

Abstract
Activation of Sirtuin1 (Sirt1), an nicotinamide adenine dinucleotide oxidized-dependent deacetylase, by natural or synthetic compounds like resveratrol, SRT2104, or SRT3025 attenuates the loss of bone mass caused by ovariectomy, aging, or unloading in mice. Conversely, Sirt1 deletion in osteoclast progenitors increases osteoclast number and bone resorption. Sirt1 deacetylates forkhead box protein (Fox) O1, FoxO3, and FoxO4, and thereby modulates their activity. FoxOs restrain osteoclastogenesis and bone resorption. Here, we tested the hypothesis that the antiresorptive effects of Sirt1 are mediated by FoxOs. We report that Sirt1 activation by SRT2104 and SRT3025 inhibited murine osteoclast progenitor proliferation and reduced osteoclastogenesis. The effect of Sirt1 stimulators on osteoclastogenesis was abrogated in cells lacking FoxO1, FoxO3, and FoxO4. FoxO1 acetylation was increased by knocking down Sirt1 or addition of receptor activator of nuclear factor kappa-B ligand, the critical cytokine for osteoclast differentiation. Furthermore, acetylation inhibited, whereas deacetylation promoted, FoxO-mediated transcription. SRT3025 increased the expression of the FoxO-target genes catalase and hemeoxygenase-1 (HO-1) in osteoclast progenitors, in a FoxO-dependent manner. HO-1 catabolizes heme and attenuates mitochondrial oxidative phosphorylation and ATP production in macrophages. HO-1 levels were strongly reduced and ATP levels increased by Receptor activator of nuclear factor kappa-B ligand. In contrast, SRT3025 and FoxOs decreased ATP production, and the effect of SRT3025 was mediated by FoxOs. These findings reveal that the antiosteoclastogenic actions of Sirt1 are mediated by FoxOs and result from impaired mitochondria activity. Along with earlier findings that the osteoblastogenic effects of Sirt1 are also mediated by FoxOs, these results establish that the dual antiosteoporotic efficacy of Sirt1 stimulators (ie, decreasing bone resorption and promoting bone formation) is mediated via FoxO deacetylation.
A decline in the levels and activity of Sirtuin1 (Sirt1), an nicotinamide adenine dinucleotide oxidized (NAD+)-dependent class III deacetylase, has been linked to the onset of aging and aging-associated diseases, including diabetes, neurodegeneration, cancer, sarcopenia, and osteoporosis (1, 2). The antiaging actions of Sirt1 result from the deacetylation of histone and nonhistone proteins including the forkhead box protein (Fox) O transcription factors. We have shown earlier that FoxOs inhibit osteoclastogenesis by attenuating osteoclast progenitor proliferation (3). These effects result from the stimulation of catalase and thereby catabolism of H2O2, the most abundant form of reactive oxygen species (ROS). Furthermore, we had shown that generation of mitochondrial H2O2 contributes to osteoclast formation and bone resorption, and is indispensable for skeletal homeostasis under physiological and pathological conditions. Most ROS are generated in the mitochondria as byproducts of ATP production via oxidative phosphorylation (OXPHOS) (4). During this process, high-energy electrons derived from the metabolic fuels are passed through a series of protein complexes (I–IV) embedded in the inner-mitochondrial membrane, collectively known as the electron transport chain. Mitochondrial ROS is generated from the escape of electrons at complexes I and III (5). In line with the importance of mitochondrial H2O2 for osteoclast generation, receptor activator of nuclear factor kappa-B ligand (RANKL) increases mitochondria biogenesis and OXPHOS in osteoclast progenitors (6, 7). Furthermore, disruption of complex I in osteoclast progenitors decreases osteoclast formation and bone resorption (8).
Similar to FoxOs, Sirt1 inhibits bone resorption via direct actions in cells of the osteoclast lineage (9, 10). Importantly, stimulation of Sirt1 with the natural polyphenol resveratrol, or with synthetic compounds like SRT2140 or SRT3025, attenuates the loss of bone mass caused by ovariectomy, hind limb unloading, or aging in mice (11–15). Additionally, resveratrol or SRT2140 also inhibit osteoclast formation in vitro (15–18). It has been proposed that Sirt1 attenuates osteoclastogenesis by inhibiting nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling. Under inflammatory conditions Sirt1 attenuates NF-κB activity in cultured bone marrow macrophages (BMMs) by deacetylating V-rel avian reticuloendotheliosis viral oncogene homolog A/nuclear factor NF-kappa-B p65 subunit (p65) (10, 19). However, in the absence of inflammatory stimuli the levels of acetylated RelA/p65 are very low in BMM and indistinguishable in the presence or absence of Sirt1. Thus, in conditions other than inflammation, the mechanisms responsible for the antiosteoclastogenic actions of Sirt1 remain unclear.
Sirt1-mediated FoxOs deacetylation causes an increase or decrease in FoxO activity depending on the cell type (20). For example, Sirt1 attenuates muscle atrophy via FoxO deacetylation and attenuation of FoxO-mediated transcription of muscle RING-finger protein-1 and atrogin 1 (21, 22). On the other hand, Sirt1 protects the heart from ischemia/reperfusion injury by deacetylating FoxO1 and thereby stimulating the expression of the antioxidant gene manganese superoxide dismutase (23). FoxOs are acetylated by histone acetyltransferases, including cAMP-response element-binding protein-binding protein and E1A binding protein p300 (p300) at Lys residues K242, K245, K259, K262, K271, and K291 (in FoxO1) (24, 25). We found earlier that Sirt1 promotes bone formation via deacetylation of FoxOs (26). Specifically, in osteoblast progenitors FoxOs bind to β-catenin and prevent β-catenin/T cell factor-mediated transcription, thereby restraining Wnt-induced proliferation and osteoblastogenesis. Deacetylation of FoxO by Sirt1 inhibits the binding of FoxOs to β-catenin and thus increases osteoblast progenitor proliferation, the number of osteoblasts, and bone formation. Here, we examined the contribution of FoxO deacetylation to the antiosteoclastogenic actions of Sirt1.
Materials and Methods
Animal experimentation
The FoxO1,3,4ΔLysM mice were generated as described previously (3). Sirt1 floxed (f/f) mice (C57BL/6 genetic background) were obtained from The Jackson Laboratory. Bone marrow cells were collected from the tibiae and femora of 3- to 6-month-old mice. All procedures involving mice were approved by the Institutional Animal Care and Use Committees of the University of Arkansas for Medical Sciences and the Central Arkansas Veterans Healthcare System.
