

Abstract
ISLET1 is a homeodomain transcription factor necessary for development of the pituitary, retina, motor neurons, heart, and pancreas. Isl1-deficient mice (Isl1−/−) die early during embryogenesis at embryonic day 10.5 due to heart defects, and at that time, they have an undersized pituitary primordium. ISL1 is expressed in differentiating pituitary cells in early embryogenesis. Here, we report the cell-specific expression of ISL1 and assessment of its role in gonadotropes and thyrotropes. Isl1 expression is elevated in pituitaries of Cga−/− mice, a model of hypothyroidism with thyrotrope hypertrophy and hyperplasia. Thyrotrope-specific disruption of Isl1 with Tshb-cre is permissive for normal serum TSH, but T4 levels are decreased, suggesting decreased thyrotrope function. Inducing hypothyroidism in normal mice causes a reduction in T4 levels and dramatically elevated TSH response, but mice with thyrotrope-specific disruption of Isl1 have a blunted TSH response. In contrast, deletion of Isl1 in gonadotropes with an Lhb-cre transgene has no obvious effect on gonadotrope function or fertility. These results show that ISL1 is necessary for maximal thyrotrope response to hypothyroidism, in addition to its role in development of Rathke's pouch.
The pituitary gland is a master organ that controls the endocrine organs of the body. The anterior lobe secretes 6 major hormones that regulate various processes including growth, lactation, stress response and reproduction, whereas the posterior lobe regulates mainly water homeostasis (1). Pituitary organogenesis is a complex process requiring temporal and spatial regulation of several signaling pathways and transcription factors. Four LIM homeodomain transcription factors are required for normal pituitary development: LHX2, LHX3, LHX4, and ISL1. The LIM name comes from the names of the proteins in which the zinc finger protein interaction domains were first discovered: Lin11, Isl1, and Mec3. LHX2 has a role in pituitary posterior lobe development (2). Lhx3 and Lhx4 are crucial in the early steps of pituitary ontogenesis and have dosage sensitive, partially overlapping functions in development of the anterior pituitary primordium called Rathke's pouch (3–6). Loss of Lhx3 or Lhx4 expression in mice results in pituitary hypoplasia, increased cell death, cell differentiation defects, and ectopic ISL1 expression (4–7). Humans with loss-of-function LHX3 or LHX4 mutations have similar pituitary phenotypes to the mouse models. Patients homozygous for LHX3 mutations have variable pituitary defects that include hypopituitarism, combined pituitary hormone deficiency, and a hypoplastic anterior pituitary gland. Patients can also exhibit other phenotypes that include hearing loss and reduced neck rotation (8–11). Patients heterozygous for LHX4 mutations also have variable pituitary phenotypes that include combined pituitary hormone deficiency and a hypoplastic anterior pituitary gland, and in some cases, ectopic posterior pituitary gland (12–15).
Isl1 was identified more than 20 years ago in a cDNA library derived from insulin producing cells (16). Since then, roles for ISL1 have been defined in the development, proliferation, function and maintenance of specialized cells in the pancreas, retina, inner ear, motor neurons, and heart (17, 18). Isl1 is required for pancreatic ISLET cell differentiation and is highly expressed in mesenchymal stem cells of human pancreas (19–21). Isl1 expression in the eye is necessary for retinal ganglion cell differentiation (22, 23). Cardiovascular progenitors expressing ISL1 give rise to the cardiomyocyte, pacemaker, smooth muscle and endothelial cell lineages (24, 25). ISL1 is indispensable for the craniocervical and spinal motor neuron formation in the neural tube and for interneuron differentiation (18). ISL1 is also expressed in various regions of the hypothalamus. It is necessary for melanocortin neuron cell fate and for estrogen receptor-α expression (26, 27). In all these organs, ISL1 is expressed in the early steps of organogenesis, and it is crucial for the differentiation process leading to a mature and functional organ.
Isl1 is expressed early in pituitary development and is required for development of Rathke's pouch (28). Isl1 mutants have a small, rudimentary pouch, and the affected embryos die at e10.5 due to heart defects. Thus, the role of Isl1 has not been assessed at later stages of pituitary growth and development, and the cell specificity of expression has not been fully analyzed. In mice, Isl1 is expressed in the oral ectoderm that gives rise to the pouch at embryonic day (e)9.5 and then is expressed in differentiating cells (29). A great portion of these cells express the α-subunit of the pituitary glycoprotein hormones (officially known as chorionic gonadotropin-α [CGA]) and give rise to POU1F1-independent rostral tip thyrotropes (30). In sheep and chick, ISL1 is expressed in thyrotropes and gonadotropes (31, 32), which suggests a role for ISL1 in these cell types. Gene expression analysis in immortalized cell lines revealed that ISL1 is enriched in gonadotrope-like cells and can regulate gonadotropin releasing hormone receptor expression, but it is not detected in corticotrope or somatomammotrope-like cells (33).
Here, we report a detailed analysis of Isl1 expression in pituitaries of normal and hypothyroid mice and test the requirement for Isl1 in pituitary cell maintenance and function. We detect ISL1 in thyrotropes and gonadotropes during development. We discovered that levels of ISL1 transcripts are substantially elevated under conditions of thyrotrope hypertrophy and hyperplasia. Inactivation of Isl1 in thyrotropes causes mild hypothyroidism, reduced pituitary TSH response to hypothyroidism, and reduced growth. In contrast, mice with ISL1-deficient gonadotropes have normal fertility. In addition to its established role in early pituitary organogenesis, these results suggest that ISL1 has a role in thyrotrope function after birth.
Materials and Methods
Mice
All mice were maintained at the University of Michigan under the guidelines of the Unit for Laboratory Animal Medicine and the University Committee for Care and Use of Animals. Tg(Pou1f1-cre) mice were donated by Paul LeTissier (34). Tg(Lhb-cre) mice were previously described in Charles et al (35). Tg(Tshb-cre) mice were previously described in Castinetti et al (36). The Isl1loxp/loxp were donated by Lin Gan (37). The Isl1+/− mice were generated by mating the Isl1loxp/loxp mice to B6.C-Tg(CMV-cre)1Cgn/J mice (stock number 006054; The Jackson Laboratory). The B6.129S4-Gt(ROSA)26Sortm1Sor/J mice (R26R/R26R) were purchased from The Jackson Laboratory (stock number 003474) and maintained as homozygotes.
Tissue preparation and histology
All tissues were fixed in 4% paraformaldehyde in 1× PBS. Pituitaries and thyroids were collected at 4 and 8 weeks of age and fixed for 1 hour. Embryos collected at e11.5 were fixed for 30 minutes, and later stages, e12.5, e13.5, and e14.5, were fixed for 1 hour, and e16.5 embryos were fixed for 2 hours. Heads collected from postnatal day (P)1, P2, and P7 neonates were fixed for 4 hours. Tissues were rinsed in 1× PBS, dehydrated with series of graduated ethanols, and embedded in paraffin. The frozen tissues were rinsed in 1× PBS, then embedded in Optimum Cutting Temperature Compound (Tissue-Tek), frozen on dry ice, and stored at −80°C. Adult pituitaries and heads from neonates were sectioned in the coronal plane at 5-μm thickness for paraffin and at 16-μm thickness for frozen sections. Embryos collected at e11.5, e12.5, e13.5, e14.5, e16.5, and adult thyroid glands were sectioned in the sagittal plane at 5-μm thickness for paraffin and at 16-μm thickness for frozen.
At least 3 animals of each age and genotype were analyzed by immunostaining and hematoxylin and eosin (H&E) staining. The most midsagittal and midcoronal sections were used from embryos and pituitaries, respectively. The sections presenting the greatest area of thyroid tissue were used for quantifying follicular area. Slides used for immunohistochemistry were dewaxed in xylene and then rehydrated through 100% and 95% ethanols before washing in PBS. Please see Table 1 for specifics for each experiment, including antigen retrieval, endogenous peroxidase inactivation, blocking, antibody source and concentration, and detection methods. After immunohistochemistry, 4',6-diamidino-2-phenylindole counterstaining was performed for 5 minutes. After 3 washes in PBS, slides were mounted with fluorescent mounting media, and images were captured using a Leica DMRB fluorescent microscope.
Table 1.
