

Abstract
An inflammatory response is instrumental in the physiological process of parturition but the upstream signals initiating inflammation are undefined. Because endogenous ligands for Toll-like receptor 4 (TLR4) are released in late gestation, we hypothesized that on-time labor requires TLR4 signaling, to trigger a cytokine and leukocyte response and accelerate the parturition cascade. In pregnant TLR4-deficient (Tlr4−/−) mice, average gestation length was extended by 13 hours and increased perinatal mortality was seen compared with wild-type controls. Quantification of cytokine and uterine activation gene expression showed that late gestation induction of Il1b, Il6, Il12b, and Tnf expression seen in control placenta and fetal membranes was disrupted in Tlr4−/− mice, and accompanied by a transient delay in expression of uterine activation genes, including prostaglandin F receptor, oxytocin receptor, and connexin-43. Leukocyte populations were altered before birth in TLR4-deficient females, with fewer neutrophils and macrophages in the placenta, and fewer dendritic cells and more regulatory T cells in the myometrium. Administration of TLR4 ligand lipopolysaccharide to pregnant wild-type mice induced cytokine expression and fetal loss, whereas Tlr4−/− pregnancies were protected. The small molecule TLR4 antagonist (+)-naloxone increased mean duration of gestation by 16 hours in wild-type mice. Collectively, these data demonstrate that TLR4 is a key upstream regulator of the inflammatory response acting to drive uterine activation and control the timing of labor. Because causal pathways for term and preterm labor converge with TLR4, interventions to manipulate TLR4 signaling may have therapeutic utility for women at risk of preterm labor, or in postterm pregnancy.
Parturition, the process of birth, involves events characteristic of inflammation (1), with progressively accelerating proinflammatory cytokine synthesis and leukocyte influx into the fetal and maternal reproductive tissues (2, 3) culminating in uterine contractions and expulsion of the fetus. Precocious inflammation is a key feature of preterm labor (PTL), defined as birth at less than 37 weeks of gestation (4). PTL affects 5%–13% of pregnancies worldwide (4, 5) and accounts for 28% of all neonatal deaths globally (6) and major health consequences for surviving children (7). Devising interventions to prevent preterm birth requires understanding of the physiological pathway underpinning normal on-time birth.
In late gestation there is an increase in maternal systemic and local inflammatory activity (1), with evidence of altered activation phenotypes in peripheral blood leukocytes (8–10) and elevated expression of Il1b, Il6, and Tnf and a range of chemokines within the uterus, cervix, placenta, and fetal membranes (2, 3). Studies in rodent and primate models indicate that inflammatory cytokines are elevated before active labor commences, indicating a shift to an inflammatory state begins in the preparation phase of parturition (11, 12) and accelerates during active labor and into the early postpartum phase (13–15). In mice, this shift is accompanied by withdrawal of the protective antiinflammatory actions of progesterone (16).
Cytokine and chemokine expression causes recruitment of leukocytes, predominantly macrophages and neutrophils, into the fetal membranes, maternal decidua, myometrium and cervix (2). Leukocytes accumulating in gestational tissues release an array of proinflammatory factors to amplify and accelerate feed-forward mechanisms. Nuclear factor kappa B (NFκB)-driven expression of proinflammatory cytokines, including Il1b, Il6, and Tnf in leukocytes and other cell lineages (17), promotes expression of uterine activation genes (UAGs) that include prostaglandin synthase genes and prostaglandin receptors, activating uterine contractility to expel the fetus (18, 19). Additional UAGs, notably gap junction protein α1 (Gja1) (commonly known as connexin-43) and the oxytocin receptor (Oxtr), advance myometrial contractile activity (20, 21). Inhibition of UAGs by pharmacological agents in women or using genetically manipulated mice causes parturition to be delayed (22, 23). Prominent among these leukocytes are macrophages and neutrophils, which produce a range of matrix metalloproteinases presumed to assist in cervical ripening and rupture of the fetal membranes in the active phase of delivery (24, 25), and implicated also in postpartum tissue remodeling and repair (13, 14, 26).
Inflammation induced by uterine infection, or noninfectious stress or injury, causes aberrant expression of UAGs resulting in PTL (1, 27). Defining the extent of intersection between the pathways mediating preterm and term labor and how their regulatory mechanisms converge is a priority (28). The Toll-like receptors (TLRs) are strong candidates for activating inflammation in both term and PTL (1). Study of infection-initiated PTL reveals important roles for TLR2, which binds bacterial lipoprotein, and TLR4, which binds bacterial lipopolysaccharide (LPS) (29, 30). Elevated expression of both TLR2 and TLR4 is evident in chorioamniotic membranes in PTL (31). In C3H/HeJ mice, a spontaneous TLR4 mutation protects against PTL induced by Escherichia coli or LPS to model bacterial infection (32, 33). Not all PTL involves infection (5), and TLR4 might reasonably contribute to mediating effects of sterile stressors such as stretch or ischemic injury, because tissue damage causes release of endogenous TLR4 ligands called damage-associated molecular patterns (DAMPs) (34).
TLR4 is well positioned to prime the inflammatory cascade underpinning normal term parturition, because it is expressed in a wide range of gestational tissues and up-regulated in late gestation in the lower uterine segment in mice (33) and in women commencing labor (31). TLR4 ligation is upstream of signaling molecules, including myeloid differentiation factor 88 (MyD88), which triggers NFκB, and proinflammatory cytokine and prostaglandin production (32, 35). TLR4 signaling regulates recruitment and/or activation of leukocytes, including dendritic cells (36), neutrophils (37), and T cells (38). TLR4 activation might reasonably occur through release of endogenous DAMPs from maternal tissues under progressively increasing physical stress as gestation advances, or DAMPs such as lung surfactants released from the maturing fetus (28, 39–41).
Thus we hypothesize that TLR4 and its associated MyD88-dependent signaling are a point of convergence for the physiological inflammation of normal on-time labor, and the precocious inflammation of PTL. In the current study we have used mice with Tlr4 or Myd88 null mutation and a novel TLR4 pharmacological manipulation to investigate the role of TLR4 signaling in on-time labor in the absence of infection, and in a model of infection-associated PTL, using the bacterial mimetic LPS. Our experiments show that TLR4 advances the proinflammatory response and UAG expression required to achieve on-time parturition and labor, through inflammatory pathways that are comparable with those seen in PTL induced by infection-associated TLR4 ligation.
Materials and Methods
Mice and mating protocols
BALB/c and C57Bl/6 (B6) mice were obtained from Laboratory Animal Services, University of Adelaide or the Animal Resource Centre. Mice with null mutations in the Tlr4 gene (Tlr4−/− mice) and Myd88 gene (Myd88−/− mice) back-crossed onto BALB/c for more than 10 generations were sourced from Professor Akira (Osaka University, Osaka, Japan) and supplied by Professor Paul Foster (University of Newcastle, New South Wales, Australia). Mice were housed and maintained in the specific pathogen-free University of Adelaide Medical School Animal House with a 12-hour light, 12-hour dark cycle. Food and water were provided ad libitum, and animals were used according to the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes with approval from the University of Adelaide Animal Ethics Committee.
One to 3 virgin female mice of 8–14 weeks of age were housed with a proven fertile male of the same genotype and checked daily between 8 and 10 am for vaginal plugs, as evidence of mating. The morning of vaginal plug detection was designated gestational day (gd) 0.5. Mated females were removed from the male and housed individually.
Treatments and pregnancy outcome analysis
For analysis of gestation length pregnant Tlr4−/− and BALB/c mice were monitored for the time of parturition and the number of pups born. The viability of pups was recorded and pups were weighed at 12–24 hours after delivery and at 7 days postpartum, to determine viable pups born and percentage postnatal mortality (proportion of pups lost between 12–24 h and 7 d of age). At 21 days postpartum, the number, sex, and weight of surviving pups were recorded. For quantitative PCR (qPCR), pregnant mice were killed by cervical dislocation on gd 16.5, 17.5, 18.5, or 19.5, and the uterine myometrium (from between implantation sites) and entire uterine decidua (at placental attachment site), placenta, fetal membranes, and fetal head were dissected and snap frozen in liquid N2, then stored at −80°C. For flow cytometry, pregnant mice were killed at gd 18.5, and the myometrium, decidua, placenta, fetal membrane, and paraaortic lymph node (PALN) were analyzed. All tissues from pregnant mice were recovered before onset of labor, and tissues from 2 implantation sites per dam were pooled.
In some experiments pregnant Tlr4−/− and BALB/c mice were administered 10 μg of LPS (Salmonella typhimurium [Sigma-Aldrich], in 200-μL PBS + 0.1% BSA, ip) or vehicle alone, at 11 am on gd 16.5, and dams were killed by cervical dislocation at 10 am to 12 pm on gd 18.5. Total implantation sites were counted and classified as viable (presence of healthy fetus and placenta), dead (fetus anemic, malformed, or severely growth retarded), resorbed (small necrotic mass), or delivered (fresh implantation scar with absent fetus and placenta, or placenta but no fetus). Additional pregnant Tlr4−/− and BALB/c mice were given LPS or vehicle on gd 16.5 were killed 4 hours later, and the uterus (combined decidua and myometrium), placenta, and fetal head were recovered for analysis of cytokine expression.
In some experiments, B6 mice were administered TLR4 antagonist (+)-naloxone (60 mg/kg in PBS, ip) (42) or vehicle control at gd 16.5, 17.0, 17.5, and 18.0, then monitored for the time of parturition and the number of pups born.