BMM and osteoclast cultures
BMMs were obtained as described previously (3). Whole bone marrow cells were plated in α-MEM complete media (10% fetal bovine serum, 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin) after removing red blood cells by ammonium-chloride-potassium buffer for 24 hours in the presence of 10 ng mL−1 macrophage colony-stimulating factor (M-CSF). Nonadherent cells were cultured in Petri dishes with 30 ng mL−1 M-CSF for 3–4 days to generate BMMs. To generate preosteoclasts and mature osteoclasts, BMMs were cultured in α-MEM complete media with 30 ng mL−1 M-CSF and 30 ng mL−1 RANKL (R&D Systems) for 2 and 4 days, respectively. Cells were fixed with 10% neutral-buffered formalin for 15 minutes and stained for tartrate-resistant acid phosphatase (TRAP), using the Leukocyte Acid Phosphatase Assay kit, following the manufacturer's instructions (Sigma-Aldrich). A preosteoclast was defined as a round mononuclear TRAP-positive cell and an osteoclast as a multinuclear TRAP-positive cell. For all assays, cells were plated in triplicate. Proliferation of BMMs was quantified using bromodeoxyuridine incorporation kit from Roche according to the manufacturer's protocol, as described previously (3).
Quantitative real-time PCR analysis
BMMs were cultured in 6-well plates and total RNA extracted using TRIzol reagent (Invitrogen). cDNA was obtained from 2 μg of total RNA extract using the High-Capacity cDNA Archive kit (Applied Biosystems) according to the manufacturer's instructions. TaqMan quantitative real-time PCR was performed using the next primers from Applied Biosystems: catalase (Mm00437992_m1), FoxO1 (Mm00490672_m1), hemeoxygenase-1 (HO-1) (Mm00516005_m1), TNF (Mm00443258_m1), and Fas ligand (Mm00438864_m1). All reactions were run in triplicate, and target gene expression was calculated by normalizing to the housekeeping gene ribosomal protein S2 (Mm00475528_m1) using the delta threshold cycle method (27). Mitochondrial DNA copy number in osteoclast precursors was quantitated as described previously (28).
Western blotting
Cells were washed twice with ice-cold PBS and lysed with a buffer containing 20mM Tris-HCL, 150mM NaCl, 1% Triton X-100, protease inhibitor mixture, and phosphatase inhibitor cocktail (Sigma-Aldrich). After incubation on ice for 30 minutes, the cell lysates were centrifuged at 13 200 rpm for 15 minutes at 4°C. Protein concentration of cell lysates were determined using the DC Protein Assay kit (Bio-Rad). The extracted protein (20–40 μg per sample) was subjected to 8%–12% SDS-PAGE gels and transferred electrophoretically onto polyvinyl difluoride membranes. The membranes were blocked in 5% fat-free milk/Tris-buffered saline for 90 minutes and incubate with each primary antibody followed by secondary antibodies conjugated with horseradish peroxidase. Monoclonal antibodies against HO-1 (1:5000, ab13248; Abcam), OXPHOS-Complex (1:10 000, 457999; Invitrogen), Flag (1:10 000, F1804; Sigma), Sirt1 (1:1000, 8469; Cell Signaling), phosphorylated (p)-Inhibitor of kappa B (1:1000, 9246; Cell Signaling), p-c-Jun N-terminal kinase (Jnk) (1:1000, 9255; Cell Signaling), cyclinD1 (1:500, sc-753; Santa Cruz Biotechnology, Inc), Nuclear factor of activated T-cells, cytoplasmic 1 (NFATc1) (1:5000, sc-7294; Santa Cruz Biotechnology, Inc), p-Erk (1:500, sc-7383; Santa Cruz Biotechnology, Inc), Jnk (1:500, sc-1648; Santa Cruz Biotechnology, Inc), and β-actin (1:2000, sc-81178; Santa Cruz Biotechnology, Inc) were used to detect their corresponding protein levels. We determined FoxO1, FoxO3, acetylated (Ac) histone 3 (H3), phosphorylated mitogen-activated protein kinase (p38), and Akt levels in cultured cell lysates using rabbit monoclonal antibodies for FoxO1 (1:1000, 2880; Cell Signaling), FoxO3 (1:1000, 9467; Cell Signaling), acetylated-H3 (1:1000, 9649; Cell Signaling), p-p38 (1:1000, 9215; Cell Signaling), and p-Akt (1:1000, 4058; Cell Signaling). We also used rabbit polyclonal antibodies for Ac-FoxO1 (1:500, sc-81178; Santa Cruz Biotechnology, Inc), catalase (1:500, sc-50508; Santa Cruz Biotechnology, Inc), IκB (1:500, sc-847; Santa Cruz Biotechnology, Inc), Erk (1:500, sc-94; Santa Cruz Biotechnology, Inc), p-p65 (1:1000, 3039; Cell Signaling), p38 (1:1000, 9212; Cell Signaling), and Akt (1:1000, 9272; Cell Signaling) to analyze their protein levels. The membranes were subjected to Western blot analysis with enhanced chemiluminescence reagents (Millipore). Quantification of the intensity of the bands in the autoradiograms was performed using a VersaDoc imaging system (Bio-Rad).
Transient transfections and luciferase assay
Raw264.7 cells were seeded in 6-well plates 24 hours before transfection. Cells were incubated with Flag-Sirt1 plasmid (1791; Addgene) for 20 minutes at room temperature in serum-free medium containing LipofectAMINE plus reagent (Invitrogen) according to the manufacturer's instruction. Cells were then washed and incubated in α-MEM complete media for 24 hours followed by addition of 30 ng mL−1 RANKL. To perform the FoxO activity assay, Raw264.7 cells were plated on a 48-well plate and 12 hours later transfected with 200 ng/well of reporter plasmid FoxO-luciferase (luc) and then cotransfected with 200 ng/well of Flag-FoxO1-wild type (WT) and Flag-FoxO-6KQ (12148 and 17562; Addgene) using LipofectAMINE plus reagent. Luciferase activity was measured using the dual-luciferase assay system (Promega) in a Dynex luminometer.
Adenoviral and retroviral transduction of BMMs
Adenoviral infection of BMMs was performed as described previously (29). In short, BMMs from Sirt1f/f mice or WT mice were incubated in α-MEM complete media containing recombinant adenovirus encoding Cre recombinase (Ad-Cre) (Vector Biolabs) at a multiplicity of infection of 100 for 24 hours. Cells were then washed twice with α-MEM and plated on a 6-well plate. Cell lysates were collected 2 days later for Western blot analysis.
