

Abstract
The RIG-I-like helicase melanoma differentiation-associated protein 5 (MDA5) is an innate immune receptor for double-stranded viral RNA (dsRNA) that, upon activation, induces a Type I interferon (IFN)-driven immune response. In the present study, we demonstrate that human und murine pancreatic cancer cells express functional MDA5 and are highly sensitive to MDA5-induced cell death. Activation of MDA5 by cytosolic delivery of the synthetic dsRNA analog poly(I:C) led to phosphorylation of the transcription factor IRF3, IFNβ production and upregulation of MHC-I expression. MDA5 signaling also induced tumor cell apoptosis via the intrinsic pathway and sensitized tumor cells toward extrinsic, Fas-mediated apoptosis. Systemic treatment of orthotopic pancreatic cancer-bearing mice with the MDA5 ligand resulted in activated CD8+ T cell tumor infiltration, an increased frequency of tumor antigen-specific CD8+ T cells and an immunogenic cytokine milieu in the tumor microenvironment. These effects were paralleled by MDA5-induced pronounced tumor cell death in situ and significantly prolonged survival in two different mouse models for pancreatic cancer, an immunotherapeutic response dependent on CD8+ T cells. Treated mice were further protected from subsequent tumor challenge. In summary, we identified MDA5 as a novel therapeutic target for overcoming apoptosis resistance and tumor-mediated immunosuppression in pancreatic cancer. MDA5 ligands link innate with adaptive immune mechanisms for effective tumor control.
Keywords: Immunotherapy, MDA5, pancreatic cancer, poly(I:C), RIG-I-like helicases, Type I IFN
Abbreviations
- CTL
cytotoxic T lymphocyte
- CXCL10
chemokine (C-X-C) motif ligand 10
- DC
dendritic cell
- IFN
interferon
- IRF3
IFN regulatory factor 3
- MDA5
melanoma differentiation-associated protein 5
- MHC-I
major histocompatibility complex Class I
- PEI
polyethylenimine
- poly(I:C)
polyinosinic:polycytidylic acid
- RIG-I-retinoic
acid-inducible gene 1
- RLH
RIG-I-like helicases
- dsRNA
double-stranded RNA
- TLR
Toll-like receptor
Introduction
Patients diagnosed with pancreatic cancer face a dismal prognosis due to late diagnosis and limited treatment benefits offered by chemotherapy or irradiation.1 Apoptosis resistance and a highly immunosuppressive tumor microenvironment are two major disease hallmarks underlying the pressing need for new compounds for pancreatic cancer treatment. Immunotherapy aims at counteracting tumor immune evasion and has shown promising results in preclinical and in early clinical studies.2,3 Strategies tested so far include vaccination with tumor antigens,4-6 modulation of antigenpresenting cells,7 restoration of impaired T cell effector function (e.g., via immune checkpoint inhibitors), and targeting inhibitory leukocyte populations such as regulatory T cells, myeloid-derived suppressor cells or tumor-associated macrophages (e.g., via CD40 activating monoclonal antibodies).8-10 However, in contrast to immunogenic tumors, such as melanoma and renal cell carcinoma, these approaches have so far translated into limited clinical success in patients with pancreatic cancer.
The extensive resistance to apoptosis-inducing agents in pancreatic cancer is associated with expression of multiple pro-survival proteins of the extrinsic and intrinsic apoptosis signaling cascades.11 To tackle apoptosis resistance, novel strategies to induce tumor cell death, such as therapy with oncolytic viruses, have been explored and successfully translated in early clinical trials in patients suffering from pancreatic cancer.12 A similar but more subtle strategy is to mimic viral infection of tumor cells using synthetic RNA molecules that activate cytosolic immune receptors for viral RNA species, the RIG-I-like helicases (RLH).13 Of particular interest for tumor therapy are the two RLHs retinoic acid-inducible gene I (RIG-I) and interferon induced with helicase C domain (IF1H1) –better known as melanoma differentiation-associated protein 5 (MDA5)– which recognize 5′-ppp-RNA and the synthetic dsRNA analog polyinosinic:polycytidylic acid (poly(I:C)), respectively.14-17 Upon RNA binding RLHs initiate a signaling cascade mediated by interferon (IFN) regulatory factor 3 and 7 (IRF-3 and IRF-7) as well as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), leading to production of type I IFN, pro-inflammatory cytokines and chemokines, as well as other innate immune response genes.18,19 In addition, RLH activation leads to an endosomal stress program inducing autophagy, which is mediated by pro-apoptotic mitochondrial proteins and culminates in intrinsic, caspase-9-mediated apoptosis.20-22 Tumor cells are highly sensitive to this type of RLH-induced apoptosis, whereas nonmalignant cells are protected via endogenous expression of the anti-apoptotic protein Bcl-xL, providing a therapeutic window for RLH-based tumor therapy.22,23 We previously demonstrated that pancreatic cancer cells express functional RIG-I and are sensitive to RIG-I-mediated tumor cell apoptosis irrespective of their p53 mutational status.24 These findings provide a rationale for exploiting the RLH pathway to a) circumvent apoptosis resistance to conventional cytotoxic drugs and b) to induce a Type I IFN-driven immune response.25
In the present study, we evaluated MDA5 as a potential target for pancreatic cancer therapy. We investigated MDA5 expression in human patient tumor specimens as well as in human and murine pancreatic cancer cells. We further examined the functional consequences of MDA5 signaling in the context of pancreatic cancer, such as MDA5-induced cytokine production, malignant cell phenotypic changes, and apoptosis induction via intrinsic and extrinsic pathways. Finally, we assessed the therapeutic efficacy and mode of action of MDA5-based immunotherapy in two different murine pancreatic cancer models.
