vs. Saline + Aβ (1–40), #P < 0.05 or ##P < 0.01; CA3: 25 or 50 mg/kg gintonin + Aβ (1–40) vs. Saline + Aβ (1–40), ##P < 0.01; dentate gyrus (DG): 50 mg/kg gintonin + Aβ (1–40) vs. Saline + Aβ (1–40), #P < 0.05] (Fig. 4, lower panel), and these effects were comparable to those of donepezil (1 mg/kg) (Fig. 4). These results are consistent with the results of above-mentioned biochemical analyses on gintonin-mediated protection of cholinergic systems against Aβ.
Fig. 4.
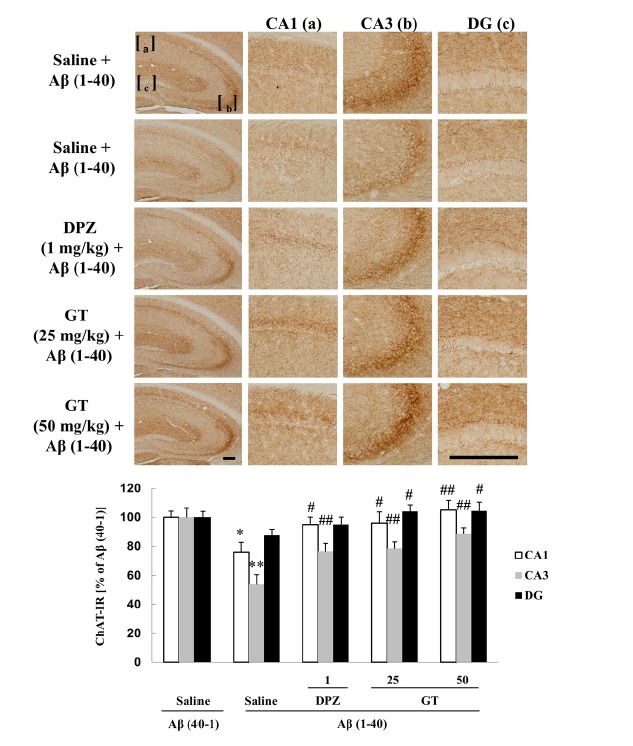
Effect of gintonin (GT; 25 or 50 mg/kg, p.o.) on the Aβ (1–40)-induced decrease in choline acetyltransferase-immunoreactivity (ChAT-IR) in the hippocampus of the mouse. Donepezil (DPZ; 1 mg/kg, i.p.) was used as a reference drug. Each value is the mean ± S.E.M of five mice. *P < 0.05, **P < 0.01 vs. Sal + Aβ (40-1), #P < 0.05, ##P < 0.01 vs. Sal + Aβ (1–40) (one-way ANOVA followed by Fisher’s PLSD test). Scale bar = 200 μm.
Long-term oral administration of gintonin improves cholinergic function in APPswe/PSEN-1 double Tg mice
The long-term effects of gintonin on the level of acetylcholine and activities of AChE and ChAT in the APPswe/PSEN-1 double Tg mice were examined according to the procedure described in Fig. 5A. Chronic treatment with gintonin (25 or 50 mg/kg, p.o., thrice a week for 3 months) significantly increased acetylcholine levels (25 or 50 mg/kg gintonin vs. Sal, #P < 0.05 or ##P < 0.01) (Fig. 5B) and ChAT activity (25 or 50 mg/kg gintonin vs. Sal, #P < 0.05 or ##P < 0.01), but significantly decreased AChE activity in the hippocampus of the APPswe/PSEN-1 double Tg mice (25 or 50 mg/kg gintonin vs. Sal, #P < 0.05 or ##P < 0.01) (Fig. 5C), indicating that gintonin improves the hippocampal cholinergic function in the AD model mice. The effect of gintonin on the hippocampal cholinergic function of APPswe/PSEN-1 double Tg mice was comparable to that of donepezil (1 mg/kg, thrice a week for 3 months), and these results are in accordance with those observed in the acute Aβ (1–40)-treated mice (Fig. 5).
Fig. 5.