Quantitative PCR
Tissues were homogenized using ceramic beads (Mo Bio) in TRIzol (Ambion RNA). RNA was deoxyribonuclease-treated using Ambion DNA-free kit according to the manufacturer's instructions. First-strand cDNA was reverse transcribed from 1 μg of the total extracted RNA using Superscript II ribonuclease H reverse transcriptase kit (90 min at 43°C; Invitrogen). Primer pairs specific for published cDNA sequences were designed using Primer Express software (Applied Biosystems). Each reaction contained 2 μL of cDNA (10 ng/μL) and 18 μL of master mix consisting of Power SYBR Green PCR Master Mix (Life Technologies), 0.5μM–1μM 5′ and 3′ primers (listed in Supplemental Table 1), and ribonuclease-free water. qPCR was performed in a C1000 Touch Thermal Cycler (Bio-Rad) for 2 minutes at 50°C, 10 minutes at 95°C, followed by 40 cycles of 20 seconds at 95°C, and 1 minute at 60°C. Data were normalized to β-actin mRNA expression and expressed as delta delta cycle threshold using the formula mRNA level = Log2 − (CtBactin − Cttarget gene).
Progesterone assay
Serum prepared from retro-orbital blood collected from pregnant Tlr4−/− and BALB/c mice on gd 16.5, 17.5, and 18.5 was stored at −80°C until assay using an ELISA kit (ALPCO Diagnostics) according to the manufacturer's instructions.
Flow cytometry
Decidua, placenta, and fetal membranes from all implantation sites were pooled for each pregnant female. Tissues were placed in RPMI 1640 containing 2% FCS (RPMI-fetal calf serum [FCS]; Life Technologies) plus 1 μg/mL collagenase from Clostridium histolyticum Type IA (Sigma-Aldrich) and 4 U/mL of deoxyribonuclease I (Sigma-Aldrich), finely macerated with scissors and incubated at 37°C with agitation for 1 hour. PALN were crushed between 2 frosted ends of glass slides in RPMI-FCS. After removal of a fraction for later count bead analysis, cell suspensions were incubated with Fc block (clone 2.4G2, 10 μg/mL, 10 min on ice; Becton Dickinson). Three aliquots were analyzed using antibody panels 1–3 (Supplemental Table 2) in PBS-BSA buffer (0.1% BSA and 0.05% sodium azide in PBS). For panels 1 and 2, cells were then incubated with biotinylated streptavidin-V500 before addition of 4′,6-diamidino-2-phenylindole solution (4 μg/mL; Sigma-Aldrich) and analysis by flow cytometry. For panel 3, cells were labeled with fixable viability dye V450 (Becton Dickinson), washed, and then fixed, permeabilized, and labeled with anti-forkhead box P3 (FOXP3) antibody using the FOXP3 staining kit (eBioscience) according to the manufacturer's instructions. Samples for count bead analysis were labeled with antibodies against cluster of differentiation (CD) 11C, the macrophage glycoprotein F4/80, CD4, and CD45 (APC; eBioscience) before addition of CountBright Absolute Counting Beads (Molecular Probes, Invitrogen) and 4′,6-diamidino-2-phenylindole (see Table 1). The ratio between beads and labeled cells in each sample was used to calculate the total number of cells in the whole myometrium and PALN, and the total number of cells per implantation site in fetal membrane, placenta and decidua.
Table 1.
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| CD11B | M1/70 | eBioscience | Rat | 1:100 | |
| F4/80 | BM8 | eBioscience | Rat | 1:100 | |
| Ly6G | A8 | BD | Rat | 1:100 | |
| MHCII | M5/114.15.2 | eBioscience | Rat | 1:100 | |
| CD11C | N418 | eBioscience | Rat | 1:100 | |
| CD80 | 6-10A1 | eBioscience | Rat | 1:100 | |
| CD86 | GL1 | eBioscience | Rat | 1:100 | |
| CD3 | 17A2 | eBioscience | Rat | 1:100 | |
| CD4 | RM4-5 | eBioscience | Rat | 1:100 | |
| CD25 | PC61.5 | eBioscience | Rat | 1:100 | |
| FOXP3 | FKJ-16s | eBioscience | Rat | 1:100 | |
| IL-17AR | PAJ-17R | eBioscience | Rat | 1:100 |
Statistical analysis
All statistical analysis was conducted using SPSS for Windows, version 20.0 software (SPSS, Inc). Data were tested for normality using a Shapiro-Wilk test. ANOVA and post hoc Sidak t tests were used when data were normally distributed. Kruskal-Wallis and Mann-Whitney U test were used when data were not normally distributed. Categorical data were compared by χ2 analysis. Differences between groups were considered significant when P < .05.
Results
TLR4 deficiency delays term labor and causes perinatal loss
To investigate whether disruption of TLR4 signaling impacts pregnancy outcome, Tlr4−/− and control BALB/c females were mated with genotyped-matched males. Gestation length was extended in the Tlr4−/− females, with mean time of birth 13.2 hours later than in control mice (mean ± SEM, 20.05 ± 0.09 d in Tlr4−/− mice vs 19.50 ± 0.08 d in wild-type mice; P < .001) (Figure 1A). The mean number of viable pups born was reduced by 26% in Tlr4−/− mice compared with control (P = .017) (Figure 1B). Later delivery in Tlr4−/− mice was associated in a 7.8% increase in pup weight at 12–24 hours after birth compared with controls (P = .002) (Figure 1C). Tlr4−/− pups exhibited a 3.1-fold higher rate of postnatal death (P < .001) (Figure 1D), resulting in a 35% reduction in litter size by weaning (P = .018) (Figure 1E). These data show that TLR4 is essential for normal on-time birth in mice, and in the absence of TLR4, litter size at birth and pup viability in the postnatal period are reduced.
Figure 1.
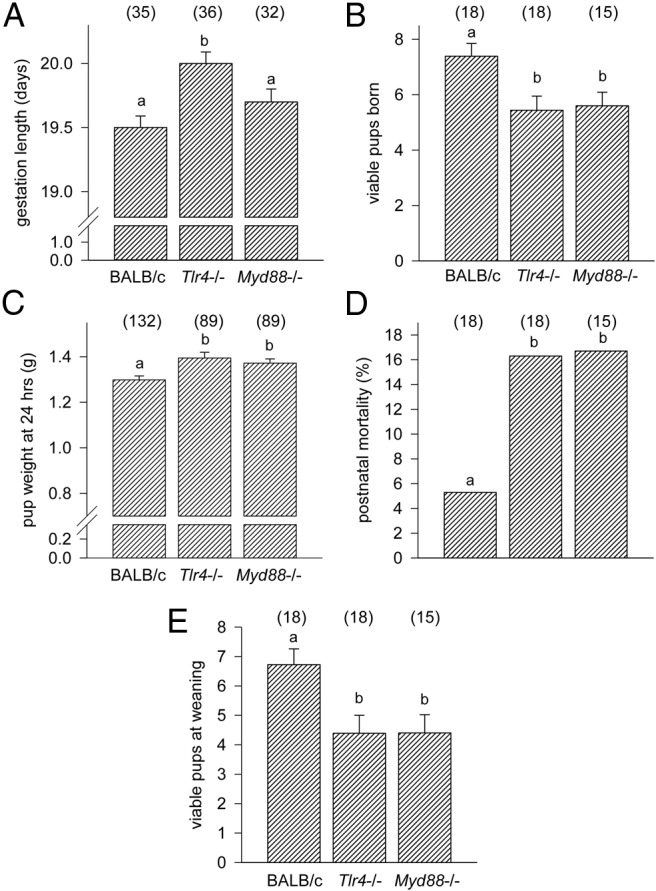
Term labor is delayed in TLR4-deficient, but not MyD88-deficient, mice. Tlr4−/−, MyD88−/−, or wild-type control mice were mated to males of the same genotype. Detection of a vaginal plug was designated gd 0.5, and females were monitored from gd 18.0 mice to record the length of gestation (A) and the number of viable pups born (B). Pups were weighed at 12–24 hours after birth (C). Pup survival was recorded and the percentage of pups lost in the first 7 days (D), as well as viable pups per litter at weaning (E) calculated. Data shown as mean ± SEM (A–C and E) were analyzed by ANOVA and Sidak t test to determine effect of genotype. Categorical data (D) was compared by χ2 analysis. The number of dams or pups per group is given in parentheses. a and b, different letters indicate significant difference between genotypes, P < .05.
Delay of term labor with TLR4 deficiency does not depend on MyD88 adaptor
To determine the requirement for MyD88 adaptor protein in TLR4 regulation of gestation length, perinatal outcomes were also measured in mice deficient in MyD88, a key adaptor molecule immediately downstream of TLR4 ligation in the TLR4 signaling cascade. In Myd88−/− females mated with Myd88−/− males, no increase in gestation length was seen (Figure 1A) compared with wild-type controls. However, like Tlr4−/− mice, Myd88−/− mice exhibited a 24% decrease in litter size (P = .044) (Figure 1B), a 6.1% increase in pup weight (P = .014) (Figure 1C), a 3.2-fold higher rate of postnatal death (P < .002) (Figure 1D), and an overall 34% reduction in litter size at weaning (P = .027) (Figure 1E) compared with controls. This result indicates that MyD88 is not essential for the downstream signaling through which TLR4 regulates the timing of birth, but that MyD88-mediated TLR signaling exerts effects on litter size and pup viability that are additional to any consequences of extended gestation.
TLR4 deficiency inhibits inflammatory cytokine gene expression in fetal tissues before term labor
To examine the effect of TLR4 deficiency on expression of inflammatory cytokines induced in the parturition cascade in mice, Tlr4−/− and wild-type control mice were mated to males of the same genotype, then placenta, fetal membranes, and fetal head were recovered at time points over the course of late gestation on gd 16.5, 17.5, 18.5, or 19.5 for analysis of Il1b, Il6, Il12, Tnfa, and Il10 expression by qPCR. In the placenta, proinflammatory Il1b, Il6, Tnf, and antiinflammatory Il10 mRNA levels all increased 1.6- to 4.3-fold from gd 16.5 to 19.5 (all P < .05) (Figure 2, A–E). Expression was altered in placenta from Tlr4−/− mice, with mean expression 50% lower for Tnf at gd 16.5 (Figure 2D), 69% lower Il10 at gd 18.5 (Figure 2E), 31% lower Il1b at gd 18.5 (Figure 2A), and 46% lower Il6 at gd 19.5 (Figure 2B) compared with placenta from control mice (all P < .05). Although Il12b did not increase in either genotype, expression was reduced in Tlr4−/− mice at gd 16.5 and 18.5 (both P < .05) (Figure 2C).