Results
Human pancreatic cancer cells express functional MDA5
We first assessed MDA5 expression in human pancreatic cancer by immunohistochemistry using tissue sections of six primary tumors and one metastasis of patients with pancreatic cancer (5 adenocarcinoma, one solid carcinoma and one acinar cell carcinoma). All tumors showed a cytosolic MDA5 expression pattern (Fig. S1), with the strongest expression observed in adenocarcinomas (Grade 2), as indicated by increased MDA5 staining intensity as compared to that of Grade 3–4 tumors. Next, we evaluated MDA5 as a potential target for the treatment of pancreatic cancer by assessing MDA5 expression in four human tumor cell lines (IMIM-PC-1, SUIT-007, MiaPaCa-2 and PANC-1) by Western blot under basal culture conditions and following stimulation with Type I IFN. Expression levels of MDA5, which belongs to the group of IFN-stimulated genes, were rapidly upregulated in response to IFNα in all cell lines (Fig. 1A). Transfection of tumor cells with the MDA5 ligand poly(I:C) complexed to lipofectamine (poly(I:C)c), but not control RNA, led to phosphorylation of interferon regulatory factor 3 (IRF3) and increased MDA5 expression, indicative of intact MDA5 signaling (Fig. 1B–C).
Figure 1.
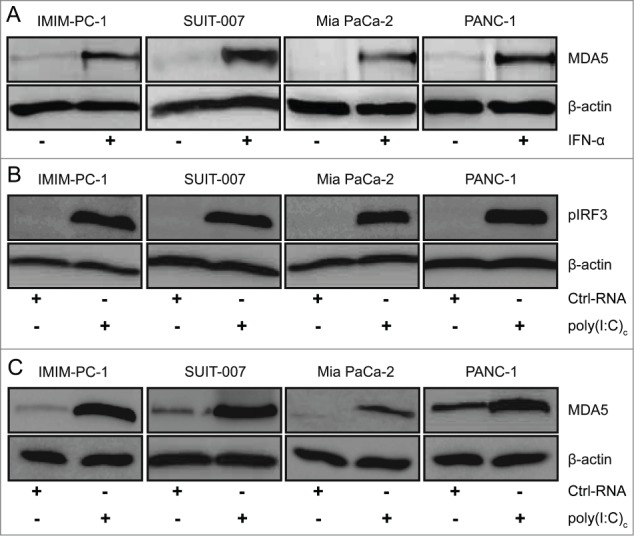
Human pancreatic cancer cells express functional MDA5. (A) Tumor cells were cultured in the presence or absence of 100 U/mL interferon α (IFNα) for 24 h and melanoma differentiation-associated protein 5 (MDA5) protein expression was determined by Western blot. (B–C) Phosphorylation of IFN regulatory factor 3 (IRF3) and MDA5 expression in tumor cells transfected with 400 ng/mL control RNA or poly(I:C) complexed with lipofectamine (poly(I:C)c) for 3 h and 24 h, respectively, was determined by Western blot. Representative data of 2 independent experiments are shown.
To further assess functional consequences of MDA5 activation we next analyzed cytokine and chemokine production along with MHC-I expression in tumor cells. All tumor cell lines, with the exception of Mia PaCa-2 which is known to have a deletion of the IFNβ gene,26 upregulated IFNβ mRNA expression in response to transfection with poly(I:C)c, but not control RNA (Fig. 2A). Treated pancreatic cancer cells also upregulated MHCI surface expression (Fig. 2B). To control for a potential contribution of Toll-like receptor 3 (TLR3), another pattern recognition receptor recognizing dsRNA, we treated tumor cells with poly(I:C) without transfection agent. Poly(I:C) only treatment had no influence on IFNβ or MHC-I expression levels, indicating that MDA5 and not TLR3 is responsible for the observed effects (Fig. 2A–B). RNAimediated gene silencing experiments further confirmed the role of MDA5 as opposed to TLR3 or RIG-I in poly(I:C)-mediated increase in IFNβ transcript levels (Fig. 2C).
Figure 2.
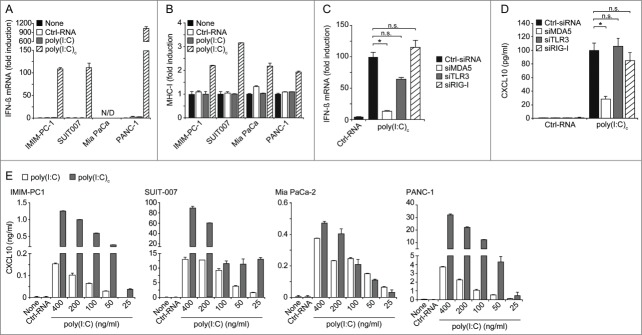
MDA5 activation leads to upregulation of type I IFN, MHC-I and CXCL10 in human pancreatic cancer cells. (A) IFNβ mRNA expression levels in indicated tumor cell lines in response to stimulation with RNAs (200 ng/mL each) was assessed by qRT-PCR after 12 h. (N/D, not detectable). (B) MHC-I surface expression of RNA treated tumor cells was determined by immunofluorescence staining and cytofluorimetric analysis after 24 h. (C, D) IMIM-PC-1 cells were transfected with siRNA specific for MDA5, TLR3, RIG-I or control RNA (Ctrl) for 24 h and subsequently transfected with poly(I:C) for further 24 h. Expression of IFNβ mRNA (C) and secretion of CXCL10 (D) were determined via qRT-PCR and ELISA, respectively. (E) CXCL10 levels in supernatants of tumor cell lines, treated with the indicated concentrations of RNA for 24 h, were measured by ELISA. Control RNA (Ctrl-RNA) was used at 400 ng/mL. Poly(I:C)c refers to poly(I:C) complexed with Lipofectamine. A, B, C: data are depicted as fold induction as compared to untreated controls. (A, B, E) data represent means ± SD of triplicates, one representative of at least 2 independent experiments is shown. (C, D) pooled data ± SD of 3 independent experiments are shown. CXCL10, chemokine (C-X-C) motif ligand 10; IFN, interferon; MDA5, melanoma differentiation-associated protein 5; poly(I:C), polyinosinic:polycytidylic acid; RIG-I, retinoic acid-inducible gene 1; TLR3, Toll-like receptor 3.