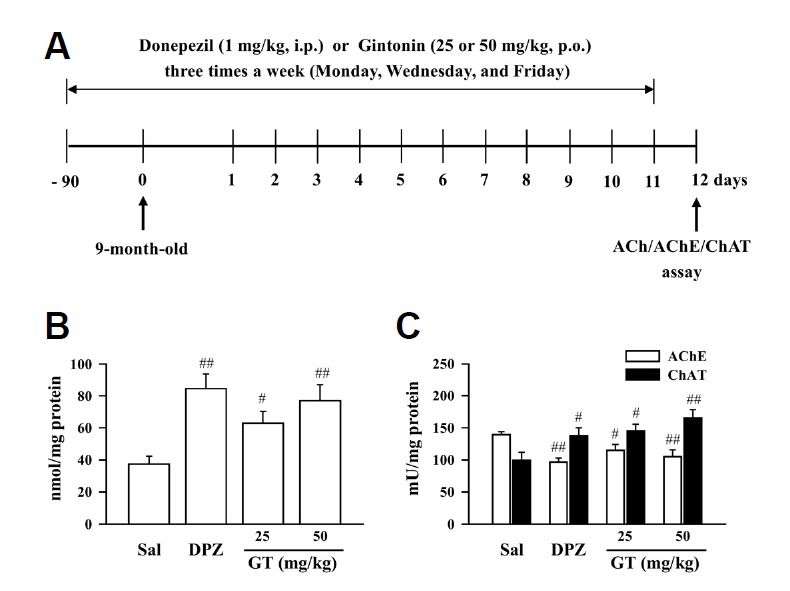
Effect of gintonin (GT; 25 or 50 mg/kg, p.o.) on cholinergic system changes. (A) Experimental schedule to evaluate the long-term effect of gintonin on cholinergic system impairment in the APPswe/PSEN-1 double Tg mouse. (B) Acetylcholine level, (C) acetylcholinesterase activity, and choline acetyltransferase activity in the hippocampus of the APPswe/PSEN-1 double Tg mouse. Donepezil (DPZ; 1 mg/kg, i.p.) was used as a reference drug. Sal = saline. Each value is the mean ± S.E.M of four mice. #P < 0.05, ##P < 0.01 vs. Sal (one-way ANOVA followed by Fisher’s PLSD test).
Long-term oral administration of gintonin attenuates the decrease in ChAT-IR in the hippocampus of APPswe/PSEN-1 double Tg mice
Accumulating evidences showed that dystrophy and reorganization of cholinergic terminals are observed in the hippocampus and cortex of APPswe/PSEN-1 double Tg mice without significant change in cholinergic neuronal bodies of basal fore-brain system, including medial septum and nucleus basalis (Jaffar et al., 2001; Perez et al., 2007; Wong et al., 1999). In these studies, cholinergic axonal dystrophy in the hippocampus could be observed as early as 2–3 months of age, that is prior to the formation of amyloid deposition, while there was no significant change in cholinergic neuronal bodies in the medial septum and nucleus basalis of 10–16-month-old APPswe/PSEN-1 double Tg mice with extensive amyloid deposition. Thus, we have focused on the changes in ChAT-IR in the hippocampus. Hippocampal ChAT-IR was significantly lower in the APPswe/PSEN-1 double Tg mice than that in the non-Tg mice (CA1 and CA3: **P < 0.01 vs. non-Tg mice treated with saline; DG: *P < 0.05 vs. non-Tg mice treated with saline) (Fig. 6, upper panel). Long-term treatment with gintonin (25 or 50 mg/kg, p.o., thrice a week for 3 months) significantly increased hippocampal ChAT-IR [CA1 and CA3: 25 or 50 mg/kg gintonin vs. Saline, ##P < 0.01; DG: 50 mg/kg gintonin vs. Saline, #P < 0.05] in the APPswe/PSEN-1 double Tg mice, and these effects were comparable to those of donepezil (1 mg/kg) (Fig. 6, lower panel). These results are consistent with the results above-mentioned biochemical analyses on gintonin-mediated protection of cholinergic systems in the APPswe/PSEN-1 double Tg mice.
Fig. 6.
