

Abstract
Local immune-activating therapies seek to improve the presentation of tumor antigen, thereby promoting the activation of antitumor CD8+ T cells and delaying tumor growth. Surprisingly, little is known about the ability of these therapies to stimulate antitumor CD4+ T cells. We examined tumor-specific CD4+ T cell responses after peri-tumoral administration of the TLR3 agonist polyinosinic-polycytidylic acid (poly I:C), or the danger signal monosodium urate crystals in combination with Mycobacterium smegmatis (MSU + Msmeg) in mice. Both treatments delayed tumor growth, however, only MSU + Msmeg induced proliferation of tumor-specific CD4+ T cells in the draining lymph node (dLN). In line with the proliferation data, administration of MSU + Msmeg, but not poly I:C, enhanced the infiltration of CD4+FoxP3− T cells into the tumor, increased their capacity to produce IFNγ and TNF-α, and decreased PD-1 expression on tumor-infiltrating CD8+ T cells. Induction of CD4+ T cell proliferation by treatment with MSU + Msmeg required IL-1βR signaling, as it was blocked by administration of the IL-1βR antagonist Anakinra. In addition, treatment with Anakinra or with anti-CD4 also reversed the increased survival after tumor challenge in MSU + Msmeg treated mice. Thus, peri-tumoral treatment with MSU + Msmeg results in IL-1βR-dependent priming of antitumor CD4+ T cells in the LN, with consequent superior activation of CD4+ and CD8+ T cells within the tumor, and sustained antitumor activity.
Keywords: CD4+ T cells, IL-1β, monosodium urate crystals, mycobacteria, PD-1, poly I:C, regulatory T cells, tumor immunotherapy
Abbreviations
- CTL
cytotoxic T lymphocyte
- dLN
draining lymph node
- IFNγ
interferon γ
- IL-1βR
interleukin 1β receptor
- LPS
lipopolysaccharide
- MHC II
major histocompatibility complex II
- Msmeg
Mycobacterium smegmatis
- MSU
monosodium urate crystals
- NK
natural killer
- PBS
phosphate buffered saline
- PD-1
programmed cell death 1
- poly I:C
polyinosinic-polycytidylic acid
- TLR
toll-like receptor
- TNF-α
tumor necrosis factor α
- Treg
regulatory T cells
Introduction
Most current tumor immunotherapy approaches focus on CD8+ T cells, as these cells can directly and effectively eliminate tumor cells. However, CD4+ T cells are also central to optimal antitumor immunity.1 Co-transfer of CD4+ and CD8+ T cells enhances CD8+ effector function and therapeutic efficacy in a lymphopenic environment.2 Similarly, combined vaccination with CD4+ and CD8+ T-cell epitopes augments CD8+ T cell responses and improves clinical outcomes.3 These effects of CD4+ T cells are mediated through a variety of mechanisms. First, CD4+ T cells provide help for the priming of CD8+ T cell responses in lymphoid organs. Activated CD4+ T cells license antigen-presenting cells, enabling CD8+ T cells to become fully functional cytotoxic T lymphocytes (CTL).4 Subsequently, tumor-specific CD4+ T cells facilitate accumulation of CD8+ T cells at the effector site and enhance their antitumor activity.5 CD4+ T cells are also required for the maintenance of CD8+ T cell numbers, cytotoxic function and cytokine production within the tumor.6 Finally, CD4+ T-cell help can rescue CTL from activation-induced cell death, promoting effector function and memory formation.7
In addition to their prominent role in the induction and maintenance of CTL responses, CD4+ T cells can also mediate CD8+ T cell-independent antitumor activity. CD4+ T cells can directly recognize and kill tumor targets,8 and cytokines produced by CD4+ T cells in tumors, especially IFNγ, can promote the antitumor activity of both hematopoietic and non-hematopoietic cells.9,10 These cytokine-mediated effects can occur independently of expression of MHCII on tumor cells and therefore do not require direct tumor cell recognition by the CD4+ T cells.9
Several studies have shown that microbial products, including bacteria and synthetic or natural TLR agonists, promote antitumor immune responses and exhibit antitumor activity in experimental11-13 and clinical (reviewed in ref. 14) settings. The activity of these products may be further increased by combination with endogenous danger signals.15 Treatments that turn the tumor into a site of immune activation are highly attractive as they could be easily applied in the clinic and, if effective, activate antitumor immune responses that are tailored to the patient and of broad specificity. Several of these treatments are known to activate antitumor CD8+ T cells without requiring CD4+ responses11,13 whereas for others the capacity to activate CD4+ T cells is unknown.12 Recruiting the activity of both CD8+ and CD4+ T cells to the tumor should increase the impact of these immunotherapies.
We recently compared the antitumor activity of a range of immune stimulators, and found that effective treatments delayed tumor growth by activating both CD8+ T cells and NK cells.16 We now report that the same treatments differed in their ability to prime antitumor CD4+ T cell responses, and that this was due to their differential ability to engage IL-1βR-dependent signaling. Increased CD4+ T cell priming in the LN was associated with increased tumor infiltration of effector CD4+ T cells and a concomitant decrease in regulatory T cells (Treg), reduced CD8+ T cell exhaustion at the tumor site, and sustained retardation of tumor growth after cessation of therapy. Conversely, blocking IL-1βR or depleting CD4+ T cells abrogated the antitumor activity of MSU + Msmeg. Thus, local immunostimulatory treatments can successfully activate tumor-specific CD4+ T cells and favorably impact on the duration of antitumor CD8+ T cell responses.
Results
Local treatment with poly I:C or MSU + Msmeg delays the growth of B16.OVA tumors
We previously compared a range of immune activating agents for the ability to delay the growth of murine tumors including the B16-F1 melanoma and the 4T1 mammary carcinoma. Treatment of established tumors with poly I:C or a combination of MSU + Msmeg delayed primary tumor growth and reduced lung metastases, whereas several other agents were ineffective.16 Here, we evaluated the effect of these treatments on the OVA-expressing melanoma B16.OVA. Four local treatments with either poly I:C or MSU + Msmeg significantly reduced the size of B16.OVA tumors at the end of experiment (Fig. 1). In contrast, treatments with LPS, LPS and MSU (both not shown), MSU or Msmeg alone (Fig. 1) did not alter tumor growth compared to PBS controls. Thus, the antitumor activity of poly I:C and MSU + Msmeg on B16.OVA melanomas was similar to that observed in B16-F1 and 4T1 tumors.
Figure 1.
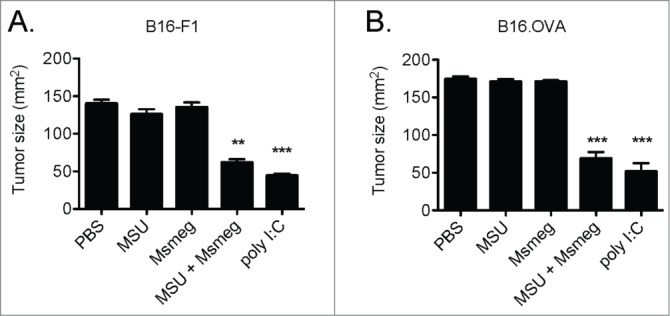
Peri-tumoral treatment with poly I:C or a combination of MSU + Msmeg delays the growth of B16-F1 and B16.OVA tumors. C57BL/6 mice bearing palpable s.c. B16-F1 or B16.OVA tumors were given four peri-tumoral injections of the indicated treatments, starting on day 7 (B16-F1) or day 9 (B16.OVA) after tumor challenge and every second day thereafter. Bar graphs show tumor sizes 2 days after the fourth treatment and include all mice that underwent treatment. Bar graphs show mean + SEM for combined data from two independent experiments, each with 5 mice per group. p values refer to the comparison to the PBS group; ***, p < 0.001; **, p < 0.01; p values larger than 0.05 are not shown.
