

Abstract
The poor survival rates of refractory/relapsed acute myeloid leukemia (AML) patients after haematopoietic cell transplantation (HCT) requires the development of additional immune therapeutic strategies. As the elicitation of tumor-antigen specific cytotoxic T lymphocytes (CTLs) is associated with reduced relapses and enhanced survival, enhanced priming of these CTLs using an anti-AML vaccine may result in long-term immunity against AML. Cord blood (CB), as allogeneic HCT source, may provide a unique setting for such post-HCT vaccination, considering its enhanced graft-versus-leukemia (GvL) effects and population of highly responsive naïve T cells. It is our goal to develop a powerful and safe immune therapeutic strategy composed of CB-HCT followed by vaccination with CB CD34+-derived dendritic cells (DCs) presenting the oncoprotein Wilms Tumor-1 (WT1), which is expressed in AML-blasts in the majority of patients. Here, we describe the optimization of a clinically applicable DC culture protocol. This two-step protocol consisting of an expansion phase followed by the differentiation toward DCs, enables us to generate sufficient cord blood-derived DCs (CBDCs) in the clinical setting. At the end of the culture, the CBDCs exhibit a mature surface phenotype, are able to migrate, express tumor antigen (WT1) after electroporation with mRNA encoding the full-length WT1 protein, and stimulate WT1-specific T cells.
Keywords: cord blood, dendritic cells, transplantation, vaccination, WT1
Abbreviations
- AML
acute myeloid leukemia
- APC
antigen presenting cell
- CB
cord blood
- CBDC
cord blood-derived DC
- CTL
cytotoxic T lymphocytes
- DC
dendritic cell
- GvHD
graft-versus-host disease
- GvL
graft versus leukemia
- HCT
haematopoietic cell transplantation
- MLR
mixed leukocyte reaction
- moDC
monocyte-derived DC
- WT1
Wilms Tumor 1
Introduction
Survival rates of pediatric patients with acute leukemia have improved significantly over the last decades. However, patients suffering from AML still have a poor prognosis with an estimated 5-y probability of overall survival (pOS) rate of only ∼50%, even after the last and only potentially curative treatment option, HCT.1-5 Hence, there is clear medical need for additional therapies for these patients.
As relapse remains the main obstacle even after HCT, novel immunotherapeutic strategies are being developed aimed at preventing relapses after HCT.6-9 CB is an emerging allogeneic stem cell source with important advantages over conventional stem cell sources (bone marrow or mobilized peripheral blood), including reported enhanced GvL 4,10 in addition to lower graft-versus-host disease (GvHD). It has been recognized that the fast generation of tumor-specific CTLs early after HCT (i.e., in the period of minimal residual disease) may be crucial for the GvL effects.11 As such, enhanced priming of these CTLs using an anti-AML vaccine may result in long-term immunity against AML, further reducing the relapse rates and enhancing survival after HCT. Furthermore, an additional anti-AML activity can be achieved due to better selection of donors taking NIMA (non-inherited maternal antigen: available in most of the CB grafts) and anti-IPA (inherited paternal antigen) immunity into account, nicely described by van Rood et al. and commented by Burlinghame.12,13 This makes the use of CB as cell source for HCT (and adjuvant immunotherapies) especially attractive.
In vivo T cell priming and proliferation requires the presentation of antigens and activation signals provided by a specialized group of antigen presenting cells (APC); in particular DCs. The inefficiency of host DCs to initiate antitumor immunity led to the development of vaccination strategies, where ex vivo-generated and expanded DCs are loaded ex vivo with specific antigens that are expressed by tumor cells. Accordingly, these DCs are given back to the patients, thus directing the targets for the in vivo-generated CTLs.14 In either bone marrow or mobilized peripheral blood HCT settings, the donor can provide the source of DCs needed for vaccination, either via primary DCs15 or CD14+ monocytes.16 In the CB-HCT setting, the amount of donor material is however limited. As such, we will use the CD34+ cell population, containing the haematopoietic stem cells, to generate our CBDC vaccine. Using the CD34+ population will allow for a massive expansion of the myeloid precursors17 before differentiating them into DCs, which is required for yielding sufficient CBDCs for vaccination.
Hence, it is our goal to develop a powerful and safe immune therapeutic strategy composed of CB-HCT followed by vaccination with CBDCs presenting the oncoprotein WT1, recently ranked the number one cancer vaccine target antigen.18 This zinc finger transcription factor is overexpressed in the majority of leukemias (∼90% in AML) as well as various solid tumors, while very low expression is seen in normal tissues.19 With regard to antigen loading, using a full-length protein overcomes HLA-peptide restrictions allowing the same therapy to be used irrespective of HLA-type. Using mRNA electroporation to express full length WT1 protein warrants cytoplasmic expression and optimal class I presentation of WT1 peptides.20 This approach was previously used by Van Tendeloo et al. who observed clinical responses as well as WT1-specific CTLs in patients with AML.16 We hypothesize that increased WT1 antigen presentation provided by a CBDC vaccine combined with the intrinsic increased proliferative capacity of the T cells in the CB graft 21,22 will result in fast differentiation and proliferation of WT1-specific CTLs early after HCT.
Here we describe the optimization of a GMP (good manufacturing practice) applicable culture protocol to generate sufficient CBDCs from a limited amount of CD34+ cells, that are mature, able to migrate and express tumor antigen (WT1) to stimulate specific T cells.
Results
Generation of CBDCs from CD34+ stem cells
The first aim was to develop a culture protocol to generate sufficient CBDCs for multiple vaccinations, which would require a 250–500-fold expansion of CD34+ cells. The requirement for GMP-grade culture conditions excludes the use of animal-derived components, for instance fetal calf serum, as serum supplement. We tested human AB serum, which has been used before in various clinical trials using DCs, platelet lysate, or TGF-β to supplement differentiation media. Platelet lysate and TGF-β resulted in poor CBDC induction compared with AB serum (data not shown), which was used for all further experiments.
In the one-step protocol CD34+ cells were cultured for one week by GM-CSF, SCF and TNFα (GST) in 5% AB serum (Fig. 1A). The two-step protocol consisted of a one-week culture of CD34+ cells in expansion medium containing Flt3L, SCF, IL-3 and IL-6 (FS36), followed by a one-week culture in differentiation medium containing 5% human AB serum, further supplemented with GM-CSF, Flt3L, SCF, and IL-4/L15 (GFS4 or GFS15) or GM-CSF and IL-4 (G4) (Fig. 1A). The one-step protocol (GST) induced an immediate differentiation of the CD34+ population as depicted by expression of CD11c+, HLA-DR+ and CD1a+ DCs after the first week (Fig. S1A). The expansion potential averaged 6.5 times over one week of culture (Fig. 1B). As this resulted in insufficient cell numbers for multiple vaccinations, this protocol was excluded in further studies.