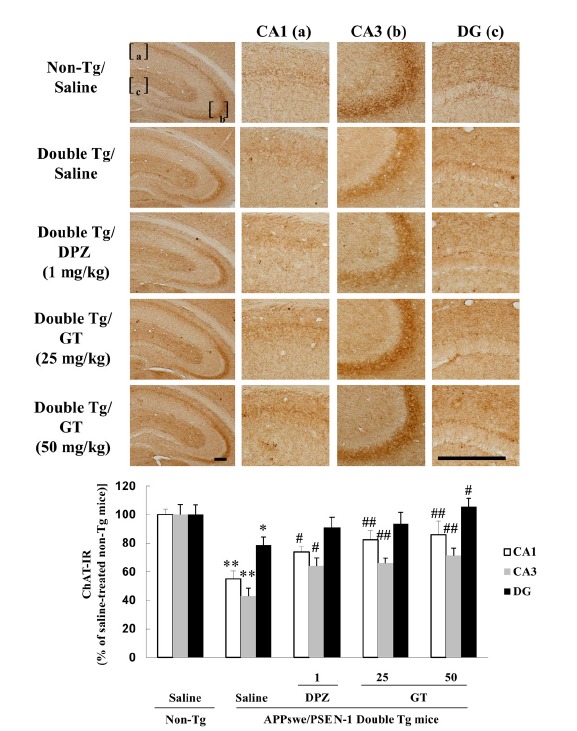
Effect of gintonin (GT; 25 or 50 mg/kg, p.o.) on the decrease in ChAT-IR in the hippocampus of APPswe/PSEN-1 double Tg mice. Donepezil (DPZ; 1 mg/kg, i.p.) was used as a reference drug. Each value is the mean ± S.E.M for five mice. *P < 0.05, **P < 0.01 vs. Saline-treated non-Tg mice, #P < 0.05, ##P < 0.01 vs. Saline-treated APPswe/PSEN-1 double Tg mice (one-way ANOVA followed by Fisher’s PLSD test). Scale bar = 200 μm.
DISCUSSION
Human AD is a progressive neurodegenerative disease associated with age (Alzheimer, 1907). As the most common form of dementia, the number of AD patients is increasing almost exponentially with the aged population (Yankner, 1996). Two important characteristics of AD are the formation of senile plaques containing Aβ and dysfunctions of the cholinergic system in the brain (Vassar et al., 1999). Accumulating evidence shows that Aβ causes imbalances of the cholinergic system by increasing AChE and decreasing ChAT activity and by subsequently showing selective toxicity to cholinergic neurons (Bales et al., 2006; Bartus et al., 1982; Maurice et al., 1996). Therefore, AChE inhibitors, agents or drugs acting on the cholinergic system, or muscarinic agonists, which target the m1 muscarinic receptor, are being investigated for AD prevention and therapeutics (Davie et al., 2013). However, little is known about whether activation of the G protein-coupled LPA receptor could also modulate the cholinergic system and further ameliorate scopolamine-induced or acute or long-term Aβ-induced dysfunctions of the cholinergic system.
In this study, we examined whether gintonin affects in vitro and in vivo cholinergic functions in mouse hippocampal NPCs, scopolamine-treated mice, Aβ-infused mice, and a transgenic AD mouse model. We could obtain three major findings that gintonin-mediated activation of LPA receptors could be associated with the cholinergic system regulations. First, gintonin-mediated [Ca2+]i transient is coupled to acetylcholine release in cultured hippocampal NPCs (Fig. 1) and also increased the expression of ChAT in hippocampal NPCs as well as in the wild-type mouse hippocampus (Supplementary Figs. S1 and S2). Second, gintonin-enriched fraction significantly attenuates scopolamine-induced cognitive dysfunctions (Fig. 2). Third, gintonin has a protective effect on the cholinergic system against the dysfunction induced by Aβ-infusion and in the transgenic AD mouse model (Figs. 3–6). Thus, short-term oral administration of gintonin attenuated Aβ-induced dysfunctions of the hippocampal cholinergic system by inhibiting AChE and by increasing ChAT and acetylcholine concentrations (Figs. 3 and 4). Long-term oral administration of gintonin also attenuated hippocampal cholinergic dysfunctions in the AD mouse model (Figs. 5 and 6). These results indicate that activation of G protein-coupled LPA receptors by gintonin could induce beneficial effects on the cholinergic system in hippocampus under infusion of Aβ and in the transgenic AD mouse model.
There could be a question what are the molecular mechanisms underlying the gintonin-mediated protective effects on the cholinergic system against scopolamine-induced memory impairments and Aβ-infused cholinergic dysfunction in wild-type mice and in the long-term transgenic AD mouse model. Gintonin could act in three ways in this study. Firstly, previous reports showed that acute treatment of cortical neuroblast cells with LPA induces an increased conductance that consists of non-selective cation currents (Dubin et al., 1999) and that LPA also induces dopamine and glutamate release in PC12 cells and hippocampal glutamatergic neurons, respectively (Shiono et al., 1993; Trimbuch et al., 2009). Thus, gintonin might also act as a neurotransmitter, and acute gintonin treatment could increase neurotransmitter release and enhance synaptic transmission in the hippocampus via N-methyl-d-aspartate (NMDA) receptor activation (Hwang et al., 2015; Park et al., 2015; Shin et al., 2012). In present study, we further demonstrated that gintonin stimulates acetylcholine release in an intracellular Ca2+-sensitive manner through the LPA receptor in NPCs and also increased brain acetylcholine concentration under Aβ infusion and in transgenic AD mouse models. Thus, the additional gintonin-mediated regulations of cholinergic system might contribute to restorations from cholinergic dysfunctions by scopolamine-induced memory impairments and Aβ-infused cholinergic dysfunction in wild-type mice.