Immunohistochemistry Experiments
| Experiment | Citric Acid Boil | CH3OH: H2O2 | Block | Primary Antibody | Secondary Antibody and Detection | Biotin Block | Other Primary Antibody | Other Secondary and Detection |
|---|---|---|---|---|---|---|---|---|
| ISL1 | 10 min-embryo 15 min-postnatal | Yes | M.O.M.,a TNBb | 1:600 anti-ISL1, DSHBc | Antirat biotin, TSA FITC | |||
| ISL1 and POU1F1g | 10 min-embryo 15 min-postnatal | Yes | M.O.M., TNB | 1:600 anti-ISL1, DSHB | Antirat biotin, TSA FITC | Yes | 1:500 anti-POU1F1, gift from S. Rhodes | Antirabbit biotin, TSA TRITC |
| ISL1 and NR5A1g | 10 min | Yes | M.O.M., TNB | 1:600 anti-ISL1, DSHB | Antirat biotin, TSA FITC | Yes | 1:1000 anti-NR5A, gift from G. Hammer | Antirabbit biotin, TSA TRITC |
| ISL1 and TBX19g | 10 min | Yes | M.O.M., TNB | 1:600 anti-ISL1, DSHB | Antirat biotin, TSA FITC | Yes | 1:200 anti-TBX19, gift from J. Drouin | Antirabbit biotin, TSA TRITC |
| CGA and ISL1 | 10 min | Yesf | M.O.M., TNB | 1:600 anti-ISL1, DSHB | Antirat biotin, TSA FITC | Yes | 1:1000, anti-CGA, NHPPd | Antirabbit biotin, TSA TRITC |
| TSHb and ISL1 | 15 mine | Yesf | M.O.M., TNB | 1:1000 anti-TSHb, NHPP | Antirabbit biotin, TSA TRITC | Yes | 1:600 anti-ISL1, DSHB | Antirat biotin, TSA FITC |
| LHb and ISL1 | 15 mine | Yesf | M.O.M., TNB | 1:500 anti-LHb, NHPP | Antiguinea pig, TSA TRITC | Yes | 1:600 anti-ISL1, DSHB | Antirat biotin, TSA FITC |
| GH | none | Yes | NGSh | 1:1000 anti-GH,j NHPP | Antihuman biotin, SA Cy2i | |||
| TSH | none | Yes | NGS | 1:1000, anti-TSH, NHPP | Antirabbit biotin, SA Cy2i | |||
| TSH and POU1F1 | 10 mine | Yesf | NGS M.O.M. | 1:1000, anti-TSH, NHPP | Antiguinea pig, TSA TRITC | Yes | 1:500, anti-POU1F1 | Antirabbit biotin, TSA FITC |
| TSH and NR5A1 | 10 mine | Yesf | NGS M.O.M. | 1:1000, anti-TSH, NHPP | Antiguinea pig, TSA TRITC | Yes | 1:1000, anti-NR5A1 | Antirabbit biotin, TSA FITC |
Mouse kit from Vector Labs.
Block from PerkinElmer Tyramide Signal Amplification TSA Fluorescein isothiocyanate FITC and Tetramethylrhodamine (TRITC) kit.
Developmental Studies Hybridoma Bank.
National Hormone and Pituitary Program.
Citrate boiling done after primary antibody.
Second CH2OH:H2O2 block done before second primary antibody.
Primary antibodies incubated at same time.
5% normal goat serum, 3% BSA in 1× PBS.
Streptavidin cyanine 2.
Anti-GH antibody left on for 1 hour.
For H&E (Sigma-Aldrich) staining the paraffin embedded tissue sections were dewaxed, and rehydrated, soaked in hematoxylin for 3 minutes, rinsed in distilled water, “blued” using a combination of Differentiation Solution (Sigma-Aldrich) and Scott's Tap Water (Sigma-Aldrich) with a distilled water rinse in between, soaked in eosin for 20 seconds, and rinsed in distilled water. The slides were gradually dehydrated in a series of ethanols of increasing concentration, soaked in xylene and mounted with xylene:permount 1:2 (Fisher) media. X-gal (5 bromo-4-chloro-3-indolyl-b-D-galactopyranoside) staining and neutral red staining were performed on frozen tissue as previously described (35, 38).
Thyroid follicle measurement
Adult thyroid glands were collected at 4 and 8 weeks of age. H&E-stained thyroid sections were analyzed using the ImageJ software (National Center for Biotechnology Information). The area of individual follicles was measured in 3 sections taken from the middle of the gland that were 5 sections apart (n = 3 for control, n = 4 for Isl1loxP/loxP;Tg(Tsh-cre)). A range of 627–729 follicles was counted in 3 Isl1loxp/loxp controls, and a range of 466–895 follicles was counted in 4 Isl1loxP/loxP;Tg(Tsh-cre) mutants. The average follicle area was determined by dividing the total follicle area by the number of follicles measured. The Mann-Whitney U test was used for analysis. P value was set to .05.
Real-time PCR
8-week old pituitaries and livers were dissected and stored in RNALater at −20°C. Four pituitaries and 4 livers from Isl1loxP/loxP;Tg(Tshb-cre) and Isl1+/loxP or Isl1loxP/loxP mice were used. RNA was isolated and deoxyribonuclease I treated using the protocols provided in the RNAqueous 4 PCR kit (Life Technologies). The cDNA was generated using the Superscript II reverse transcription system from Invitrogen (Life Technologies). Specifically, RNA and oligo dT primers were denatured at 70°C for 10 minutes and then placed on ice; 100mM dithiothreitol, 10mM deoxynucleotide triphosphates 5× first strand buffer, and 1-U Superscript II were added to the RNA primer mix. Samples were incubated at 42°C for 50 minutes, then heat inactivated for 10 minutes at 70°C. Samples without reverse transcriptase were included as negative controls. The quality of the cDNA reaction and effectiveness of the DNase I treatment of the RNA was tested using PCR with primers to the housekeeping gene, hypoxanthine guanine phosphoribosyl transferase. Quantitative PCR (qPCR) was carried out using TaqMan gene expression assays for Igf1 (Mm00439559_m1), Gh (Mm00433590_g1), Tshb (Mm00437190_m1), and Gapdh (4308316). The qPCR was set up in triplicate with 50-ng cDNA per reaction using TaqMan Universal PCR Master Mix and default run parameters on the ABI 7500 Real-Time PCR instrument. Results were expressed as fold changes compared with Gapdh using the delta cycle threshold method as described earlier (22). Triplicates containing the Master Mix and water with the TaqMan primer sets were used as a negative controls. The fold change of expression in Isl1loxP/loxP;Tg(Tshb-cre) (n = 4) compared with Isl1+/loxP and Isl1loxP/loxP (n = 4) mice was calculated as described previously (39). All reagents and instruments were obtained from Life Technologies.
Cell counts
Immunopositive cells from of 3–4 sections spanning the pituitary from Isl1+/loxP;Tg(Tshb-cre) (n = 4) and Isl1loxP/loxP;Tg(Tshb-cre) (n = 3) mice were quantitated using ImageJ software (Leica). The number of cells coimmunostained for TSH and ISL1 were compared with the total number of cells immunopositive for TSH. The average percentage of double immunopositive cells is displayed. Statistical analysis was performed using the Student's t test, and significance level was set to 0.05.
Serum hormone measurements
After euthanasia with CO2, blood was collected by cardiac puncture while the heart was still beating. After clotting, the blood was stored at 4°C for 24 hours and was centrifuged at 8000g for 10 minutes. The serum was stored at −20°C until use. A total of 5 μL of serum was used to analyze the total T4 concentration (MP Biomedicals), and 20 μL of serum were used to analyze the TSH concentration (Millipore). The T4 measure was performed in triplicates, whereas the TSH level was a single measurement.
Hypothyroidism challenge
Low iodine diet enriched in propylthiouracil (PTU) (0.15%) (Harlan Lab) was given to 3- to 8-week-old Isl1loxP/loxP;Tg (Tshb-cre) and control mice for 4 weeks (40). Blood collection and hormone measurements were performed as described above.
Statistical analysis of hormone measurements and body weights
Data are presented as mean ± SD. Student's t test, ANOVA, or Mann-Whitney U test were used for statistical analysis where appropriate. Data were analyzed with SPSS version 17.0. ImageJ software, and Excel version 14.3.9. P < .05 was accepted as significant.