WT- and acetylation mimic-FoxO1 cDNA were subcloned from their respective Addgene plasmids into the pMX-IRES-BSR retrovirus vector and the resulting constructs were verified by DNA sequencing. Retroviral particles were generated by transfecting Plat E packaging cells with the above generated retroviral constructs in presence of TransIT-LT1 transfection reagent (Fisher Scientific) as described previously (30). Supernatants containing viral particles were collected 48 hours after transfection, filtered through a 0.45-μm filter, and either used immediately or stored at −80°C. Subconfluent BMMs were transduced with viral particles for 24 hours in α-MEM complete media containing M-CSF and 20 μg/mL of protamine.
ATP production
ATP levels were measured by a luciferin-luciferase based assay using an ENLITEN ATP assay system bioluminescence detection kit (Promega) according to the manufacturer's protocol. In brief, culture medium was removed, BMMs cultured in 12-well plates were washed with PBS and extracted with 100 μL of 0.5% trichloroacetic acid in ATP-free water. Each extracted sample (10 μL) was neutralized by adding a Tris-acetate buffer (90 μL) adjusted to pH 7.75. The luciferase reagent (25 μL) was added immediately into the neutralized samples (10 μL) before measurement using a luminometer. The standard curve for intracellular ATP was obtained with a series of dilution of the ATP standards and ATP-free water provided in the kit. ATP was normalized for protein concentration.
NAD+/nicotinamide adenine dinucleotide assay
BMMs (4 × 104 cells/well) were plated on 24-well plates with 30 ng mL−1 M-CSF and 30 ng mL−1 RANKL in α-MEM complete media. After 48 hours, cells were lysed with ice-cold NAD extraction buffer (100 μL; BioAssay Systems). NAD+/nicotinamide adenine dinucleotide levels were determined using the EnzyFluo Assay kit (EFND-100; BioAssay Systems) according to the manufacturer's instructions.
Statistical analysis
Group mean values were compared, as appropriate, by Student's 2-tailed t test or two-way ANOVA with Tukey's test, after determining that the data were normally distributed and exhibited equivalent variances. P ≤ .05 was considered significant for all statistical comparisons.
Results
Sirt1 activation inhibits osteoclastogenesis via a FoxO-dependent mechanism
In agreement with our earlier findings that SRT2104 inhibits osteoclastogenesis (15), we found that SRT2104 inhibited RANKL-induced osteoclast formation in BMM (Figure 1, A and B). Similarly, SRT3025 dose-dependently inhibited osteoclastogenesis in bone marrow-derived cells or the macrophage-like cell line Raw264.7 (Figure 1, A–C). To determine whether the antiosteoclastogenic actions of SRT3025 were mediated by Sirt1, we used BMM from Sirt1f/f mice and deleted Sirt1 in vitro using Ad-Cre. Cells from WT littermate mice were used as control. Deletion of Sirt1 increased osteoclast formation (Figure 1D). Furthermore, the suppressive effect of SRT3025 was prevented in the absence of Sirt1, demonstrating the specificity of this compound. In line with the antiosteoclastogenic actions of Sirt1 activators, overexpression of Sirt1 in Raw264.7 cells inhibited osteoclast formation (Figure 1, E–G).
Figure 1.

Sirt1 inhibits osteoclast formation. Representative pictures (A) and number (B) of TRAP-positive multinucleated osteoclasts derived from BMMs cultured with M-CSF (30 ng/mL), RANKL (30 ng/mL), and the indicated doses of Sirt1 activators for 4.5 days. C, Raw264.7 cells cultured with RANKL (50 ng/mL) and the indicated doses of SRT3025 for 5 days. D, BMMs from WT or Sirt1f/f mice, infected with Ad-Cre to delete Sirt1, were cultured with RANKL (30 ng/mL) in the presence or absence of SRT3025 (10μM) for 5 days. E–G, Raw264.7 cells were transfected with empty vector or with a Flag-tagged Sirt1 plasmid and cultured with RANKL (50 ng/mL) for 5 days. E, Sirt1 levels detected by Western blotting. Representative pictures (F) and number (G) of TRAP-positive multinucleated osteoclasts. Bars represent mean ± SD; *, P < .05 vs untreated cells or empty vector by Student's t test; #, P < .05 by two-way ANOVA.
Next, we examined whether the effects of the Sirt1 activators were mediated by FoxOs. To this end, we used BMM obtained from mice lacking FoxO1, FoxO3, and FoxO4 in the myeloid lineage (designated FoxO1,3,4ΔLysM) or from littermate control mice (FoxO1,3,4f/f) (3). As we had shown earlier, FoxO1,3,4 deletion increased osteoclast generation and BMM proliferation (Figure 2, A and B). The inhibitory actions of SRT2104 or SRT3025 on osteoclastogenesis, seen in cells from FoxO1,3,4f/f control mice, were prevented in cells from FoxO1,3,4ΔLysM mice. In addition, SRT3025 greatly attenuated the stimulatory effects of M-CSF on cyclinD1 protein levels and macrophage number in cells from FoxO1,3,4f/f control mice (Figure 2, C and D). All these effects of SRT3025 were greatly attenuated in cells lacking FoxO1,3,4.
Figure 2.

Sirt1 activation decreases osteoclastogenesis via a FoxO-dependent mechanism. Representative pictures (A) and number (B) of TRAP-positive multinucleated osteoclasts derived from BMMs of mice of the indicated genotypes cultured with M-CSF (30 ng/mL) and RANKL (30 ng/mL) in the presence or absence of Sirt1 activators (5μM SRT2104, 10μM SRT3025) for 4 days. C, Western blotting of BMMs, which were pretreated with vehicle (DMSO) or SRT3025 (10μM) for 30 minutes, followed by M-CSF (30 ng/mL) for 8 hours. D, Macrophage cell number in cultures derived from mice of the indicated genotypes in the presence of M-CSF (30 ng/mL) for 2 days. Bars represent mean ± SD; #, P < .05 vs untreated cells within the same genotype; *, P < .05 vs untreated cells from FoxO1,3,4f/f by two-way ANOVA.