Figure 1.

Generation of CBDCs from CB-derived CD34+ stem cells. (A) Different protocols for generating DCs from CB-derived CD34+ cells. (B) Expansion of CD34+ cells for the different protocols. (C–E) Phenotype of DCs for the G4, GFS4 and GFS15 protocols. (C) Gating strategy with flow cytometry and cytospins of DCs stained with May–Grünwald–Giemsa. (D) Expression of CD1c, CD1a, CD141, CLEC9A, CD207, CD209, BDCA2, CD123, CD14, CD16, CD64 and CD56 on DCs compared to isotype control. (E) Number of DCs generated from CD34+ cells. Data represent at least three independent experiments.
The two-step protocols produced an average expansion of 600 fold for FS36/GFS4, 300 fold for FS36/GFS15 and 150 fold for FS36/G4 (Fig. 1B). Moreover, depending on the total number of CD34+ cells at the start, this expansion phase could be repeated for at least two more weeks (with an additional 10 times expansion each week) without affecting final DC differentiation (Fig. S1B). Flow cytometric analysis of the cells after the expansion phase showed that all cells were of myeloid lineage (CD33+), showed no differentiation into CD11c+ DC, and that some CD34+ CB cells remained (Fig. S1C). This suggests that the culture consists of a pool of myeloid precursors at the end of the expansion phase. In the second week these myeloid precursors were stimulated to differentiate into DCs using the various differentiation media. After differentiation, DCs were identified based on CD11c+ HLA-DR+ expression, and typical dendrites were seen with May–Grünwald staining after flow sorting (Fig. 1C). Differentiation with GFS4 induced the highest percentage of CD11c+ HLA-DR+ CBDCs. Further analysis of the expression levels of various DC markers (CD1a, CD1c, CD141, CLEC9A, CD207, BDCA2, CD123, CD16) showed no clear differences between the different protocols, except for the CD209 expression that was absent after differentiation with GFS15 (Fig. 1D). Although CD141 (BDCA3) was expressed on the majority of the CBDCs, no CLEC9A expression was detected on the CBDCs. Based on the absence of BDCA2 and CD123 expression on the CD11c− HLA-DR+ population, we concluded that no pDC were present in these CBDC cultures (Fig. S1D).
Based on the total cell expansion data (Fig. 1B) combined with the flow cytometry analyses of the percentage CD11c+ HLA-DR+ CBDCs, the number of CD11c+ HLA-DR+ CBDCs generated per CD34+ CB cell was calculated (Fig. 1E). Based on these data the GFS4 protocol was selected for further studies, since this protocol showed the highest potential to generate sufficient numbers of CBDCs.
Antigen uptake and processing by CBDCs
Antigen uptake and processing are essential features of DCs. In order to further analyze the functionality of the CBDCs, we first assessed the antigen uptake and processing capacity. Uptake of dextran-FITC (70 kD) increased dose-dependently in the CD11c+ HLA-DR+ population, while there was minimal uptake in the CD11c− HLA-DR+ CBDCs (Fig. 2A). Pulse/chase experiments using DQ-BSA, a protein that becomes fluorescent upon unfolding as a consequence of proteolysis, however showed maximal fluorescence irrespective of the different HLA-DR+ CBDCs. Within one day, all DQ-BSA was completely degraded as seen by a decline in DQ+ signal (Fig. 2B). The differences within the uptake and processing could be mediated due to different uptake mechanisms. Dextran-FITC is taken up by mannose-receptor mediated endocytosis, while DQ-BSA enters the cell via macropinocytosis.23 In conclusion, the CD11c+ HLA-DR+ population has the capacity of antigen uptake and processing.
Figure 2.
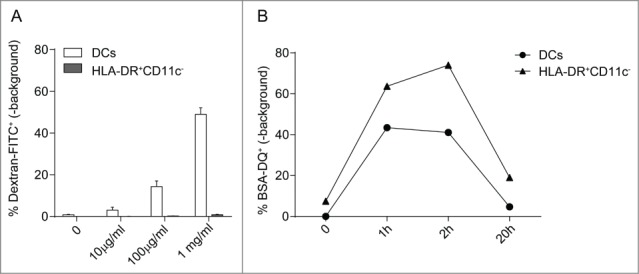
Antigen uptake and processing by CBDCs. (A) Uptake of increasing doses of FITC-dextran by DCs and HLA-DR+CD11c− populations. The percentage of FITC+ signal taken up at 37°C is shown minus the FITC+ signal detected at 4°C. (B) The processing of BSA-DQ is shown by DCs or HLA-DR+CD11c− population at indicated time points. The DQ-signal seen at 4°C is subtracted from the DQ+ cells at 37°C. Data represent three independent experiments.
Maturation and Migration of CBDCs
The next step was the optimization of CBDC activation/maturation. In this regard we tested the effect of a GMP-grade available cytokine mixture (IL-1β, IL-6, TNFα and PGE2) on CBDCs that has been regularly used in clinical DC vaccination trials.24,25 The effect of this “CYTOMIX” was compared to that of an agonistic anti-CD40 antibody known for its stimulatory effect on Mo-DC maturation. Both CYTOMIX and anti-CD40 induced maturation of CBDCs as shown by the upregulation of CD80, CD86, CD83 and CCR7 (Fig. 3A). All mature cells were CD11c+ HLA-DR+ supporting the notion that these cells are differentiated further into primary DC, whereas the CD11c− fraction seems still in a myelo/monocytic developmental stage. Combined results from 30 individual experiments/donors (Fig. 3B) showed a highly significant increase in the percentage of CD83-expressing cells. The increase in percentages of CD86 and CCR7 positive cells were smaller and not significant since for both markers the control samples sometimes already expressed these markers. The exact reason for this is not known but may relate to handling of the individual cultures.26 In these experiments CD83 is therefore the most specific and discriminative marker for CBDC maturation.
Figure 3.

Maturation and migration of CBDCs. (A) CBDCs are matured for 24 h with either anti-CD40 or CYTOMIX containing IL-1β, IL-6, TNFα and PGE2. Live cells are further gated on HLA-DR and CD11c to analyze CD80, CD86, CD83 and CCR7 expression or isotype as a control (solid gray graph). (B) Plots are shown of whole culture followed by CD86, CCR7 and CD83 expression on DCs after maturation for 24 h with CYTOMIX. (C) Migration assay in a transwell system with the whole culture matured for 24 h with CYTOMIX or medium as a control in the upper compartment. In the lower compartment CCL19 was added, or medium as a control for 2 h. One representative FACS plot is shown and a bar graph of three independent experiments. (D) CBDCs are matured for 24 h with CYTOMIX, washed and subsequently stimulated for another 24 h using anti-CD40 or medium control. CBDC supernatants were analyzed for a variety of cytokines using Luminex. Data represent three (C and D) or more (A, B) independent experiments. Error bars represent the SEM. *, P < 0.05.