Results
ISL1 is expressed during pituitary development
We evaluated the expression of ISL1 in the mouse pituitary during development and at early postnatal age by ISL1 immunostaining (Figure 1). From e11.5 to e13.5, ISL1 immunoreactivity is detected in Rathke's pouch among the cells in the differentiation zone but not cells in the proliferative zone (41). At later stages, e14.5, e16.5, and P3, there are scattered ISL1-expressing cells in the anterior lobe. There are traces of ISL1 immunostaining in the intermediate lobe at P3, and none in the posterior lobe. The ventral hypothalamus, located rostral to the pituitary gland, exhibits robust ISL1 staining throughout development.
Figure 1.

ISL1 is expressed in Rathke's pouch during development. ISL1 immunostaining (green) and DAPI counterstaining (blue) was carried out in sections from developing pituitary glands collected from e11.5 to P3. The photographs of sagittal e11.5, e12.5, and e13.5 sections were taken at ×200, and the later ages, sagittal e14.5, e16.5, and coronal P3, at ×100. Scale bars, 100 μm. White box inset of P3 photo was taken at ×630. RP, Rathke's pouch; VD, ventral diencephalon; AL, anterior lobe; IL, intermediate lobe; PL, posterior lobe; VH, ventral hypothalamus.
To determine which pituitary cell types express ISL1, we carried out immunohistochemistry for ISL and lineage-specific transcription factors at e16.5 and evaluated nuclear colocalization (Figure 2, A–H). ISL1 colocalized with a few POU1F1+ cells (Figure 2, A and B), but most of the immunopositive cells expressed either ISL1 or POU1F1 but not both. ISL1 immunostaining did not colocalize with any of the TBX19+ cells, which mark future melanotropes and corticotropes (Figure 2, C and D) (42). ISL1 colocalized with about half of the NR5A1+ cells, which differentiate into gonadotropes (43), at e16.5 (Figure 2, E and F), but less colocalization was observed at P7 (Figure 2I). ISL1 colocalized with many of the CGA+ cells at e16.5 (Figure 2, G and H) and with many TSHb cells at P7 (Figure 2J). These data suggest that ISL1 might be involved in thyrotrope function and in the initiation of the gonadotrope differentiation but not maintenance.
Figure 2.
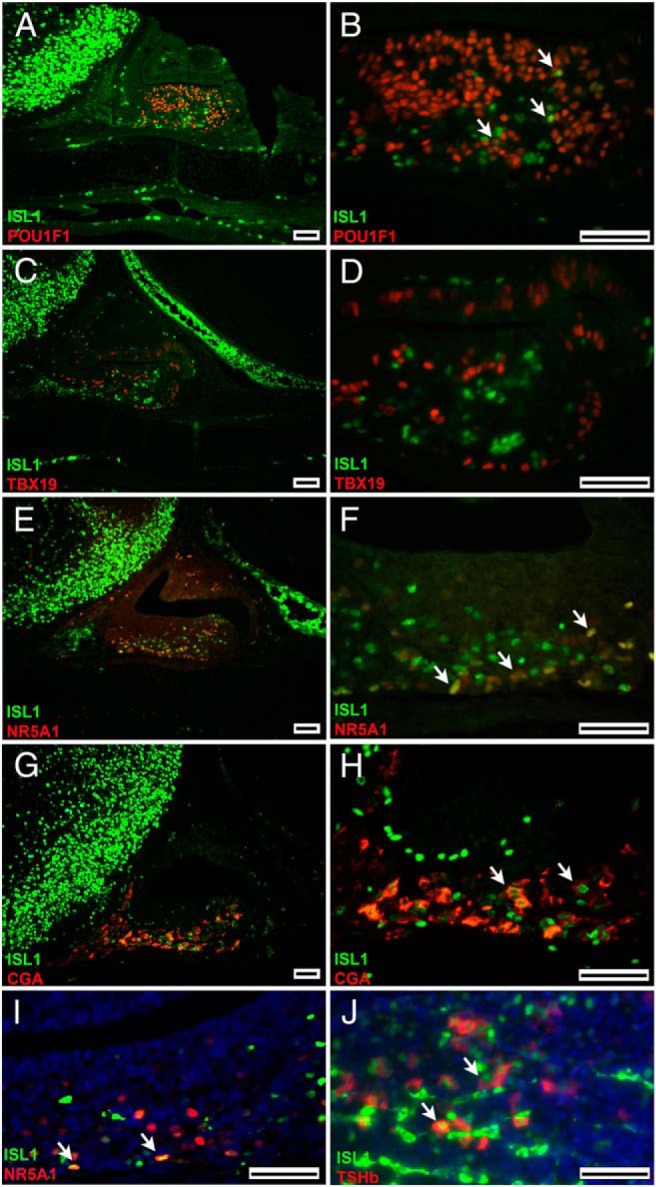
ISL1 is expressed in early thyrotropes and gonadotropes. A–H, ISL1 immunostaining (green) was carried out together with lineage-specific transcription factor staining (red) and the common subunit to thyrotropes and gonadotropes (CGA) on sections from e16.5 embryos. A and B, Coimmunostaining of ISL1 (green) and POU1F1 (red) is represented by the yellow nuclei (left panels at low [×100] and right high [×630] magnification). C and D, No colocalization was detected for nuclear ISL1 and TBX19. E–H, Colocalization was detected for ISL1 and NR5A1 (yellow nuclei) as well as for ISL1 (nuclear, green) and CGA (cytoplasmic, red). I, The bottom left panel shows limited coimmunostaining of ISL1 and NR5A2 postnatally (P1). J, The bottom right panel shows coimmunostaining of ISL1 and TSH postnatally (P7). White arrows point to colocalized cells. Both bottom panel images were taken at ×630. Scale bars for ×100 images are 100 μm and for ×630 images are 50 μm.
To explore the role of ISL1 in thyrotropes, we quantified Isl1 transcripts in Cga−/− mice (44). These mutants are hypothyroid and hypogonadal due to the inability to secrete bioactive glycoprotein hormones and have an ancillary deficiency in GH and PRL (44). The pituitary glands of these mice undergo profound thyrotrope hypertrophy and hyperplasia, and have very few somatotropes and lactotropes (38, 45–47). Pituitaries were collected from 8-week-old mutant and wild-type mice (3 males and 3 females of each genotype, 12 samples total) and analyzed for Isl1 expression by qPCR using TaqMan assay. Isl1 transcripts were elevated 2.6-fold (SEM, 0.7; P = .001) in mutant pituitaries, suggesting a role in thyrotrope hypertrophy and hyperplasia.
To understand the specific roles of ISL1 in the anterior pituitary gland, we inactivated Isl1 using a Pou1f1-specific cre recombinase transgenic line (Tg[Pou1f1-cre]) (34), a gonadotrope-specific cre line (Tg[Lhb-cre]) (35), and a thyrotrope-specific cre line (Tg[Tshb-cre]) (36).
Deletion of Isl1 using Pouf1-cre had no effect on pituitary development
To characterize the developmental onset and pituitary specificity of cre activity, we crossed mice carrying cre recombinase under the control of the Pou1f1 promoter, Tg(Pou1f1-cre) (34), with a cre reporter strain consisting of a floxed stop sequence, which upon cre mediated excision permits lacZ expression (R26R/R26R) (48). We assessed the efficiency and penetrance of cre-mediated recombination by X-gal staining of frozen sections (Supplemental Figure 1A). Pou1f1-cre activity is first evident at e14.5 in the developing anterior lobe as indicated by the blue X-gal staining. Not many of the cells appear X-gal positive at e14.5. At 4 weeks, the X-gal staining appears throughout the anterior lobe, indicating cre activity beyond the cell types expected for the Pou1f1 lineage, consistent with previous reports (49). The Pou1f1-cre also exhibits unexpected activity in the thyroid gland in adult mice.