Secondly, LPA is a phospholipid growth factor and affects diverse cell functions via LPA receptors (Tabuchi et al., 2000). Gintonin, as an exogenous LPA receptor ligand, could exert its effect to increase of ChAT expression and the gintonin-induced ChAT expression might be coupled to the increase of acetylcholine synthesis and might help to ameliorate hippocampal cholinergic dysfunctions under Aβ infusion and in transgenic AD mouse models. In addition, it is known that sAPPα exhibits the neuroprotective and neurotrophic effects (Hasebe et al., 2013). In previous report we demonstrated that gintonin stimulates sAPPα release, while gintonin inhibits Aβ formation via non-amyloidogenic pathway (Hwang et al., 2012b). Activation of m1 muscarinic receptor by acetylcholine is also coupled to sAPPα release (Kim et al., 2006). Acetylcholine that is released by gintonin via LPA receptor activation might also indirectly contribute to sAPPα release rather than Aβ formation (Fig. 1F). Thus, direct gintonin-mediated increase of ChAT expression and sAPPα release and indirect sAPPα release via m1 muscarinic receptor by acetylcholine might be together coupled to protection of the cholinergic system induced by Aβ insult and in the transgenic AD mouse model.
The last possibility is that gintonin-mediated LPA receptor activation is coupled to dual signaling pathways. Gintonin-mediated activations of LPA receptor could induce acetylcholine release and increase acetylcholine synthesis as role of a lipid-derived neurotransmitter and/or tropic factor. Gintonin-mediated activations of LPA receptor could be also coupled to the stimulation of neurotrophic and neuroprotective sAPPα release in the hippocampus. Therefore, both gintonin-mediated maintenance of acetylcholine levels by restoring cholinergic systems under Aβ or scopolamine insult and gintonin-mediated beneficial sAPPα formation in nervous system might contribute to conserve cholinergic systems against Aβ and in the transgenic AD mouse model. Figure 7 shows a schematic diagram of how gintonin-mediated LPA receptor activations are coupled to anti-AD actions via stimulations of cholinergic systems and sAPPα formation.
Fig. 7.
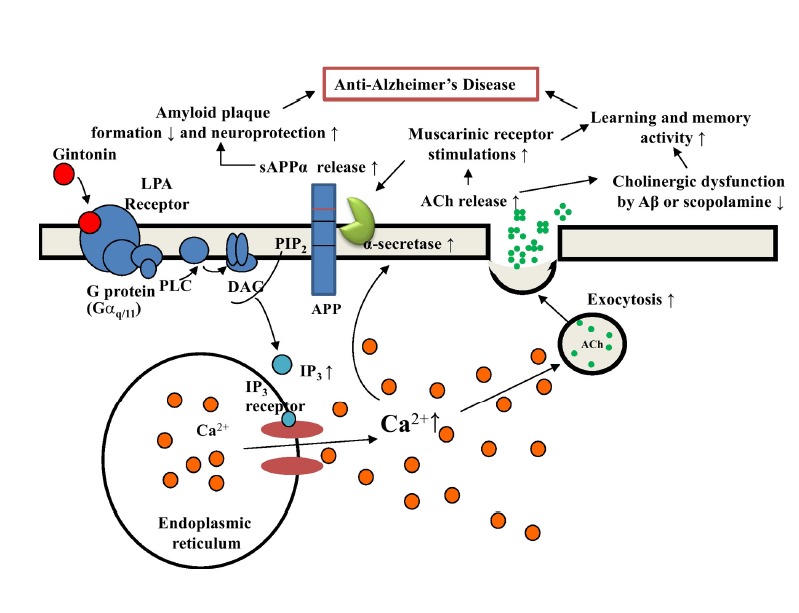
Schematic diagram of the involvement of the cholinergic system in gintonin-mediated anti-Alzheimer’s disease (AD) through LPA receptor activation. In our previous report, we showed that gintonin attenuated amyloid plaque formation and ameliorated learning and memory dysfunctions (Hwang et al., 2012). In this study, we found that gintonin stimulates the cholinergic systems by acetylcholine release and the increase of ChAT expression and protective effects against acute Aβ-induced and long-term cholinergic dysfunctions in the transgenic AD animal model. Gintonin-mediated activation of LPA receptors could be coupled to anti-AD effects via dual actions of the non-amyloidogenic pathway and modulations of the cholinergic system in the brain according to the described pathways.