We further assessed production of the interferon-inducible chemokine (C-X-C) motif ligand 10 (CXCL10), a chemoattractant for T and natural killer (NK) cells to sites of viral infections.27,28 Transfection with poly(I:C)c resulted in dose-dependent CXCL10 secretion by all tumor cell lines. Significantly lower levels were produced by tumor cells treated with non-complexed poly(I:C) (Fig. 2E). Again, RNAi-mediated gene silencing experiments confirmed the role of MDA5 in poly(I:C)mediated CXCL10 secretion, as opposed to TLR3 or RIG-I (Fig. 2D). Together, these findings demonstrate that MDA5 signaling is intact in pancreatic tumor cells and that poly(I:C)-mediated effects are independent of TLR3 or RIG-I signaling.
MDA5 ligands induce tumor cell apoptosis and sensitize toward FAS-mediated killing
Next, we assessed if MDA5 activation can lead to apoptosis in human pancreatic cancer cells, as previously described for melanoma cells.22 Transfection with poly(I:C)c induced pronounced apoptosis in a dose-dependent manner, whereas uncomplexed poly(I:C) or transfection with a control RNA had no effect on tumor cell viability (Fig. 3A). MDA5 but not TLR3 or RIG-I silencing with siRNA significantly reduced poly(I:C)-induced cell death (Fig. 3B). Characterization of MDA5-induced cell death revealed cleavage of caspase-3, caspase-9, as well as PARP-1 by Western blot in treated tumor cells (Fig. 3C–E). In addition, tumor cell death was prevented by the pan-caspase inhibitor zVAD-fmk, indicative of a caspase-mediated apoptotic pathway (Fig. 3F). These findings are in line with activation of the intrinsic apoptosis pathway, as previously reported for RLH ligands in other tumor entities.22,24
Figure 3.

Human pancreatic cancer cells are highly sensitive toward MDA5-induced intrinsic and extrinsic apoptosis. (A) Tumor cells were treated for 48 h with RNAs at the indicated concentrations or left untreated. Control RNA (Ctrl-RNA) and uncomplexed poly(I:C) were used at 400 ng/mL. Viability is expressed as annexin V/propidium iodide (PI) double-negative cell fraction analyzed by flow cytometry. (B) IMIM-PC-1 cells were transfected with MDA5-, TLR3- or RIG-I-specific or control siRNA 24 h prior to stimulation with 100 ng/mL of indicated RNAs. Viability was measured by annexin V/PI staining after an additional 24 h. (C–E) Cleavage of caspase-3 (C), caspase-9 (D) and PARP1 (E) was assessed by Western blot analysis in IMIM-PC-1 cells treated with 200 ng/mL RNAs for 24 h. (F) Tumor cells were incubated with 20 µM of the caspase inhibitor zVAD-FMK 1 h prior to stimulation with 200 ng/mL RNAs for further 24h. Apoptosis was assessed by annexin V/PI staining. (G) CD95 (Fas) expression of annexin V/PI negative tumor cells stimulated with 200 ng/mL RNAs for 24 h. (H) Tumor cells were stimulated with 25 ng/mL RNA for 12 h and subsequently incubated with 1 µg/mL of a Fas-activating mAb (clone CH11) for additional 24 h. Poly(I:C)c refers to poly(I:C) complexed with Lipofectamine. (A, F, H) data represent means ± SD of triplicates, one representative of 3 independent experiments is shown. (B) pooled data ± SD of 3 independent experiments are shown. (C, D, E, G) one representative WB/FACS blot out of 3 is shown. MDA5, melanoma differentiation-associated protein 5; poly(I:C), polyinosinic:polycytidylic acid; RIG-I, retinoic acid-inducible gene 1; TLR3, Toll-like receptor 3.
Engagement of the death receptor Fas (CD95) with its ligand FasL leads to initiation of the extrinsic apoptosis pathway. We previously reported that activation of RIG-I stimulated tumor cells to upregulate CD95 expression.23,29 In line with this finding, we observed a significant upregulation of CD95 surface expression in human pancreatic cancer cells transfected with poly(I:C)c (Fig. 3G). To assess the functional consequence of enhanced CD95 expression, we transfected tumor cells with low doses of the MDA5 ligand and subsequently incubated them with an agonistic anti-Fas monoclonal antibody. Tumor cells were strongly sensitized toward Fas-mediated killing (Fig. 3H). Together, these findings show that MDA5 activation promotes pancreatic tumor cell apoptosis via both the intrinsic and extrinsic apoptotic pathways.
Murine pancreatic cancer cells express a functional MDA5 signaling pathway
To evaluate the potential of MDA5 ligands for tumor therapy in preclinical in vivo models, we assessed MDA5 expression in three different murine pancreatic carcinoma cell lines. Panc02 is a chemically-induced cell line, whereas T110299 and T510479 cell lines were generated from pancreatic tumors of genetically engineered mice with targeted expression of Kras mutation with or without additional p53 mutation (KPC and KC mice, respectively).30 Similarly to human tumor cells, murine cells upregulated MDA5 expression in response to IFNα stimulation (Fig. 4A). Transfection of poly(I:C)c led to phosphorylation of IRF3 (Fig. 4B), upregulation of IFNβ mRNA expression and CXCL10 secretion (Fig. 4C–D), increased levels of MHC-I and CD95 (Fas) surface expression (Fig. 4E–F), and dose-dependent tumor cell apoptosis in all three tumor cell lines (Fig. 4G). As for human cells, uncomplexed poly(I:C) was ineffective in this respect, ruling out a contribution of TLR3. In conclusion, murine tumor models appear to be suitable for evaluating the in vivo efficacy of MDA5-based immunotherapy against pancreatic cancer.
Figure 4.