Mice heterozygous for Isl1 (Isl1+/−) were mated with Tg(Pou1f1-cre) to produce Isl1+/−;Tg(Pou1f1-cre) mice. We crossed Isl1+/−;Tg(Pou1f1-cre) mice with mice homozygous for a floxed Isl1 allele (Isl1loxP/loxP) to generate the conditional knockout Isl1−/loxp;Tg(Pou1f1-cre) mice which are designated as mutant. Isl1−/loxp, Isl1+/loxp, and Isl1+/loxp;Tg(Pou1f1-cre) were used as control mice. We analyzed the pituitaries of neonates to confirm that ISL1 was effectively ablated in the POU1F1 cells (Supplemental Figure 1B). At P2, about one third of the ISL1+ cells normally express POU1F1 (yellow arrows), and there are many POU1F1 cells that do not express ISL1 (pink arrow). No cells coexpressing ISL1 and POU1F1 were detected in the P2 pituitaries of Isl1−/loxP;Tg(Pou1f1-cre) mice, indicating efficient ablation of ISL1. As expected, ISL1+, POU1F1− cells are present (green arrow). No difference was detected in TSH immunostaining in the pituitary gland at P1 between the control and mutant groups (n = 3 for each genotype) (data not shown). The weight of control and mutant mice were the same at 5 and 8 weeks of age, suggesting that the pituitary production of TSH and GH was sufficient to support normal growth (Supplemental Figure 1C). As expected, males of both genotypes were larger than females, suggesting that sexually dimorphic regulation of GH secretion was intact. We used immunohistochemistry to quantify the number of POU1F1 cells that are TSH+. Surprisingly, only about half of the TSH cells were also POU1F1+ (Supplemental Figure 1D, white arrow). Some of the TSH+/POU1F1− cells are NR5A1+, but only a few cells expressed both proteins (Supplemental Figure 1D). These results suggest that thyrotropes are heterogeneous and that effective deletion of ISL1 in thyrotropes could be better achieved with a Tshb-cre line.
Thyrotrope-specific Isl1 knockout mice are smaller than normal
Mice carrying cre recombinase under the control of the Tshb promoter, Tg(Tshb-cre), exhibit strong cre activity at e14.5 and excellent thyrotrope specificity (36). Mice with a floxed Isl1 allele (Isl1loxP/loxP) were mated with Tg(Tshb-cre) mice, to produce Isl1loxP/+; Tg(Tshb-cre) mice. We crossed Isl1loxP/+;Tg(Tshb-cre) mice with Isl1loxP/loxP mice to obtain Isl1loxP/loxP;Tg(Tshb-cre) mice. Isl1loxP/loxP;Tg(Tshb-cre) mice are mutant and Isl1loxP/+ and Isl1loxP/loxP were used as controls.
At P3, ISL1 colocalizes with more than half of the TSH immunopositive cells in the pituitary gland (white arrowhead) (Figure 3A). In the Isl1loxP/loxP;Tg(Tshb-cre) mice there is little or no detectable coimmunostaining of ISL1 and TSH (n = 3). We quantified the number of cells with colocalized ISL1 and TSH out of the total number of TSH immunopositive cells. The 55% decrease in colocalization of ISL1+/TSH+ cells in the mutant mice is significant (P = .02). In controls, 58 ± 8% of the TSH cells coexpressed ISL1, but only 26 ± 11% did in mutants (Figure 3B). The efficiency of deleting ISL1 with Tshb-cre was less than observed for PITX2 or a cre reporter strain (36), which is not unusual (50). Nevertheless, it is clear that Isl1-deficient thyrotrope cells can survive and maintain Tshb expression.
Figure 3.

Isl1loxP/loxP;Tg(Tshb-cre) mice present a significant growth insufficiency. A, Coronal pituitary sections from P3 Isl1loxP/loxP;Tg(Tshb-cre) and control mice were immunostained with ISL1 (green nuclear stain) and TSH (red cytoplasmic stain). Cells with both ISL1 and TSH are present in the control sections (white arrowheads). There are very few cells with ISL1 and TSH in the Isl1loxP/loxP;Tg(Tshb-cre) mice (white arrowheads). Photos were taken at ×630. Scale bar, 50 μm. B, The percentage of TSH cells that also express ISL1 (dICC, double immunopositive) was calculated for the Isl1loxP/loxP;Tg(Tshb-cre) mutant and control mice. The mutant mice have significantly fewer colocalized ISL1+, TSH+ cells in their pituitaries compared with controls (*, P < .05). C, Average weights of male and female Isl1loxP/loxP;Tg(Tshb-cre) and control mice at 5 and 8 weeks. There is a significant difference between both male and females at both ages. At 5 weeks, Isl1loxP/loxP;Tg(Tshb-cre) males are 17.2 ± 3.2 g (n = 9), control males are 21.8 ± 1.0 g (n = 8), Isl1loxP/loxP;Tg(Tsh-bcre) females are 13.5 ± 1.6 g (n = 10), and control females are 17.2 ± 1.5 g (n = 14). At 8 weeks, Isl1loxP/loxP;Tg(Tshb-cre) males are 21.9 ± 2.6 g (n = 6), control males are 25.6 ± 1.4 g (n = 10), Isl1loxP/loxP;Tg(Tshb-cre), and control females are 20.9 ± 1.5 g (n = 10). Statistical significance: ***, P < .001; **, P < .01.
We found that male and female Isl1loxP/loxP;Tg(Tshb-cre) mice are significantly smaller than their normal littermates at 5 and 8 weeks of age (Figure 3C). At 5 weeks, the male and female Isl1loxP/loxP;Tg(Tshb-cre) mice are 21% smaller when compared with control mice and at 8 weeks the male and female Isl1loxP/loxP;Tg(Tshb-cre) mice are 14% smaller when compared with control mice. The weight differences at these times are statistically significant in females and males (P < .01, n = 6 or greater for each sex and genotype). This growth deficiency is consistent with expectations for moderate hypothyroidism. For both genotypes, males are larger than females, which is consistent with normal sexually dimorphic secretion patterns of GH.
Isl1loxP/loxP;Tg(Tshb-cre) mice have reduced response to hypothyroidism
If ISL1 is necessary for thyrotrope differentiation and/or maintenance, then Isl1loxP/loxP;Tg(Tshb-cre) pituitaries might have fewer thyrotropes than controls, or mutant thyrotropes might undergo hypertrophy in an effort to maintain homeostasis. To investigate these possibilities, we used immunohistochemistry to detect TSH in the adult Isl1loxP/loxP;Tg(Tshb-cre) and control pituitary glands. There was no difference in the number or morphology of thyrotropes between the transgenic and control mice (Figure 4A).
Figure 4.

Blunted TSH response to hypothyroidism in Isl1loxP/loxP;Tg(Tshb-cre) mice. A, There is no obvious difference in TSHβ staining in Isl1loxP/loxP;Tg(Tshb-cre) pituitary glands at 8 weeks. TSHβ staining was performed on 5 pituitary sections at regular intervals for each transgenic and control mouse (n = 3/genotype). Photos were taken at ×100. Scale bar, 100 μm. B, qPCR was used to determine the mRNA expression fold change in liver Igf1 and pituitary Gh of Isl1loxP/loxP;Tg(Tshb-cre) relative to controls. C, qPCR was used to determine the fold change in pituitary Tshb mRNA expression in Isl1loxP/loxP;Tg(Tshb-cre) mice compared with controls in both basal and hypothyroid challenge conditions. Serum levels of TSH were measured in Isl1loxP/loxP;Tg(Tshb-cre) and control females under basal and hypothyroid conditions. There was no significant difference in basal levels between Isl1loxP/loxP;Tg(Tshb-cre) mice (297 ± 99 pg/mL, n = 6) and the controls (234 ± 114 pg/mL, n = 12), respectively. After hypothyroidism challenge, the transgenics responded with less dramatic elevation in TSH relative to controls: 27 × 103 ± 13 × 103 pg/mL, n = 4 vs 165 × 103 ± 94 × 103 pg/mL, n = 10. Note the logarithmic scale. **, P < .01. D, Total serum T4 levels were measured in Isl1loxP/loxP;Tg(Tshb-cre) and control male mice under basal and hypothyroid conditions. Under basal conditions, T4 levels were significantly different (6.7 ± 1.3 ng/mL, n = 14, vs 5.1 ± 0.93 ng/mL, n = 7; *, P < .05). After hypothyroid challenge, the total T4 levels in serum were reduced compared with basal levels (1.4 ± 0.57 ng/mL, n = 10, and 1.1 ± 0.29 ng/mL, n = 5).