Sirt1 deacetylates FoxO1 in osteoclast precursors
Lysine acetylation alters the transcriptional activity of FoxOs (31). The levels of FoxO1 acetylated at Lys259, Lys262, and Lys271 were higher in BMM lacking Sirt1 as compared with control cells (Figure 3A). The increased FoxO1 acetylation was associated with reduced levels of FoxO1 protein, as well as reduced levels of the FoxO-target gene catalase. Moreover, FoxO1 acetylation was increased in preosteoclasts and mature osteoclasts when compared with BMM (Figure 3B). However, Sirt1levels were unchanged during osteoclast differentiation. As expected, the levels of NFATc1, the critical transcription factor for osteoclastogenesis, were greatly increased with osteoclast differentiation. SRT3025 prevented RANKL-induced acetylation of FoxO1 (Figure 3C) as well as acetylation of histone 3 (Figure 3D), an established target of Sirt1. We next determined whether a decrease in the Sirt1 cofactor NAD+ could be responsible for the increase in FoxO acetylation with osteoclast differentiation. The levels of NAD+ were modestly higher in preosteoclasts when compared with BMM (Figure 3E), suggesting that the increase in FoxO1 acetylation is independent from changes in Sirt1 activity.
Figure 3.
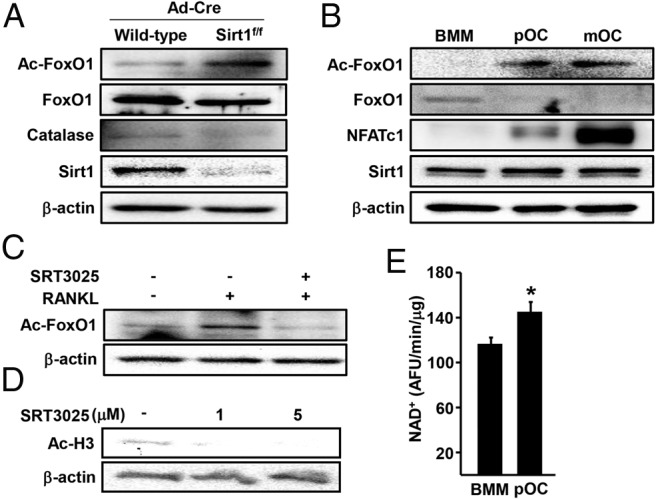
Sirt1 deacetylates FoxO1 in BMMs. A–D, Whole-cell lysates were subjected to Western blotting with indicated antibodies. A, Sirt1 was deleted in BMMs as described in Figure 1. B, Acetylated FoxO1 at Lys259, Lys262, and Lys271 in BMMs, preosteoclast and mature osteoclast cultures. C, BMMs were pretreated with vehicle (DMSO) or SRT3025 (10μM) for 1 hour and then stimulated with RANKL (30 ng/mL) for 48 hours. D, BMMs were cultured with M-CSF (30 ng/mL) at the indicated dose of SRT3025 for 24 hours. E, NAD+ levels in BMMs and preosteoclasts. Bars represent mean ± SD; *, P < .05 by Student's t test.
Acetylation inhibits and deacetylation by Sirt1 promotes FoxO-mediated transcription and its antiosteoclastogenic effects
The results detailed above suggested that acetylation inhibits the actions of FoxOs in osteoclastic cells. To further investigate the role of acetylation, we used a FoxO1 mutant construct, designated KQ-FoxO1, in which the 6 lysine acetylation sites are mutated to glutamine to mimic acetylation (32). The KQ-FoxO1 mutant, WT-FoxO1, or the empty vector were introduced into BMM from FoxO1,3,4ΔLysM mice via viral infection. Both WT- and KQ-FoxO1 were expressed at high levels in the respective cells (Figure 4A). WT-FoxO1 abrogated the increased osteoclastogenesis seen in BMM lacking FoxO1,3,4 (Figure 4B). The KQ-FoxO1 mutant also attenuated osteoclastogenesis but less potently than WT-FoxO1. Furthermore, WT-FoxO1 greatly attenuated both cyclinD1 and osteoclast progenitor proliferation induced by FoxO1,3,4 deletion (Figure 4, A and C). In contrast, the KQ-FoxO1 mutant had no effect on cyclinD1 levels or proliferation. We next examined the expression of the FoxO-target catalase in macrophages from FoxO1,3,4ΔLysM mice infected with WT-FoxO1 or with KQ-FoxO1 mutant. As expected, WT-FoxO1 increased the protein levels of catalase. In contrast, KQ-FoxO1 had no effect on the level of this protein. To further examine the effect of acetylation on FoxO-mediated transcription, we transfected Raw264.7 macrophages with a FoxO-luciferase (luc) reporter construct along with WT-FoxO1 or KQ-FoxO1 mutant. In agreement with the findings in primary BMMs, WT-FoxO1 stimulated FoxO-luc activity, whereas KQ-FoxO1 had no effect (Figure 4D).
Figure 4.

Deacetylation by Sirt1 promotes FoxO-mediated transcription. A–C, BMMs were transduced with retroviruses expressing empty vector (EV), WT-FoxO1, or KQ-FoxO1. A, Western blotting of whole-cell lysates. B, Number of TRAP-positive multinucleated osteoclasts. C, Proliferation by BrdU labeling in BMM cultures. D, Luciferase activity in Raw264.7 cells transfected with a FoxO-luc reporter construct and with EV, WT-FoxO1, or KQ-FoxO1 plasmid and cultured for 24 hours. E, mRNA levels in BMMs cultured with or without SRT3025 (10μM) for 24 hours. Bars represent mean ± SD; *, P < .05 vs EV and †, P < .05 vs WT by Student's t test; #, P < .05 vs untreated cells of the same genotype and ‡, P < .05 vs equivalent treatment in cells from FoxO1,3,4f/f mice by two-way ANOVA.
We then examined whether deacetylation promotes FoxO-mediated transcription. The mRNA levels of catalase were increased by addition of SRT3025 to BMM (Figure 4E), as was the expression of HO-1 and FoxO1. The effect of SRT3025 on the expression of catalase, HO-1 and FoxO1 was attenuated in BMM from FoxO1,3,4ΔLysM mice.
Sirt1 does not alter RANKL-induced NF-κB or MAPK activation
As mentioned earlier, attenuation of NF-κB might contribute to the antiosteoclastogenic actions of Sirt1 (9, 10, 33). To test this possibility, we first examined whether acetylation of p65 was altered in macrophages lacking Sirt1. We found that the levels of p65 acetylated at Lys310 are very low and indistinguishable in the presence or absence of Sirt1 (Figure 5A). In addition, SRT3025 did not alter the RANKL-induced phosphorylation of Ikb or p65 nor it affected the expression of the NF-κB target genes TNF and FasL (Figure 5, B–D). Stimulation of MAPK and Akt by RANKL, as shown by the phosphorylation of Erk, p38, Jnk, and Akt, was also unaffected by SRT3025 (Figure 5E).