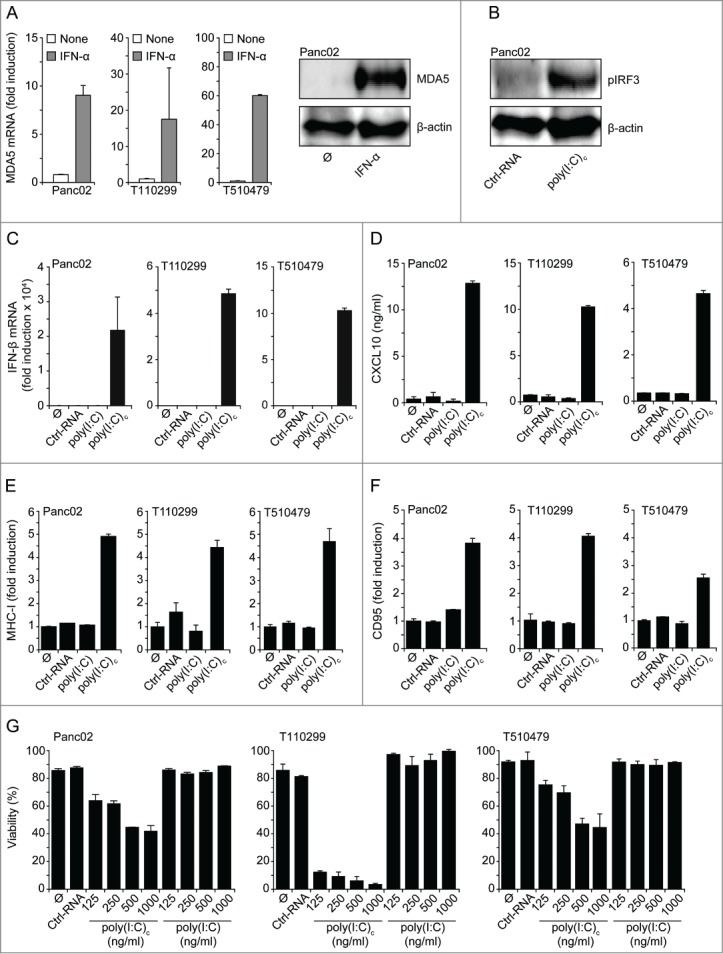
Murine pancreatic carcinoma cell lines express functional MDA5. (A) Tumor cells were incubated with 1000 U/mL IFNα for 24 h and MDA5 expression levels were assessed by qRT-PCR and Western blot. (B) Panc02 cells were transfected with RNAs for 3 h and IRF3 phosphorylation was measured by Western blot. (C-F) Panc02, T110299 or T510479 cells were treated for 24 h with RNAs (1 µg/mL) or left untreated. (C) Expression levels of IFNβ mRNA were determined with qRT-PCR, (D) CXCL10 levels in cell culture supernatants by ELISA, (E) MHC-I and (F) CD95 surface expression by flow cytometry. (G) Viability of tumor cells treated with the indicated concentrations of RNA was measured via annexin V/propidium iodide (PI) staining. Control RNA (Ctrl-RNA) and uncomplexed poly(I:C) were used at 1000 ng/mL. Poly(I:C)c refers to poly(I:C) complexed with Lipofectamine. A, C, E, F: data are shown as fold induction as compared to untreated controls. Data represent means + SD of triplicates, one representative out of 3 independent experiments are shown. CXCL10, chemokine (C-X-C) motif ligand 10; IFN, interferon; IRF3, IFN regulatory factor 3; MDA5, melanoma differentiation-associated protein 5; poly(I:C), polyinosinic:polycytidylic acid.
Poly(I:C)-PEI leads to systemic immune activation in mice with pancreatic tumors
First, we assessed systemic signs of immune activation in mice carrying orthotopic Panc02 tumors after i.v. treatment with poly(I:C), either uncomplexed or formulated with polyethylenimine (poly(I:C)-PEI). Serum levels of IFNα and CXCL10 were significantly increased in poly(I:C)-treated mice, irrespective of the formulation indicative of in vivo activation of TLR3, MDA or both17 (Fig. 5A). Similarly, B, NK, CD4+ and CD8+ T cells in spleens of poly(I:C)-treated mice upregulated expression of the activation marker CD69 (Fig. 5B). This was paralleled by upregulated expression of the costimulatory molecules CD80 and CD86 in CD8α+ and CD8α− DC populations, with highest expression levels found in mice treated with poly(I:C)-PEI (Fig. 5C).
Figure 5.

Systemic treatment with poly(I:C)-PEI stimulates immune activation, a Type I IFN cytokine profile, T-cell recruitment/activation and pancreatic tumor cell apoptosis. (A) Mice with orthotopic Panc02 tumors were injected i.v. with phosphate buffered saline (PBS), poly(I:C) or poly(I:C)-PEI (each 25 µg) on day 8 after tumor induction. Serum levels of IFNα and CXCL10 were quantified by ELISA after 6 h. (B-F) 24 h after a second treatment (day 12), mice were sacrificed, spleens and tumors were removed and processed for FACS analysis, mRNA extraction and immunohistochemistry. (B) CD69 expression on B cells, NK cells and T cells, and (C) CD80 and CD86 expression on CD8α+ and CD8α− CD11c+ DC populations were analyzed by FACS analysis. (D) Induction of tumor cell death was assessed by TUNEL staining. Fluorescence microscopic pictures of representative tumor sections are shown. (E) Expression levels of IFNβ, MDA5, CXCL10, IFNγ and IL-5 mRNA were quantified with qRT-PCR and the IFNγ/IL-5 transcript ratio calculated as marker for Th1/Th2 polarization. (F) Tumor sections were stained for CD3ε and CD8 expression and total CD3+ and CD3+/CD8+ T cells per high power field (HPF) were counted. (G, H) Mice treated as in B were sacrificed 12 h after the second RNA injection and CD8+ T cells were isolated from tumor tissue by immunostaining and cell sorting via flow cytometry. (G) Expression of CD69, IFNγ and FasL was measured with flow cytometry. (H) Granzyme B and perforin expression levels were quantified with qRT-PCR and are depicted as fold induction relative to PBS treatment. Data represent mean ± SEM of 3–4 individually analyzed mice per group. CXCL10, chemokine (C-X-C) motif ligand 10; IFN, interferon; MDA5, melanoma differentiation-associated protein 5; NK, natural killer cell; PEI, polyethylenimine; poly(I:C), polyinosinic:polycytidylic acid; Th, T helper cell.