Treatment with MSU + Msmeg, but not poly I:C, primes tumor-specific CD4+ T cells in the dLN in an IL-1βR-dependent manner
To determine the effect of poly I:C and MSU + Msmeg treatment on CD4+ T cell responses, we examined the proliferation of CFSE-labeled OTII T cells adoptively transferred into B16.OVA-bearing mice. As previously reported for untreated mice,17 no OTII proliferation was detected in the dLN of PBS-treated or poly I:C-treated mice (Fig. 2A). In contrast, MSU + Msmeg treatment induced significant proliferation of OTII T cells, leading to a higher percentage and number of total and divided OTII cells compared to poly I:C (Figs. 2A–E). Proliferation could not be induced by treating with M. smegmatis alone (not shown), and was restricted to the tumor-dLN, suggesting that tumor antigen was necessary for proliferation.
Figure 2.
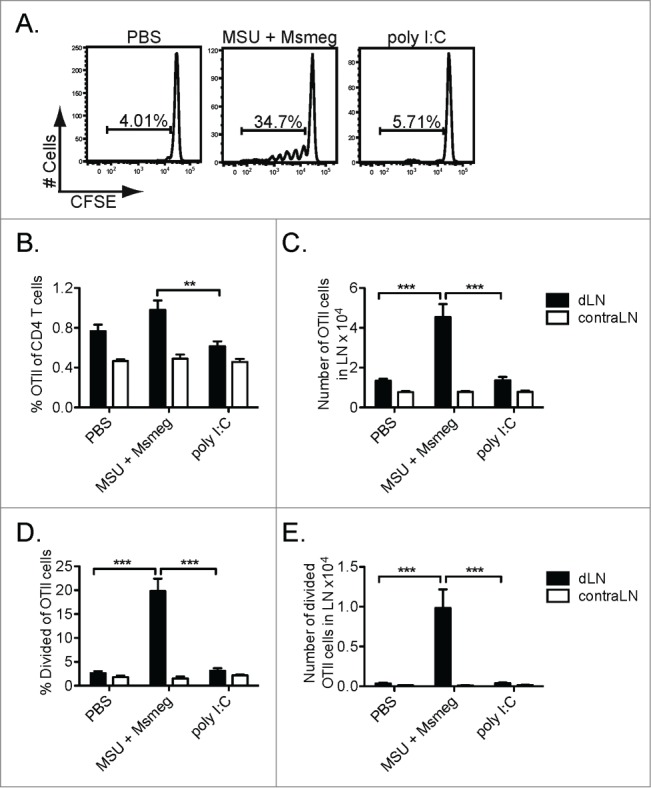
Peri-tumoral treatment with MSU + Msmeg induces the proliferation of tumor-specific CD4+ T cells in dLNs. CFSE-labeled CD45.1+ OTII T cells were adoptively transferred into C57BL/6 mice bearing day 8 B16.OVA tumors. Mice were treated at the tumor site with poly I:C, MSU + Msmeg or PBS vehicle on day 9, and OTII proliferation was assessed in the tumor draining (d) and contralateral (contra) LN on day 14. OTII T cells were identified as single live CD45.1+ CD4+ Vα2+ cells. (A) Representative histograms showing CFSE dilution profiles for OTII cells in tumor-dLN. (B) Frequency and (C) number of OTII cells in LN. (D) Frequency and (E) number of divided OTII cells in LN. Bar graphs show mean + SEM for combined data from 2 independent experiments, each with 5 mice/group.
Our previous work showed that treatment with MSU + Msmeg induced detectable amounts of IL-1β in the serum, while poly I:C did not.16 As IL-1β is known to enhance CD4+ T cell responses,18 we used the IL-1βR antagonist Anakinra to establish whether IL-1β signaling was critical for the ability of MSU + Msmeg to induce proliferation of tumor-specific CD4+ T cells in vivo. As shown in Figs. 3A–D, treatment with Anakinra decreased OTII proliferation by more than half. This suggested that IL-1β was important for the ability of MSU + Msmeg to stimulate CD4+ T cell proliferation.
Figure 3.
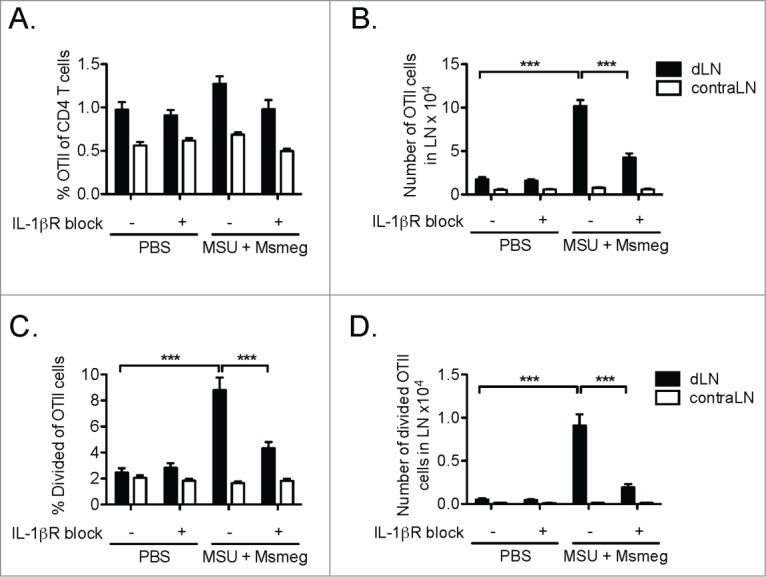
The proliferation of tumor-specific CD4+ T cells in MSU + Msmeg-treated mice requires responsiveness to IL-1β. CFSE-labeled OTII T cells were adoptively transferred into C57BL/6 mice bearing day 8 B16.OVA tumors. Mice were treated at the tumor site with MSU + Msmeg or PBS vehicle on day 8 and 10. The IL-1βR antagonist Anakinra was injected i.p. daily from day 8 through to the end of experiment. OTII proliferation was assessed in draining (d) and contralateral (contra) LN on day 13 using the strategy described in the legend to Fig. 2. (A) Frequency and (B) number of OTII cells in LN. (C) Frequency and (D) number of divided OTII cells in LN. Bar graphs show mean + SEM for combined data from 2 independent experiments, each with 5 mice/group.
Treatment with MSU + Msmeg enhances tumor infiltration by effector CD4+ T cells and reduces Treg
To assess whether increased proliferation of CD4+ T cells in the dLN of MSU + Msmeg-treated mice translated into enhanced effector CD4+ T cell responses in the tumor, we compared the phenotype of tumor-infiltrating CD4+ T cells in treated and untreated mice. Compared to PBS, treatment with MSU + Msmeg significantly increased the frequency of CD4+ T cells in the tumor (Fig. 4A). These CD4+ T cells showed an increased capacity to produce IFNγ and TNF-α, as assessed by intracellular staining without restimulation (Figs. 4B & C), and increased expression of the activation markers CD69 and CD25 in the absence of FoxP3 (Figs. 4D & E). In contrast, poly I:C treatment did not significantly change any of these parameters compared to PBS controls (Fig. 4).
Figure 4.