Figure 2.

Late gestation induction of proinflammatory cytokines in placental and fetal tissue is retarded in TLR4-deficient mice. Wild-type control (white bars) or Tlr4−/− (hatched bars) mice were mated to males of the same genotype. Detection of a vaginal plug was designated gd 0.5, and females were killed at gd 16.5, 17.5, 18.5, or 19.5 when placenta, fetal membranes, and fetal head were recovered. Relative expression of Il1b (A, F, K), Il6 (B, G, L), Il12b (C, H, M), Tnf (D, I, N), and Il10 (E, J, O) mRNAs were determined in each tissue by qPCR and normalized to Actb. Data are shown as mean ± SEM relative gene expression in tissue pooled from 2 implantation sites, with n = 6–10 dams/group. The effect of genotype and gestation time was analyzed by Kruskal-Wallis and Mann-Whitney U test. §, significant difference compared with wild-type at equivalent time point, P < .05; *, significant difference compared with gd 16.5 for same genotype, P < .05.
In fetal membranes from control mice, expression of Il1b and Il6 was low until both increased dramatically on gd 19.5 (P < .05) (Figure 2, F and G) and a trend towards similar increases in Tnf and Il10 was evident (P < .1) (Figure 2, I and J). However, induction of all 4 genes failed to occur in Tlr4−/− mice, with expression at gd 19.5 reduced by 60%–85% compared with controls (P < .05), and no change relative to gd 16.5 (Figure 2, F–J). In control fetal head, induction of Il1b, Il6, Il12b, Tnf, and Il10 peaked from gd 16.5 to 18.5 and then declined on gd 19.5. In each case, cytokine expression in control tissues at gd 18.5 was higher than in Tlr4−/− mice (all P < .05) (Figure 2, K–O).
TLR4 deficiency does not disrupt inflammatory cytokine gene expression in maternal uterus before term labor
To examine the effect of TLR4 deficiency on inflammatory cytokine expression in the maternal compartment of the gestational tissues, uterine decidua and uterine myometrium were also analyzed by qPCR. In decidua of wild-type control mice, there was an increase in Il1b and Il10 expression at gd 19.5 compared with gd 16.5 (P < .05) and a trend towards increased Il6 and Il12b (P < .1) (Figure 3, A–E). Il10 expression was 60% less in decidua from Tlr4−/− mice at gd 19.5 (P = .009) (Figure 3E). No significant differences in proinflammatory cytokine expression were detected in decidua of Tlr4−/− mice, despite a trend towards lower Il1b and Il12b in late gestation compared with wild-type mice (P < .1) (Figure 3, A and C).
Figure 3.

Late gestation induction of proinflammatory cytokines in uterine decidua and uterine myometrium is retarded in TLR4-deficient mice. Wild-type control (white bars) or Tlr4−/− (hatched bars) mice were mated to males of the same genotype. Detection of a vaginal plug was designated gd 0.5, and females were killed at gd 16.5, 17.5, 18.5, or 19.5 when decidua and myometrium were recovered. Relative expression of Il1b (A, F), Il6 (B, G), Il12b (C, H), Tnf (D, I), and Il10 (E, J) mRNAs were determined in each tissue by qPCR and normalized to Actb. Data are shown as mean ± SEM relative gene expression in tissue pooled from 2 implantation sites for decidua, and total uterine myometrium, with n = 6–10 dams/group. The effect of genotype and gestation time was analyzed by Kruskal-Wallis and Mann-Whitney U test. *, significant difference compared with gd 16.5 for same genotype, P < .05; §, significant difference compared with wild type at equivalent time point, P < .05.
In the myometrium of wild-type control mice, Il1b and Il6 mRNA increased from gd 16.5 to 19.5, but no differences were evident between control and Tlr4−/− females (Figure 3, F and G). Il12b, Tnf, and Il10 were not differentially expressed in the myometrium across the later stages of gestation, irrespective of genotype (Figure 3, H and I).
TLR4 deficiency transiently delays expression of UAGs before term labor
To examine the effect of TLR4 deficiency on the uterine activation cascade, decidua and myometrium were analyzed in control wild-type and Tlr4−/− mice by qPCR for expression of UAGs. In decidua of control mice, Ptgfr mRNA encoding PGF2α receptor (PTGFR) progressively increased over late gestation, peaking at gd 19.5. Mean expression in Tlr4−/− mice was 71% and 39% less at gd 17.5 and 18.5 (P < .05) (Figure 4A), before comparable expression at gd 19.5, suggesting delayed induction. In the myometrium Ptgfr surged at gd 19.5 in both wild-type and Tlr4−/− mice, with a trend to reduced expression compared with controls (P = .096) (Figure 4E).
Figure 4.
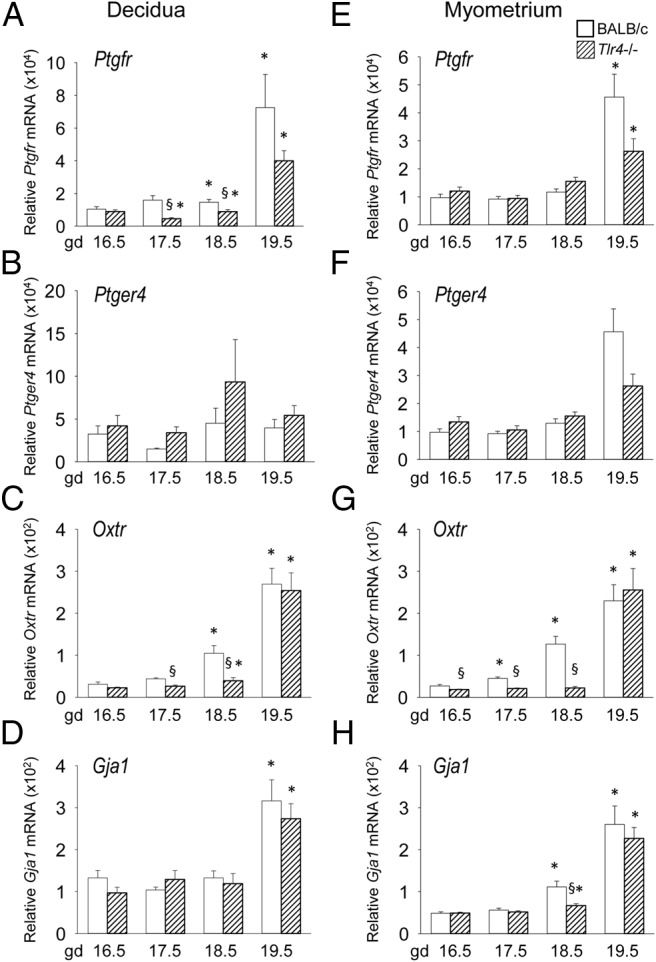
Late gestation induction of UAGs in uterine decidua and uterine myometrium is retarded in TLR4-deficient mice. Wild-type control (white bars) or Tlr4−/− (hatched bars) mice were mated to males of the same genotype. Detection of a vaginal plug was designated gd 0.5, and females were killed at gd 16.5, 17.5, 18.5, or 19.5 when decidua and myometrium were recovered. Relative expression of Ptgfr (A, E), Ptger4 (B, F), Oxtr (C, G), and Gja1 (D, H) mRNAs were determined in each tissue by qPCR and normalized to Actb. Data are shown as mean ± SEM relative gene expression in tissue pooled from 2 implantation sites for decidua, and total uterine myometrium, with n = 6–10 dams/group. The effect of genotype and gestation time was analyzed by Kruskal-Wallis and Mann-Whitney U test. *, significant difference compared with gd 16.5 for same genotype, P < .05; §, significant difference compared with wild type at equivalent time point, P < .05.
Expression of Oxtr progressively increased in both the decidua and myometrium, peaking at gd 19.5 (P < .05) (Figure 4C). Although Oxtr reached comparable levels in the decidua and myometrium of control and Tlr4−/− mice at gd 19.5, it was reduced by 40%–83% in both tissues in Tlr4−/− mice at gd 17.5 and 18.5, indicating delayed induction (P < .05) (Figure 4, C and D). Myometrial Gja1 was reduced by 40% in Tlr4−/− mice at day 18.5 (P = .011), whereas decidual Gja1 was not different between control and Tlr4−/− mice (Figure 4, D and H).
Ptger4 encoding the PGE receptor (subtype EP4) was not differentially expressed between control and Tlr4−/− mice (Figure 4, B and F). In wild-type mice, the expected increase in uterine prostaglandin synthase Ptgs1 (Cox1) expression was seen in late gestation, whereas Ptgs2 (Cox2) remained stable until a decline on gd 19.5. Ptgs1 was not differentially expressed between the 2 genotypes, whereas Ptgs2 expression was comparable at time points other than at gd 16.5, when expression was 32% higher in Tlr4−/− mice (Supplemental Figure 1).
TLR4 deficiency reduces leukocyte recruitment and activation before term labor
To determine whether leukocyte populations were affected by TLR4 deficiency, Tlr4−/− and wild-type control mice were mated to males of the same genotype and on gd 18.5, the decidua and myometrium were recovered, together with placenta and fetal membranes and PALN, for analysis of leukocyte populations by flow cytometry. The percentage of total viable cells comprised by F4/80+CD11B+ macrophages in the fetal membranes and placenta of Tlr4−/− mice was reduced by 46% and 53%, respectively (P < .005) (Figure 5A). The placental macrophages also had a less activated phenotype, with 18% fewer macrophages expressing major histocompatibility antigen II (MHCII) in placenta of Tlr4−/− mice (P < .001) (Figure 5E). Neutrophils comprised a 33% and 39% smaller proportion of total viable cells in the placenta and myometrium, respectively, of Tlr4−/− mice compared with controls (both P < .04) (Figure 5B).