Figure 5.

Sirt1 activation does not affect NF-κB and MAPK pathway. A, Sirt1 was deleted in BMMs as described in Figure 1. B, BMMs cultured in medium containing 0.2% FBS for 3 hours, pretreated with vehicle (DMSO) or SRT3025 (10μM) for 1 hour, followed by RANKL (30 ng/mL) for the indicated times. C, BMMs pretreated with vehicle (DMSO) or SRT3025 (10μM) for 30 minutes, followed by RANKL (30 ng/mL) or TNF (50 ng/mL) for 3 hours. D, BMMs cultured with or without SRT3025 (10μM) for 24 hours. E, Western blotting of BMMs cell lysates cultured as in B. Bars represent mean ± SD.
Sirt/FoxO activation increases HO-1 expression and attenuates ATP generation
Stimulation of OXPHOS by RANKL is critical for osteoclastogenesis (7, 8). In liver cells, FoxO1 attenuates OXPHOS by increasing HO-1 (34). We, therefore, examined whether the increase in HO-1 by Sirt1/FoxO activation was accompanied by changes in mitochondria activity. In line with the decline in FoxO1, FoxO3, and FoxO4 levels with osteoclast differentiation (3), the mRNA and protein levels of HO-1 were greatly decreased in preosteoclasts and mature osteoclast when compared with BMM (Figure 6, A and B). A decline in HO-1 was seen as early as 24 hours after incubation with RANKL and this effect was specific because incubation with TNF did not change the levels of HO-1 (Figure 6C). The low HO-1 levels seen in the presence of RANKL were associated with increased levels of proteins of complexes II (succinate dehydrogenase complex, subunit A, flavoprotein), III (ubiquinol-cytochrome c reductase core protein I), IV (mitochondrially encoded cytochrome C oxidase 1), and V (ATP synthase, H+ transporting, mitochondrial F1 complex, alpha subunit 1) of the electron transport chain (Figure 6D). Consistent with these changes, the levels of mtDNA and ATP were increased in response to RANKL (Figure 6, E and F). Deletion of Sirt1 or FoxOs decreased HO-1 protein levels (Figures 5A and 6G). BMMs from FoxO1,3,4 null mice exhibited higher ATP production (Figure 6H). SRT3025 decreased ATP production in BMM from control mice but had no effect in cells from FoxO1,3,4ΔLysM mice, indicating that the inhibitory effect of Sirt1 stimulation on ATP production was mediated by FoxOs. However, the levels of proteins of complexes II–V were unaffected by FoxOs (Figure 6I), suggesting that FoxOs inhibit ATP production independently of changes in the levels of proteins of the electron transport chain.
Figure 6.
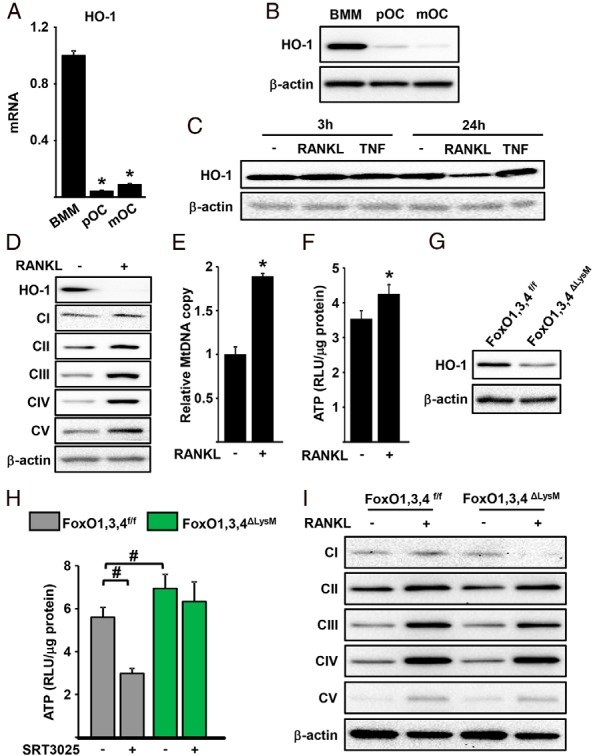
Sirt1 activation inhibits RANKL-induced mitochondrial function. HO-1 mRNA (A) and protein (B) levels in BMMs, preosteoclast (pOC), and mature osteoclast (mOC) cultures. C, HO-1 protein levels of BMMs treated with RANKL (30 ng/mL) or TNF (50 ng/mL) for the indicated time. Mitochondria electron transport chain protein levels (D), mitochondrial DNA copy number (E), and ATP levels (F) in BMMs treated with RANKL (30 ng/mL) for 48 hours. RLU, relative luminescence units. G, Western blotting in BMMs cell lysates. H, ATP levels in BMMs treated without or with SRT3025 (10μM) in the presence of RANKL (30 ng/mL) for 24 hours. I, Mitochondria electron transport chain protein levels in BMMs treated with RANKL (30 ng/mL) for 48 hours. *, P < .05 by Student's t test and #, P < .05 by two-way ANOVA.
Discussion
In cells of the myeloid lineage, Sirt1 and FoxOs act to attenuate osteoclastogenesis and bone resorption (3, 9). We have now found that Sirt1 activation attenuated BMM proliferation and osteoclastogenesis by deacetylating and, thereby, stimulating FoxO activity (Figure 7). In contrast, a mutant form of FoxO1 that is constitutively acetylated had very limited or no effect on BMM proliferation and osteoclastogenesis. These findings establish that deacetylation of FoxO1 is critical for the antiosteoclastogenic effects of this transcription factor.
Figure 7.