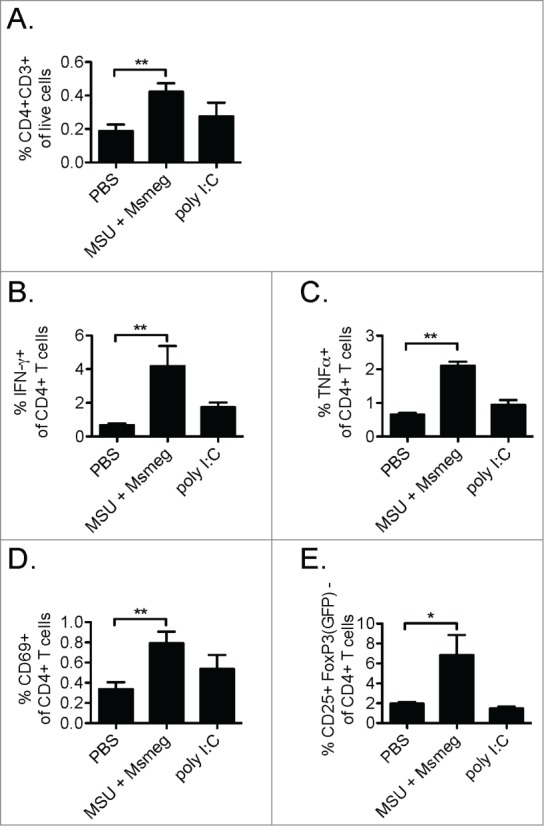
Treatment with MSU + Msmeg increases CD4+ T cell infiltration into the tumor and enhances effector CD4+ T cell activation. C57BL/6 (A–D) or Foxp3GFP (E) mice bearing established B16-F1 tumors were treated with PBS as a vehicle control, MSU + Msmeg or poly I:C on days 7, 9, 11, and 13 after tumor challenge. Tumor cell suspensions were analyzed by flow cytometry one day after the last treatment. (A) Percentages of CD3+CD4+ T cells in the total live tumor cell population. (B & C) Percentages of IFNγ+ (B) and TNF-α+ (C) CD4+ T cells in the tumor, as determined by intracellular staining without restimulation. (D) Frequency of intratumoral CD4+ T cells expressing the activation maker CD69. (E) Frequency of intratumoral CD4+FoxP3(GFP)- cells expressing the activation marker CD25. Bar graphs show mean + SEM for combined data from 2–3 independent experiments, each with 3–5 mice/group.
As total CD4+ T cells encompass not only effector cells but also Treg, we analyzed the CD4+ T cell infiltrate in B16-F1 melanomas from Foxp3GFP mice. These mice express a FoxP3-GFP reporter which identifies tumor-infiltrating CD4+ T cells with regulatory function in vitro.19 In PBS-treated tumors, about 20% of the CD4+ T cells were FoxP3-GFP+. After MSU + Msmeg treatment, the frequency of Treg was nearly halved, but remained unchanged after poly I:C administration (Figs. 5A & B). The reduction in the frequency of Treg in MSU + Msmeg vs. PBS-treated tumors was still apparent when calculated as percentage of total live cells in the tumor, suggesting that it was not simply a consequence of the increased proportion of CD4+ effectors (Fig. 5C). Therefore, treatment with MSU + Msmeg, but not poly I:C, altered the CD4+ infiltrate in tumors, increasing CD4+ effector T cells, and at the same time reducing the proportion of CD4+ Treg.
Figure 5.
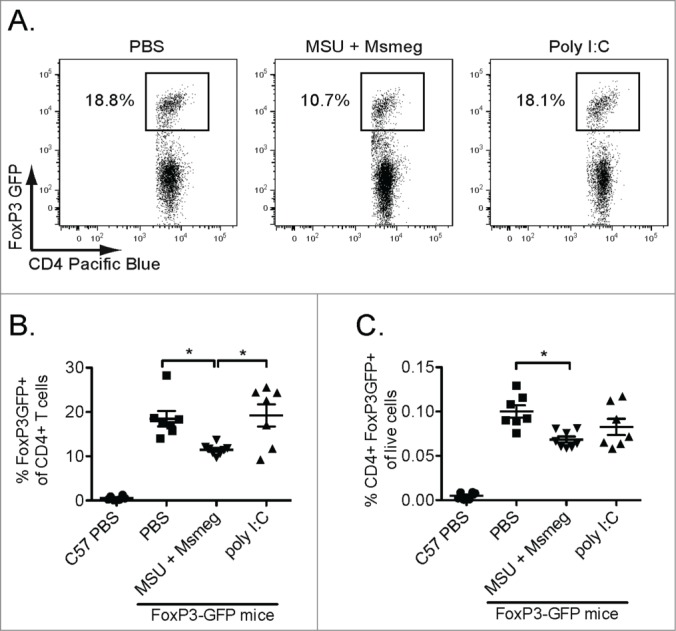
Treatment with MSU + Msmeg reduces the frequency of tumor-infiltrating CD4+FoxP3+ Treg. C57BL/6 or Foxp3-GFP mice bearing established B16-F1 tumors were treated as in Fig. 2. One day after the last treatment, tumor cell suspensions were prepared and analyzed by flow cytometry. (A) Representative gating of CD4+FoxP3-GFP+ Treg in the CD4+ T cell population. Only single live CD45+CD4+ events are shown. (B) Frequency of CD4+FoxP3-GFP+ Treg in the intratumoral CD4+ T cell population. Each symbol corresponds to one mouse. (C) Frequency of CD4+FoxP3-GFP+ Treg in the total live tumor cell population. Each symbol corresponds to one mouse. Data are pooled from two independent experiments, each with 3–5 mice/group. Means and SEM are shown for each scatter plot.
Enhanced CD4+ T cell responses after MSU + Msmeg treatment reduce exhaustion of tumor-infiltrating CD8+ T cells
To assess whether the enhanced CD4+ T cell response in MSU + Msmeg-treated mice influenced the quality of tumor-infiltrating T cells, we examined the expression of the exhaustion marker PD-1 on intratumoral CD4+ and CD8+ T cells. In PBS-treated controls, the proportion of intratumoral CD4+ and CD8+ T cells that expressed high levels of PD-1 increased over time (Figs. 6A & B, PBS controls, 1 treatment vs. 4 treatments). A similar increase was observed in poly I:C-treated tumors. In contrast, in MSU + Msmeg-treated tumors, the percentage of T cells expressing PD-1 remained stable after 4 treatments and was significantly lower compared to both the PBS and poly I:C-treated groups (Figs. 6A & B).
Figure 6.