Gintonin is a unique form of herbal-medicine LPA, as it consists of LPAs-ginseng protein complexes. Recent study showed how GLP151 protein as a protein component of gintonin binds to LPA through the elucidation of three-dimensional structure of GLP151. The phosphate group of LPA binds to the imidazole ring of histidine residues at C-terminal of GLP151 with hydrogen bonds and acyl-chain of LPA interacts with other amino acids though hydrophobic interactions of GLP151 (Choi et al., 2015b). GLP151 protein was identified as a first plant-derived carrier or transporter of LPA, and deliver LPA to cognitive LPA receptors (Choi et al., 2015b). Ginsenoside is also another component of ginseng with various effects in biological system. However, the representative ginsenosides such as Rb1 or Rg1 had no effects on [Ca2+]i transients in hippocampal NPCs (data not shown) and ginsenoside Rg1 did not induce acetylcholine release and ginsenoside Rb1 rather inhibited acetylcholine release in hippocampal NPCs (Fig. 1F). Thus, we provide additional information that gintonin but not ginsenosides is the main active ingredient of ginseng for acetylcholine release in hippocampal NPCs.
In conclusion, we showed that gintonin increases acetylcholine release and ChAT expression in hippocampal NPCs through LPA receptors. We also showed that gintonin attenuates scopolamine-induced memory impairment and Aβ-induced cholinergic impairments in wild type and transgenic AD mouse models. Finally, we suggest that, in addition to gintonin-mediated non-amyloidogenic pathway activation, the anti-AD effect of gintonin might be achieved via its boosting effects on the cholinergic system. These actions of gintonin might be an additional molecular basis for the neuroprotective effects of ginseng on AD-related neuropathies.
Acknowledgments
This work was supported by the Basic Science Research Program (NRF-2014R1A1A2054538), which is funded by the Ministry of Education, Science, and Technology, and by the BK21 Plus Project fund to S.-Y. Nah.
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Alzheimer A. Uber eine eigenartige Erkrankung der Hirnrinde. Allg. Zeitschrife Psychiatr. 1907;64:146–148. [Google Scholar]
- Bales K.R., Tzavara E.T., Wu S., Wade M.R., Bymaster F.P., Paul S.M., Nomikos G.G. Cholinergic dysfunction in a mouse model of Alzheimer disease is reversed by an anti-A beta antibody. J. Clin. Invest. 2006;116:825–832. doi: 10.1172/JCI27120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartus R.T., Dean R.L., Beer B., Lippa A.S. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217:408–414. doi: 10.1126/science.7046051. [DOI] [PubMed] [Google Scholar]
- Braak H., Braak E., Grundke-Iqbal I., Iqbal K. Occurrence of neuropil threads in the senile human brain and in Alzheimer’s disease: a third location of paired helical filaments outside of neurofibrillary tangles and neuritic plaques. Neurosci. Lett. 1986;65:351–355. doi: 10.1016/0304-3940(86)90288-0. [DOI] [PubMed] [Google Scholar]
- Castilla-Ortega E., Sánchez-López J., Hoyo-Becerra C., Matas-Rico E., Zambrana-Infantes E., Chun J., De Fonseca F.R., Pedraza C., Estivill-Torrús G., Santin L.J. Exploratory, anxiety and spatial memory impairments are dissociated in mice lacking the LPA1 receptor. Neurobiol. Learn. Mem. 2010;94:73–82. doi: 10.1016/j.nlm.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilla-Ortega E., Hoyo-Becerra C., Pedraza C., Chun J., Rodríguez De Fonseca F., Estivill-Torrús G., Santín L.J. Aggravation of chronic stress effects on hippocampal neurogenesis and spatial memory in LPA1 receptor knockout mice. PLoS One. 2011;6:e25522. doi: 10.1371/journal.pone.0025522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilla-Ortega E., Pedraza C., Chun J., de Fonseca F.R., Estivill-Torrús G., Santín L.J. Hippocampal c-Fos activation in normal and LPA-null mice after two object recognition tasks with different memory demands. Behav. Brain Res. 2012;232:400–405. doi: 10.1016/j.bbr.2012.04.018. [DOI] [PubMed] [Google Scholar]