Next, we investigated the effects of poly(I:C) treatment on pancreatic cancer cell apoptosis and tumor cytokine profiles. Orthotopic Panc02 tumors were surgically excised 24 h after treatment with PBS or poly(I:C) formulations and analyzed for cytokine mRNA expression levels by qRT-PCR. Immunohistochemistry of excised tumors revealed pronounced tumor cell death in poly(I:C)-PEI treated mice, as assessed by TUNEL staining (Fig. 5D). No pathological findings were observed in adjacent normal pancreatic tissue (data not shown). In addition, the tumors expressed increased levels of IFNβ, MDA5 and CXCL10 mRNA, indicative of a Type I IFN signature. This was paralleled by an increased ratio of IFNγ to IL-5, pointing toward a Th1 response (Fig. 5E) and corresponding to increased recruitment of CD8+ T cells into the tumor tissue (Fig. 5F; Fig. S2). CD8+ T cells isolated from tumors of poly(I:C)-PEI-treated mice exhibited an activated phenotype, as assessed by expression of CD69 and IFNγ, as well as markers associated with lytic function, such as FasL and perforin (Fig. 5G–H). These observations indicate the induction of a potent antitumor immune response in poly(I:C)-PEI treated mice, which was superior to uncomplexed poly(I:C), in line with our in vitro findings that therapeutic efficacy is mediated via MDA5 and not TLR3.
Immunotherapy with poly(I:C)-PEI prolongs survival in murine pancreatic cancer models
The promising in vivo findings on cytokine milieu and tumor cell death prompted us to investigate the efficacy of MDA5-based immunotherapy on tumor control and survival. Since uncomplexed poly(I:C) was shown to be inferior to poly(I:C)-PEI (Fig. 5C–H), we performed the following experiments with poly(I:C)-PEI only. Mice with orthotopic Panc02 tumors were treated with poly(I:C)-PEI or PBS starting 8 d after surgical tumor induction (average tumor size of 5–8 mm diameter).24 RNA injections were repeated twice weekly for three weeks. Treatment with poly(I:C)-PEI significantly prolonged survival of Panc02 tumor-bearing mice (Fig. 6A). In the PBS group, median survival was 30 days, as compared to 54 d for poly(I:C)-PEI treated mice (P < 0.0001). Some mice completely rejected their tumor with no signs for residual tumor mass at necropsy after an observation period of 100 d (Fig. 6A and C). Therapeutic efficacy was next assessed in a second tumor model. Mice with orthotopically transplanted T110229 tumors derived from KPC mice were treated as above (Fig. 6B). In the PBS group, median survival was 24 days, as compared to 42 d for poly(I:C)-PEI treated mice (P < 0.0001). Thus, systemic MDA5-based therapy led to significant tumor control in two different murine pancreatic carcinoma models. To explore the role of specific immune effector cells for the observed anti-tumor effect we injected mice with αCD8 or αNK1.1 antibodies during poly(I:C)-PEI treatment to deplete CD8+ T cells or NK cells, respectively. These experiments revealed that treatment efficacy was completely abrogated in CD8+ T cell-depleted mice, whereas NK cells were dispensable (Fig. 6C). To assess the induction of a memory T cell response, survivors of the poly(I:C)-PEI + IgG or αNK1.1 treatment groups were rechallenged s.c. with Panc02 tumor cells and tumor growth was monitored. Whereas tumors progressed in naïve mice, all survivors were protected from tumor outgrowth, indicative of an antitumor-directed T cell memory induction (Fig. 6D).
Figure 6.
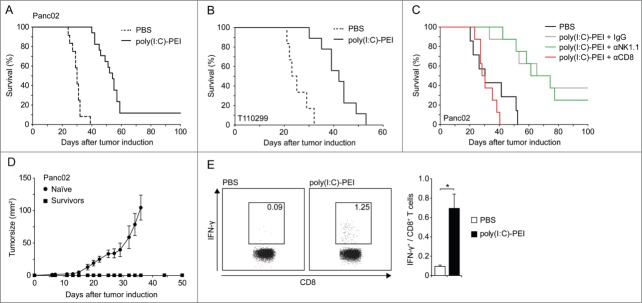
Systemic treatment with poly(I:C)-PEI leads to efficient tumor control in mice with pancreatic cancer. (A, B) Mice with orthotopic Panc02 (A) or T110299 tumors (B) were treated i.v. with 25 µg each poly(I:C) complexed to polyethylenimine (PEI) twice weekly for 3 weeks starting on day 8 after tumor implantation. Survival was monitored. (C) Mice with orthotopic Panc02 tumors were treated with phosphate buffered saline (PBS) or poly(I:C)-PEI as described in (A). CD8+ T cells or natural killer (NK) cells were depleted by i.p. injection with anti-CD8 or -NK1.1 monoclonal antibody (mAb) one day prior to poly(I:C)PEI treatment. IgG served as control. Survival was monitored. Statistical analysis was performed by Student's t test; P = 0.002 for PBS vs. poly(I:C)-PEI+IgG, P = 0.988 for poly(I:C)-PEI+IgG vs. poly(I:C)PEI+αNK1.1 and P < 0.001 for poly(I:C)-PEI+IgG vs. poly(I:C)-PEI+αCD8. (D) Naïve mice and mice that had survived their tumors in experiment C (>100 days) were re-challenged s.c. with Panc02 cells and tumor growth was monitored. (E) Poly(I:C)-PEI-treated mice that had survived the initial orthotopic tumor challenge for 100 d were re-challenged with Panc02 tumor cells and analyzed for tumor antigen (p15E)-specific CD8+ T cells in peripheral blood by ex vivo intracellular IFNγ staining after p15E peptide stimulation. Experiments with (A) n = 12 − 17, (B) n = 6 − 9, (C) n = 8 and (D, E) n = 5 mice per group are shown.