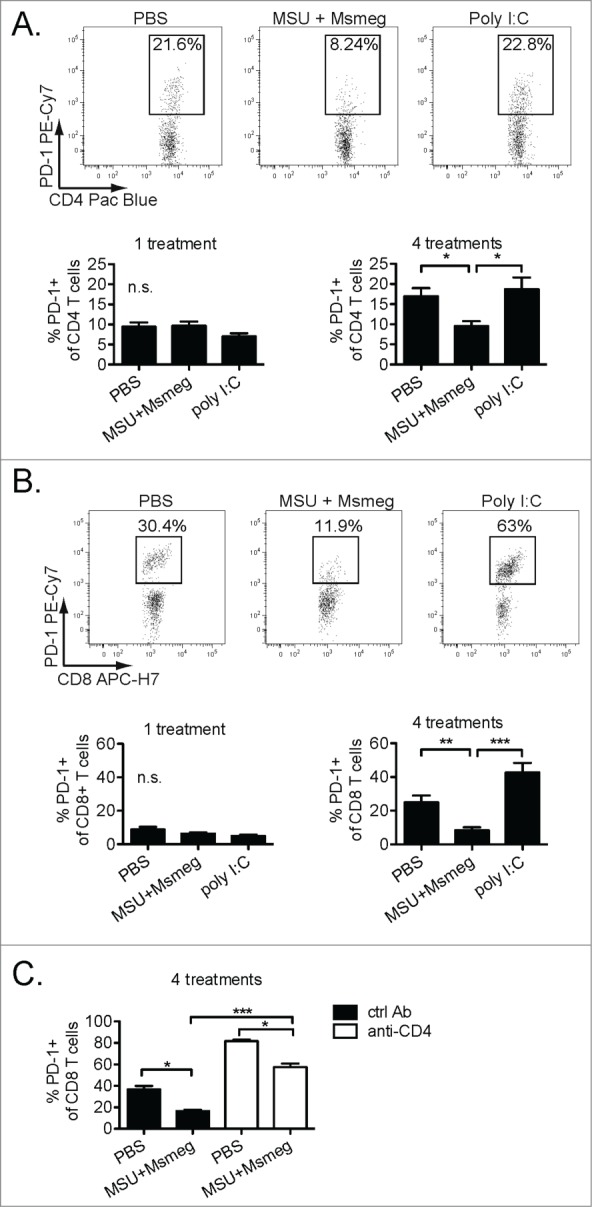
Treatment with MSU + Msmeg results in decreased PD-1 expression on tumor-infiltrating CD4+ and CD8+ T cells. C57BL/6 mice bearing established B16 melanomas were treated with PBS, MSU + Msmeg or poly I:C as indicated; treatment was given either once (one treatment) or four times every other day (four treatments). Mice were sacrificed one day after the last treatment, and expression of the exhaustion marker PD-1 on intratumoral T cells was assessed by flow cytometry. (A) Representative gating of PD-1-expressing CD4+ T cells in tumors is shown at the top; the percentages of PD-1-expressing intratumoral CD4+ cells are shown at the bottom. (B) As in A, except that CD8+ T cells are shown. (C) Impact of CD4+ T cell depletion by administration of anti-CD4 antibodies 36 and 12 h before the first peri-tumoral treatment on the expression of PD-1 on intratumoral CD8+ T cells. Bar graphs show mean + SEM for combined data from two independent experiments, each with 5 mice/group.
To further assess the role of CD4+ T cells in controlling expression of PD-1 on CD8+ T cells, we examined intra-tumoral CD8+ T cells in mice depleted of CD4+ T cells by anti-CD4+ antibody treatment. Compared to CD4+-sufficient mice, mice treated with anti-CD4 one day before the start of immunotherapy had much higher frequencies of PD-1+ CD8+ T cells in their tumors (Fig. 6C), indicating that CD4+ T cells have a large impact on PD-1 expression by CD8+ T cells. However, even after CD4+ T cell depletion, treatment with MSU + Msmeg reduced PD-1 expression on CD4+ T cells compared to PBS controls, suggesting that MSU + Msmeg can also exert additional, CD4+ T cell-independent effects that help prevent CD8+ T cell exhaustion (Fig. 6C).
MSU + Msmeg treatment induces sustained antitumor immunity in a CD4+ T cell and IL-1βR dependent manner
Activation of CD4+ T cell responses and the consequent reduction of CD8+ T cell exhaustion may result in prolonged antitumor activity. To investigate this possibility, we switched from the B16 melanoma model to the more immunogenic E.G7-OVA and EL-4 thymomas, as these tumor models might enable a more sensitive detection of immune responses at the later stages of tumor growth. Repeated peri-tumoral administrations of both poly I:C and MSU + Msmeg similarly delayed tumor growth during the course of treatment (day 4–14, Fig. 7A). However, after cessation of treatment, poly I:C-treated tumors resumed growth at a rate comparable to that of PBS-treated controls. In contrast, MSU + Msmeg-treated tumors continued to grow at a reduced rate (Fig. 7A), and MSU + Msmeg-treated mice survived significantly longer than poly I:C-treated mice (Fig. 7B). Depletion of CD4+ T cells just before the commencement of treatment completely abrogated the MSU + Msmeg induced antitumor activity (Figs. 7C, D). In contrast, tumor growth and survival of PBS control and poly I:C treated groups was unchanged by CD4+ depletion (Figs. 7C, D). Depletion of CD4+ T cells after the fourth of 6 peri-tumoral MSU + Msmeg treatments led to faster tumor growth and worse survival compared to the non-depleted MSU + Msmeg treated group (Figs. 7C, D, day 10 depleted groups). These results suggest that CD4+ T cells play a critical ongoing role in MSU + Msmeg induced antitumor immunity that extends beyond the priming phase of CD8+ T cells.
Figure 7.
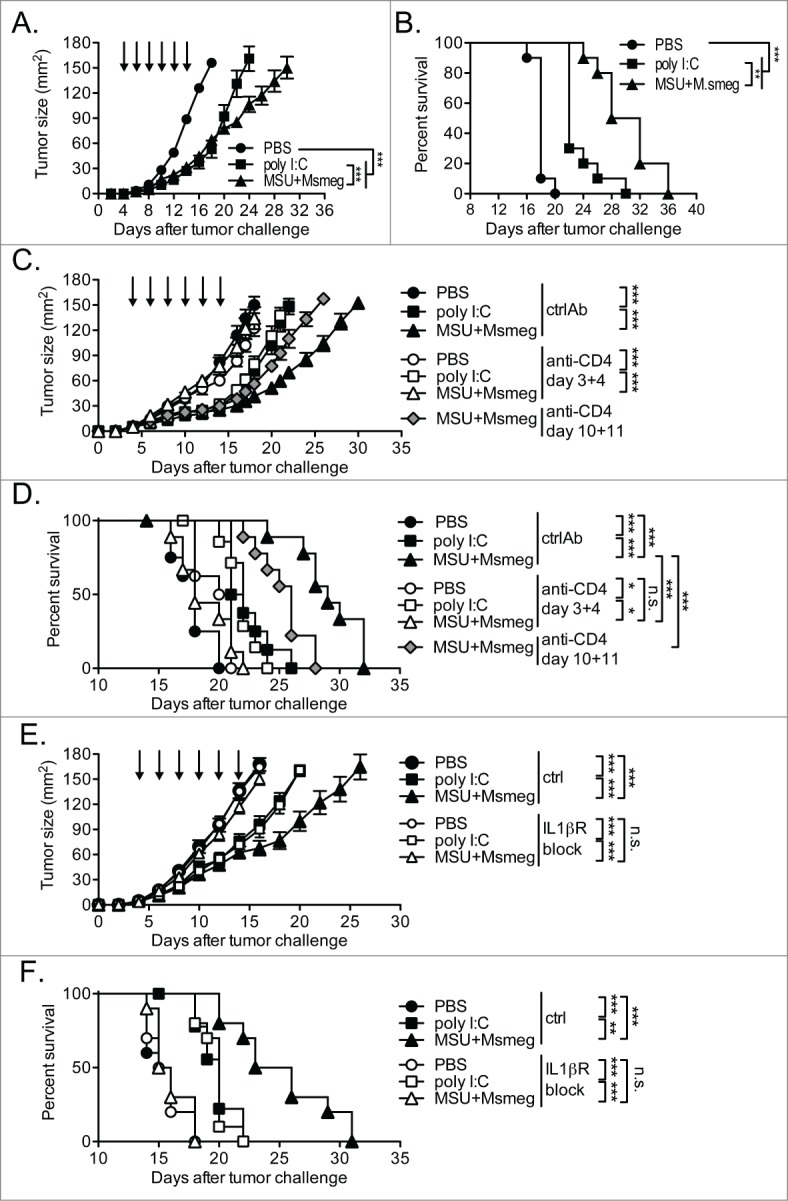
(see previous page). CD4+ T cells and IL-1βR responsiveness are required for sustained tumor protection after treatment with MSU + Msmeg in a thymoma tumor model. C57BL/6 mice were challenged s.c. with E.G7-OVA (A, B) or EL4 thymoma cells (C–F) and given peri-tumoral treatments as indicated by the arrows. (A) Tumor growth and (B) survival of treated E.G7-OVA bearing mice is depicted. (C) Tumor growth and (D) survival of treated EL-4 bearing mice depleted of CD4+ T cells by i.p. injection of anti-CD4 antibodies on either days 3 and 4 or days 10 and 11 as indicated. (E) Tumor growth and (F) survival of treated EL-4 bearing mice given daily i.p. applications of the IL-1βR antagonist Anakinra from days 3–14. All tumor growth data are shown as mean ± SE and analyzed by 2-way ANOVA with Bonferroni's post-test. Survival was evaluated by Log-rank test with Bonferroni's correction for multiple testing. Data are pooled from 2 independent experiments, each with 5 mice/group (A, B, E, F) or 3–5 mice/group (C, D).