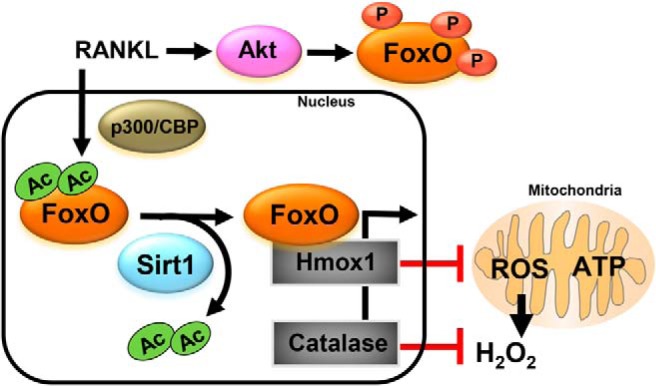
Model of the role of Sirt1/FoxOs in cells of the osteoclast lineage. RANKL-induced phosphorylation of FoxOs retains FoxOs in the cytoplasm and prevents FoxO transcriptional activity (3). RANKL also prevents FoxO activity via acetylation. Sirt1 deacetylates and stimulates FoxO-mediated transcription of catalase and HO-1. HO-1 decreases mitochondria activity which, along with catalase, causes attenuation of H2O2 levels. Both low ATP and H2O2 levels contribute to the antiosteoclastogenic effects of Sirt1/FoxO signaling.
Sirt1 deletion increased FoxO1 acetylation and decreased the levels of FoxO1 and its target genes catalase and HO-1. Likewise, RANKL increased the acetylation of FoxO1 and, thereby, reduced FoxO1 transcriptional activity. Acetylation regulates the function of FoxOs by altering their DNA binding activity, as well as by increasing their susceptibility to phosphorylation by Akt, thereby, promoting FoxOs retention in the cytoplasm (35, 36). We have previously shown that RANKL promotes the phosphorylation and inactivation of FoxO1 and FoxO3 by Akt (3). It is, therefore, possible that RANKL-induced phosphorylation and acetylation cooperate to inactivate FoxOs in osteoclasts. Our present results also suggest that the increase in FoxO1 acetylation by RANKL is not due to loss of Sirt1 activity because the levels of Sirt1 or the Sirt1 cofactor NAD+ were not decreased during osteoclastogenesis. Alternatively, the increased acetylation of FoxO1 might be due to the enhanced acetyltransferase activity caused by RANKL (Figure 7) (18, 37). For example, NFATc1 is acetylated by both p300 and p300/cAMP-response element-binding protein-binding protein-associated factor.
We had also shown earlier that similar to FoxO1, FoxO3, and FoxO4 levels decrease during osteoclastogenesis and that RANKL inhibits the transcriptional activity of FoxOs. Because all 3 FoxOs are targets of Sirt1 (24, 25, 38), it is likely that deacetylation of FoxO3 and FoxO4 also promotes their activity in osteoclasts. The evidence that FoxO1, similar to FoxO3 (3), inhibits osteoclastogenesis are in contrast with a recent report by Wang et al, suggesting that FoxO1 promotes osteoclastogenesis (39). However, consistent with our contention that FoxO1, FoxO3, and FoxO4 are functionally redundant in inhibiting osteoclastogenesis (3), Wang et al found that deletion of FoxO1 using LysM-Cre does not alter bone mass or osteoclast number under physiological conditions.
Sirt1 in cells of the myeloid lineage attenuates lipopolysaccharide- or TNF-induced NF-κB signaling via deacetylation of p65 (19, 40). However, similar to findings by others (10, 19, 40), we show here that macrophages lacking Sirt1 exhibit no changes in acetylated p65 levels, in the absence of inflammatory signals. Furthermore, we have examined whether SRT3025 altered the stimulatory actions of RANKL on NF-κB but found no effect. Instead, our results indicate that SIRT3025 inhibits osteoclastogenesis, at least in part, by attenuating the proliferation of BMMs in a FoxO-dependent manner. Similar antiproliferative actions of Sirt1/FoxO signaling have been described in different cell types (24, 38). Nevertheless, it remains possible that targets other than FoxOs contribute to the suppressive effects of Sirt1 on osteoclastogenesis.
The results of the present work indicate that RANKL increases the levels of proteins of the electron transport chain and ATP production in BMM, in line with earlier reports (6, 7). Optimal function of the electron transport chain is critical for osteoclast formation as evidenced by the lower osteoclast number in mice with disrupted complex I in BMM (8). In addition, we found that the inhibitory actions of Sirt1/FoxO signaling on osteoclastogenesis are associated with increased expression of HO-1 and decreased levels of ATP. FoxO1 in the liver attenuates mitochondria activity and ATP production via stimulation of HO-1 (34); and HO-1 in macrophages (expressing LysM-Cre) inhibits mitochondrial metabolism and ATP production (41). HO-1 is the rate-limiting first step of heme degradation leading to generation of biliverdin, CO, and free iron. Heme is an essential cofactor of mitochondrial oxidation, as it facilitates electron transport and ensures the function of complexes III and IV (42, 43). In contrast to Sirt1/FoxO, RANKL decreases the expression of HO-1, as previously seen by others (44). These findings suggest that HO-1 mediates the actions of RANKL and Sirt/FoxOs on mitochondrial activity in osteoclast (Figure 7). Importantly, studies in mice with global deletion of HO-1 indicate that this enzyme decreases the number of osteoclasts and bone resorption (44), strengthening the contention that HO-1 contributes to the inhibitory effects of Sirt1/FoxOs on osteoclastogenesis.
RANKL increases ROS levels in osteoclast progenitors which in turn stimulates osteoclast differentiation and survival. We have shown that the increase in ROS by RANKL is due, at least in part, to the inhibition of FoxO-mediated transcription of catalase (3). Furthermore, H2O2 generated in the osteoclast mitochondria is critical for bone resorption and skeletal homeostasis. Accordingly, stimulation of FoxO activity in osteoclasts attenuates H2O2 production by RANKL and inhibits bone resorption. The studies of the present report indicate that, in addition to stimulating catalase, Sirt1/FoxO activity might decrease H2O2 levels via HO-1-mediated repression of mitochondrial activity (Figure 7). Support for this contention is provided by the findings that HO-1 decreases H2O2 in BMMs (41, 44).
In recent years, results obtained from murine models of loss or gain of Sirt1 function or pharmacologic studies with natural or synthetic Sirt1 activating compounds have provided compelling evidence that Sirt1 delays aging-related diseases and can promote longevity. The findings of the present report support the premise that common mechanism of aging affect different tissues and that single molecules can counteract these mechanisms and extend health and lifespan (2, 45). We have previously shown that administration of SRT2104 prolongs lifespan of male mice and preserves bone and muscle mass (15). SRT2104 also provides neuroprotection in a murine model of Huntington's disease (46). Furthermore, Artsi et al have shown that SRT3025 attenuates the loss of bone mass caused by estrogen deficiency in female mice (12). In view of the evidence that the loss of bone mass with sex steroid deficiency is due to increased osteoclast generation and bone resorption (47), it is likely that the antiosteoclastogenic effects described in the present report contribute to the beneficial actions of SRT3025 on the skeleton. Notably, SRT3025 also provides protection against atherosclerosis, hyperglycemia, and neurodegeneration (48–50).