The migratory capacity of DCs is an important functional feature to ensure traveling to lymph nodes and subsequent T cell activation. As depicted in Figure 3C, maturation of the CBDC culture with CYTOMIX induced a strong increase in CCL19 specific migration of CD11c+ HLA-DR+ CBDCs in an established in vitro assay for CCR7-dependent migration.20,27
Next, we investigated the cytokine production by the matured CBDC vaccine after activation of CD40 signaling using an agonistic anti-CD40 antibody. Overall the cytokines produced after the CD40 stimulation show a proinflammatory profile and suggest a Th17 inducing capacity as depicted by enhanced IL-6 and IL-23p19 production (Fig. 3D). Very low levels of IL-12p70 were detected (Fig. 3D) which is in line with earlier studies showing low release of IL-12p70 from DCs using PGE2 containing CYTOMIX.28 As such, we tested whether addition of IFNγ, alone or combined with R848, to the CYTOMIX was able to increase the levels of IL-12p70 after CD40 stimulation as show previously for monocyte-derived DCs (moDCs).29 IL-12p70 was rapidly induced after CD40 stimulation when CYTOMIX was combined with IFNγ, and levels were further enhanced by R848 (Fig. S2B). These data show that CBDCs have the capacity to produce IL12p70 in this CD4+ help (CD40L-CD40 and IFNγ) mimicking condition.
To address the T cell stimulatory capacity of the matured CBDC culture, we first performed an allo-MLR (mixed leukocyte reaction). CBDCs were able to stimulate allogeneic T cell proliferation compared to responder cells alone (histogram), which did not show any proliferation. Both CD4+ (Fig. 4A) as well as CD8+ T cells (Fig. 4B) are stimulated by CBDCs. These data confirm that CBDC express the necessary signals to stimulate CD8+ and CD4+ cells in a non-antigen driven manner without specifically enhancing CD3+CD4+CD25+CD127− regulatory T cells (data not shown). Analyzing the MLR culture supernatants showed a TNFα/IFNγ skewed response, whereas IL-10 and IL-17 remained low (Fig. S3).
Figure 4.
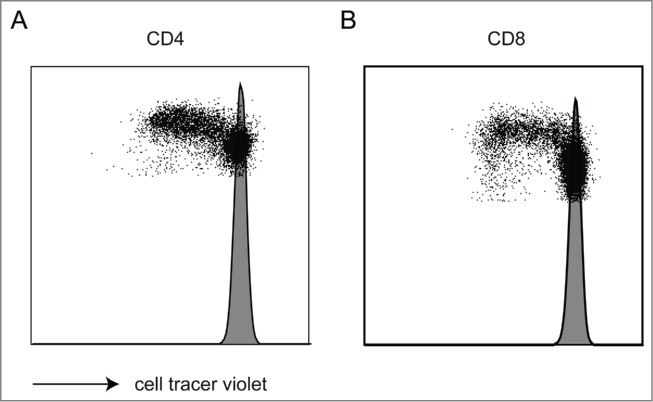
Allogeneic-mixed leukocyte reaction. (A, B) An allo-MLR reaction of CBDCs and CB CD3 lymphocytes was performed. Cell proliferation was studied within the CD4 (A) and CD8 (B) population after 3 d by cell trace violet dilution. Shaded histogram represents the unstimulated CD3 lymphocytes. Data represent four independent experiments.
Characterization of HLA-DR negative population in the CBDC culture
Using the whole heterogeneous CBDC culture as vaccine, without any need for further purification steps, is attractive for clinical application. This however requires a full characterization of the cells in this culture, especially the population of cells negative for HLA-DR. All cells in the CBDC cultures express CD33 and lacked the expression of CD3, CD19, CD20, and CD56 (data not shown) indicating that all cells are of the myeloid lineage. Using the combined expression of CD11b and CD66b, four different subsets could be detected within the HLA-DR− population with the double negative and double positive representing the major populations (Fig. 5A and B). Since similar markers are used to identify myeloid-derived suppressor cells,30,31 we tested whether HLA-DR− cells could suppress T cell proliferation using a T cell suppression assay. In summary, none of the populations inhibited the anti-CD3 induced T cell proliferation but rather stimulated the proliferation of T cells in a dose dependent manner (Fig. 5C and D). In addition, the non-DC fraction did not induce increased levels of the inhibitory cytokine IL-10 or affect the production of TNFα during an MLR compared to the CBDC vaccine culture or sorted CBDCs (data not shown).
Figure 5.

HLA-DR negative population in the CBDC culture. (A–D) Characterization of HLA-DR−CD11c− population. (A) Gating strategy used for identifying different subsets. (B) Percentage of different subsets. (C) Suppression assay with cell trace violet labeled effector cells, activated by anti-CD3. Different amounts of sorted cell populations are added as indicated, Tregs were sorted as a positive control. After 4 d of co-culture, effector T cell proliferation is assessed by cell trace violet dilution as shown with FACS plots or (D) in a bar graph. Data represent at least three independent experiments.
CBDCs stimulate WT1-specific T cells
There are several possibilities to load DCs with tumor antigens, all of which may have specific advantages or disadvantages for the induction CD4+ and CD8+ T cell responses. For WT1, electroporation with the full-length WT1 mRNA has been shown to induce intracellular expression, processing and presentation in moDCs.32 In addition, this technique has been used in several clinical trials without severe side effects and thus is considered safe.16 An optimal concentration of mRNA during electroporation of CBDC was determined using different amounts of EGFP mRNA (Fig. 6A). Electroporation of 20 μg WT1 mRNA per 200 μL of cells resulted in WT1 protein expression in CBDC cultures from various donors as observed by Western blot (Fig. 6B).
Figure 6.

CBDCs stimulate WT1-specific T cells. RNA-electroporation of CBDCs. (A) GFP expression 4 h after increasing doses of GFP-electroporation measured by FACS. (B) WT1 Western blot of three different donors 4 h after electroporation with WT1 or sham as a control. K562 cell-line was used as a positive control. (C, D) Cytokine production by WT1 specific T cell clone after 4 h co-culture with CBDCs of seven different donors (dots) after WT1 electroporation. Sham electroporated DCs with (squares) or without (triangles) WT1 peptide serve as controls. (C) IFNγ and (D) TNFα production. Data represent at least three independent experiments.
We next electroporated CYTOMIX-matured CBDCs with WT1 or sham as control and, after an incubation step to allow protein expression, processing and MHC loading, co-cultured the CBDCs with a WT1126–134 peptide-specific T cell clone. Notwithstanding a large donor-to-donor variation, four out of seven donors showed elevated levels of IFNγ (Fig. 6C) and/or TNFα production (Fig. 6D) confirming the presentation of WT1126–134 peptides in the HLA-A2 molecules after electroporation with full-length WT1 mRNA.