We also assessed the effect of blocking IL-1βR on tumor growth and the effect of immunotherapy. In line with our observation that IL-1βR activity was necessary for the priming of tumor-specific CD4+ T cells (Fig. 3), blocking IL-1βR during MSU + Msmeg treatment of EL-4 thymomas abolished the antitumor effect (Figs. 7E, F). In contrast, tumor growth and survival in poly I:C treated groups were not affected by IL-1βR blockade (Figs. 7E, F).
These data show that the sustained antitumor activity conferred by treatment with MSU + Msmeg requires IL-1βR signaling and the ongoing presence of CD4+ T cells, thus conferring a significant host survival benefit compared to treatments, such as polyI:C, that fail to support CD4+ T cell priming.
Discussion
In this study we compare different immune activating agents for ability to support the priming of CD4+ T cell responses to tumor-derived antigens. Previous work demonstrated that the OVA-expressing tumors B16.OVA and E.G7-OVA17,20 are unable to induce the proliferation of OVA-specific OTII CD4+ T cells in vivo, suggesting a defective function of tumor-associated DC. Here we report that treatment with MSU + Msmeg, but not poly I:C, can restore CD4+ T cell priming and increase tumor homing and intratumoral cytokine production by CD4+ T cells. We also show that increased CD4+ T cell priming by MSU + Msmeg was associated with decreased proportions of intratumoral Treg, and reduced expression of PD-1 on CD4+ and CD8+ T cells in tumors. Therefore, treatment with selected immune activating agents can enhance presentation of tumor antigen, and restore immune reactivity in the tumor context.
We previously reported that MSU + Msmeg and poly I:C can both delay tumor growth by a mechanism which requires intact CD8+ and NK cell responses.16 However, as shown in this paper, only MSU + Msmeg supports CD4+ T cell priming and requires CD4+ T cells for its antitumor effect. In contrast, CD4+ T cell responses were not involved in the antitumor effect of poly I:C, as also described in the literature using a malignant mesothelioma tumor model.11 The differential capacity to induce CD4+ T cell priming can be explained by the selective ability of MSU + Msmeg to elevate the levels of IL-1β in serum,16 as blocking IL-1β signaling in vivo using the IL-1βR antagonist Anakinra significantly reduced CD4+ T cell proliferation and selectively abrogated the MSU + Msmeg but not the poly I:C induced antitumor effect. IL-1β is known to enhance CD4+ T cell responses18 and promotes induction of antitumor immunity,21,22 however, its effects on CD4+ T cell responses have not been examined in a tumor context. IL-1β can enhance CD4+ T cell priming both via effects on antigen-presenting cells23 and through direct signaling in CD4+ T cells, leading to increased proliferation and cytokine production.24 Furthermore, IL-1β can activate MyD88 signaling in CD4+ T cells, rendering them refractory to Treg-mediated suppression and increasing their effector function.25 Any of these IL-1β effects could be contributing to the improved CD4+ T cell response we report here.
In addition to increasing the proportion of effector CD4+ T cells in tumors, MSU + Msmeg treatment also reduced the frequency of Treg. This lower frequency may have been purely due to reduced Treg accumulation in the tumor. It is also possible that MSU + Msmeg treatment maintained T effector function and reduced conversion of effector T cells to Treg. Alternatively, MSU + Msmeg treatment may reprogram induced Treg to an effector-like phenotype with downregulation of FoxP3-GFP expression. Downregulation of FoxP3 by IL-1β and IL-6 has been reported in an elegant study using reporter mice, and could also be induced by vaccination with antigen in CFA.26 Therefore, the induction of IL-1β by MSU + Msmeg, possibly in combination with other pro-inflammatory signals also elicited by this treatment, may be a contributing factor in the reprogramming of Treg to CD4+ effector cells. Recent evidence suggests that in some situations reprogrammed Treg may be essential for providing help for antitumor CD8+ T cell responses and effective vaccination.27
The concomitant increase in tumor-infiltrating effector T cells and decrease in Treg in MSU + Msmeg-treated mice resulted in an increased T effector/Treg ratio in tumors. It has been shown that the ratio of T effectors to Treg, rather than just activation of effector cells, is critical for improved antitumor immunity in mice.28 In addition, clinical studies in different types of cancer report improved survival with increased T effector to Treg ratios29-31 even when the absolute counts of Treg or T effector cells were not indicative of outcome.32 Thus the relative presence of effector cells over suppressive cells in tumors is critical for successful antitumor immunity, and treatments such as MSU + Msmeg, which affect both effector and Treg, offer a particularly promising approach to tumor immunotherapy.
We have previously shown that MSU + Msmeg and poly I:C could both stimulate tumor-specific CD8+ T cell proliferation and CD8+ T cell infiltration into tumors.16 However, poly I:C was unable to stimulate CD4+ T cell priming and its antitumor activity was unaltered by CD4+ T cell depletion. As also proposed by others,11,13 these observations suggest that poly I:C is capable of inducing some level of CD4-independent CD8+ T cell priming. The extent of CD8+ T cell priming and tumor infiltration, however, was more pronounced in response to MSU + Msmeg,16 and in this case depletion of CD4+ T cells before or during treatment led to a complete or partial loss of antitumor activity, respectively. These findings are consistent with a report by Marzo et al., who found that CD4+ T cells were dispensable for CD8+ T cell priming, but were required for maintenance of the CD8+ response including tumor infiltration and cytokine production.6
The sustained antitumor activity of CD8+ T cells in the presence of CD4+ T cells might result, at least in part, from decreased expression of exhaustion markers such as PD-1 on CD8+ T cells.7 In our experiments on untreated mice, the proportion of PD-1-expressing tumor-infiltrating CD8+ T cells increased substantially with tumor progression. This increase was not changed after poly I:C treatment, but was significantly reduced in response to MSU + Msmeg. Depletion of CD4+ T cells greatly increased the percentage of PD-1-expressing intratumoral CD8+ T cells, indicating that CD4+ T cells indeed play a critical role in preventing PD-1 upregulation on CD8+ effectors. However, in CD4+-depleted mice, MSU + Msmeg-treated tumors still showed a reduced frequency of PD-1high CD8+ T cells compared to PBS controls. This suggests that MSU + Msmeg can maintain CD8+ T cell function through additional mechanisms that do not require CD4+ T cells, possibly including the effects of cytokines and/or IL-1β on DC33,34 or directly on CD8+ T cells.35 Consistent with an improved maintenance of antitumor CD8+ T cell activity, we observed sustained antitumor activity against murine thymomas only after MSU + Msmeg, but not poly I:C, treatment, and this was CD4+ T cell dependent. Similarly, other authors reported increased antitumor CD8+ responses in mice treated with poly I:C and anti-PD-L1 antibodies.36
In parallel to its effect on CD8+ T cells, MSU + Msmeg treatment also kept the proportion of PD-1-expressing CD4+ T cells low throughout tumor progression. Although the association of PD-1 expression with exhaustion is not as firmly established in CD4+ T cells as in CD8+ T cells, several studies have now linked functional impairment of CD4+ T cells with high levels of PD-1 expression.37-39 For example, in a model of recurrent melanoma, blocking the PD-1 ligand PD-L1 in combination with Treg depletion restored CD4+ effector function and resulted in tumor eradication in the absence of CD8+ T cells.39 These findings illustrate the functional importance of PD-1 expression on tumor-infiltrating CD4+ T cells. Recent successes in clinical trials blocking PD-1 or its ligands further underscore the significance of this pathway for the activation of effective antitumor immunity.40,41
In conclusion, we show that treatment with MSU + Msmeg leads to the activation of a substantial CD4+ T cell response, reduced Treg infiltration in tumors, improved CD8+ T cell activity, and reduced T cell exhaustion resulting in sustained antitumor immunity. MSU has been found to be safe as a vaccine adjuvant for use in humans.42 Mycobacteria such as Bacillus Calmette-Guérin are already used in the clinic for the treatment of bladder cancer,43 and their safety in immune-compromised cancer patients can be further improved by replacing with non-pathogen derived species such as Msmeg44 or by heat-killing.16 We conclude that, on the basis of its efficacy and safety profile, MSU + Msmeg would be a useful basis for immune stimulatory treatments, also in combination with other immunotherapies or chemotherapy, to promote and sustain anticancer immunity.