We previously reported that RLH activation in tumor cells induces immunogenic cell death leading to protective antitumor immunity in the situation of tumor re-challenge.29 For determining whether systemic MDA5-based immunotherapy of mice with viable tumors leads to a tumor-specific T cell response, we measured the frequency of p15E-specific cytotoxic T lymphocytes (CTLs) in the peripheral blood of treated mice that had rejected their tumor. These T cells recognize an H2‐Kb restricted gp70 epitope expressed by Panc02 tumor cells.31 In poly(I:C)-PEI but not PBS treated mice we found an increased frequency of p15Especific CTL, indicative of the emergence of an adaptive antitumor immune response (Fig. 6E). These findings are in agreement with our hypothesis that MDA5 activation leads to immunogenic cell death in vivo with the emergence of tumorreactive CD8+ T cells that control tumor growth.
Discussion
Mimicking a viral infection with synthetic RLH ligands is a promising strategy for tumor immunotherapy to overcome apoptosis resistance and immunosuppressive networks in cancer tissue.20-22,24 In the current study, we provide evidence for MDA5 expression and intact signaling in human pancreatic cancer cells. Immunohistochemistry of human tissue specimen revealed MDA5 staining in 7 out of 7 patients. Interestingly, expression levels appear to correlate with the degree of differentiation (high expression in G2 tumors vs. low expression in G3/4 tumors), which has to be confirmed in a larger cohort. In cell lines, the synthetic MDA ligand poly(I:C) induced IRF3 phosphorylation, Type I IFN production, upregulation of MHC-I expression and pronounced apoptotic tumor cell death, which are typical antiviral immune-defense mechanisms. We previously reported that similar effects can be achieved via RIG-I activation with 5′-ppp-RNA.24 However, poly(I:C)-PEI-induced cell death was far more effective requiring significantly lower RNA concentrations. Another group recently reported that human pancreatic cancer cells are sensitive to cell death induced by poly(I:C)-PEI and linked this sensitivity to repressed XIAP and survivin expression.32 Gene silencing experiments revealed that poly(I:C)-PEI mediated effects on tumor cells were mediated by MDA5 and not by other RNA sensors, such as TLR3 or RIG-I (Figs. 2C and D, and 3B and data not shown). This high sensitivity to MDA5 activation offers a broad therapeutic window since others and our own group have previously shown that tumor cells are particularly susceptible to RLH-induced cell death as compared to non-malignant cells.21-23,32 In addition, among diverse human and murine pancreatic cancer cell lines MDA5-induced cell death was independent of p53 mutational status. Furthermore, there was no evidence of apoptosis resistance to MDA5 ligands in any tumor cell line tested, which is in sharp contrast to the occurrence of resistance to chemotherapeutic drugs or radiation. Another interesting observation and similar to the cytological responses of murine pancreatic cancer cells, MDA5 activation in human tumor cells induced expression of the death receptor Fas and sensitized tumor cells toward FasL-mediated apoptosis.29 Thus, both intrinsic and extrinsic apoptosis pathways may contribute to the high vulnerability of pancreatic tumor cells toward MDA5 ligands in vivo. A limitation of our in vivo tumor model is that we cannot discriminate whether the therapeutic effect is mediated by MDA5 activation in tumor cells, immune cells or both. Furthermore, a contribution of TLR3 signaling is possible, although the lack of apoptosis and Type I IFN responses in tumors of mice treated with uncomplexed poly(I:C) argue against a role for TLR3 in the pancreatic cancer cell responses observed here. Together, these findings make MDA5 a promising target for pancreatic cancer therapy.
Importantly, our data indicate that local and systemic immune suppression, which can be viewed as a hallmark of pancreatic cancer,3 is effectively counteracted by MDA5-based immunotherapy. Systemic administration of poly(I:C)-PEI induced high serum levels of Type I IFN and activation of leukocyte populations, such as DCs, T cells, B cells and NK cells. Furthermore, the tumor micro-milieu was changed into an immune-permissive state with high levels of Type I and Type II IFNs and IFN-inducible genes. Type I IFN decreases the suppressive function of inhibitory immune cell populations, such as regulatory T cells and myeloid-derived suppressor cells abundant in pancreatic cancer.33,34 Type I IFN also favors Th1 polarization and promotes antigen-presentation by DCs.13 Thus, MDA5 activation in tumors has the potential to break tumor-induced immunosuppression and to re-activate tumor-specific T cells that are functionally defective in the tumor microenvironment. In fact, we observed the emergence of tumor-reactive CD8+ T cells in blood of treated mice and accumulation of CD8+ T cells in the tumor tissue. Tumor-resident CD8+ T cells expressed an activated phenotype, IFNγ and markers of enhanced cytolytic function, such as FasL and perforin.