Discussion
The poor survival rates of refractory/relapsed AML patients after HCT warrant the development of additional immunotherapeutic treatments strategies.1–5 As such, we optimized strategies to generate sufficient amounts of CBDCs with the capacity to initiate activation and proliferation of AML antigen-specific CTLs in a pre-clinical setting. The materials and tools used are all GMP-available which makes quick translation to a clinical trial feasible.
Since only about 20% of the CB graft will be available for the preparation of the DC vaccine (80% is used for the actual CBT) extensive cell expansion is an essential part of the culture strategy to generate sufficient CBDCs. The present data show that a two-step protocol consisting of a massive expansion of myeloid precursors from the hematopoietic stem cells followed by the differentiation of part of these precursors toward DCs induced an average of 600 CBDC culture cells for each initial CD34+ cell. Even when considering the worst case scenario using the lowest amount of cells in the 20% fraction of the CB-unit of about 500,000 CD34+ cells, resulting in about 250,000 cells after CD34+ CliniMACS (personal observations), and taking into account a loss of about 25% of the cells during the remainder of the protocol, this protocol will still generate up to 90 × 106 cells. This amount should be enough for three vaccinations using 1 × 106 cells/kg in a 30 kg pediatric patient. However, since the CB graft is, in addition to the regular selection criteria, also selected based on a minimal number of CD34+/kg infused per CB graft, the odds are that higher numbers will be generated.
Maturation of the CBDC culture using CYTOMIX induced upregulation of maturation markers and allowed specific migration toward a CCL19 chemokine gradient comparable with previous reports with other DC sources.20,27 The CYTOMIX-treated CBDC culture stimulated CD4+ and CD8+ proliferation in an allogeneic MLR setting and electroporation with full-length WT1 mRNA led to expression of the protein and presentation of a HLA-A2-specific WT1 peptide. As such, the proposed protocol fulfills the requirement for application as a DC vaccine for treatment of pediatric AML patients.
Although this optimal culture strategy resembles to some extend the protocol described by Poulin et al.,33 we were unable to generate substantial amounts of CD141+CLEC9A+ DC. This could be due to the use of human AB serum instead of FCS, which failed to induce this subset even when the remainder of the protocol was unchanged. In addition, CD141+CLEC9A+ DC can only be generated using 96-well plates instead of flasks, which strongly reduced the viability of the CBDC cultures (data not shown). The addition of human AB serum is essential for the differentiation of DC in this presented system, since possible alternatives for AB serum like platelet lysate or TGFβ induced less DC differentiation (data not shown). It has been reported that the use of IL-15 instead of IL-4 may result in the generation of more powerful DC, mostly moDC.34,35 In contrast to moDCs, IL-15 failed to noticeably modify the CD56 expression CBDC (not shown). CD209 (DC-SIGN) expression was absent in the IL-15 differentiated CBDC, which fit with the observations that IL-4 is the inducer of CD209 expression in moDC cultures.36
Cumulative data (n = 20) showed that about 35% (+/−10%) of our CBDC culture consisted of CD11c+ MHCII+ expressing DCs after the described protocol. We are currently investigating whether the expression of specific markers or genes at the end of the expansion period could predict the differentiation of a certain subset of cells toward DCs. Although small adaptations like the addition of low levels of TNFα to the differentiation medium did increase the percentage of DCs,37 we ended up with a smaller amount of DCs because of its detrimental effect on viability (not shown).
With regard to the antigen uptake, a clear difference was observed between CD11c+ HLA-DR+ and the CD11c− HLA-DR+ population, with the latter being unable to take up 70 kD dextran molecules probably because of a lack of a specific uptake receptor.38 Although this difference in uptake may not be important in our current setting using mRNA electroporation for loading the DCs with tumor antigen, we are currently further exploring the expression of different uptake receptors that may be useful for different antigen loading strategies, like antibody-mediated targeting or uptake of apoptotic or necrotic tumor cells.39 Both CYTOMIX and agonistic anti-CD40, an antibody developed for use in cancer immune therapy, provided strong maturation of CBDC. In addition, newly developed methods for antigen loading and DC maturation, like the use of long peptides linked with TLR ligands or loading this compounds in PLGA particles 40,41 may in the future provide alternative clinical-grade available maturation and antigen-loading strategies.
With regard to the presentation of WT1 peptides after electroporation of the CBDC, using mRNA encoding the full-length protein, we found highly variable induction of generally low levels of IFNγ expression by a WT1126–134 peptide-specific T cell clone. These data are in line with those obtained using moDC electroporated with the same mRNA construct followed by co-culture with a similar WT1126–134 peptide-specific T cell clone.42 Considering that the T cell clone used here is only responsive to one of the many possible peptides in the WT1 proteins the low percentage of responding T cells is not surprising.43 The clinical potential of our CBDC WT1 vaccine is further supported by a clinical trial using an moDC vaccine electroporated with the same WT1 mRNA construct.16 In this trial WT1-specific tetramer-positive CD8 cells were enhanced in some of the HLA-A2 positive patients in this trial and this was associated with induction of clinical and molecular (WT1 mRNA levels) remission.16 Variation between the donors was not related to the amount of DCs generated in the culture or the success of electroporation (generally between 80–95%). Another attractive explanation may simply be that the variability between donors in protein degradation and processing leads to different WT1 peptides being loaded into MHC molecules.
In summary we describe the pre-clinical development of a WT1 mRNA-electroporated DC vaccine with feasibility to generate sufficient amounts of cells for clinical application from the limited number of CD34+ CB stem cells. In addition, we provided proof-of-principle for the capacity of these cells to present antigen to WT1-specific T cells. The next step in the bench to bedside approach is the translation of our pre-clinical protocol toward generation of this DC-vaccine under GMP conditions, apply for ethical approval and study the above-described concept in a phase II clinical study. Early specific WT1 immunity in an allogeneic setting may have impact on the tumor load and survival.
Materials and Methods
CB collection and CD34 isolation
Umbilical CB was collected after informed consent was obtained according to the Declaration of Helsinki. The ethics committee of the University Medical Center Utrecht approved these collection protocols. CB mononuclear cells were isolated from human umbilical CB by density centrifugation over Ficoll-Paque solution (GE Healthcare Bio-Sciences AB). CD34+ cells were isolated from fresh CB using magnetic bead separation (Miltenyi Biotec) resulting in a 80–95% pure CD34+ population after running two columns, as determined with flow cytometry. The CD34− population was frozen and stored at −80°C for use in MLR experiments.