Materials and Methods
Mice
All mice were bred at the Malaghan Institute of Medical Research Biomedical Research Unit and were age and sex-matched within experiments. C57BL/6J mice were originally from Jackson Laboratories, USA; B6.SJL-Ptrprca mice were from the Animal Resource Centre, Canning Vale, Western Australia. Foxp3GFP mice45 and OTII mice expressing a transgenic TCR specific for I-Ab+OVA323–33946 were originally provided by Drs. A Rudensky and F Carbone, respectively. All experimental protocols were approved by the Victoria University of Wellington Animal Ethics Committee.
Tumor cell lines and tumor challenge
The B16-F1 melanoma and E.G7 OVA-expressing thymoma47 were obtained from the American Type Culture Collection, Manassas, VA. The B16.OVA melanoma48 was kindly provided by Dr Roslyn Kemp, Trudeau Institute. EL-4 thymoma cells were a gift from Prof. Michael Berridge, Malaghan Institute of Medical Research. All cell lines were maintained in complete Iscove's modified Dulbecco medium as described.19,49 Extended in vitro passaging was avoided. Mice were injected with either 105 (B16-F1, B16.OVA) or 5 × 105 (E.G7-OVA, EL-4) cells s.c. into the flank. Tumor size and survival were calculated as described.19
Peri-tumoral treatments
Treatments of 50 µg poly I:C (LMW, Invivogen) or 2 × 106 CFU Mycobacterium smegmatis (Msmeg mc2155) combined with 250 µg MSU (endotoxin < 0.01 EU/10 mg) were given in a total volume of 100 µl PBS injected in the area directly adjacent to the tumors as described.16 PBS (Invitrogen) was used as a vehicle control. Treatment was started once the tumor became palpable (day 4–9, depending on tumor and experiment) and given every second day for 4 to 6 times. This schedule was chosen on the basis of preliminary experiments comparing tumor size and survival after treatment.
Flow cytometry
Lymph nodes (LN) or tumors were digested using DNase I and Liberase TL (Roche). Single cell preparations were resuspended in Flow buffer (PBS with 10mM EDTA, 2% FBS and 0.01% NaN3) and pretreated with anti-mouse CD16/32 (2.4G2) before staining with antibodies specific for the following markers: CD45 (30F11), CD45.1 (A20), CD4 (RM4–5), CD3ϵ (2C11), CD8α (53–6.7), CD25 (PC61), CD69 (H1.2F3), Vα2 (B20.1) (all BD Biosciences), PD-1 (RMP1–30) from Biolegend and CD4 (GK1.5) prepared in-house. Streptavidin-PE or PE-Texas-Red (BD Biosciences) were used to reveal biotinylated antibodies. Dead cells were identified by DAPI or Live/Dead Fixable blue (Invitrogen) exclusion. For intracellular cytokine staining, cell suspensions were incubated for 6 h in Golgi Stop without restimulation. Cells were then stained for surface markers, followed by intracellular staining with anti-IFNγ (XMG1.2) and anti-TNF-α (MP6-XT22) antibodies, or isotype controls (R3–34 or EBRG1), using the BD Cytofix/Cytoperm kit (all from BD Biosciences). Acquisition was performed on a BD LSRII SORP (Becton Dickinson) and data were analyzed using FlowJo version 9.7.4 (Tree Star).
In vivo T cell proliferation assay
Anti-CD4 microbeads (Miltenyi Biotech) were used to positively select CD4+ T cells from spleens and LNs of OTII x B6.SJL-Ptprca mice. CD4+ T cells were subsequently labeled with CFSE as described50 and 1 – 2 × 106 cells were injected i.v. into C57BL/6J hosts bearing established B16.OVA tumors. OTII proliferation was assessed in tumor-dLNs 5 d later.
CD4+ T cell depletion
CD4+ T cells were depleted by i.p. injection of 2 × 250µg of purified anti-CD4 (GK1.5) antibody as indicated in the figures. Depletion of CD4+ T cells was assessed in blood during the experiment and in some tumors on day 15, and was consistently >92%.
IL-1βR blocking
For IL-1βR blockade, mice were given daily i.p. injections of 100 mg/kg of the IL-1βR antagonist Anakinra (Swedish Orphan Biovitrum AB, Stockholm, Sweden) in sterile PBS on the days indicated in the figures.
Statistics
Statistical analyses were performed using Prism 5.0 software (GraphPad). Means ± SE are shown in all graphs. Unless otherwise indicated, data were compared using the Kruskal–Wallis test with Dunn's multiple comparison test. Tumor growth data was analyzed by 2-way-ANOVA with Bonferroni's post test. Survival data was analyzed using the log-rank test with Bonferroni's correction for multiple testing. Differences of p < 0.05 were deemed significant (*), p < 0.01 very significant (**) and p < 0.001 extremely significant (***).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The Authors wish to thank all colleagues at the Malaghan Institute for discussion and suggestions. In particular, the expert support of the Hugh Green Cytometry Core and Biomedical Research Unit staff is gratefully acknowledged.
Funding
This work was funded by research grants from the Cancer Society of NZ, NZ Lottery Health and the Wellington Medical Research Foundation to FR. SK was supported by a PhD Scholarship from Victoria University of Wellington and in part by a Deutscher Akademischer Austauschdiesnt Doktorandenstipendium.