To ensure that the growth defect is not due to disruption in the GH axis, we used qPCR to measure liver Igf1 expression and pituitary Gh expression of 8-week-old Isl1loxP/loxP;Tg(Tshb-cre) and control mice. We saw no difference in expression of Igf1 or Gh (Figure 4B). We also used immunohistochemistry to examine GH protein in the mutant and control mice and saw no difference in the intensity or pattern of GH staining (Supplemental Figure 2).
The circulating levels of total T4 provide one read-out of thyroid function. The Isl1loxP/loxP;Tg(Tshb-cre) male mice have significantly lower basal levels of total T4 compared with the control mice: 6.7 ± 1.3 ng/mL in controls (n = 14) vs 5.1 ± 0.93 ng/mL in transgenics (n = 7), P < .05, although the level is within the normal range (Figure 4D).
To assess the function of the thyrotropes, we measured serum levels of TSH and the amount of Tshb transcripts in the pituitary gland in female control and Isl1loxP/loxP;Tg(Tshb-cre) mice at 8 weeks of age. We found no significant difference in the basal circulating levels of TSH (Figure 4C) or Tshb transcripts in the pituitary. We hypothesized that hypofunction of ISL1-deficient thyrotropes could cause the small body size of the mutants and low circulating T4 levels. To test this idea we challenged the thyrotropes to determine whether they can respond to low thyroid hormone levels with elevated TSH. We induced hypothyroidism in female transgenic mice and controls (n = 6 transgenics vs 8 controls) by feeding them a low iodine diet enriched in a goiterogen, 0.15% PTU, for 4 weeks. To confirm that the treatment effectively induced hypothyroidism, we analyzed circulating total T4 of transgenic and control mice treated with PTU. T4 levels were dramatically reduced to less than 2 ng/mL (Figure 4D). We measured Tshb transcript levels and serum TSH levels in the PTU treated mice and found that although both transgenics and controls responded with elevated Tshb transcripts and serum TSH, the response in transgenics was 6-fold lower than in the controls: 27 × 103 ± 13 × 103 pg/mL in transgenics vs 165 × 103 ± 94 × 103 pg/mL in controls, P < .01 (Figure 4C). This demonstrates that Isl1loxP/loxP;Tg(Tshb-cre) mutants exhibit a blunted response to hypothyroidism.
Isl1loxP/loxP;Tg(Tshb-cre) mice have reduced thyroid function
Thyroid gland growth and function is directly affected by the action of thyrotropes. TSH stimulates the thyroid epithelial cells to produce thyroglobulin. The size of the colloid-filled follicle is a measure of thyroid activity. We examined H&E-stained sections of the thyroid gland in 8-week-old transgenic and control mice. The thyroid glands of Isl1loxP/loxP;Tg(Tshb-cre) mice appear less developed than the control ones (n = 4, Isl1loxP/loxP;Tg(Tshb-cre) and n = 3, Isl1loxP/loxP) (Figure 5A). The overall size of the gland is smaller and the range of follicle sizes is smaller. To quantify the difference in thyroid gland morphology we measured the area of active thyroid follicles. The average follicular area of the Isl1loxP/loxP;Tg(Tshb-cre) mice was 32% less than that of the control mice (P = .002) (Figure 5B). To ensure that the Tg(Tshb-cre) had no ectopic effect on the thyroid we examined ISL1 immunoreactivity in the thyroid glands. There was no difference in the number of ISL1+ cells in the Isl1loxP/loxP;Tg(Tshb-cre) thyroid glands compared with controls (Figure 5C), suggesting that the reduced thyroid function is secondary to ISL1 deletion in the pituitary gland.
Figure 5.
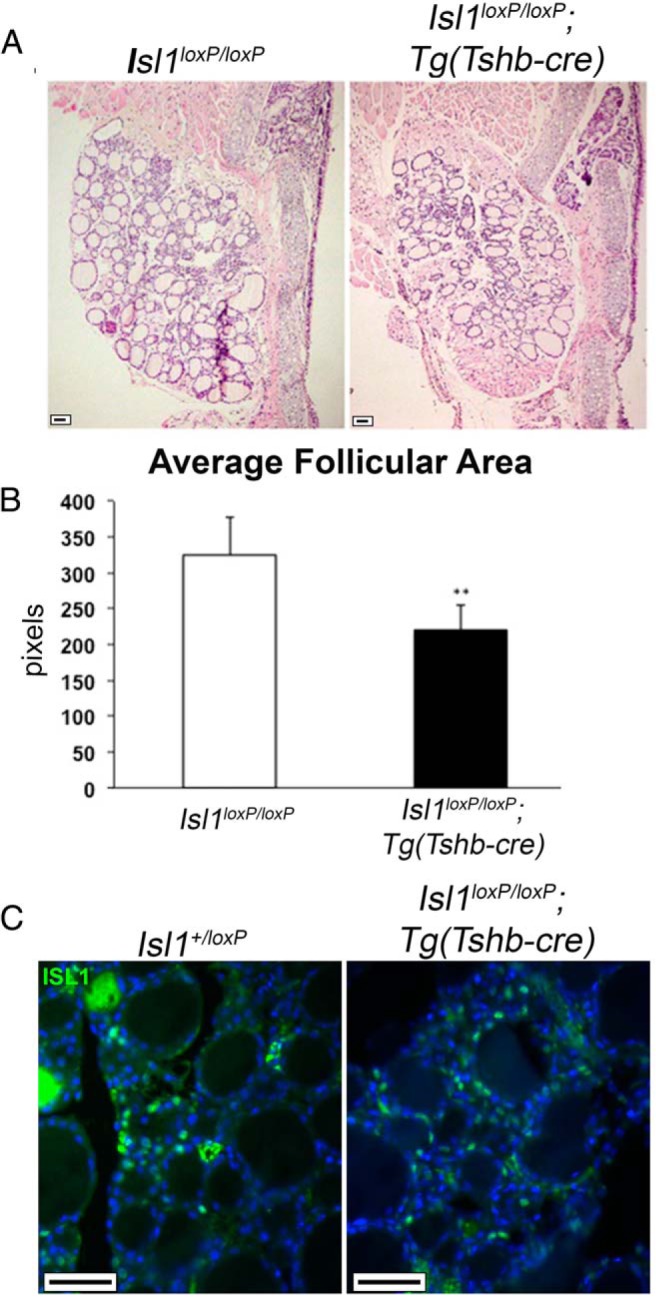
Isl1loxP/loxP;Tg(Tshb-cre) mice have impaired thyroid function. A, H&E stain of thyroid gland sections from 8-week-old Isl1loxP/loxP;Tg(Tshb-cre) mice and controls. Photos were taken at ×100. Scale bar, 100 μm. B, The average follicular area in Isl1loxP/loxP;Tg(Tshb-cre) was 221 ± 35 pixels (n = 4) vs control mice 325 ± 53 pixels (n = 3); **, P < .001 according to Mann-Whitney U test. C, ISL1 immunostaining (green) on thyroid gland sections from 8-week-old mice. Photos taken at ×630. Scale bar, 50 μm.
ISL1 is dispensable for gonadotrope maintenance and function
ISL1 is coexpressed with NR5A1 during development (Figure 2, E and F), but postnatally, ISL1 is not expressed in many gonadotropes (Supplemental Figure 3). To test the importance of ISL1 in gonadotrope function, we crossed Isl1loxP/loxP mice with the previously described gonadotrope-specific cre strain: Tg(Lhb-cre) (35). The resulting Isl1loxP/+;Tg(Lhb-cre) progeny were crossed with Isl1loxP/loxP mice to obtain Isl1loxP/loxP;Tg(Lhb-cre) mice with a conditional inactivation of Isl1 in differentiated gonadotropes. Isl1loxP/loxP;Tg(Lhb-cre) male mice are the same size as normal males and larger than females, suggesting normal gonadal steroid effects on GH pulsatility. The Isl1loxP/loxP;Tg(Lhb-cre) females have normal puberty as evaluated by vaginal openings, and both males and females have normal fertility (data not shown). This suggests that ISL1 is dispensable for maintaining mature, basal gonadotrope function.