CBDC culture
In the single-step protocol, CD34+ cells are cultured in X-VIVO 15 supplemented with GM-CSF (100 ng/mL), SCF (25 ng/mL) and TNFα (2.5 ng/mL) and 5% human AB serum (Sanquin) for 7 d.44 The two-step protocol consists of an expansion and differentiation phase. In the expansion phase 5 × 104 CD34+ cells/mL are cultured in X-VIVO 15 supplemented with Flt3L (50 ng/mL), SCF (50 ng/mL), IL-3 (20 ng/mL) and IL-6 (20 ng/mL) for 7 d. After washing, the cells are differentiated at 2 × 105 cells/mL in X-VIVO 15 containing 5% human AB serum and supplemented with Flt3L (100 ng/mL), SCF (20 ng/mL), GM-CSF (20 ng/mL) and IL-4 (20 ng/mL) for another 7 d.33 In indicated experiments the IL-4 was replaced with IL-15 (100 ng/mL) during the differentiation.45 Recombinant human GM-CSF, SCF, Flt3L, IL-1beta, IL-3 and IL-6 were all obtained from Miltenyi Biotec. Recombinant IL-4, IL-15 and TNFα from Immunotools. To induce maturation an agonistic anti-CD40 (Bioceros), or CYTOMIX, a combination of IL-1β, IL-6 and TNFα (all used at 10 ng/mL) and PGE2 (1 μg/mL) from Pfizer, was added to the DCs for 24 h. In some experiments the CYTOMIX (with the addition of IFNγ +/− R848) matured CBDC cultures were washed and stimulated with the agonistic anti-CD40 for 24 h. Supernatants were analyzed using multiparameter Luminex (BIO-RAD) or IL-12p70 ELISA (e-bioscience).
Flow cytometry
Anti-CD1c (L161), anti-CD3 (UCHT1), anti-CD16 (3G8), anti-CD64 (10.1), anti-CD207 (10E2), anti-CD209 (9E9A8), anti-DNGR-1 (CLEC9a/ 8F9), anti-HLA-DR (L243) and anti- TNFα (Mab11) were purchased from Biolegend. Anti-BDCA-2/CD303 (AC144) and anti-BDCA-3/CD141 (AD5–14 H 12) were obtained from Miltenyi Biotec. Anti-CD1a (HI149), anti-CD8α (RPA-T8), anti-CD11c (B-ly6), anti-CD14 (M5E2), anti-CD56 (B159), anti-CD80 (L307.4), anti-CD83 (HB15e), anti-CD86 (IT2.2), anti-CD123 (7G3) and anti-IFNγ (4SB4) were purchased from BD Bioscience. Anti-CD4 (RPA-T4), anti-CD40 (5C3) and isotype-matched controls antibodies were purchased from eBioscience. Anti-CCR7/CD197 (FR11-11E8) was purchased from R&D systems.
Cells were incubated on ice with mouse serum (Jackson immunoresearch) to block Fc receptors, and stained with appropriate antibody combinations. Multiparameter analysis was performed on a FACS Canto II (BD) flow cytometer. Dead cells were excluded by scatter gating. Analysis was performed using FlowJo software (Tree Star, Inc.).
Sorting and histochemical staining CBDC
CBDC cultures were stained with Abs directed against CD11c and HLA-DR. CD11c+HLA-DR+ cells were sorted using a FacsARIA II cytometer (purity > 95%). May–Grünwald–Giemsa staining was used to analyze DC cultures. Cytospins were prepared from 5 × 104 sorted CBDCs and were fixed in methanol for 5–10 min. After fixation cytospins were stained in May–Grünwald (J.T.Baker, The Netherlands) for 5 min, rinsed in buffered water (pH 6.8), and the nuclei were counterstained with Giemsa solution (Merck kGaA, Darmstadt, Germany) for 15 min.
Uptake and processing assay
For uptake, CBDC culture was incubated with 10, 100 μg/mL or 1 mg/mL dextran-FITC (m.w. 70.000; Sigma) for 30 min at 4°C to measure non-specific binding or at 37°C to measure specific uptake. Cells were then washed extensively with ice-cold PBS, 0.1% FCS, and 0.05% NaN3 and labeled on ice with appropriate antibodies. The actual uptake was determined as the percentage of FITC+ cells incubated at 37°C minus the percentage of FITC+ cells incubated at 4°C.
For analysis of processing by CBDC cultures we used DQ Green BSA, a self-quenched dye conjugate of BSA. CBDCs were incubated with 0.5 μg/mL DQ Green BSA at 4 or 37°C for 10 min. After extended washes CBDCs were stained with the appropriate antibodies at indicated time points.
Transwell migration assay
In vitro migration assays were performed using 24 transwell (3 μm pore size) plates (Greiner). In brief, 400.000 CBDCs in 200 μL culture medium (X-VIVO 15 with 5% human AB serum) were plated in the upper compartment. Culture medium, either alone or supplemented with 250 ng/mL CCL19 (R&D systems), was added to the lower compartment. After 2 h cells were collected from the lower compartment and analyzed using flow cytometry.
Mixed leukocyte reaction
CD3 cells purified from allogenic CD34− cells using anti-CD3 magnetic microbeads (Miltenyi). These responder CD3 cells (1 × 106/mL) were then labeled with cell trace violet (5 µM; Invitrogen), and co-cultured with matured CBDCs (2 × 105/mL) as stimulator cells. Unstimulated cell trace violet-labeled cells served as negative control. After 3 and 5 d, cells were stained with CD3, CD4, CD8 and analyzed using a FACS Canto II (BD). T cell proliferation was assessed by quantifying the percent of cell trace violet-diluted (Pacific bluelow) cells within the CD3+CD4+ or CD3+CD8+ gate after background subtraction. MLR supernatants were analyzed using multiparameter Luminex (BIO-RAD).
T cell suppression assay
From the CD34− cells, one part is labeled with 2 µM cell trace violet for 7 min at 37°C and extensively washed. 25,000 responder cells were plated into anti-CD3-coated wells (OKT-3, 1.5 µg/mL). The other part is used for sorting regulatory T cells (Tregs) as a positive control for suppression. First, the CD4+ cells are enriched using a CD4 T Lymphocyte Enrichment Set (BD Biosciences). The magnetic cell isolations were performed according to the manufacturer's instructions. Subsequently, CD4+CD25+CD127− T cells were sorted as Treg from the CD4+ cells by FACS Aria (BD Biosciences) and added in different ratios to the labeled responder cells. Fractions of the CBDCs, based on their CD11c and HLA-DR expression representing CD11c−HLA-DR+, CD11c+HLA-DR+ (DCs) en CD11c−HLA-DR− (Neg), were sorted by FACS and added in different ratios. Cells were cultured for 4 d and proliferation was measured by flow cytometry on a FACS Canto (BD Biosciences). All data were analyzed using FlowJo software.