Next to the Panc02 model, which forms highly aggressive tumors and a potent immunosuppressive network in the host, we studied efficacy of MDA5-based immunotherapy in mice with orthotopic tumors induced by implantation of T110299 cells, a cell line derived from a genetically engineered mouse model with targeted expression of mutated KrasG12D and Trp53fl/R172H(KPC mice).30 This tumor model closely reflects the biology of human pancreatic cancer in regards to genetic instability, desmoplastic stroma reaction and therapy resistance.30,35,36 In both tumor models we could show that systemic poly(I:C)-PEI therapy was well tolerated and significantly prolonged survival of mice. We also found that therapeutic efficacy was completely dependent on CD8+ T cells, whereas NK cells were dispensable. We recently described that RLH activation induces the release of tumor antigen in an immunogenic context favoring DC activation via Type I IFN and presentation of tumor antigen to CD8+ T cells, culminating in protective antitumor immunity.29 Long-lasting T cell memory could be confirmed in this study, as long-term surviving mice that had been systemically treated with poly(I:C)-PEI were protected from subsequent tumor challenge. These findings are characteristic of the immunogenic cell death previously described for certain chemotherapeutic drugs.37 Together, these findings show that MDA5-based immunotherapy combines innate and adoptive immune effector mechanisms for efficient tumor control in vivo. MDA5 ligands may prove useful for “autovaccination” against tumor antigens, for example via intratumoral injection into tumors destined for surgical removal or for the generation of tumor vaccine ex vivo.
In conclusion, our study demonstrates that MDA5-based immunotherapy tackles both tumor-mediated immunosuppression and apoptosis resistance in pancreatic cancer. Therapeutic efficacy could be shown in two different murine pancreatic cancer models in vivo, corroborating the concept of immunogenic tumor cell death induced by RLH ligands. Further advances can be expected by designing new delivery systems for selective transport of the therapeutic RNA into the tumor while limiting potential systemic toxicity38 and by combination with cytotoxic agents or irradiation, which is the focus of ongoing studies. MDA5-based immunotherapy could represent a new treatment option for pancreatic cancer patients and this approach warrants further investigation.
Material and Methods
Cell lines
PANC-1 was obtained from CLS cell lines service (Eppelheim, Germany) and MIAPaCa-2 cells from American Type Culture Collection (ATCC). Cells were used within 6 months after resuscitation. IMIM-PC1 and SUIT-007 cells were kindly provided by Prof. Patrick Michl (University of Marburg, Germany). The murine Panc02 cell line has been described.39 The tumor cell lines T510479 and T110299 were generated from primary pancreatic tumors of Ptf1a-Cre; LSL-KrasG12D and Ptf1a-Cre; LSL-KrasG12D; LSL-Trp53fl/R172H mice, respectively, that were back-crossed on a C57BL/6 background. Tumor cells were cultured in DMEM with 10% fetal calf serum (FCS; Gibco BRL, Berlin, Germany), 2 mM L-glutamine, 100 U/L penicillin and 0.1 mg/mL streptomycin (PAA, Pasching, Austria). Cell lines were routinely tested for mycoplasma contamination.
Reagents
Poly(I:C) (HMW) VacciGrade was purchased from Invivogen (Toulouse, France). Control RNA (5-GCG CUA UCC AGC UUA CGU ATT-3), siRNA against human MDA5 (5GUAUCGUGUUAUUGGAUUATT-3) and TLR3 (5GGUUGGUAAGGAUUCCUUU GCTT-3) were designed according to published guidelines and purchased from Eurofins MWG Operon (Ebersberg, Germany). In vitro transfection of cell lines with RNA was performed using Lipofectamine RNAiMax (Invitrogen, Darmstadt, Germany). For in vivo administration, poly(I:C) was complexed with in vivo-jetPEI (Peqlab, Erlangen, Germany) at an N/P ratio of 6 in 5% glucose solution. IFNα and zVAD-fmk were from Merck Millipore (Darmstadt, Germany), ELISA for CXCL10 from R&D Systems (Wiesbaden, Germany) and for IFNα from PBL Interferon source (Lörrach, Germany). The peptide p15E604611 was synthesized by Jerini Peptide Technologies (Berlin, Germany).
Mice, tumor engraftment and treatment
C57BL/6 mice were from Janvier (St Berthevin, France). Mice were at least 8 weeks old at the onset of experiments. Animal studies were approved by the local regulatory agency (Regierung von Oberbayern). Orthotopic tumors were induced by surgical implantation of 2 × 105 tumor cells into the pancreas as described.24 For in vivo administration, 25 µg of poly(I:C) was complexed (or not) with in vivo-JetPEI for tail vein injection. Therapy started on day 8 after tumor induction and was administered twice weekly over 3 weeks. Tumor growth and behavior of mice was monitored daily and distressed mice were sacrificed. For depletion of CD8+ T cells or NK cells mice were injected i.p. with 250 µg of anti-CD8 (clone YTS169.4) or anti-NK1.1 (PK136) mAb (BioXCell, Hölzel Diagnostika GmbH, Cologne, Germany) twice weekly starting one day prior to RNA injection. Surviving mice were used for tumor re-challenge experiments. These and control mice were injected with 0.5 × 106 Panc02 tumor cells and tumor growth was monitored for >50 d Mice were sacrificed when tumor size exceeded 100 mm2.
Western blot
Cells were lysed in Laemmli buffer, heated at 95°C for 5 min and SDS-PAGE was performed. Protein was transferred onto a nitrocellulose membrane and blocked with 3% non-fat dry milk in Tris-buffered saline supplemented with 0.05% Tween. Blots were incubated with the following antibodies: rabbit anti-human MDA5 or rabbit anti-mouse MDA5 (Enzo Life Science, Lörrach, Germany), rabbit anti-human phospho-IRF-3 or rabbit anti-mouse IRF-3, rabbit anti-human caspase-3, rabbit anti-human caspase-9 (all Cell Signaling, Frankfurt am Main, Germany) or mouse anti-mouse PARP1 (clone C2–10). HRP-conjugated goat anti-rabbit IgG and goat anti-mouse IgG (Santa Cruz Biotechnology, Heidelberg, Germany) served as secondary antibodies. For loading control, HRP-conjugated β-actin IgG (Santa Cruz) was used. Visualization was performed with ECL substrate (Fisher Scientific, Schwerte, Germany).