Discussion
During development we detected coexpression of ISL1 with the gonadotrope lineage transcription factor NR5A1 but not the corticotrope marker TBX19. There was limited coexpression of ISL1 and POU1F1. This is consistent with reports of enrichment of ISL1 in gonadotrope-like cell lines, αT3-1 and LβT2, relative to corticotrope or somatolactotrope cell lines (33), and the reports on cell-specific expression in sheep and chick (31, 32). It is intriguing that ISL1 is required for development of pro-opiomelanocortin neurons, but it is not expressed in pro-opiomelanocortin cells of the pituitary (27). Colocalization of LH and NR5A1 with ISL1 was markedly reduced after birth. Despite the ability of ISL1 to act synergistically with NR5A1 and LHX3 to promote GnRH receptor expression in cell culture (33), we discovered that mice with Isl1 deleted in gonadotropes had normal puberty and fertility, suggesting that ISL1 is dispensable for mature gonadotrope function. ISL1 may have a role earlier in gonadotrope differentiation because it is coexpressed with NR5A1 at e14.5, and Lhb transcription is not detected until e17.5, which is the earliest time that Lhb-cre could act to delete ISL1. It is also possible that ISL1 has such a role in gonadotrope function that is compensated for by other LIM homeodomain transcription factors like LHX3, LHX4, or ISL2.
We present several lines of evidence that support a role for ISL1 in thyrotrope function. First, ISL1 expression colocalizes with most TSH cells in the postnatal pituitary. Secondly, Isl1 transcripts are increased in the Cga−/− mice, which have extensive thyrotrope hypertrophy and hyperplasia (44). Third, deletion of ISL1 in thyrotropes using the Tg(Tshb-cre) line causes reduced body size, reduced circulating T4 and reduced thyroid gland development. Moreover, these mutants exhibit a blunted thyrotrope response to hypothyroidism.
The efficiency of deleting Isl1 with Tshb-cre was less than observed for PITX2 or a cre reporter strain (36). This target site variability in cre efficiency is not unusual, and it can be confounded if there is a selective advantage for survival or proliferation of cells that escape the effects of the transgene (50, 51). Because residual ISL1+, TSH+ cells were present, ISL1 may be more important for thyrotrope function than suggested by the moderate growth insufficiency phenotype we observed.
The normal circulating TSH but low T4 levels in Isl1loxP/loxP, Tshb-cre mice is consistent with mild central hypothyroidism. The mutant pituitaries should have responded to the lower T4 level with a rise in TSH, but they did not. This is consistent with the lower than expected rise in TSH in response to severe hypothyroidism. In the basal state, the slight challenge of modestly reduced T4 levels did not elicit a rise in TSH, and although the challenge of severe hypothyroidism did provoke a response, the fact that it is 7-fold lower provides further support for the importance of ISL1 in pituitary-thyroid axis regulation. The phenotype of the Isl1loxP/loxP, Tshb-cre mice is similar to the central hypothyroidism in TRHR and TRH mutant mice, which also present with low T4 levels, relatively normal TSH levels, and impaired growth (52, 53).
POU1F1 is accepted as a critical transcription factor for specifying thyrotrope cell fate. Loss of function mutations result in severe TSH deficiency and the absence of TSH producing cells, except for a few transient TSH+, POU1F1− cells that arise in the rostral tip of the developing gland (30, 54). POU1F1 activates Tshb gene expression together with GATA-binding protein 2 (55), and the spatial and temporal expression patterns of Pou1f1 and Tshb overlap. Pou1f1 transcripts first appear in cells located in the caudal medial region of the gland at e15.5, coincident with the appearance of Tshb transcripts. Thus, we were surprised that immunostaining revealed so many POU1F1− thyrotropes in normal neonates. In contrast, most of the neonatal thyrotropes expressed ISL1, and some expressed NR5A1. Thus, there are clearly distinct, subpopulations of thyrotropes. In support of this, cells that express FSH, TSH, NR5A1, and LH receptor have been detected in pituitaries during late gestation and the neonatal period (56). These “thyro-gonadotrope” cells convert to functional gonadotropes in response to paracrine LH secretion, which triggers gonadotropin-releasing hormone receptor production and extinguishes TSH expression.
Pou1f1-cre mediated deletion of ISL1 did not have an obvious effect on growth or thyrotrope function. This could be attributable to less robust activity of Pou1f1-cre at e14.5–e16.5 compared with Tshb-cre (36). Another explanation is that there are many more ISL1+, TSH+ cells than ISL1+, POU1F1+ cells during early development and neonatal life. Perhaps the ISL1+, POU1F1− thyrotropes are progenitors that progress to ISL1−, POU1F1+ thyrotropes. This would be consistent with the marked elevation in Isl1 expression that we observed in the pituitaries of mice with genetically engineered hypothyroidism, the Cga knockout mice.
Several transcription factors are necessary for thyrotrope function and normal growth. These include POU1F1, GATA-binding protein 2, PITX2, and ISL1 (1). The degree of growth insufficiency in both male and female Isl1loxP/loxP;Tg(Tshb-cre) mice is modest compared with the severe dwarfism of Cga knockout mice, which do not secrete any bioactive TSH. However, the Isl1loxP/loxP;Tg(Tshb-cre) mice have a more pronounced growth defect than we observed in the pituitary-specific knockout of Gata2 (57). Those Gata2-deficient mice had a modest growth defect in males but not in females, and the defect was only transient. The growth of Isl1loxP/loxP;Tg(Tshb-cre) mice is also more impaired than that of mice with a conditional inactivation of Pitx2 in thyrotropes. The Pitx2flox/−;Tg(Tshb-cre) mice have unchanged TSH and thyroid hormone levels in basal conditions, and a 2-fold reduction in TSH response after hypothyroidism challenge compared with controls (36). Taken together, these data indicate roles for multiple transcription factors in thyrotrope function, and they suggest a strong feedback mechanism to maintain normal homeostasis.
In the ganglion cells of the retina, ISL1 interacts synergistically with a POU homeodomain transcription factor, brain-specific homeobox transcription factor 3B, to promote cell differentiation (37). Because POU1F1 is the prototype POU homeodomain transcription factor expressed in the pituitary, and it is necessary for thyrotrope differentiation, we considered the possibility that ISL1 could interact with POU1F1 during pituitary development. Our preliminary results do not confirm this hypothesis, however. ISL1 did not interact synergistically with POU1F1 to increase transcription of either the Pou1f1 or Tshb promoters in HeLa cells (data not shown). There are many changes in transcription factor expression in Cga knockout mice, which, together with ISL1, could be involved in regulating the pituitary response to hypothyroidism (P. Gergics, unpublished data).
During development, several LIM domain transcription factors are necessary for a proper pituitary cell proliferation and differentiation, and elimination of one can affect the spatial and temporal regulation of the other factors. ISL1 and LHX3 expression overlaps early in pituitary development (e9.5–e10.5) but becomes mutually exclusive after that (29). Lhx3 null embryos have a transient lack of ISL1 expression at e12.5, a time that is critical for restricting ISL1 expression to the prospective anterior lobe (2, 6). However, ISL1 expression recovers in Lhx3 mutant pituitaries later in development (6). This suggests that LHX3 expression is necessary for ISL1 expression at e12.5. We hypothesize that delayed ISL1 expression contributes to the lack of thyrotropes in Lhx3 null mice.
In humans there are several known genetic causes of TSH deficiency, including mutations of genes coding for leptin receptor, TRH receptor, TSHβ subunit (58), IGSF1 (59) and multiple transcription factors (POU1F1, LHX3, LHX4, prophet of Pit1, and homeobox gene expressed in embryonic stem cells 1) (1). The ISL1 defect in mouse thyrotropes induces modest hypothyroidism and reduced growth. Therefore, mutations in Isl1 may contribute to pituitary thyroid axis dysfunction in humans. Individuals with homozygous loss of function mutations in ISL1 are expected to be nonviable due to the fact that ISL1 is involved in the development of several mouse organs (heart, pancreas, retina, motor neurons), and homozygous inactivation in mice is responsible for early death during embryogenesis. Hypomorphic mutations or those with a dominant negative effect might be compatible with life but be associated with pituitary based growth insufficiency. Patients with hypothyroidism and associated heart defects or diabetes may be most likely to have defects in ISL1. To date no systemic screening for ISL1 mutations has been carried out in patients with these features (60).