- Chao L.P., Wolfgram F. Spectrophotometric assay for choline acetyltransferase. Anal. Biochem. 1972;46:114–118. doi: 10.1016/0003-2697(72)90401-0. [DOI] [PubMed] [Google Scholar]
- Choi S.H., Jung S.W., Kim H.S., Kim H.J., Lee B.H., Kim J.Y., Kim J.H., Hwang S.H., Rhim H., Kim HC., et al. A brief method for the preparation of gintonin-enriched fraction from ginseng. J. Ginseng Res. 2015a doi: 10.1016/j.jgr.2015.05.002. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S.H., Hong M.K., Kim H.J., Ryoo N., Rhim H., Nah S.Y., Kang L.W. Crystal structure of ginseng major latex-like protein 151 and its proposed lysophosphatidic acid-binding mechanism. Acta Crystallogr. D Biol. Crystallogr. 2015b;71:1039–1050. doi: 10.1107/S139900471500259X. [DOI] [PubMed] [Google Scholar]
- Colom L.V., Castaneda M.T., Hernandez S., Perry G., Jaime S., Touhami A. Intrahippocampal amyloid-β (1–40) injections injure medial septal neurons in rats. Curr. Alzheimer Res. 2011;8:832–840. doi: 10.2174/156720511798192763. [DOI] [PubMed] [Google Scholar]
- Cui H.L., Qiao J.T. Promotive action of lysophosphatidic acid on proliferation of rat embryonic neural stem cells and their differentiation to cholinergic neurons in vitro. Sheng Li Xue Bao. 2006;58:547–555. [PubMed] [Google Scholar]
- Dash P.K., Orsi S.A., Moody M., Moore A.N. A role for hippocampal Rho-ROCK pathway in long-term spatial memory. Biochem. Biophys. Res. Commun. 2004;322:893–898. doi: 10.1016/j.bbrc.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Davie B.J., Christopoulos A., Scammells P.J. Development of M1 mAChR allosteric and bitopic ligands: prospective therapeutics for the treatment of cognitive deficits. ACS Chem. Neurosci. 2013;4:1026–1048. doi: 10.1021/cn400086m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin A.E., Bahnson T., Weiner J.A., Fukushima N., Chun J. Lysophosphatidic acid stimulates neurotransmitter-like conductance changes that precede GABA and L-glutamate in early, presumptive cortical neuroblasts. J. Neurosci. 1999;19:1371–1381. doi: 10.1523/JNEUROSCI.19-04-01371.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Flier W.M., Van Den Heuvel D.M.J., Weverling-Rijnsburger A.W.E., Spilt A., Bollen E.L.E.M., Westendorp R.G.J., Middelkoop H.A.M., Van Buchem M.A. Cognitive decline in AD and mild cognitive impairment is associated with global brain damage. Neurology. 2002;59:874–879. doi: 10.1212/wnl.59.6.874. [DOI] [PubMed] [Google Scholar]
- Hasebe N., Fujita Y., Ueno M., Yoshimura K., Fujino Y., Yamashita T. Soluble β-amyloid Precursor Protein Alpha binds to p75 neurotrophin receptor to promote neurite outgrowth. PLoS One. 2013;8:e82321. doi: 10.1371/journal.pone.0082321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht J.H., Weiner J.A., Post S.R., Chun J. Ventricular zone gene-1 (vzg-1) encodes a lysophosphatidic acid receptor expressed in neurogenic regions of the developing cerebral cortex. J. Cell Biol. 1996;135:1071–1083. doi: 10.1083/jcb.135.4.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo J.H., Lee S.T., Chu K., Oh M.J., Park H.J., Shim J.Y., Kim M. An open-label trial of Korean red ginseng as an adjuvant treatment for cognitive impairment in patients with Alzheimer’s disease. Eur. J. Neurol. 2008;15:865–868. doi: 10.1111/j.1468-1331.2008.02157.x. [DOI] [PubMed] [Google Scholar]