Figure 5.
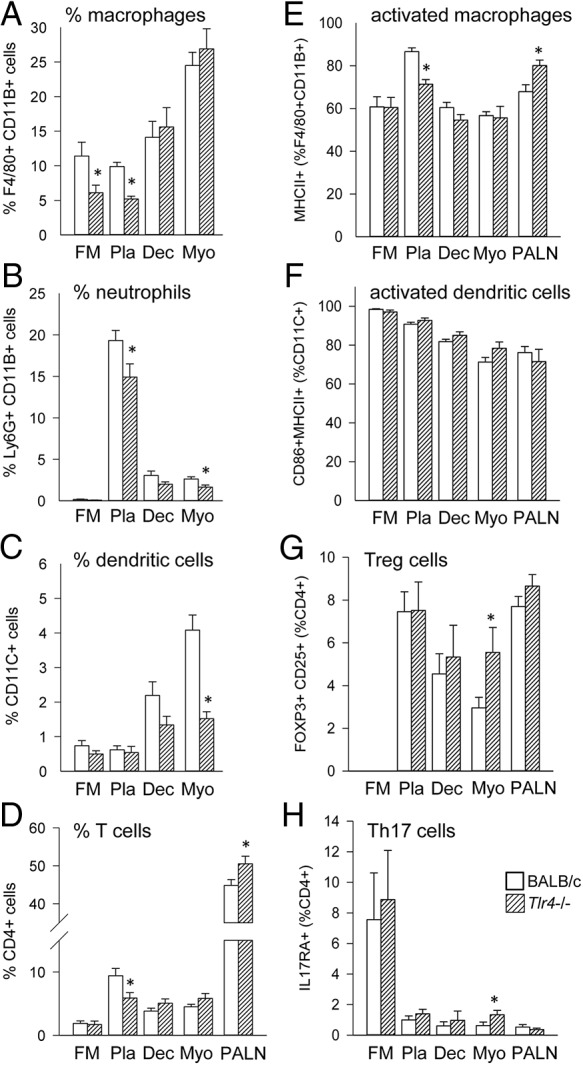
Leukocytes in gestational tissues are fewer in number before term labor in TLR4-deficient mice. Wild-type control (white bars) or Tlr4−/− (hatched bars) mice were mated to males of the same genotype. Detection of a vaginal plug was designated gd 0.5, and females were killed at gd 18.5 when fetal membrane (FM), placenta (Pla), decidua (Dec), uterus (Ut), and PALN were recovered, and F4/80+CD11B+ macrophages (A), Ly6G+CD11B+ neutrophils (B), CD11C+ dendritic cells (C), and CD4+ T cells (D) were analyzed by flow cytometry to determine percentage of viable cells. In addition, the expression of MHCII by F4/80+CD11B+ macrophages (activated macrophages) (E), expression of CD80, CD86, and MHCII by CD11C+ dendritic cells (activated dendritic cells) (F), expression of Foxp3 by CD4+ T cells (regulatory T cells) (G), and expression of IL-17RA by CD4+ T cells (Th17 cells) (H) was determined. Data are shown as mean ± SEM cell number per implantation sites for FM, Pla, and Dec, or for entire uterine myometrium and PALN, with n = 6–11 dams/group. The effect of genotype was analyzed by Kruskal-Wallis and Mann-Whitney U test. The absolute numbers of leukocytes in each tissue are shown in Supplemental Figure 1. *, significant difference compared with same tissue from wild type, P < .05.
CD11C+ dendritic cells were reduced by 63% in Tlr4−/− myometrium (P < .001), and a similar trend was seen in the decidua (P = .076) (Figure 5C). There was no change in the activation status of dendritic cells (DCs), as assessed by CD86 and MHCII expression, between wild-type and Tlr4−/− mice (Figure 5F).
CD3+CD4+ T cells were 38% fewer in the placenta of Tlr4−/− mice compared with controls (P = .025) (Figure 5D). The proportion of myometrial CD4+ T cells expressing the regulatory T-cell marker FOXP3 was increased from 2.9% in control mice to 5.6% in Tlr4−/− mice (P = .039) (Figure 5G). An increase in Th17 cells, from 0.6% to 1.3% of CD4+ T cells as indicated by interleukin 17 receptor A (IL-17RA) expression, occurred in the myometrium of Tlr4−/− mice (P = .044) (Figure 5H). When leukocytes were quantified as absolute cell number, similar patterns with reduced total neutrophils and dendritic cells were seen (Supplemental Figure 2).
TLR4 deficiency does not alter systemic progesterone before term labor
To investigate the impact of Tlr4 null mutation on circulating progesterone in late gestation, progesterone was measured in serum recovered on gd 16.5, 17.5, and 18.5 from wild-type control and Tlr4−/− females mated to males of the same genotype. The expected decline in plasma progesterone was seen in wild-type mice between gd 17.5 and 18.5, and although Tlr4−/− mice showed greater variability in plasma progesterone at gd 18.5, there was no significant difference compared with controls (Supplemental Figure 3).
TLR4-deficient mice are protected from LPS-induced PTL
To investigate the impact of Tlr4 null mutation on the responsiveness of mice to LPS-induced preterm birth, wild-type control and Tlr4−/− females mated to males of the same genotype were administered 10 μg of LPS or PBS (vehicle) via ip injection on gd 16.5, and pregnancy outcomes were measured on gd 18.5.
Administration of LPS induced fetal loss attributable to preterm birth and/or fetal death in utero of all fetuses in 57% (8/14) of control pregnancies, whereas Tlr4−/− mice were protected with none of 10 mice experiencing fetal loss (Figure 6A). In total, a 70% reduction in viable fetuses was seen in wild-type mice given LPS (P < .01) compared with PBS control, whereas there was no significant reduction in viable fetuses in Tlr4−/− mice administered the same LPS dose (Figure 6B). This difference was attributable to late gestation fetal loss as opposed to reduced implantations, as the total number of implantation sites per litter was not different between genotypes (Figure 6C).
Figure 6.
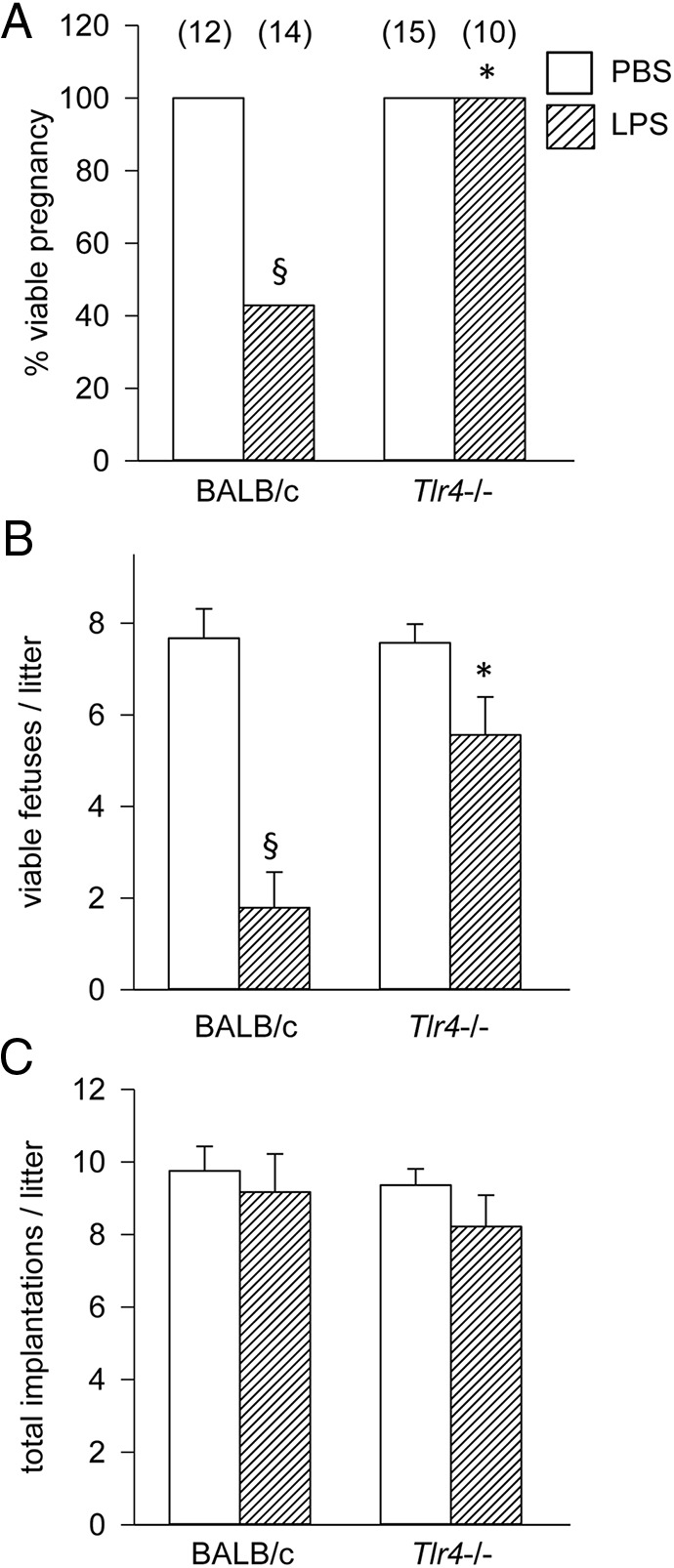
TLR4-deficient mice are protected from LPS-induced preterm loss. Tlr4−/− or control wild-type mice were mated to males of the same genotype. Detection of a vaginal plug was designated gd 0.5, and females were administered 10 μg of LPS ip on gd 16.5 (hatched bars) or PBS vehicle control (white bars), and pregnancy outcomes were measured at gd 18.5 to record viable pregnancy (at least 1 viable implantation site) (A), the number of viable fetuses per litter (B), and total implantation sites per litter. Categorical data (A) were compared by χ2 analysis, and data shown as mean ± SEM (B and C) were analyzed by ANOVA and Sidak t test to determine effect of genotype. The number of dams or pups per group is given in parentheses. §, significant difference compared with PBS treatment in same genotype, P < .05; *, significant difference compared with LPS treatment in wild-type controls, P < .05.