To conclude, our study shows that ISL1 is necessary for normal growth, pituitary thyrotrope response to hypothyroidism function, in addition to its previously known role in expansion of Rathke's pouch.
Acknowledgments
We thank Dr Jun Z. Li and his lab members; Dr Richard Miller, James Harper, and members of the Miller Lab; and members of the Camper lab for their help. We also thank Paul LeTissier for providing the Pou1f1-cre mice. P.G. is a participant of the International Endocrine Scholars Program of The Endocrine Society.
This work was supported by Center for Genetics in Health and Medicine, University of Michigan (F.C.); National Institutes of Health Grants R37HD30428 and R01HD34283 (to S.A.C.); and by Association pour le Développement de la Recherche Médicale au Centre Hospitalier Universitaire de Marseille grants (F.C. and T.B.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- CGA
- chorionic gonadotropin-α
- e
- embryonic day
- H&E
- hematoxylin and eosin
- ISL1
- ISLET1
- LHX
- LIM/homeobox proteins
- NR5A1
- nuclear receptor subfamily 5, group A, member 1
- P
- postnatal day
- PITX2
- paired-like homeodomain transcription factor 2
- POU1F1
- POU domain class 1 transcription factor 1
- PTU
- propylthiouracil
- qPCR
- quantitative PCR
- X-gal
- 5 bromo-4-chloro-3-indolyl-b-D-galactopyranoside.
References
- 1. Kelberman D, Rizzoti K, Lovell-Badge R, Robinson IC, Dattani MT. Genetic regulation of pituitary gland development in human and mouse. Endocr Rev. 2009;30:790–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhao Y, Morales DC, Hermesz E, Lee WK, Pfaff SL, Westphal H. Reduced expression of the LIM-homeobox gene Lhx3 impairs growth and differentiation of Rathke's pouch and increases cell apoptosis during mouse pituitary development. Mech Dev. 2006;123:605–613. [DOI] [PubMed] [Google Scholar]
- 3. Sheng HZ, Zhadanov AB, Mosinger B, Jr, et al. Specification of pituitary cell lineages by the LIM homeobox gene Lhx3. Science. 1996;272:1004–1007. [DOI] [PubMed] [Google Scholar]
- 4. Sheng HZ, Moriyama K, Yamashita T, et al. Multistep control of pituitary organogenesis. Science. 1997;278:1809–1812. [DOI] [PubMed] [Google Scholar]
- 5. Raetzman LT, Ward R, Camper SA. Lhx4 and Prop1 are required for cell survival and expansion of the pituitary primordia. Development. 2002;129:4229–4239. [DOI] [PubMed] [Google Scholar]
- 6. Ellsworth BS, Butts DL, Camper SA. Mechanisms underlying pituitary hypoplasia and failed cell specification in Lhx3-deficient mice. Dev Biol. 2008;313:118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mullen RD, Colvin SC, Hunter CS, et al. Roles of the LHX3 and LHX4 LIM-homeodomain factors in pituitary development. Mol Cell Endocrinol. 2007;265–266:190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Savage JJ, Hunter CS, Clark-Sturm SL, Jacob TM, Pfaeffle RW, Rhodes SJ. Mutations in the LHX3 gene cause dysregulation of pituitary and neural target genes that reflect patient phenotypes. Gene. 2007;400:44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rajab A, Kelberman D, de Castro SC, et al. Novel mutations in LHX3 are associated with hypopituitarism and sensorineural hearing loss. Hum Mol Genet. 2008;17:2150–2159. [DOI] [PubMed] [Google Scholar]
- 10. Kriström B, Zdunek AM, Rydh A, Jonsson H, Sehlin P, Escher SA. A novel mutation in the LIM homeobox 3 gene is responsible for combined pituitary hormone deficiency, hearing impairment, and vertebral malformations. J Clin Endocrinol Metab. 2009;94:1154–1161. [DOI] [PubMed] [Google Scholar]
- 11. Bonfig W, Krude H, Schmidt H. A novel mutation of LHX3 is associated with combined pituitary hormone deficiency including ACTH deficiency, sensorineural hearing loss, and short neck-a case report and review of the literature. Eur J Pediatr. 2011;170:1017–1021. [DOI] [PubMed] [Google Scholar]
- 12. Machinis K, Pantel J, Netchine I, et al. Syndromic short stature in patients with a germline mutation in the LIM homeobox LHX4. Am J Hum Genet. 2001;69:961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Romero CJ, Pine-Twaddell E, Radovick S. Novel mutations associated with combined pituitary hormone deficiency. J Mol Endocrinol. 2011;46:R93–R102. [DOI] [PubMed] [Google Scholar]
- 14. Dateki S, Fukami M, Uematsu A, et al. Mutation and gene copy number analyses of six pituitary transcription factor genes in 71 patients with combined pituitary hormone deficiency: identification of a single patient with LHX4 deletion. J Clin Endocrinol Metab. 2010;95:4043–4047. [DOI] [PubMed] [Google Scholar]
- 15. Tajima T, Yorifuji T, Ishizu K, Fujieda K. A novel mutation (V101A) of the LHX4 gene in a Japanese patient with combined pituitary hormone deficiency. Exp Clin Endocrinol Diabetes. 2010;118:405–409. [DOI] [PubMed] [Google Scholar]
- 16. Karlsson O, Thor S, Norberg T, Ohlsson H, Edlund T. Insulin gene enhancer binding protein Isl-1 is a member of a novel class of proteins containing both a homeo- and a Cys-His domain. Nature. 1990;344:879–882. [DOI] [PubMed] [Google Scholar]
- 17. Zhuang S, Zhang Q, Zhuang T, Evans SM, Liang X, Sun Y. Expression of Isl1 during mouse development. Gene Expr Patterns. 2013;13:407–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pfaff SL, Mendelsohn M, Stewart CL, Edlund T, Jessell TM. Requirement for LIM homeobox gene Isl1 in motor neuron generation reveals a motor neuron-dependent step in interneuron differentiation. Cell. 1996;84:309–320. [DOI] [PubMed] [Google Scholar]
- 19. Du A, Hunter CS, Murray J, et al. Islet-1 is required for the maturation, proliferation, and survival of the endocrine pancreas. Diabetes. 2009;58:2059–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Habener JF, Kemp DM, Thomas MK. Minireview: transcriptional regulation in pancreatic development. Endocrinology. 2005;146:1025–1034. [DOI] [PubMed] [Google Scholar]
- 21. Baertschiger RM, Bosco D, Morel P, et al. Mesenchymal stem cells derived from human exocrine pancreas express transcription factors implicated in β-cell development. Pancreas. 2008;37:75–84. [DOI] [PubMed] [Google Scholar]
- 22. Elshatory Y, Everhart D, Deng M, Xie X, Barlow RB, Gan L. Islet-1 controls the differentiation of retinal bipolar and cholinergic amacrine cells. J Neurosci. 2007;27:12707–12720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Elshatory Y, Deng M, Xie X, Gan L. Expression of the LIM-homeodomain protein Isl1 in the developing and mature mouse retina. J Comp Neurol. 2007;503:182–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bu L, Jiang X, Martin-Puig S, et al. Human ISL1 heart progenitors generate diverse multipotent cardiovascular cell lineages. Nature. 2009;460:113–117. [DOI] [PubMed] [Google Scholar]
- 25. Moretti A, Caron L, Nakano A, et al. Multipotent embryonic isl1+ progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell. 2006;127:1151–1165. [DOI] [PubMed] [Google Scholar]
- 26. Gay F, Anglade I, Gong Z, Salbert G. The LIM/homeodomain protein islet-1 modulates estrogen receptor functions. Mol Endocrinol. 2000;14:1627–1648. [DOI] [PubMed] [Google Scholar]
- 27. Nasif S, de Souza FS, González LE, et al. Islet 1 specifies the identity of hypothalamic melanocortin neurons and is critical for normal food intake and adiposity in adulthood. Proc Natl Acad Sci USA. 2015;112:E1861–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Takuma N, Sheng HZ, Furuta Y, et al. Formation of Rathke's pouch requires dual induction from the diencephalon. Development. 1998;125:4835–4840. [DOI] [PubMed] [Google Scholar]