- Hooijmans C.R., Rutters F., Dederen P.J., Gambarota G., Veltien A., van Groen T., Broersen L.M., Lütjohann D., Heerschap A., Tanila H., et al. Changes in cerebral blood volume and amyloid pathology in aged Alzheimer APP/PS1 mice on a docosahexaenoic acid (DHA) diet or cholesterol enriched Typical Western Diet (TWD) Neurobiol. Dis. 2007;28:16–29. doi: 10.1016/j.nbd.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Hwang S.H., Shin T.J., Choi S.H., Cho H.J., Lee B.H., Pyo M.K., Lee J.H., Kang J., Kim H.J., Park C.W., et al. Gintonin, newly identified compounds from ginseng, is novel lysophosphatidic acids-protein complexes and activates G protein-coupled lysophosphatidic acid receptors with high affinity. Mol. Cells. 2012a;33:151–162. doi: 10.1007/s10059-012-2216-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S.H., Shin E.J., Shin T.J., Lee B.H., Choi S.H., Kang J., Kim H.J., Kwon S.H., Jang C.G., Lee J.H., et al. Gintonin, a ginseng-derived lysophosphatidic acid receptor ligand, attenuates alzheimer’s disease-related neuropathies: Involvement of non-amyloidogenic processing. J. Alzheimer’s Dis. 2012b;31:207–223. doi: 10.3233/JAD-2012-120439. [DOI] [PubMed] [Google Scholar]
- Hwang S.H., Lee B.H., Choi S.H., Kim H.J., Jung S.W., Kim H.S., Shin H.C., Park H.J., Park K.H., Lee M.K., et al. Gintonin, a novel ginseng-derived lysophosphatidic acid receptor ligand, stimulates neurotransmitter release. Neurosci. Lett. 2015;584:356–361. doi: 10.1016/j.neulet.2014.11.007. [DOI] [PubMed] [Google Scholar]
- Jaffar S., Counts S.E., Ma S.Y., Dadko E., Gordon M.N., Morgan D., Mufson E.J. Neuropathology of mice carrying mutant APP(swe) and/or PS1(M146L) transgenes: alterations in the p75(NTR) cholinergic basal forebrain septohippocampal pathway. Exp. Neurol. 2001;170:227–243. doi: 10.1006/exnr.2001.7710. [DOI] [PubMed] [Google Scholar]
- Jin C.H., Shin E.J., Park J.B., Jang C.G., Li Z., Kim M.S., Koo K.H., Yoon H.J., Park S.-J., Choi W.-C., et al. Fustin flavonoid attenuates beta-amyloid (1–42)-induced learning impairment. J. Neurosci. Res. 2009;87:3658–3670. doi: 10.1002/jnr.22159. [DOI] [PubMed] [Google Scholar]
- Jung B.D., Shin E.J., Nguyen XK.T., Jin C.H., Bach J.H., Park S.J., Nah S.Y., Wie M.B., Bing G., Kim H.C. Potentiation of methamphetamine neurotoxicity by intrastriatal lipopolysaccharide administration. Neurochem. Int. 2010;56:229–244. doi: 10.1016/j.neuint.2009.10.005. [DOI] [PubMed] [Google Scholar]
- Kennedy D.O., Scholey A.B. Ginseng: potential for the enhancement of cognitive performance and mood. Pharmacol. Biochem. Behav. 2003;75:687–700. doi: 10.1016/s0091-3057(03)00126-6. [DOI] [PubMed] [Google Scholar]
- Kim J.H., Choi S., Jung J.E., Roh E.J., Kim H.J. Capacitative Ca2+ entry is involved in regulating soluble amyloid precursor protein (sAPPalpha) release mediated by muscarinic acetylcholine receptor activation in neuroblastoma SH-SY5Y cells. J. Neurochem. 2006;97:245–254. doi: 10.1111/j.1471-4159.2006.03734.x. [DOI] [PubMed] [Google Scholar]
- Kim B.-W., Yang S., Lee C.H., Son H. A critical time window for the survival of neural progenitor cells by HDAC inhibitors in the hippocampus. Mol. Cells. 2011;31:159–164. doi: 10.1007/s10059-011-0019-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E.J., Jung I.H., Van Le T.K., Jeong J.J., Kim N.J., Kim D.H. Ginsenosides Rg5 and Rh3 protect scopolamine-induced memory deficits in mice. J. Ethnopharmacol. 2013;146:294–299. doi: 10.1016/j.jep.2012.12.047. [DOI] [PubMed] [Google Scholar]
- Lee S.T., Chu K., Sim J.Y., Heo J.H., Kim M. Panax ginseng enhances cognitive performance in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2008;22:222–226. doi: 10.1097/WAD.0b013e31816c92e6. [DOI] [PubMed] [Google Scholar]
- Liu J.X., Cong W.H., Xu L., Wang J.N. Effect of combination of extracts of ginseng and ginkgo biloba on acetylcholine in amyloid beta-protein-treated rats determined by an improved HPLC. Acta Pharmacol. Sin. 2004;25:1118–1123. [PubMed] [Google Scholar]
- Maurice T., Lockhart B.P., Privat A. Amnesia induced in mice by centrally administered beta-amyloid peptides involves cholinergic dysfunction. Brain Res. 1996;706:181–193. doi: 10.1016/0006-8993(95)01032-7. [DOI] [PubMed] [Google Scholar]