LPS-induced cytokine synthesis is attenuated in TLR4-deficient mice
To define the TLR4-dependent cytokine response to LPS in PTL and examine its relationship with the TLR4-dependent cytokine response in term labor, pregnant Tlr4−/− and control wild-type BALB/c mice were treated with LPS or PBS control on gd 16.5, and 4 hours later the fetus, uterus and placenta were harvested for mRNA expression analysis. Wild-type mice administered LPS showed strong 2.61- to 50.0-fold up-regulation of proinflammatory Il1a, Il6, Il12, and Tnf in the uterine myometrium (all P < .02) (Figure 7, A–D). Expression of antiinflammatory Il10 mRNA was 6.22-fold higher (P = .047) (Figure 7E). When Tlr4−/− mice were challenged with LPS, there was no change in cytokine synthesis relative to wild-type or Tlr4−/− mice given LPS, and cytokine synthesis was substantially lower than in LPS-treated wild-type mice (all P < .05) (Figure 7, A–E). Similar patterns of cytokine expression were seen in the placenta, where Il1a, Il6, Il12, Tnf, and Il10 mRNA were all induced in wild-type mice, but not in Tlr4−/− mice, by LPS treatment (Figure 7, F–J). Collectively, the TLR4-dependent mRNA cytokine profile induced by LPS in myometrial and placental tissue showed similar patterns to tissues of wild-type mice during on-time labor (Figures 2 and 3).
Figure 7.
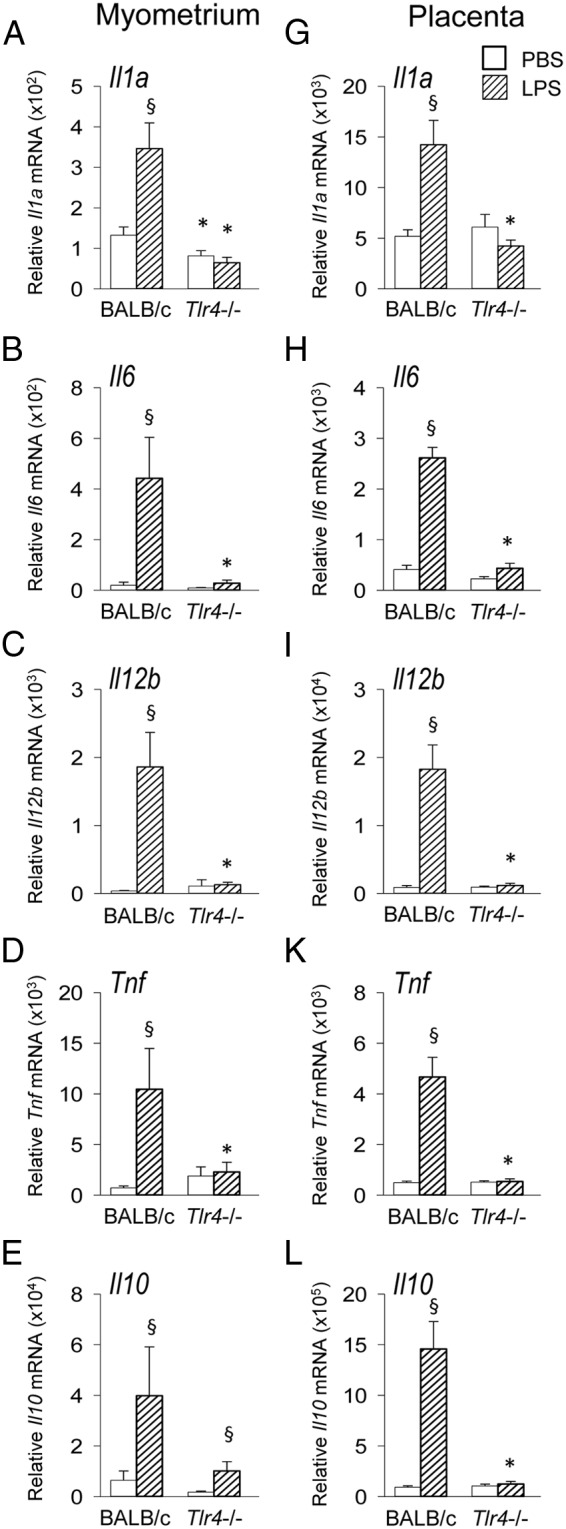
TLR4-deficient mice are resistant to LPS-mediated induction of proinflammatory cytokines in uterine myometrium and placenta. Tlr4−/− or control wild-type mice were mated to males of the same genotype. Detection of a vaginal plug was designated gd 0.5, and females were administered 10 μg of LPS ip on gd 16.5 (hatched bars) or PBS vehicle control (white bars), and 4 hours later, uterine myometrium and placenta were recovered. Relative expression of Il1a (A, G), Il6 (B, H), Il12b (C, I), Tnf (D, K), and Il10 (E, L) mRNAs were determined in each tissue by qPCR and normalized to Actb. Data are shown as mean ± SEM relative gene expression in tissue pooled from 2 implantation sites for placenta and total uterine myometrium, with n = 6–11 dams/group. The effect of genotype and LPS was analyzed by Kruskal-Wallis and Mann-Whitney U test. §, significant difference compared with PBS treatment in same genotype, P < .05; *, significant difference compared with same treatment in wild-type controls, P < .05.
The TLR4 antagonist (+)-naloxone delays term labor
The TLR4 antagonist (+)-naloxone (42) was employed to further investigate the role of TLR4 ligation in the initiation of labor. Wild-type B6 females mated to B6 males were administered (+)-naloxone or PBS vehicle at 12-hour intervals from gd 17.5 to 19.0, and perinatal outcomes were recorded. B6 mice were used as pilot studies demonstrated strong efficacy of TLR4 antagonism in this strain (data not shown), associated with higher levels of Tlr4 expression (43). Treatment with (+)-naloxone extended mean gestation length by 16.8 hours, compared with females given PBS control (Figure 8A). Treatment did not alter the number of viable pups born (Figure 8B), the incidence of postnatal death, pup weight at birth, or survival to weaning (data not shown).
Figure 8.
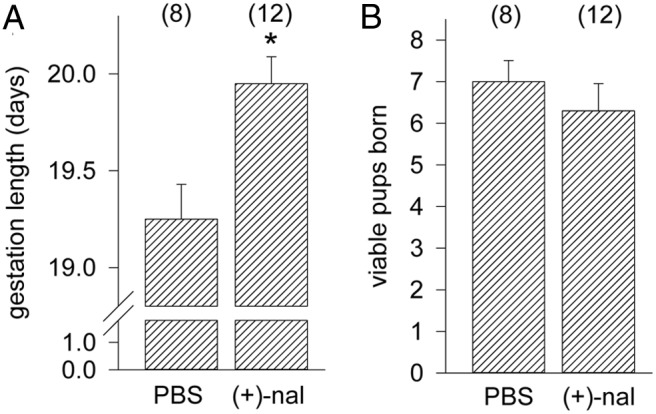
TLR4 antagonist (+)-naloxone delays term labor. Wild-type dams were mated to wild-type males, and detection of a vaginal plug was designated gd 0.5. Pregnant mice were given 60 mg/kg of (+)-naloxone or PBS vehicle on gd 16.5, 17.0, 17.5, and 18.0, and females were monitored from gd 18.0 mice to record the length of gestation (A) and the number of viable pups born (B). Data shown as mean ± SEM were analyzed by ANOVA and Sidak t test to determine effect of (+)-naloxone. The number of dams or pups per group is given in parentheses. *, significant difference compared with PBS control group, P < .05.
Discussion
The normal physiological events of parturition are accompanied by a progressive transition in cytokine expression and leukocyte populations in the reproductive and gestational tissues from an antiinflammatory to a proinflammatory state (2, 3). Activation of TLR4 by bacterial components is a known trigger of precocious inflammation leading to PTL (30, 31). In this study, we show that TLR4 is involved not only in PTL, but also in regulating normal on-time parturition. Without TLR4, parturition is delayed by 13 hours and pup survival is compromised, which demonstrates that TLR4 influences the timing and successful progression of the birth cascade. Gestational tissues from TLR4-deficient pregnancies exhibit reduced proinflammatory cytokines Il1b, Il6, Il12b, and Tnf, delayed expression of the UAGs Ptgfr, Oxtr, and Gja1, reduced neutrophil, macrophage and dendritic cell populations and increased regulatory T cells. Collectively, these data show that TLR4 is a limiting factor in the inflammatory response that regulates the timing of birth in healthy, infection-free pregnancy.
TLR4 is well placed to contribute to parturition by virtue of its wide pattern of expression in the uterus and placental tissues, by trophoblasts and other nonleukocytic lineages (44–46) as well as resident leukocytes in the reproductive tissues (9, 10). In the mouse and human, expression of Tlr4 increases across gestation in the maternal and fetal compartments, then declines postpartum (31, 46). Our findings that such a wide range of inflammatory regulators and labor-associated genes and leukocytes are disrupted in Tlr4−/− mice positions TLR4 signaling as a key upstream trigger of the labor-inducing cascade. TLR4 ligation is a potent activator of NFκB, a master regulator of the labor cascade, which in turn activates proinflammatory cytokines and labor-associated genes (17). Antiinflammatory Il10, another cytokine controlled by NFκB (47), was also reduced in Tlr4−/− mice. IL-10 acts to modulate labor-associated inflammation, and IL-10-deficient mice are prone to poor perinatal outcomes (48).