- 29. Ericson J, Norlin S, Jessell TM, Edlund T. Integrated FGF and BMP signaling controls the progression of progenitor cell differentiation and the emergence of pattern in the embryonic anterior pituitary. Development. 1998;125:1005–1015. [DOI] [PubMed] [Google Scholar]
- 30. Lin SC, Li S, Drolet DW, Rosenfeld MG. Pituitary ontogeny of the Snell dwarf mouse reveals Pit-1-independent and Pit-1-dependent origins of the thyrotrope. Development. 1994;120:515–522. [DOI] [PubMed] [Google Scholar]
- 31. Liu J, Liu Z, Yi S, Cui S. Islet-1 expression and its colocalization with luteinising hormone, thyroid-stimulating hormone and oestrogen receptor α in the developing pituitary gland of the sheep foetus. J Neuroendocrinol. 2005;17:773–780. [DOI] [PubMed] [Google Scholar]
- 32. Liu J, He Y, Wang X, Zheng X, Cui S. Developmental changes of Islet-1 and its co-localization with pituitary hormones in the pituitary gland of chick embryo by immunohistochemistry. Cell Tissue Res. 2005;322:279–287. [DOI] [PubMed] [Google Scholar]
- 33. Granger A, Bleux C, Kottler ML, Rhodes SJ, Counis R, Laverrière JN. The LIM-homeodomain proteins Isl-1 and Lhx3 act with steroidogenic factor 1 to enhance gonadotrope-specific activity of the gonadotropin-releasing hormone receptor gene promoter. Mol Endocrinol. 2006;20:2093–2108. [DOI] [PubMed] [Google Scholar]
- 34. Olson LE, Tollkuhn J, Scafoglio C, et al. Homeodomain-mediated β-catenin-dependent switching events dictate cell-lineage determination. Cell. 2006;125:593–605. [DOI] [PubMed] [Google Scholar]
- 35. Charles MA, Mortensen AH, Potok MA, Camper SA. Pitx2 deletion in pituitary gonadotropes is compatible with gonadal development, puberty, and fertility. Genesis. 2008;46:507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Castinetti F, Brinkmeier ML, Gordon DF, et al. PITX2 AND PITX1 regulate thyrotroph function and response to hypothyroidism. Mol Endocrinol. 2011;25:1950–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pan L, Deng M, Xie X, Gan L. ISL1 and BRN3B co-regulate the differentiation of murine retinal ganglion cells. Development. 2008;135:1981–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brinkmeier ML, Gordon DF, Dowding JM, et al. Cell-specific expression of the mouse glycoprotein hormone α-subunit gene requires multiple interacting DNA elements in transgenic mice and cultured cells. Mol Endocrinol. 1998;12:622–633. [DOI] [PubMed] [Google Scholar]
- 39. Brinkmeier ML, Davis SW, Carninci P, et al. Discovery of transcriptional regulators and signaling pathways in the developing pituitary gland by bioinformatic and genomic approaches. Genomics. 2009;93:449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Weiss RE, Murata Y, Cua K, Hayashi Y, Seo H, Refetoff S. Thyroid hormone action on liver, heart, and energy expenditure in thyroid hormone receptor β-deficient mice. Endocrinology. 1998;139:4945–4952. [DOI] [PubMed] [Google Scholar]
- 41. Bilodeau S, Roussel-Gervais A, Drouin J. Distinct developmental roles of cell cycle inhibitors p57Kip2 and p27Kip1 distinguish pituitary progenitor cell cycle exit from cell cycle reentry of differentiated cells. Mol Cell Biol. 2009;29:1895–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu J, Lin C, Gleiberman A, et al. Tbx19, a tissue-selective regulator of POMC gene expression. Proc Natl Acad Sci USA. 2001;98:8674–8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhao L, Bakke M, Krimkevich Y, et al. Steroidogenic factor 1 (SF1) is essential for pituitary gonadotrope function. Development. 2001;128:147–154. [DOI] [PubMed] [Google Scholar]
- 44. Kendall SK, Samuelson LC, Saunders TL, Wood RI, Camper SA. Targeted disruption of the pituitary glycoprotein hormone α-subunit produces hypogonadal and hypothyroid mice. Genes Dev. 1995;9:2007–2019. [DOI] [PubMed] [Google Scholar]
- 45. Brinkmeier ML, Stahl JH, Gordon DF, et al. Thyroid hormone-responsive pituitary hyperplasia independent of somatostatin receptor 2. Mol Endocrinol. 2001;15:2129–2136. [DOI] [PubMed] [Google Scholar]
- 46. Kulig E, Camper SA, Kuecker S, Jin L, Lloyd RV. Remodeling of hyperplastic pituitaries in hypothyroid us-subunit knockout mice after thyroxine and 1713-estradiol treatment: role of apoptosis. Endocr Pathol. 1998;9:261–274. [DOI] [PubMed] [Google Scholar]
- 47. Stahl JH, Kendall SK, Brinkmeier ML, et al. Thyroid hormone is essential for pituitary somatotropes and lactotropes. Endocrinology. 1999;140:1884–1892. [DOI] [PubMed] [Google Scholar]
- 48. Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. [DOI] [PubMed] [Google Scholar]
- 49. Xiao R, Sun Y, Ding JH, et al. Splicing regulator SC35 is essential for genomic stability and cell proliferation during mammalian organogenesis. Mol Cell Biol. 2007;27:5393–5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vooijs M, Jonkers J, Berns A. A highly efficient ligand-regulated Cre recombinase mouse line shows that LoxP recombination is position dependent. EMBO Reports. 2001;2:292–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sandgren EP, Palmiter RD, Heckel JL, Daugherty CC, Brinster RL, Degen JL. Complete hepatic regeneration after somatic deletion of an albumin-plasminogen activator transgene. Cell. 1991;66:245–256. [DOI] [PubMed] [Google Scholar]
- 52. Yamada M, Saga Y, Shibusawa N, et al. Tertiary hypothyroidism and hyperglycemia in mice with targeted disruption of the thyrotropin-releasing hormone gene. Proc Natl Acad Sci USA. 1997;94:10862–10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rabeler R, Mittag J, Geffers L, et al. Generation of thyrotropin-releasing hormone receptor 1-deficient mice as an animal model of central hypothyroidism. Mol Endocrinol. 2004;18:1450–1460. [DOI] [PubMed] [Google Scholar]
- 54. Li S, Crenshaw EB, 3rd, Rawson EJ, Simmons DM, Swanson LW, Rosenfeld MG. Dwarf locus mutants lacking three pituitary cell types result from mutations in the POU-domain gene pit-1. Nature. 1990;347:528–533. [DOI] [PubMed] [Google Scholar]
- 55. Gordon DF, Lewis SR, Haugen BR, et al. Pit-1 and GATA-2 interact and functionally cooperate to activate the thyrotropin β-subunit promoter. J Biol Chem. 1997;272:24339–24347. [DOI] [PubMed] [Google Scholar]
- 56. Wen S, Ai W, Alim Z, Boehm U. Embryonic gonadotropin-releasing hormone signaling is necessary for maturation of the male reproductive axis. Proc Natl Acad Sci USA. 2010;107:16372–16377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Charles MA, Saunders TL, Wood WM, et al. Pituitary-specific Gata2 knockout: effects on gonadotrope and thyrotrope function. Mol Endocrinol. 2006;20:1366–1377. [DOI] [PubMed] [Google Scholar]
- 58. Yamada M, Mori M. Mechanisms related to the pathophysiology and management of central hypothyroidism. Nat Clin Pract Endocrinol Metab. 2008;4:683–694. [DOI] [PubMed] [Google Scholar]
- 59. Sun Y, Bak B, Schoenmakers N, et al. Loss-of-function mutations in IGSF1 cause an X-linked syndrome of central hypothyroidism and testicular enlargement. Nat Genet. 2012;44:1375–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Balasubramanian M, Shield JP, Acerini CL, et al. Pancreatic hypoplasia presenting with neonatal diabetes mellitus in association with congenital heart defect and developmental delay. Am J Med Genet A. 2010;152A:340–346. [DOI] [PubMed] [Google Scholar]