In vitro mRNA transcription, mRNA electroporation and protein expression analysis
In vitro transcribed mRNA was produced from linearized pGEM4Z/WT1/A64 and pGEM4Z/EGFP/A64 plasmids using the T7 mMessage mMachine Ultra kit (Ambion, Life Technologies, NY, USA). CBDCs from HLA-A*0201+ individuals were loaded with these in vitro transcribed WT1-encoding mRNA or GFP-encoding mRNA as a control by electroporation as previously described,46 with minor modifications. Briefly, 5–10 × 106 cells in 200 μL Optimem media were transferred to a 4 mm electroporation cuvette (Bio-Rad, Hercules, CA, USA) and electroporated with 10–30 µg RNA by an exponential decay pulse of 300 V for 7 ms using the Gene Pulser Xcell device (Bio-Rad). To evaluate protein expression, the EGFP expression was assessed using flow cytometry, Western blot was used to determine the expression of WT1 protein after mRNA electroporation. WT1 electroporated CBDCs, sham electroporated, to serve as a negative control or K562 cell line for a positive control, were lysed by adding laemmli buffer (2% SDS, 10% glycerol, 62.5 mM Tris pH 6.8) and samples are boiled for 10 min at 95°C. Aliquots of 25 μg of protein were electrophoresed on 10% SDS-polyacrylamide gels, transferred to polyvinylidene difluoride membranes and probed with mouse antibodies specific for WT1 (Dako; 0.5 μg/mL) or goat polyclonal-actin (Santa Cruz; 1:5000). Horseradish peroxidase-conjugated rabbit anti-goat or rabbit anti-mouse (Dako, 1:5000) were used as secondary antibody. The fluorescent intensity was measured on the ChemiDoc system (Bio-Rad).
WT1 antigen presentation
WT1 mRNA or sham-electroporated CBDCs (50,000) were co-cultured with an HLA-A2–restricted WT1-specific T cell clone recognizing the WT1126–134 epitope (50,000 T cells) at a DC-to-T cell ratio of 1:1 for 4 h in the presence of Golgi-stop (1/1500; BD Bioscience). Sham-electroporated CBDCs loaded with/without WT1 peptide (Think Peptides) were used as a positive control. The T cells were subsequently stained for surface markers and, after fixation and permeabilization with the BD fix/perm (BD Biosciences), labeled with anti-IFNγ and anti-TNFα antibodies (BD Biosciences), followed by flow cytometry-based analysis.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Klusmann J-H, Reinhardt D, Zimmermann M, Kremens B, Vormoor J, Dworzak M, Creutzig U, Klingebiel T. The role of matched sibling donor allogeneic stem cell transplantation in pediatric high-risk acute myeloid leukemia: results from the AML-BFM 98 study. Haematologica 2012; 97:21-9; PMID:21933851; http://dx.doi.org/ 10.3324/haematol.2011.051714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beier R, Albert MH, Bader P, Borkhardt A, Creutzig U, Eyrich M, Ehlert K, Gruhn B, Greil J, Handgretinger R et al.. Allo-SCT using BU, CY and melphalan for children with AML in second CR. Bone Marrow Transplant 2013; 48:651-6; PMID:23103678; http://dx.doi.org/ 10.1038/bmt.2012.204 [DOI] [PubMed] [Google Scholar]
- 3.Sander A, Zimmermann M, Dworzak M, Fleischhack G, Neuhoff von C, Reinhardt D, Kaspers GJL, Creutzig U. Consequent and intensified relapse therapy improved survival in pediatric AML: results of relapse treatment in 379 patients of three consecutive AML-BFM trials. Leukemia 2010; 24:1422-8; PMID:20535146; http://dx.doi.org/ 10.1038/leu.2010.127 [DOI] [PubMed] [Google Scholar]
- 4.Eapen M, Rubinstein P, Zhang M-J, Stevens C, Kurtzberg J, Scaradavou A, Loberiza FR, Champlin RE, Klein JP, Horowitz MM et al.. Outcomes of transplantation of unrelated donor umbilical cord blood and bone marrow in children with acute leukaemia: a comparison study. The Lancet 2007; 369:1947-54; PMID:17560447; http://dx.doi.org/ 10.1016/S0140-6736(07)60915-5 [DOI] [PubMed] [Google Scholar]
- 5.Kaspers GJL, Zimmermann M, Reinhardt D, Gibson BES, Tamminga RYJ, Aleinikova O, Armendariz H, Dworzak M, Ha SY, Hasle H et al.. Improved outcome in pediatric relapsed acute myeloid leukemia: results of a randomized trial on liposomal daunorubicin by the international BFM study group. J Clin Oncol 2013; 31:599-607; PMID:23319696; http://dx.doi.org/ 10.1200/JCO.2012.43.7384 [DOI] [PubMed] [Google Scholar]
- 6.Levenga H, Schaap N, Maas F, Esendam B, Fredrix H, Greupink-Draaisma A, de Witte T, Dolstra H, Raymakers R. Partial T cell-depleted allogeneic stem cell transplantation following reduced-intensity conditioning creates a platform for immunotherapy with donor lymphocyte infusion and recipient dendritic cell vaccination in multiple myeloma. Biol Blood Marrow Transplant 2010; 16:320-32; PMID:19835972; http://dx.doi.org/ 10.1016/j.bbmt.2009.10.006 [DOI] [PubMed] [Google Scholar]
- 7.Rapoport AP, Aqui NA, Stadtmauer EA, Vogl DT, Fang H-B, Cai L, Janofsky S, Chew A, Storek J, Akpek G et al.. Combination immunotherapy using adoptive T-cell transfer and tumor antigen vaccination on the basis of hTERT and survivin after ASCT for myeloma. Blood 2011; 117:788-97; PMID:21030558; http://dx.doi.org/ 10.1182/blood-2010-08-299396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frumento G, Zheng Y, Aubert G, Raeiszadeh M, Lansdorp PM, Moss P, Lee SP, Chen FE. Cord blood T cells retain early differentiation phenotype suitable for immunotherapy after TCR gene transfer to confer EBV specificity. Am J Transplant 2013; 13:45-55; PMID:23016879; http://dx.doi.org/ 10.1111/j.1600-6143.2012.04286.x [DOI] [PubMed] [Google Scholar]