References
- 1.Kim HJ, Cantor H. CD4 T-cell subsets and tumor immunity: the helpful and the not-so-helpful. Cancer Immunol Res 2014; 2:91-8; PMID:24778273; http://dx.doi.org/ 10.1158/2326-6066.CIR-13-0216 [DOI] [PubMed] [Google Scholar]
- 2.Church SE, Jensen SM, Antony PA, Restifo NP, Fox BA. Tumor-specific CD4+ T cells maintain effector and memory tumor-specific CD8+ T cells. Eur J Immunol 2014; 44:69-79; PMID: 24114780; http://dx.doi.org/23087058 10.1002/eji.201343718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aarntzen EHJG, de Vries IJM, Lesterhuis WJ, Schuurhuis D, Jacobs JFM, Bol K, Schreibelt G, Mus R, De Wilt JHW, Haanen JBAG et al.. Targeting CD4+ T helper cells improves the induction of anti-tumor responses in dendritic cell based vaccination. Cancer Res 2013; 73:19-29; PMID:23087058; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-1127 [DOI] [PubMed] [Google Scholar]
- 4.Ossendorp F, Mengedé E, Camps M, Filius R, Melief CJ. Specific T helper cell requirement for optimal induction of cytotoxic T lymphocytes against major histocompatibility complex class II negative tumors. J Exp Med 1998; 187:693-702; PMID: 9480979; http://dx.doi.org/11086036 10.1084/jem.187.5.693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wong SBJ, Bos R, Sherman LA. Tumor-specific CD4+ T cells render the tumor environment permissive for infiltration by low-avidity CD8+ T cells. J Immunol 2008; 180:3122-31; PMID: 18292535; http://dx.doi.org/11086036 10.4049/jimmunol.180.5.3122 [DOI] [PubMed] [Google Scholar]
- 6.Marzo AL, Kinnear BF, Lake RA, Frelinger JJ, Collins EJ, Robinson BW, Scott B. Tumor-specific CD4+ T cells have a major “post-licensing” role in CTL mediated anti-tumor immunity. J Immunol 2000; 165:6047-55; PMID:11086036; http://dx.doi.org/ 10.4049/jimmunol.165.11.6047 [DOI] [PubMed] [Google Scholar]
- 7.Kamphorst AO, Ahmed R. CD4 T-cell immunotherapy for chronic viral infections and cancer. Immunotherapy 2013; 5:975-87; PMID:23998732; http://dx.doi.org/ 10.2217/imt.13.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, Blasberg R, Yagita H, Muranski P, Antony PA et al.. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med 2010; 207:637-50; PMID: 20156971; http://dx.doi.org/10894167 10.1084/jem.20091918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The central role of CD4(+) T cells in the antitumor immune response. J Exp Med 1998; 188:2357-68; PMID: 9858522; http://dx.doi.org/10894167 10.1084/jem.188.12.2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qin Z, Blankenstein T. CD4+ T cell–mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFN gamma receptor expression by nonhematopoietic cells. Immunity 2000; 12:677-86; PMID:10894167; http://dx.doi.org/ 10.1016/S1074-7613(00)80218-6 [DOI] [PubMed] [Google Scholar]
- 11.Currie AJ, van der Most RG, Broomfield SA, Prosser AC, Tovey MG, Robinson BWS. Targeting the effector site with IFN-alphabeta-inducing TLR ligands reactivates tumor-resident CD8 T cell responses to eradicate established solid tumors. J Immunol 2008:1535-44; PMID: 18209049; http://dx.doi.org/24083080 10.4049/jimmunol.180.3.1535 [DOI] [PubMed] [Google Scholar]
- 12.Kawarada Y, Ganss R, Garbi N, Sacher T, Arnold B, Hämmerling GJ. NK- and CD8(+) T cell-mediated eradication of established tumors by peritumoral injection of CpG-containing oligodeoxynucleotides. J Immunol 2001; 167:5247-53; PMID: 11673539; http://dx.doi.org/24083080 10.4049/jimmunol.167.9.5247 [DOI] [PubMed] [Google Scholar]
- 13.Broomfield SA, van der Most RG, Prosser AC, Mahendran S, Tovey MG, Smyth MJ, Robinson BWS, Currie AJ. Locally administered TLR7 agonists drive systemic antitumor immune responses that are enhanced by anti-CD40 immunotherapy. J Immunol 2009; 182:5217-24; PMID: 19380767; http://dx.doi.org/24083080 10.4049/jimmunol.0803826 [DOI] [PubMed] [Google Scholar]
- 14.Vacchelli E, Eggermont A, Sautes-Fridman C, Galon J, Zitvogel L, Kroemer G, Galluzzi L. Trial Watch: Toll-like receptor agonists for cancer therapy. Oncoimmunology 2013; 2:e25238; PMID:24083080; http://dx.doi.org/ 10.4161/onci.25238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Escamilla-Tilch M, Filio-Rodríguez G, García-Rocha R, Mancilla-Herrera I, Mitchison NA, Ruiz-Pacheco JA, Sánchez-García FJ, Sandoval-Borrego D, Vázquez-Sánchez EA. The interplay between pathogen-associated and danger-associated molecular patterns: an inflammatory code in cancer? Immunol Cell Biol 2013; 91:601-10; PMID: 24100386; http://dx.doi.org/18566380 10.1038/icb.2013.58 [DOI] [PubMed] [Google Scholar]
- 16.Kuhn S, Hyde EJ, Yang J, Rich FJ, Harper JL, Kirman JR, Ronchese F. Increased Numbers of Monocyte-Derived Dendritic Cells during Successful Tumor Immunotherapy with Immune-Activating Agents. J Immunol 2013; 191:1984-92; PMID: 23858033; http://dx.doi.org/18566380 10.4049/jimmunol.1301135 [DOI] [PubMed] [Google Scholar]
- 17.Stoitzner P, Green LK, Jung JY, Price KM, Atarea H, Kivell B, Ronchese F. Inefficient presentation of tumor-derived antigen by tumor-infiltrating dendritic cells. Cancer Immunol Immunother 2008; 57:1665-73; PMID: 18311487; http://dx.doi.org/18566380 10.1007/s00262-008-0487-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pape KA, Khoruts A, Mondino A, Jenkins MK. Inflammatory cytokines enhance the in vivo clonal expansion and differentiation of antigen-activated CD4+ T cells. J Immunol 1997; 159:591-8; PMID: 9218573.18566380 [PubMed] [Google Scholar]
- 19.Ataera H, Hyde E, Price KM, Stoitzner P, Ronchese F. Murine melanoma-infiltrating dendritic cells are defective in antigen presenting function regardless of the presence of CD4CD25 regulatory T cells. PLoS ONE 2011; PMID: 21390236; 185663806:e17515; http://dx.doi.org/ 10.1371/journal.pone.0017515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerner MY, Casey KA, Mescher MF. Defective MHC class II presentation by dendritic cells limits CD4 T cell help for antitumor CD8 T cell responses. J Immunol 2008; 181:155-64; PMID:18566380; http://dx.doi.org/ 10.4049/jimmunol.181.1.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Björkdahl O, Dohlsten M, Sjögren HO. Vaccination with B16 melanoma cells expressing a secreted form of interleukin-1beta induces tumor growth inhibition and an enhanced immunity against the wild-type B16 tumor. Cancer Gene Ther 2000; 7:1365-74; PMID: 11059695; http://dx.doi.org/15048719 10.1038/sj.cgt.0242 [DOI] [PubMed] [Google Scholar]
- 22.Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E et al.. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 2009; 15:1170-8; PMID: 19767732; http://dx.doi.org/15048719 10.1038/nm.2028 [DOI] [PubMed] [Google Scholar]
- 23.Khoruts A, Osness RE, Jenkins MK. IL-1 acts on antigen-presenting cells to enhance the in vivo proliferation of antigen-stimulated naive CD4 T cells via a CD28-dependent mechanism that does not involve increased expression of CD28 ligands. Eur J Immunol 2004; 34:1085-90; PMID:15048719; http://dx.doi.org/ 10.1002/eji.200324170 [DOI] [PubMed] [Google Scholar]