In conclusion, the findings presented here, along with our earlier work showing that FoxOs mediate the actions of Sirt1 in osteoblast progenitors, indicate that Sirt1 increases osteoblastogenesis and decreases osteoclastogenesis by deacetylating FoxOs in the respective progenitors. In addition, our findings provide a mechanistic explanation for the antiosteoporotic actions of Sirt1 stimulators. Sirt1 and/or NAD+ levels decline with age in bone cells (9, 51) in line with similar evidence in other tissues (52, 53). Therefore, a decrease in Sirt1 activity might contribute to the imbalance between bone formation and resorption and the consequent loss of bone mass that occurs with aging.
Acknowledgments
We thank A. Warren for technical assistance.
This work was supported by National Institutes of Health Grants R01 AR56679 (to M.A.), and P01 AG13918 (to S.C.M.); the Biomedical Laboratory Research and Development Service of the Veteran's Administration Office of Research and Development Grant I01 BX001405 (to S.C.M.); and the University of Arkansas for Medical Sciences Tobacco Funds and Translational Research Institute Grant 1UL1RR029884. R.d.C. was supported by the Intramural Research Program of the National Institute on Aging, of the National Institutes of Health.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Ac
- acetylated
- Ad-Cre
- adenovirus encoding Cre recombinase
- BMM
- bone marrow macrophage
- FoxO
- Forkhead box protein O
- HO-1
- hemeoxygenase-1
- Jnk
- c-Jun N-terminal kinase
- M-CSF
- macrophage colony-stimulating factor
- NAD+
- nicotinamide adenine dinucleotide oxidized
- NF-kB
- nuclear factor kappa-light-chain-enhancer of activated B cells
- NFATc1
- Nuclear factor of activated T-cells, cytoplasmic 1
- OXPHOS
- oxidative phosphorylation
- p
- phosphorylated
- p300
- E1A binding protein p300
- p38
- p38 mitogen-activated protein kinase
- p65
- nuclear factor NF-kappa-B p65 subunit
- RANKL
- receptor activator of nuclear factor kappa-B ligand
- ROS
- reactive oxygen species
- Sirt1
- Sirtuin1
- TRAP
- tartrate-resistant acid phosphatase
- WT
- wild type.
References
- 1. Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baur JA, Ungvari Z, Minor RK, Le Couteur DG, de Cabo R. Are sirtuins viable targets for improving healthspan and lifespan? Nat Rev Drug Discov. 2012;11:443–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bartell SM, Kim HN, Ambrogini E, et al. FoxO proteins restrain osteoclastogenesis and bone resorption by attenuating H2O2 accumulation. Nat Commun. 2014;5:3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brand MD. The sites and topology of mitochondrial superoxide production. Exp Gerontol. 2010;45:466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ishii KA, Fumoto T, Iwai K, et al. Coordination of PGC-1β and iron uptake in mitochondrial biogenesis and osteoclast activation. Nat Med. 2009;15:259–266. [DOI] [PubMed] [Google Scholar]
- 7. Nishikawa K, Iwamoto Y, Kobayashi Y, et al. DNA methyltransferase 3a regulates osteoclast differentiation by coupling to an S-adenosylmethionine-producing metabolic pathway. Nat Med. 2015;21:281–287. [DOI] [PubMed] [Google Scholar]
- 8. Jin Z, Wei W, Yang M, Du Y, Wan Y. Mitochondrial complex I activity suppresses inflammation and enhances bone resorption by shifting macrophage-osteoclast polarization. Cell Metab. 2014;20:483–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Edwards JR, Perrien DS, Fleming N, et al. Silent information regulator (Sir)T1 inhibits NF-κB signaling to maintain normal skeletal remodeling. J Bone Miner Res. 2013;28:960–969. [DOI] [PubMed] [Google Scholar]
- 10. Hah YS, Cheon YH, Lim HS, et al. Myeloid deletion of SIRT1 aggravates serum transfer arthritis in mice via nuclear factor-κB activation. PLoS One. 2014;9:e87733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Su JL, Yang CY, Zhao M, Kuo ML, Yen ML. Forkhead proteins are critical for bone morphogenetic protein-2 regulation and anti-tumor activity of resveratrol. J Biol Chem. 2007;282:19385–19398. [DOI] [PubMed] [Google Scholar]
- 12. Artsi H, Cohen-Kfir E, Gurt I, et al. The Sirtuin1 activator SRT3025 down-regulates sclerostin and rescues ovariectomy-induced bone loss and biomechanical deterioration in female mice. Endocrinology. 2014;155:3508–3515. [DOI] [PubMed] [Google Scholar]
- 13. Pearson KJ, Baur JA, Lewis KN, et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008;8:157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Momken I, Stevens L, Bergouignan A, et al. Resveratrol prevents the wasting disorders of mechanical unloading by acting as a physical exercise mimetic in the rat. FASEB J. 2011;25:3646–3660. [DOI] [PubMed] [Google Scholar]
- 15. Mercken EM, Mitchell SJ, Martin-Montalvo A, et al. SRT2104 extends survival of male mice on a standard diet and preserves bone and muscle mass. Aging Cell. 2014;13:787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boissy P, Andersen TL, Abdallah BM, Kassem M, Plesner T, Delaissé JM. Resveratrol inhibits myeloma cell growth, prevents osteoclast formation, and promotes osteoblast differentiation. Cancer Res. 2005;65:9943–9952. [DOI] [PubMed] [Google Scholar]
- 17. He X, Andersson G, Lindgren U, Li Y. Resveratrol prevents RANKL-induced osteoclast differentiation of murine osteoclast progenitor RAW 264.7 cells through inhibition of ROS production. Biochem Biophys Res Commun. 2010;401:356–362. [DOI] [PubMed] [Google Scholar]
- 18. Shakibaei M, Buhrmann C, Mobasheri A. Resveratrol-mediated SIRT-1 interactions with p300 modulate receptor activator of NF-κB ligand (RANKL) activation of NF-κB signaling and inhibit osteoclastogenesis in bone-derived cells. J Biol Chem. 2011;286:11492–11505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schug TT, Xu Q, Gao H, et al. Myeloid deletion of SIRT1 induces inflammatory signaling in response to environmental stress. Mol Cell Biol. 2010;30:4712–4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van der Horst A, Burgering BM. Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol. 2007;8:440–450. [DOI] [PubMed] [Google Scholar]