Two distinct pathways operate downstream of TLR4 through either MyD88 or TIR-domain-containing adapter-inducing interferon-β (TRIF) adaptor molecules to activate NFκB (34, 49). MyD88 directly activates NFκB, and without MyD88, NFκB activation is delayed (50). Independently of MyD88, IL-1B (51), IL-6 (52), IL-12B (53), and TNF (54) can be induced via the TRIF-dependent pathway (49). Unlike TLR4 deficiency, genetic disruption of MyD88 did not delay parturition. This shows that the mechanism by which TLR4 activates the term birth cascade is not dependent on MyD88, although MyD88 signaling may contribute. This contrasts with PTL, where MyD88 is reported to be essential while TRIF is dispensable (35). The differential dependence on the 2 adaptor molecules presumably reflects the different structural and ligand-binding characteristics of the TLR4 ligands inducing LPS-induced PTL and term labor, including their requirement for different coreceptors (55).
Late parturition in the Tlr4−/− mice may contribute to the small but physiologically relevant increase in pup weight at birth. An alternative explanation is that TLR4 signaling impacts growth of the fetus in utero, this seems possible given that MyD88-deficient pups were also larger at birth, despite no change in birth timing. Both Tlr4−/− and Myd88−/− pups remained larger than wild-type pups at weaning (data not shown) and showed elevated postnatal mortality and a substantial reduction in litter size by weaning. Smaller litter sizes at 24 hours after birth in Tlr4−/− mice were attributed to loss of pups in late gestation and at the time of birth as opposed to earlier in gestation, because compared with wild-type mice, there was no change in overall implantation rate or viable fetuses at gd 18.5 in TLR4-deficient mice. This implies that endogenous TLR4 ligands have physiological roles in fetal development that promote perinatal viability, in addition to effects on the timing of birth. One such effect may be in maturation of tissues such as the fetal brain, where a previously unreported elevation in proinflammatory cytokines on gd 18.5, did not occur in the absence of TLR4.
It is relevant that the earliest and greatest effects of TLR4 deficiency were evident in the placenta and fetal membranes, where cytokines Tnf and Il12b were suppressed from gd 16.5, and Il10 was suppressed from gd 17.5. Decidual and myometrial cytokine gene expression was not as substantially affected by TLR4 deficiency compared with the fetal tissues in late gestation, although elevated cytokine bioactivity in the maternal compartment appears later in the parturition cascade than in fetal tissues, and may be physiologically more important in the laboring and postpartum phases (14). This can be interpreted as evidence that the first phase of TLR4 signaling required to initiate the parturition cascade occurs in the placenta and fetal membranes, as opposed to the maternal compartment. Myometrial cell culture experiments and genetic disruption of cytokines in vivo also imply that UAG induction is downstream of proinflammatory cytokine synthesis in the birth cascade (11, 18, 19).
UAGs mediate the vital actions of myometrial prostaglandin signaling to control uterine muscle contractions and cervical dilatation (22). Here, we find that although prostaglandin synthesis genes Ptgr1 and Ptgr2 are not inhibited by TLR4 disruption implying prostaglandin synthesis is not impaired, the expected increase in decidual expression of the prostaglandin receptor gene Ptgfr (56) was transiently delayed. This may be the consequence of reduced IL-1B in Tlr4−/− gestational tissues, because IL-1B is known to up-regulate Ptgfr (18). Because experiments in PTGFR-deficient mice show that this receptor is rate limiting in prostaglandin signaling and the timing of labor (23), insufficient Ptgfr expression may be a key element delaying labor in TLR4-deficient mice. PTGFR protein is similarly regulated in the myometrium of on-time laboring women (57), so is likely to be crucial in the timing of human birth.
In myometrium of wild-type mice, expression of Oxtr and Gja1 increased progressively towards parturition, as reported previously in mice (56) and in women (57). In Tlr4−/− mice, Oxtr expression was delayed, although by gd 19.5, expression was comparable with controls. Gja1 expression was also moderately diminished on gd 18.5in TLR4-deficient myometrium. This altered expression pattern is similar to that seen in IL-6-deficent mice, where delayed parturition is associated with a pronounced delay in Oxtr expression but little change in Gja1 (11). Both Oxtr (58) and Gja1 (59) are stimulated by proinflammatory cytokines in human and rodent myometrium, implying that delayed Oxtr and Gja1 expression is the consequence of reduced Il1b, Il6, Il12b, and Tnf in the absence of TLR4. Also, like IL-6-deficient mice, delayed parturition occurred regardless of unchanged kinetics of progesterone decline (11). This suggests that the most prominent roles of TLR4 in the birth cascade are distal to luteal demise, although a role for TLR4 in contributing to progesterone regulation cannot be discounted.
Genetic deficiency in TLR4 causes a reduced leukocyte response to infectious and noninfectious inflammatory stimuli in a variety of pathogenic settings (34, 36). Here, we observed a reduction in the abundance of neutrophils within the placental tissue and myometrium in TLR4-deficient mice, consistent with previous observations of reduced neutrophils during inflammation in Tlr4−/− mice (37). Recent studies showing that decidual neutrophils are increased in women with infection-driven PTL but not on-time labor (15), and that deletion of neutrophils in mice neither changes gestation length (13) or protects against LPS-triggered PTL (60), implies that neutrophils may be more important for postpartum remodeling than for facilitating delivery, so its seems unlikely that neutrophil-dependent mechanisms account for the delayed birth in Tlr4−/− mice. Macrophages are also implicated in labor-associated inflammation, increasing in the mouse as well as human decidua in late gestation pregnancy (14, 15). In the absence of TLR4, macrophages were fewer in conceptus tissues, demonstrating a crucial role for TLR4 in controlling macrophage recruitment either directly or indirectly through cytokine and chemokine expression.
In the myometrial compartment, the impact of TLR4 deficiency was to substantially reduce the number of DCs. Engagement of TLR4 is reported to induce activation and maturation of DCs (36), although we did not find evidence of altered DC phenotype. A pathway by which DCs may progress inflammation to accelerate labor is through reversing T cell-mediated immune suppression, to reinforce the direct effects of TLR4 on CD4+ T cells (38). Ligation of TLR4 on the surface of DCs can abrogate the antiinflammatory and protolerance functions of Treg cells (61). It has previously been reported that Treg cells lose potency in their suppressive function in late gestation before labor (8). Failure of DCs to remove and silence Treg cells in the absence of TLR4 signaling may explain the elevated Treg cells we observed in the uterus of Tlr4−/− mice. Excessive Treg cells would be expected to quell progression of inflammation and so may contribute to delayed delivery. TLR4 signaling can influence the production of IL-17 and other cytokines in CD4+ T cells (62), consistent with the elevated IL-17RA we observed in uterine CD4+ T cells.
Heat-killed bacteria (32, 35) and LPS (30, 48) are common models of infection-driven fetal loss and PTL in mice, so it is unsurprising that genetic TLR4-deficency protects mice from LPS-induced fetal loss. Similar experiments have previously shown that natural mutation in Tlr4 or its pharmacological inhibition, protects pregnancies from LPS-induced fetal loss or PTL (32, 63). In our model, administration of LPS to wild-type mice caused elevated Il1a, IL6, Il12b, Ifng, Tnf, and Il10 mRNA in the uterus and placenta, whereas no response was evident in Tlr4−/− mice. This clearly confirms that LPS-mediated cytokine induction in vivo is mediated through TLR4. The additional utility of this experiment in the current study is to demonstrate the similarity between the TLR4-dependent pathophysiological cytokine response of PTL, and the TLR4-dependent physiological cytokine response of on-time birth in late gestation. Our data reinforce that similar TLR4-driven pathways operate in both normal gestation and simulated preterm events.
To determine whether preventing endogenous activation of TLR4 delays on-time labor, we used the TLR4 antagonist (+)-naloxone. (+)-naloxone is a newly identified TLR4 antagonist (42), which is the nonopioid isomer of the opioid receptor antagonist (−)-naloxone, a small molecule that, in contrast to anti-TLR4 neutralizing antibodies (63), can penetrate across the placenta (64) and blood brain barrier (65). Unlike the isomer (−)-naloxone, (+)-naloxone does not have opioid receptor-binding activity and is specific for TLR4 (42). The delay in birth evident after (+)-naloxone administration strengthens our interpretation that TLR4 is a key limiting factor for parturition in wild-type mice and argues against the possibility that delayed labor without TLR4 is due to developmental defects or compensatory consequences of genetic TLR4 deficiency. Importantly, however, (+)-naloxone did not alter fetal viability, unlike in the Tlr4−/− mice, where diminished viable births were observed.
These results indicate that TLR4 is a point of convergence in the events of term and PTL. That endogenous TLR4 ligands can activate labor in the absence of infection, raises the question of the identity of the DAMPs responsible. Activation of innate inflammatory pathways is triggered by endogenous DAMPs released in the event of cell stress or cell death (34). Production of DAMPs is an expected consequence of parturition given the extensive tissue remodeling, degradation of extracellular matrix (1), and mechanical stretching of the fetal membranes, amnion cells, and myometrium (66, 67). Endogenous TLR4 ligands produced at the end of gestation include high-mobility-group box chromosomal protein 1 in the cervix of PTL women (40) and hyaluronan (41), which is known to be linked with cervical ripening and cytokine production (68). Surfactant proteins, produced by the fetus in late pregnancy, are ligands of both TLR2 and TLR4 and have been shown to elicit cytokine production and initiation of labor (39, 69, 70). Importantly, TLR4 is not the only TLR implicated in term labor, previous experiments show TLR2 deficiency leads to a similar increase in gestation length as does TLR4 deficiency (69). TLR2 and TLR4 share endogenous ligands that are produced in the later stages of gestation, including heat shock proteins, surfactant, high-mobility-group box chromosomal protein 1, and hyaluronan (71). Redundancy in the pathways activated by TLR2 and TLR4 would explain how the delay in UAG gene expression is overcome to progress delivery in Tlr4−/− mice.