- 9.Alyea EP, DeAngelo DJ, Moldrem J, Pagel JM, Przepiorka D, Sadelin M, Young JW, Giralt S, Bishop M, Riddell S. NCI first international workshop on the biology, prevention and treatment of relapse after allogeneic hematopoietic cell transplantation: report from the committee on prevention of relapse following allogeneic cell transplantation for hematologic malignancies. Biol Blood Marrow Transplant 2010; 16:1037-69; PMID:20580849; http://dx.doi.org/ 10.1016/j.bbmt.2010.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brunstein CG, Gutman JA, Weisdorf DJ, Woolfrey AE, DeFor TE, Gooley TA, Verneris MR, Appelbaum FR, Wagner JE, Delaney C. Allogeneic hematopoietic cell transplantation for hematologic malignancy: relative risks and benefits of double umbilical cord blood. Blood 2010; 116:4693-9; PMID:20686119; http://dx.doi.org/ 10.1182/blood-2010-05-285304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kapp M, Stevanović S, Fick K, Tan SM, Loeffler J, Opitz A, Tonn T, Stuhler G, Einsele H, Grigoleit GU. CD8+ T-cell responses to tumor-associated antigens correlate with superior relapse-free survival after allo-SCT. Bone Marrow Transplant 2009; 43:399-410; PMID:19139738; http://dx.doi.org/ 10.1038/bmt.2008.426 [DOI] [PubMed] [Google Scholar]
- 12.van Rood JJ, Stevens CE, Smits J, Carrier C, Carpenter C, Scaradavou A. Reexposure of cord blood to noninherited maternal HLA antigens improves transplant outcome in hematological malignancies. Proc Natl Acad Sci USA 2009; 106:19952-7; PMID:19901324; http://dx.doi.org/ 10.1073/pnas.0910310106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burlingham WJ, Nelson JL. Microchimerism in cord blood: mother as anticancer drug. Proc Natl Acad Sci USA 2012; 109:2190-1; PMID:22323582; http://dx.doi.org/ 10.1073/pnas.1120857109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity 2013; 39:38-48; PMID:23890062; http://dx.doi.org/ 10.1016/j.immuni.2013.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tel J, Aarntzen EHJG, Baba T, Schreibelt G, Schulte BM, Benitez-Ribas D, Boerman OC, Croockewit S, Oyen WJG, van Rossum M et al.. Natural human plasmacytoid dendritic cells induce antigen-specific T-cell responses in melanoma patients. Cancer Research 2013; 73:1063-75; PMID:23345163; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-2583 [DOI] [PubMed] [Google Scholar]
- 16.van Tendeloo VF, Van de Velde A, Van Driessche A, Cools N, Anguille S, Ladell K, Gostick E, Vermeulen K, Pieters K, Nijs G et al.. Induction of complete and molecular remissions in acute myeloid leukemia by Wilms' tumor 1 antigen-targeted dendritic cell vaccination. Proc Natl Acad Sci USA 2010; 107:13824-9; PMID:20631300; http://dx.doi.org/ 10.1073/pnas.1008051107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balan S, Kale VP, Limaye LS. A simple two-step culture system for the large-scale generation of mature and functional dendritic cells from umbilical cord blood CD34+ cells. Transfusion 2009; 49:2109-21; PMID:18774963; http://dx.doi.org/ 10.1111/j.1537-2995.2009.02231.x [DOI] [PubMed] [Google Scholar]
- 18.Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, Mellman I, Prindiville SA, Viner JL, Weiner LM et al.. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res 2009; 15:5323-37; PMID:19723653; http://dx.doi.org/ 10.1158/1078-0432.CCR-09-0737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Menssen HD, Renkl HJ, Rodeck U, Maurer J, Notter M, Schwartz S, Reinhardt R, Thiel E. Presence of Wilms' tumor gene (wt1) transcripts and the WT1 nuclear protein in the majority of human acute leukemias. Leukemia 1995; 9:1060-7; PMID:7596170 [PubMed] [Google Scholar]
- 20.Van Driessche A, Van de Velde ALR, Nijs G, Braeckman T, Stein B, De Vries JM, Berneman ZN, Van Tendeloo VFI. Clinical-grade manufacturing of autologous mature mRNA-electroporated dendritic cells and safety testing in acute myeloid leukemia patients in a phase I dose-escalation clinical trial. Cytotherapy 2009; 11:653-68; PMID:19530029; http://dx.doi.org/ 10.1080/14653240902960411 [DOI] [PubMed] [Google Scholar]
- 21.Cohen G, Carter SL, Weinberg KI, Masinsin B, Guinan E, Kurtzberg J, Wagner JE, Kernan NA, Parkman R. Antigen-specific T-lymphocyte function after cord blood transplantation. Biol Blood Marrow Transplant 2006; 12:1335-42; PMID:16399583; http://dx.doi.org/ 10.1016/j.bbmt.2006.08.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiesa R, Gilmour K, Qasim W, Adams S, Worth AJJ, Zhan H, Montiel-Equihua CA, Derniame S, Cale C, Rao K et al.. Omission of in vivo T-cell depletion promotes rapid expansion of naïve CD4+ cord blood lymphocytes and restores adaptive immunity within 2 months after unrelated cord blood transplant. Br J Haematol 2012; 156:656-66; PMID:22224700; http://dx.doi.org/ 10.1111/j.1365-2141.2011.08994.x [DOI] [PubMed] [Google Scholar]
- 23.Lanzavecchia A. Mechanisms of antigen uptake for presentation. Curr Opin Immunol 1996; 8:348-54; PMID:8794000; http://dx.doi.org/ 10.1016/S0952-7915(96)80124-5 [DOI] [PubMed] [Google Scholar]
- 24.Jonuleit H, Kühn U, Müller G, Steinbrink K, Paragnik L, Schmitt E, Knop J, Enk AH. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol 1997; 27:3135-42; PMID:9464798; http://dx.doi.org/ 10.1002/eji.1830271209 [DOI] [PubMed] [Google Scholar]
- 25.Rieser C, Böck G, Klocker H, Bartsch G, Thurnher M. Prostaglandin E2 and tumor necrosis factor alpha cooperate to activate human dendritic cells: synergistic activation of interleukin 12 production. J Exp Med 1997; 186:1603-8; PMID:9348319; http://dx.doi.org/ 10.1084/jem.186.9.1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vremec D, O'Keeffe M, Wilson A, Ferrero I, Koch U, Radtke F, Scott B, Hertzog P, Villadangos J, Shortman K. Factors determining the spontaneous activation of splenic dendritic cells in culture. Innate Immun 2011; 17:338-52; PMID:20501515; http://dx.doi.org/ 10.1177/1753425910371396 [DOI] [PubMed] [Google Scholar]
- 27.Hobo W, Novobrantseva TI, Fredrix H, Wong J, Milstein S, Epstein-Barash H, Liu J, Schaap N, Voort R, Dolstra H. Improving dendritic cell vaccine immunogenicity by silencing PD-1 ligands using siRNA-lipid nanoparticles combined with antigen mRNA electroporation. Cancer Immunol Immunother 2013; 62:285-97; PMID:22903385; http://dx.doi.org/ 10.1007/s00262-012-1334-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee AW, Truong T, Bickham K, Fonteneau J-F, Larsson M, Da Silva I, Somersan S, Thomas EK, Bhardwaj N. A clinical grade cocktail of cytokines and PGE2 results in uniform maturation of human monocyte-derived dendritic cells: implications for immunotherapy. Vaccine 2002; 20 Suppl 4:A8-A22; PMID:12477423; http://dx.doi.org/ 10.1016/S0264-410X(02)00382-1 [DOI] [PubMed] [Google Scholar]