- 21. Sandri M, Sandri C, Gilbert A, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee D, Goldberg AL. SIRT1 protein, by blocking the activities of transcription factors FoxO1 and FoxO3, inhibits muscle atrophy and promotes muscle growth. J Biol Chem. 2013;288:30515–30526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hsu CP, Zhai P, Yamamoto T, et al. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation. 2010;122:2170–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brunet A, Sweeney LB, Sturgill JF, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. [DOI] [PubMed] [Google Scholar]
- 25. Daitoku H, Hatta M, Matsuzaki H, et al. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc Natl Acad Sci USA. 2004;101:10042–10047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Iyer S, Han L, Bartell SM, et al. Sirtuin1 (Sirt1) promotes cortical bone formation by preventing β (β)-catenin sequestration by FoxO transcription factors in osteoblast progenitors. J Biol Chem. 2014;289:24069–24078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods. 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 28. Onal M, Piemontese M, Xiong J, et al. Suppression of autophagy in osteocytes mimics skeletal aging. J Biol Chem. 2013;288:17432–17440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ambrogini E, Almeida M, Martin-Millan M, et al. FoxO-mediated defense against oxidative stress in osteoblasts is indispensable for skeletal homeostasis in mice. Cell Metab. 2010;11:136–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou J, Ye S, Fujiwara T, Manolagas SC, Zhao H. Steap4 plays a critical role in osteoclastogenesis in vitro by regulating cellular iron/reactive oxygen species (ROS) levels and cAMP response element-binding protein (CREB) activation. J Biol Chem. 2013;288:30064–30074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Daitoku H, Sakamaki J, Fukamizu A. Regulation of FoxO transcription factors by acetylation and protein-protein interactions. Biochim Biophys Acta. 2011;1813:1954–1960. [DOI] [PubMed] [Google Scholar]
- 32. Kitamura YI, Kitamura T, Kruse JP, et al. FoxO1 protects against pancreatic β cell failure through NeuroD and MafA induction. Cell Metab. 2005;2:153–163. [DOI] [PubMed] [Google Scholar]
- 33. Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, Salminen A. Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal. 2013;25:1939–1948. [DOI] [PubMed] [Google Scholar]
- 34. Cheng Z, Guo S, Copps K, et al. Foxo1 integrates insulin signaling with mitochondrial function in the liver. Nat Med. 2009;15:1307–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Matsuzaki H, Daitoku H, Hatta M, Aoyama H, Yoshimochi K, Fukamizu A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc Natl Acad Sci USA. 2005;102:11278–11283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Qiang L, Banks AS, Accili D. Uncoupling of acetylation from phosphorylation regulates FoxO1 function independent of its subcellular localization. J Biol Chem. 2010;285:27396–27401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim JH, Kim K, Youn BU, et al. RANKL induces NFATc1 acetylation and stability via histone acetyltransferases during osteoclast differentiation. Biochem J. 2011;436:253–262. [DOI] [PubMed] [Google Scholar]
- 38. van der Horst A, Tertoolen LG, de Vries-Smits LM, Frye RA, Medema RH, Burgering BM. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1). J Biol Chem. 2004;279:28873–28879. [DOI] [PubMed] [Google Scholar]
- 39. Wang Y, Dong G, Jeon HH, et al. FOXO1 mediates RANKL-induced osteoclast formation and activity. J Immunol. 2015;194:2878–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yoshizaki T, Schenk S, Imamura T, et al. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am J Physiol Endocrinol Metab. 2010;298:E419–E428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jais A, Einwallner E, Sharif O, et al. Heme oxygenase-1 drives metaflammation and insulin resistance in mouse and man. Cell. 2014;158:25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Taillé C, El-Benna J, Lanone S, et al. Induction of heme oxygenase-1 inhibits NAD(P)H oxidase activity by down-regulating cytochrome b558 expression via the reduction of heme availability. J Biol Chem. 2004;279:28681–28688. [DOI] [PubMed] [Google Scholar]
- 43. Converso DP, Taillé C, Carreras MC, Jaitovich A, Poderoso JJ, Boczkowski J. HO-1 is located in liver mitochondria and modulates mitochondrial heme content and metabolism. FASEB J. 2006;20:1236–1238. [DOI] [PubMed] [Google Scholar]
- 44. Ke K, Safder MA, Sul OJ, et al. Hemeoxygenase-1 maintains bone mass via attenuating a redox imbalance in osteoclast. Mol Cell Endocrinol. 2015;409:11–20. [DOI] [PubMed] [Google Scholar]
- 45. Hubbard BP, Sinclair DA. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol Sci. 2014;35:146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jiang M, Zheng J, Peng Q, et al. Sirtuin 1 activator SRT2104 protects Huntington's disease mice. Ann Clin Transl Neurol. 2014;1:1047–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Manolagas SC, O'Brien CA, Almeida M. The role of estrogen and androgen receptors in bone health and disease. Nat Rev Endocrinol. 2013;9:699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Miranda MX, van Tits LJ, Lohmann C, et al. The Sirt1 activator SRT3025 provides atheroprotection in Apoe−/− mice by reducing hepatic Pcsk9 secretion and enhancing Ldlr expression. Eur Heart J. 2015;36:51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gilbert RE, Thai K, Advani SL, et al. SIRT1 activation ameliorates hyperglycaemia by inducing a torpor-like state in an obese mouse model of type 2 diabetes. Diabetologia. 2015;58:819–827. [DOI] [PubMed] [Google Scholar]
- 50. Gräff J, Kahn M, Samiei A, et al. A dietary regimen of caloric restriction or pharmacological activation of SIRT1 to delay the onset of neurodegeneration. J Neurosci. 2013;33:8951–8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li Y, He X, Li Y, et al. Nicotinamide phosphoribosyltransferase (Nampt) affects the lineage fate determination of mesenchymal stem cells: a possible cause for reduced osteogenesis and increased adipogenesis in older individuals. J Bone Miner Res. 2011;26:2656–2664. [DOI] [PubMed] [Google Scholar]
- 52. Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mouchiroud L, Houtkooper RH, Moullan N, et al. The NAD(+)/Sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 2013;154:430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]