In women, premature release of DAMPs in the event of fetal distress, uterine distension, and/or placental damage or injury (28, 40, 41) could well be a factor in the unscheduled inflammation of PTL, which is believed to occur independently of microbial infection in a large proportion of cases (5). In support of a role for TLR4 in determining sensitivity to preterm birth, women experiencing idiopathic PTL have higher levels of TLR4 on peripheral blood mononuclear cells, including monocytes (9, 10), and in the chorioamniotic membrane (31). Moreover, particular SNPs in Tlr4 are linked to increased proinflammatory cytokine production in female reproductive tissues, both in the presence and absence of infection (72) and certain Tlr4 SNPs are associated with increased risk of PTL and premature rupture of membranes (73, 74). This study raises the prospect of evaluating TLR4 antagonists as a new therapeutic option targeting a key signaling molecule of the parturition cascade, potentially offering benefits over current tocolytic strategies that target uterine contractions or prostaglandin production (19, 22).
Acknowledgments
This work was supported by National Health and Medical Research Council of Australia Grants APP1026178 and APP465423, the Canadian Institutes of Health Research Grant 2011-09-15, and the Australian Research Council Grant DP110100297. A portion of this work was supported by the National Institutes of Health Intramural Research Programs of the National Institute on Drug Abuse and the National Institute of Alcohol Abuse and Alcoholism.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- B6
- C57Bl/6
- CD
- cluster of differentiation
- DAMP
- damage-associated molecular pattern
- DC
- dendritic cell
- FOXP3
- forkhead box P3
- FCS
- fetal caif serum
- gd
- gestational day
- Gja1
- gap junction protein α1
- IL-17RA
- interleukin 17 receptor A
- LPS
- lipopolysaccharide
- MHCII
- major histocompatibility antigen II
- MyD88
- myeloid differentiation factor 88
- NFκB
- nuclear factor kappa B
- Oxtr
- oxytocin receptor
- PALN
- paraaortic lymph node
- PTGFR
- prostaglandin F2alpha receptor
- PTL
- preterm labor
- qPCR
- quantitative PCR
- TLR
- Toll-like receptor
- TRIF
- TIR-domain-containing adapter-inducing interferon-β
- UAG
- uterine activation gene.
References
- 1. Challis JR, Lockwood CJ, Myatt L, Norman JE, Strauss JF, 3rd, Petraglia F. Inflammation and pregnancy. Reprod Sci. 2009;16:206–215. [DOI] [PubMed] [Google Scholar]
- 2. Osman I, Young A, Ledingham MA, et al. Leukocyte density and pro-inflammatory cytokine expression in human fetal membranes, decidua, cervix and myometrium before and during labour at term. Mol Hum Reprod. 2003;9:41–45. [DOI] [PubMed] [Google Scholar]
- 3. Young A, Thomson AJ, Ledingham M, Jordan F, Greer IA, Norman JE. Immunolocalization of proinflammatory cytokines in myometrium, cervix, and fetal membranes during human parturition at term. Biol Reprod. 2002;66:445–449. [DOI] [PubMed] [Google Scholar]
- 4. Mattison DR, Damus K, Fiore E, Petrini J, Alter C. Preterm delivery: a public health perspective. Paediatr Perinat Epidemiol. 2001;15(suppl 2):7–16. [DOI] [PubMed] [Google Scholar]
- 5. Goldenberg RL, Culhane JF, Iams JD, Romero R. Epidemiology and causes of preterm birth. Lancet. 2008;371:75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lawn JE, Cousens S, Zupan J, Lancet Neonatal Survival Steering T. 4 million neonatal deaths: when? Where? Why? Lancet. 2005;365:891–900. [DOI] [PubMed] [Google Scholar]
- 7. Saigal S, Doyle LW. An overview of mortality and sequelae of preterm birth from infancy to adulthood. Lancet. 2008;371:261–269. [DOI] [PubMed] [Google Scholar]
- 8. Schober L, Radnai D, Schmitt E, Mahnke K, Sohn C, Steinborn A. Term and preterm labor: decreased suppressive activity and changes in composition of the regulatory T-cell pool. Immunol Cell Biol. 2012;90:935–944. [DOI] [PubMed] [Google Scholar]
- 9. Pawelczyk E, Nowicki BJ, Izban MG, et al. Spontaneous preterm labor is associated with an increase in the proinflammatory signal transducer TLR4 receptor on maternal blood monocytes. BMC Pregnancy Childbirth. 2010;10:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim J, Ko Y, Kwon K, et al. Analysis of monocyte subsets and toll-like receptor 4 expression in peripheral blood monocytes of women in preterm labor. J Reprod Immunol. 2012;94:190–195. [DOI] [PubMed] [Google Scholar]
- 11. Robertson SA, Christiaens I, Dorian CL, et al. Interleukin-6 is an essential determinant of on-time parturition in the mouse. Endocrinology. 2010;151:3996–4006. [DOI] [PubMed] [Google Scholar]
- 12. Gravett MG, Witkin SS, Haluska GJ, Edwards JL, Cook MJ, Novy MJ. An experimental model for intraamniotic infection and preterm labor in rhesus monkeys. Am J Obstet Gynecol. 1994;171:1660–1667. [DOI] [PubMed] [Google Scholar]
- 13. Timmons BC, Mahendroo MS. Timing of neutrophil activation and expression of proinflammatory markers do not support a role for neutrophils in cervical ripening in the mouse. Biol Reprod. 2006;74:236–245. [DOI] [PubMed] [Google Scholar]
- 14. Shynlova O, Nedd-Roderique T, Li Y, Dorogin A, Nguyen T, Lye SJ. Infiltration of myeloid cells into decidua is a critical early event in the labour cascade and post-partum uterine remodelling. J Cell Mol Med. 2013;17:311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hamilton S, Oomomian Y, Stephen G, et al. Macrophages infiltrate the human and rat decidua during term and preterm labor: evidence that decidual inflammation precedes labor. Biol Reprod. 2012;86:39. [DOI] [PubMed] [Google Scholar]
- 16. Mendelson CR. Minireview: fetal-maternal hormonal signaling in pregnancy and labor. Mol Endocrinol. 2009;23:947–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lappas M, Rice GE. The role and regulation of the nuclear factor κ B signalling pathway in human labour. Placenta. 2007;28:543–556. [DOI] [PubMed] [Google Scholar]
- 18. Zaragoza DB, Wilson RR, Mitchell BF, Olson DM. The interleukin 1β-induced expression of human prostaglandin F2α receptor messenger RNA in human myometrial-derived ULTR cells requires the transcription factor, NFκB. Biol Reprod. 2006;75:697–704. [DOI] [PubMed] [Google Scholar]
- 19. Challis JR, Sloboda DM, Alfaidy N, et al. Prostaglandins and mechanisms of preterm birth. Reproduction. 2002;124:1–17. [DOI] [PubMed] [Google Scholar]
- 20. Döring B, Shynlova O, Tsui P, et al. Ablation of connexin43 in uterine smooth muscle cells of the mouse causes delayed parturition. J Cell Sci. 2006;119:1715–1722. [DOI] [PubMed] [Google Scholar]
- 21. Terzidou V, Lee Y, Lindström T, Johnson M, Thornton S, Bennett PR. Regulation of the human oxytocin receptor by nuclear factor-κB and CCAAT/enhancer-binding protein-β. J Clin Endocrinol Metab. 2006;91:2317–2326. [DOI] [PubMed] [Google Scholar]
- 22. Kelly AJ, Malik S, Smith L, Kavanagh J, Thomas J. Vaginal prostaglandin (PGE2 and PGF2a) for induction of labour at term. Cochrane Database Syst Rev. 2009:CD003101. [DOI] [PubMed] [Google Scholar]
- 23. Tsuboi K, Sugimoto Y, Iwane A, Yamamoto K, Yamamoto S, Ichikawa A. Uterine expression of prostaglandin H2 synthase in late pregnancy and during parturition in prostaglandin F receptor-deficient mice. Endocrinology. 2000;141:315–324. [DOI] [PubMed] [Google Scholar]
- 24. Athayde N, Edwin SS, Romero R, et al. A role for matrix metalloproteinase-9 in spontaneous rupture of the fetal membranes. Am J Obstet Gynecol. 1998;179:1248–1253. [DOI] [PubMed] [Google Scholar]
- 25. Kelly RW. Inflammatory mediators and cervical ripening. J Reprod Immunol. 2002;57:217–224. [DOI] [PubMed] [Google Scholar]
- 26. Timmons BC, Fairhurst AM, Mahendroo MS. Temporal changes in myeloid cells in the cervix during pregnancy and parturition. J Immunol. 2009;182:2700–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gonçalves LF, Chaiworapongsa T, Romero R. Intrauterine infection and prematurity. Ment Retard Dev Disabil Res Rev. 2002;8:3–13. [DOI] [PubMed] [Google Scholar]
- 28. Romero R, Espinoza J, Kusanovic JP, et al. The preterm parturition syndrome. BJOG. 2006;113(suppl 3):17–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thaxton JE, Nevers TA, Sharma S. TLR-mediated preterm birth in response to pathogenic agents. Infect Dis Obstet Gynecol. 2010;2010pii:378472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Salminen A, Paananen R, Vuolteenaho R, et al. Maternal endotoxin-induced preterm birth in mice: fetal responses in toll-like receptors, collectins, and cytokines. Pediatr Res. 2008;63:280–286. [DOI] [PubMed] [Google Scholar]
- 31. Kim YM, Romero R, Chaiworapongsa T, et al. Toll-like receptor-2 and -4 in the chorioamniotic membranes in spontaneous labor at term and in preterm parturition that are associated with chorioamnionitis. Am J Obstet Gynecol. 2004;191:1346–1355. [DOI] [PubMed] [Google Scholar]
- 32. Wang H, Hirsch E. Bacterially-induced preterm labor and regulation of prostaglandin-metabolizing enzyme expression in mice: the role of toll-like receptor 4. Biol Reprod. 2003;69:1957–1963. [DOI] [PubMed] [Google Scholar]