- Pákáski M., Kálmán J. Interactions between the amyloid and cholinergic mechanisms in Alzheimer’s disease. Neurochem. Int. 2008;53:103–111. doi: 10.1016/j.neuint.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Park H., Kim S., Rhee J., Kim H.J., Han J.S., Nah S.Y., Chung C. Synaptic enhancement induced by gintonin via lysophosphatidic acid receptor activation in central synapses. J. Neurophysiol. 2015;113:1493–1500. doi: 10.1152/jn.00667.2014. [DOI] [PubMed] [Google Scholar]
- Pedraza C., Sánchez-López J., Castilla-Ortega E., Rosell-Valle C., Zambrana-Infantes E., García-Fernández M., Rodriguez de Fonseca F., Chun J., Santín L.J., Estivill-Torrús G. Fear extinction and acute stress reactivity reveal a role of LPA(1) receptor in regulating emotional-like behaviors. Brain Struct. Funct. 2014;219:1659–1672. doi: 10.1007/s00429-013-0592-9. [DOI] [PubMed] [Google Scholar]
- Perez S.E., Dar S., Ikonomovic M.D., DeKosky S.T., Mufson E.J. Cholinergic Forebrain Degeneration in the APPswe/PS1ΔE9 Transgenic Mouse. Neurobiol. Dis. 2007;28:3–15. doi: 10.1016/j.nbd.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyo M.K., Choi S.-H., Hwang S.H., Shin T.J., Lee B.H., Lee S.M., Lim Y.H., Kim D.H., Nah S.Y. Novel Glycolipoproteins from Ginseng. J. Ginseng Res. 2011;35:92–103. [Google Scholar]
- Shin T.J., Kim H.J., Kwon B.J., Choi S.H., Kim H.B., Hwang S.H., Lee B.H., Lee S.M., Zukin R.S., Park J.H., et al. Gintonin, a ginseng-derived novel ingredient, evokes long-term potentiation through N-methyl-D-aspartic acid receptor activation: involvement of LPA receptors. Mol. Cells. 2012;34:563–572. doi: 10.1007/s10059-012-0254-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiono S., Kawamoto K., Yoshida N., Kondo T., Inagami T. Neurotransmitter release from lysophosphatidic acid stimulated PC12 cells: involvement of lysophosphatidic acid receptors. Biochem. Biophys. Res. Commun. 1993;193:667–673. doi: 10.1006/bbrc.1993.1676. [DOI] [PubMed] [Google Scholar]
- Su C.F., Cheng J.T., Liu I.M. Increase of acetylcholine release by Panax ginseng root enhances insulin secretion in Wistar rats. Neurosci. Lett. 2007;412:101–104. doi: 10.1016/j.neulet.2006.10.044. [DOI] [PubMed] [Google Scholar]
- Sun Y., Nam J.S., Han D.H., Kim N.H., Choi H.K., Lee J.K., Rhee H.J., Huh S.O. Lysophosphatidic acid induces upregulation of Mcl-1 and protects apoptosis in a PTX-dependent manner in H19-7 cells. Cell. Signal. 2010;22:484–494. doi: 10.1016/j.cellsig.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Tabuchi S., Kume K., Aihara M., Shimizu T. Expression of lysophosphatidic acid receptor in rat astrocytes: mitogenic effect and expression of neurotrophic genes. Neurochem Res. 2000;25:573–582. doi: 10.1023/a:1007542532395. [DOI] [PubMed] [Google Scholar]
- Trimbuch T., Beed P., Vogt J., Schuchmann S., Maier N., Kintscher M., Breustedt J., Schuelke M., Streu N., Kieselmann O., et al. Synaptic PRG-1 modulates excitatory transmission via lipid phosphate-mediated signaling. Cell. 2009;138:1222–1235. doi: 10.1016/j.cell.2009.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R., Bennett B.D., Babu-Khan S., Kahn S., Mendiaz E.A., Denis P., Teplow D.B., Ross S., Amarante P., Loeloff R., et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Wang Q., Shin E.J., Nguyen X.K., Li Q., Bach J.H., Bing G., Kim W.K., Kim H.C., Hong J.S. Endogenous dynorphin protects against neurotoxin-elicited nigrostriatal dopaminergic neuron damage and motor deficits in mice. J. Neuroinflammation. 2012;9:124. doi: 10.1186/1742-2094-9-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong T.P., Debeir T., Duff K., Cuello A.C. Reorganization of cholinergic terminals in the cerebral cortex and hippocampus in transgenic mice carrying mutated presenilin-1 and amyloid precursor protein transgenes. J. Neurosci. 1999;19:2706–2716. doi: 10.1523/JNEUROSCI.19-07-02706.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankner B.A. Mechanisms of neuronal degeneration in Alzheimer’s disease. Neuron. 1996;16:921–932. doi: 10.1016/s0896-6273(00)80115-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.