- 24.Ben-Sasson SZ, Hu-Li J, Quiel J, Cauchetaux S, Ratner M, Shapira I, Dinarello CA, Paul WE. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc Natl Acad Sci USA 2009; 106:7119-24; PMID:19359475; http://dx.doi.org/ 10.1073/pnas.0902745106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schenten D, Nish SA, Yu S, Yan X, Lee HK, Brodsky I, Pasman L, Yordy B, Wunderlich FT, Brüning JC et al.. Signaling through the Adaptor Molecule MyD88 in CD4(+) T Cells Is Required to Overcome Suppression by Regulatory T Cells. Immunity 2014; 40:78-90; PMID: 24439266; http://dx.doi.org/16344461 10.1016/j.immuni.2013.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, Shah B, Chang SH, Schluns KS, Watowich SS et al.. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 2008; 29:44-56; PMID: 18585065; http://dx.doi.org/16344461 10.1016/j.immuni.2008.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma MD, Hou D-Y, Baban B, Koni PA, He Y, Chandler PR, Blazar BR, Mellor AL, Munn DH. Reprogrammed foxp3(+) regulatory T cells provide essential help to support cross-presentation and CD8(+) T cell priming in naive mice. Immunity 2010; 33:942-54; PMID: 21145762; http://dx.doi.org/16344461 10.1016/j.immuni.2010.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perret R, Sierro SR, Botelho NK, Corgnac S, Donda A, Romero P. Adjuvants that improve the ratio of antigen-specific effector to regulatory T cells enhance tumor immunity. Cancer Res 2013; 73:6597-608; PMID: 24048821; http://dx.doi.org/16344461 10.1158/0008-5472.CAN-13-0875 [DOI] [PubMed] [Google Scholar]
- 29.Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, Jungbluth AA, Frosina D, Gnjatic S, Ambrosone C et al.. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA 2005; 102:18538-43; PMID:16344461; http://dx.doi.org/ 10.1073/pnas.0509182102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu F, Lang R, Zhao J, Zhang X, Pringle GA, Fan Y, Yin D, Gu F, Yao Z, Fu L. CD8+ cytotoxic T cell and FOXP3+ regulatory T cell infiltration in relation to breast cancer survival and molecular subtypes. Breast Cancer Res Treat 2011; 130:645-55; PMID: 21717105; http://dx.doi.org/23804712 10.1007/s10549-011-1647-3 [DOI] [PubMed] [Google Scholar]
- 31.Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, Hiraoka N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer 2013; 108:914-23; PMID: 23385730; http://dx.doi.org/23804712 10.1038/bjc.2013.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Preston CC, Maurer MJ, Oberg AL, Visscher DW, Kalli KR, Hartmann LC, Goode EL, Knutson KL. The ratios of CD8+ T cells to CD4+CD25+ FOXP3+ and FOXP3- T cells correlate with poor clinical outcome in human serous ovarian cancer. PLoS ONE 2013; PMID: 24244610; 238047128:e80063; http://dx.doi.org/ 10.1371/journal.pone.0080063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gerner MY, Heltemes-Harris LM, Fife BT, Mescher MF. Cutting edge: IL-12 and type I IFN differentially program CD8 T cells for programmed death 1 re-expression levels and tumor control. J Immunol 2013; 191:1011-5; PMID:23804712; http://dx.doi.org/ 10.4049/jimmunol.1300652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pang IK, Ichinohe T, Iwasaki A. IL-1R signaling in dendritic cells replaces pattern-recognition receptors in promoting CD8(+) T cell responses to influenza A virus. Nat Immunol 2013; 14:246-53; PMID:23314004; http://dx.doi.org/ 10.1038/ni.2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ben-Sasson SZ, Hogg A, Hu-Li J, Wingfield P, Chen X, Crank M, Caucheteux S, Ratner-Hurevich M, Berzofsky JA, Nir-Paz R et al.. IL-1 enhances expansion, effector function, tissue localization, and memory response of antigen-specific CD8 T cells. J Exp Med 2013; 210:491-502; PMID:23460726; http://dx.doi.org/ 10.1084/jem.20122006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagato T, Lee YR, Harabuchi Y, Celis E. Combinatorial immunotherapy of polyinosinic-polycytidylic acid and blockade of programmed death-ligand 1 induce effective CD8 T-cell responses against established tumors. Clin Cancer Res 2014; 20:1223-34; PMID:24389326; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Antoine P, Olislagers V, Huygens A, Lecomte S, Liesnard C, Donner C, Marchant A. Functional exhaustion of CD4+ T lymphocytes during primary cytomegalovirus infection. J Immunol 2012; 189:2665-72; PMID: 22865914; http://dx.doi.org/21091799 10.4049/jimmunol.1101165 [DOI] [PubMed] [Google Scholar]
- 38.Goding SR, Wilson KA, Xie Y, Harris KM, Baxi A, Akpinarli A, Fulton A, Tamada K, Strome SE, Antony PA. Restoring immune function of tumor-specific CD4+ T cells during recurrence of melanoma. J Immunol 2013; 190:4899-909; PMID: 23536636; http://dx.doi.org/21091799 10.4049/jimmunol.1300271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang D-Y, Song SH, You S, Lee J, Kim J, Racanelli V, Son H, Shin E-C. Programmed death-1 (PD-1)-dependent functional impairment of CD4(+) T cells in recurrent genital papilloma. Clin Exp Med 2013; 14(3):305-13; PMID: 23824147; http://dx.doi.org/21091799 10.1007/s10238-013-0245-6 [DOI] [PubMed] [Google Scholar]
- 40.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al.. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. New Engl J Med 2012; 366(26):2443-54; PMID: 22658127; http://dx.doi.org/21091799 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al.. Safety and Activity of Anti-PD-L1 Antibody in Patients with Advanced Cancer. N Engl J Med 2012; 366(26):2455-65; PMID: 22658128; http://dx.doi.org/21091799 10.1056/NEJMoa1200694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sakamaki I, Inai K, Tsutani H. Safety of intradermal injection of monosodium urate crystals as a vaccine carrier in volunteers. Nucleos Nucleot Nucl 2011; 30:1077-84; PMID: 22132960; http://dx.doi.org/21091799 10.1080/15257770.2011.597368 [DOI] [PubMed] [Google Scholar]
- 43.Sylvester RJ. Bacillus Calmette-Guérin treatment of non-muscle invasive bladder cancer. Int J Urol 2011; 18:113-20; PMID:21091799; http://dx.doi.org/ 10.1111/j.1442-2042.2010.02678.x [DOI] [PubMed] [Google Scholar]
- 44.Young SL, Murphy M, Zhu XW, Harnden P, O'Donnell MA, James K, Patel PM, Selby PJ, Jackson AM. Cytokine-modified Mycobacterium smegmatis as a novel anticancer immunotherapy. Int J Cancer 2004; 112:653-60; PMID: 15382047; http://dx.doi.org/16227984 10.1002/ijc.20442 [DOI] [PubMed] [Google Scholar]
- 45.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol 2005; 6:1142-51; PMID:16227984; http://dx.doi.org/ 10.1038/ni1263 [DOI] [PubMed] [Google Scholar]
- 46.Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based α- and β-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol 1998; 76:34-40; PMID:9553774; http://dx.doi.org/ 10.1046/j.1440-1711.1998.00709.x [DOI] [PubMed] [Google Scholar]
- 47.Moore MW, Carbone FR, Bevan MJ. Introduction of soluble protein into the class I pathway of antigen processing and presentation. Cell 1988; 54: 777-85; PMID: 3261634; http://dx.doi.org/ 10.1016/S0092-8674(88)91043-4 [DOI] [PubMed] [Google Scholar]
- 48.Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol 2005; 174:7516-23; PMID: 15944250; http://dx.doi.org/ 10.4049/jimmunol.174.12.7516 [DOI] [PubMed] [Google Scholar]
- 49.Rich FJ, Kuhn S, Hyde EJ, Harper JL, Ronchese F, Kirman JR. Induction of T cell responses and recruitment of an inflammatory dendritic cell subset following tumor immunotherapy with Mycobacterium smegmatis. Cancer Immunol Immunother 2012; 61(12):2333-42; PMID: 22714285; http://dx.doi.org/ 10.1007/s00262-012-1291-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma JZ-I, Lim SN, Qin JS, Yang J, Enomoto N, Ruedl C, Ronchese F. Murine CD4+ T cell responses are inhibited by cytotoxic T cell-mediated killing of dendritic cells and are restored by antigen transfer. PLoS ONE 2012; 7:e37481; PMID: 22649530; http://dx.doi.org/ 10.1371/journal.pone.0037481 [DOI] [PMC free article] [PubMed] [Google Scholar]