- 33. Elovitz MA, Wang Z, Chien EK, Rychlik DF, Phillippe M. A new model for inflammation-induced preterm birth: the role of platelet-activating factor and Toll-like receptor-4. Am J Pathol. 2003;163:2103–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010;2010pii:672395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Filipovich Y, Lu SJ, Akira S, Hirsch E. The adaptor protein MyD88 is essential for E. coli-induced preterm delivery in mice. Am J Obstet Gynecol. 2009;200:93.e1–93.e8. [DOI] [PubMed] [Google Scholar]
- 36. Fang H, Ang B, Xu X, et al. TLR4 is essential for dendritic cell activation and anti-tumor T-cell response enhancement by DAMPs released from chemically stressed cancer cells. Cell Mol Immunol. 2014;11:150–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Andonegui G, Bonder CS, Green F, et al. Endothelium-derived Toll-like receptor-4 is the key molecule in LPS-induced neutrophil sequestration into lungs. J Clin Invest. 2003;111:1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. González-Navajas JM, Fine S, Law J, et al. TLR4 signaling in effector CD4+ T cells regulates TCR activation and experimental colitis in mice. J Clin Invest. 2010;120:570–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Condon JC, Jeyasuria P, Faust JM, Mendelson CR. Surfactant protein secreted by the maturing mouse fetal lung acts as a hormone that signals the initiation of parturition. Proc Natl Acad Sci USA. 2004;101:4978–4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dubicke A, Andersson P, Fransson E, et al. High-mobility group box protein 1 and its signalling receptors in human preterm and term cervix. J Reprod Immunol. 2010;84:86–94. [DOI] [PubMed] [Google Scholar]
- 41. Akgul Y, Holt R, Mummert M, Word A, Mahendroo M. Dynamic changes in cervical glycosaminoglycan composition during normal pregnancy and preterm birth. Endocrinology. 2012;153:3493–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hutchinson MR, Zhang Y, Brown K, et al. Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4). Eur J Neurosci. 2008;28:20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tsukamoto H, Fukudome K, Takao S, et al. Reduced surface expression of TLR4 by a V254I point mutation accounts for the low lipopolysaccharide responder phenotype of BALB/c B cells. J Immunol. 2013;190:195–204. [DOI] [PubMed] [Google Scholar]
- 44. Pioli PA, Amiel E, Schaefer TM, Connolly JE, Wira CR, Guyre PM. Differential expression of Toll-like receptors 2 and 4 in tissues of the human female reproductive tract. Infect Immun. 2004;72:5799–5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kumazaki K, Nakayama M, Yanagihara I, Suehara N, Wada Y. Immunohistochemical distribution of Toll-like receptor 4 in term and preterm human placentas from normal and complicated pregnancy including chorioamnionitis. Hum Pathol. 2004;35:47–54. [DOI] [PubMed] [Google Scholar]
- 46. Gonzalez JM, Xu H, Ofori E, Elovitz MA. Toll-like receptors in the uterus, cervix, and placenta: is pregnancy an immunosuppressed state? Am J Obstet Gynecol. 2007;197:296.e291–e296. [DOI] [PubMed] [Google Scholar]
- 47. Cao S, Zhang X, Edwards JP, Mosser DM. NF-κB1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. J Biol Chem. 2006;281:26041–26050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Robertson SA, Skinner RJ, Care AS. Essential role for IL-10 in resistance to lipopolysaccharide-induced preterm labor in mice. J Immunol. 2006;177:4888–4896. [DOI] [PubMed] [Google Scholar]
- 49. Yamamoto M, Sato S, Hemmi H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. [DOI] [PubMed] [Google Scholar]
- 50. Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. [DOI] [PubMed] [Google Scholar]
- 51. Hiscott J, Marois J, Garoufalis J, et al. Characterization of a functional NF-κ B site in the human interleukin 1 β promoter: evidence for a positive autoregulatory loop. Mol Cell Biol. 1993;13:6231–6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Son YH, Jeong YT, Lee KA, et al. Roles of MAPK and NF-κB in interleukin-6 induction by lipopolysaccharide in vascular smooth muscle cells. J Cardiovasc Pharmacol. 2008;51:71–77. [DOI] [PubMed] [Google Scholar]
- 53. Murphy TL, Cleveland MG, Kulesza P, Magram J, Murphy KM. Regulation of interleukin 12 p40 expression through an NF-κ B half-site. Mol Cell Biol. 1995;15:5258–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shakhov AN, Collart MA, Vassalli P, Nedospasov SA, Jongeneel CV. κ B-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor α gene in primary macrophages. J Exp Med. 1990;171:35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Di Gioia M, Zanoni I. Toll-like receptor co-receptors as master regulators of the immune response. Mol Immunol. 2015;63:143–152. [DOI] [PubMed] [Google Scholar]
- 56. Cook JL, Zaragoza DB, Sung DH, Olson DM. Expression of myometrial activation and stimulation genes in a mouse model of preterm labor: myometrial activation, stimulation, and preterm labor. Endocrinology. 2000;141:1718–1728. [DOI] [PubMed] [Google Scholar]
- 57. Brodt-Eppley J, Myatt L. Prostaglandin receptors in lower segment myometrium during gestation and labor. Obstet Gynecol. 1999;93:89–93. [DOI] [PubMed] [Google Scholar]
- 58. Fang X, Wong S, Mitchell BF. Effects of LPS and IL-6 on oxytocin receptor in non-pregnant and pregnant rat uterus. Am J Reprod Immunol. 2000;44:65–72. [DOI] [PubMed] [Google Scholar]
- 59. Tonon R, D'Andrea P. Interleukin-1β increases the functional expression of connexin 43 in articular chondrocytes: evidence for a Ca2+-dependent mechanism. J Bone Miner Res. 2000;15:1669–1677. [DOI] [PubMed] [Google Scholar]
- 60. Rinaldi SF, Catalano RD, Wade J, Rossi AG, Norman JE. Decidual neutrophil infiltration is not required for preterm birth in a mouse model of infection-induced preterm labor. J Immunol. 2014;192:2315–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. [DOI] [PubMed] [Google Scholar]
- 62. Cao AT, Yao S, Stefka AT, et al. TLR4 regulates IFN-γ and IL-17 production by both thymic and induced Foxp3+ Tregs during intestinal inflammation. J Leukoc Biol. 2014;96:895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li L, Kang J, Lei W. Role of Toll-like receptor 4 in inflammation-induced preterm delivery. Mol Hum Reprod. 2010;16:267–272. [DOI] [PubMed] [Google Scholar]
- 64. Dailey PA, Brookshire GL, Shnider SM, et al. The effects of naloxone associated with the intrathecal use of morphine in labor. Anesth Analg. 1985;64:658–666. [PubMed] [Google Scholar]
- 65. Ngai SH, Berkowitz BA, Yang JC, Hempstead J, Spector S. Pharmacokinetics of naloxone in rats and in man: basis for its potency and short duration of action. Anesthesiology. 1976;44:398–401. [DOI] [PubMed] [Google Scholar]
- 66. Maradny EE, Kanayama N, Halim A, Maehara K, Terao T. Stretching of fetal membranes increases the concentration of interleukin-8 and collagenase activity. Am J Obstet Gynecol. 1996;174:843–849. [DOI] [PubMed] [Google Scholar]
- 67. Kanayama N, Fukamizu H. Mechanical stretching increases prostaglandin E2 in cultured human amnion cells. Gynecol Obstet Invest. 1989;28:123–126. [DOI] [PubMed] [Google Scholar]
- 68. Obara M, Hirano H, Ogawa M, et al. Changes in molecular weight of hyaluronan and hyaluronidase activity in uterine cervical mucus in cervical ripening. Acta Obstet Gynecol Scand. 2001;80:492–496. [PubMed] [Google Scholar]
- 69. Montalbano AP, Hawgood S, Mendelson CR. Mice deficient in surfactant protein A (SP-A) and SP-D or in TLR2 manifest delayed parturition and decreased expression of inflammatory and contractile genes. Endocrinology. 2013;154:483–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ohya M, Nishitani C, Sano H, et al. Human pulmonary surfactant protein D binds the extracellular domains of Toll-like receptors 2 and 4 through the carbohydrate recognition domain by a mechanism different from its binding to phosphatidylinositol and lipopolysaccharide. Biochemistry. 2006;45:8657–8664. [DOI] [PubMed] [Google Scholar]
- 71. Erridge C. Endogenous ligands of TLR2 and TLR4: agonists or assistants? J Leukoc Biol. 2010;87:989–999. [DOI] [PubMed] [Google Scholar]
- 72. Ryckman KK, Williams SM, Krohn MA, Simhan HN. Genetic association of Toll-like receptor 4 with cervical cytokine concentrations during pregnancy. Genes Immun. 2009;10:636–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lorenz E, Hallman M, Marttila R, Haataja R, Schwartz DA. Association between the Asp299Gly polymorphisms in the Toll-like receptor 4 and premature births in the Finnish population. Pediatr Res. 2002;52:373–376. [DOI] [PubMed] [Google Scholar]
- 74. Rey G, Skowronek F, Alciaturi J, Alonso J, Bertoni B, Sapiro R. Toll receptor 4 Asp299Gly polymorphism and its association with preterm birth and premature rupture of membranes in a South American population. Mol Hum Reprod. 2008;14:555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]