- 29.Beck B, Dörfel D, Lichtenegger FS, Geiger C, Lindner L, Merk M, Schendel DJ, Subklewe M. Effects of TLR agonists on maturation and function of 3-day dendritic cells from AML patients in complete remission. J Transl Med 2011; 9:151; PMID:21910911; http://dx.doi.org/ 10.1186/1479-5876-9-151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ugel S, Delpozzo F, Desantis G, Papalini F, Simonato F, Sonda N, Zilio S, Bronte V. Therapeutic targeting of myeloid-derived suppressor cells. Curr Opin Pharmacol 2009; 9:470-81; PMID:19269896; http://dx.doi.org/ 10.1016/j.coph.2009.06.014 [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, Ochoa AC. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res 2009; 69:1553-60; PMID:19201693; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Driessche A, Gao L, Stauss HJ, Ponsaerts P, Van Bockstaele DR, Berneman ZN, Van Tendeloo VFI. Antigen-specific cellular immunotherapy of leukemia. Leukemia 2005; 19:1863-71; PMID:16121214; http://dx.doi.org/ 10.1038/sj.leu.2403930 [DOI] [PubMed] [Google Scholar]
- 33.Poulin LF, Salio M, Griessinger E, Anjos-Afonso F, Craciun L, Chen JL, Keller AM, Joffre O, Zelenay S, Nye E et al.. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8+ dendritic cells. J Exp Med 2010; 207:1261-71; PMID:20479117; http://dx.doi.org/ 10.1084/jem.20092618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harris KM. Monocytes differentiated with GM-CSF and IL-15 initiate Th17 and Th1 responses that are contact-dependent and mediated by IL-15. J Leukocyte Biol 2011; 90:727-34; PMID:21724805; http://dx.doi.org/ 10.1189/jlb.0311132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anguille S, Lion E, Tel J, de Vries IJM, Couderé K, Fromm PD, van Tendeloo VF, Smits EL, Berneman ZN. Interleukin-15-induced CD56+ myeloid dendritic cells combine potent tumor antigen presentation with direct tumoricidal potential. PLoS One 2012; 7:e51851; PMID:23284789; http://dx.doi.org/ 10.1371/journal.pone.0051851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Relloso M, Puig-Kröger A, Pello OM, Rodríguez-Fernández JL, la Rosa de G, Longo N, Navarro J, Muñoz-Fernández MA, Sánchez-Mateos P, Corbí AL. DC-SIGN (CD209) expression is IL-4 dependent and is negatively regulated by IFN, TGF-beta, and anti-inflammatory agents. J Immunol 2002; 168:2634-43; PMID:11884427; http://dx.doi.org/ 10.4049/jimmunol.168.6.2634 [DOI] [PubMed] [Google Scholar]
- 37.Encabo A, Solves P, Mateu E, Sepúlveda P, Carbonell-Uberos F, Minana MD. Selective generation of different dendritic cell precursors from CD34+ cells by interleukin-6 and interleukin-3. Stem Cells 2004; 22:725-40; PMID:15342937; http://dx.doi.org/ 10.1634/stemcells.22-5-725 [DOI] [PubMed] [Google Scholar]
- 38.Kato M, Neil TK, Fearnley DB, McLellan AD, Vuckovic S, Hart DN. Expression of multilectin receptors and comparative FITC-dextran uptake by human dendritic cells. Int Immunol 2000; 12:1511-9; PMID:11058570; http://dx.doi.org/ 10.1093/intimm/12.11.1511 [DOI] [PubMed] [Google Scholar]
- 39.Nierkens S, Tel J, Janssen E, Adema GJ. Antigen cross-presentation by dendritic cell subsets: one general or all sergeants? Trends Immunol 2013; 34:361-70; PMID:23540650; http://dx.doi.org/ 10.1016/j.it.2013.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khan S, Bijker MS, Weterings JJ, Tanke HJ, Adema GJ, van Hall T, Drijfhout JW, Melief CJM, Overkleeft HS, van der Marel GA et al.. Distinct uptake mechanisms but similar intracellular processing of two different toll-like receptor ligand-peptide conjugates in dendritic cells. J Biol Chem 2007; 282:21145-59; PMID:17462991; http://dx.doi.org/ 10.1074/jbc.M701705200 [DOI] [PubMed] [Google Scholar]
- 41.Tel J, Lambeck AJA, Cruz LJ, Tacken PJ, de Vries IJM, Figdor CG. Human plasmacytoid dendritic cells phagocytose, process, and present exogenous particulate antigen. J Immunol 2010; 184:4276-83; PMID:20304825; http://dx.doi.org/ 10.4049/jimmunol.0903286 [DOI] [PubMed] [Google Scholar]
- 42.Benteyn D, Anguille S, Van Lint S, Heirman C, Van Nuffel AM, Corthals J, Ochsenreither S, Waelput W, Van Beneden K, Breckpot K et al.. Design of an optimized Wilms' Tumor 1 (WT1) mRNA construct for enhanced WT1 expression and improved immunogenicity in vitro and in vivo. Mol Ther Nucleic Acids 2013; 2:e134; PMID:24253259; http://dx.doi.org/ 10.1038/mtna.2013.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doubrovina E, Carpenter T, Pankov D, Selvakumar A, Hasan A, O'Reilly RJ. Mapping of novel peptides of WT-1 and presenting HLA alleles that induce epitope-specific HLA-restricted T cells with cytotoxic activity against WT-1+ leukemias. Blood 2012; 120:1633-46; PMID:22623625; http://dx.doi.org/ 10.1182/blood-2011-11-394619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caux C, Massacrier C, Vanbervliet B, Dubois B, Durand I, Cella M, Lanzavecchia A, Banchereau J. CD34+ hematopoietic progenitors from human cord blood differentiate along two independent dendritic cell pathways in response to granulocyte-macrophage colony-stimulating factor plus tumor necrosis factor alpha: II. Functional analysis. Blood 1997; 90:1458-70; PMID:9207440 [PubMed] [Google Scholar]
- 45.Dubsky P, Saito H, Leogier M, Dantin C, Connolly JE, Banchereau J, Palucka AK. IL-15-induced human DC efficiently prime melanoma-specific naive CD8+ T cells to differentiate into CTL. Eur J Immunol 2007; 37:1678-90; PMID:17492620; http://dx.doi.org/ 10.1002/eji.200636329 [DOI] [PubMed] [Google Scholar]
- 46.Van Tendeloo VF, Ponsaerts P, Lardon F, Nijs G, Lenjou M, Van Broeckhoven C, Van Bockstaele DR, Berneman ZN. Highly efficient gene delivery by mRNA electroporation in human hematopoietic cells: superiority to lipofection and passive pulsing of mRNA and to electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood 2001; 98:49-56; PMID:11418462; http://dx.doi.org/ 10.1182/blood.V98.1.49 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.