T cell isolation from tumor tissue
Tumor tissue was minced and mechanically dissociated using the Miltenyi gentleMACS™ Dissociator (Miltenyi Biotech, Bergisch Gladbach, Germany). Tissue was further digested in a buffer containing 1 mg/mL collagenase (Sigma Aldrich, Steinheim, Germany) and 0.1 mg/mL DNAse (Thermo Scientific, Darmstadt, Germany) for 30 min. The cell suspension was then separated from tissue debris by filtration using 100 and 40 µm cell strainers. Cells were stained with anti-CD45 (clone: 30-F11, Pacific Blue), anti-CD3ε (clone: 145–2C11, PE/Cy7), anti-CD4 (clone: GK1.5, PE) and anti-CD8α (clone 53–6.7, PerCP) antibodies, all from BioLegend (London, UK), for 30 min on ice and processed for cell sorting (FACSAria III, BD Biosciences). Purity was checked in post-sort analyses and was > 98%. Isolated T cells were analyzed by cytofluorimetry or processed for RNA isolation using peqGOLD TriFast (Peqlab, Erlangen, Germany) following the manufacturer's protocol.
Tumor tissue RNA isolation and qPCR
Tumor tissue was snap frozen in liquid nitrogen and homogenized using mortar and pestle. Homogenate was processed for total RNA isolation using peqGOLD Total RNA Kit (Peqlab). RNA was adjusted and transcribed into cDNA with the RevertAid First Strand cDNA Synthesis Kit (Fisher Scientific). qPCR was done with the Kapa Probe Fast Universal kit (Peqlab) on the LightCycler® 480 II instrument (Roche, Mannheim, Germany) and samples were normalized to HPRT. Primers were designed with Roche's Universal Probes library.
Flow cytometry
Apoptosis was determined by staining tumor cells with APC-conjugated annexin V (Immunotools, Friesoythe, Germany) and propidium iodide (PI, Sigma, Munich, Germany) and analysis by flow cytometry. Cells staining negative for annexin V and PI were defined as viable cells. Murine tumor cells were surface stained with anti-MHC Class I (clone AF6–88.5, BioLegend) or anti-CD95 (clone Jo2, BD Biosciences) antibodies and human tumor cells with anti-HLA-A, B, C (clone G46–2.6) or anti-CD95 (clone DX2) antibodies. For assessing leukocyte activation, spleens were processed into single cell suspensions and red cells were lysed with BD Pharm Lyse lysis buffer (BD Biosciences, Heidelberg, Germany). Cell surface staining was done with flourochrome-conjugated antibodies: anti-CD3ε (clone 145–2C11), anti-CD4 (clone RM4–5), anti-CD8α (clone 53–6.7), anti-CD19 (clone 1D3), anti-NK1.1 (clone NKRP1B, NKR-P1C, all BD Biosciences, Heidelberg, Germany), and anti-CD69 (clone H1.2F3, Caltag Laboratories, Buckingham, UK). DCs were stained with anti-CD11c (clone N418), anti-CD8α (clone 53–6.7), anti-CD80 (clone 16–10A1) and anti-CD86 (clone GL-1) antibodies, all from BioLegend (London, UK). To detect tumor reactive CD8+ T cells, peripheral blood cells from tumor bearing mice were cultured ex vivo for 4 h in the presence of 2 µg/mL brefeldin A with p15E or 1 µg/mL of an irrelevant peptide. Cells were then stained with anti-CD3ε and anti-CD8α, washed, fixed and permeabilized (BD Biosciences), followed by intracellular staining with anti-IFNγ (clone XMG1.2; Caltag) antibody. Flow cytometry was performed on a FACSCanto II (BD Biosciences) and data analyzed with FloJo vX (Tree Star Inc., Ashland, OR, USA).
Fas-mediated killing
Tumor cells were seeded at a density of 5 × 104 cells per 24-well and treated with poly(I:C)c at the indicated concentrations. After 20 h, cells were incubated with 1 µg/mL anti-CD95 (clone CH11; Millipore, Schwalbach, Germany). 20 h later, cells were collected and stained with annexin V/PI for FACS analysis.
Immunohistochemistry and TUNEL staining
Cryosections of Panc02 tumors were fixed with 4% paraformaldehyde for 10 min on ice and washed with PBS. Unspecific binding sites were blocked with 5% goat serum in PBS. Sections were stained with Armenian hamster anti-mouse CD3ε (clone 500A2) and rat antimouse CD8α (clone 53–6.7) antibodies (both BD Biosciences) for 1 h. Secondary antibody Cy3-conjugated goat anti-Armenian hamster and biotin-conjugated goat anti-rat antibodies (both Dianova GmbH, Hamburg, Germany) were added for 45 min. Finally, sections were incubated with Alexa Fluor® 633-conjugated streptavidin (Invitrogen) for 20 min. For visualization of nuclei, sections were counterstained with Hoechst (Invitrogen). TUNEL staining was performed using the In Situ Cell Death Detection Kit (Roche, Mannheim, Germany) and mounted with Vectashield® w/DAPI (Vector Laboratories, Burlingame, USA) for nuclei visualization. Stained tissues were visualized by confocal fluorescence microscopy (Leica TCS SP5, Wetzlar, Germany). MDA5 staining of human tumors was performed on formalin-fixed, paraffin-embedded samples that were cut into approximately <2 µm thick slices and mounted on SuperFrost Plus microscope slides (Menzel Gläser, Braunschweig, Germany). After deparaffinization and rehydration, sections were immersed into Target Retrieval solution (Dako North America Inc., Carpinteria, USA), pH 6. Then the slides were incubated with the primary antibody (MDA5, Abcam Cat. No. ab79055), dilution 1:250, at room temperature for 60 min. Immunoreactivity was detected using MACH 3 Rabbit AP Polymer Detection (Biocare, Cat. No. M3R533H). Finally, slides were stained with Chromogen Red (Dako, taken out of Real Detection System APAAP, Cat. No. K5000) and counterstained in Hematoxylin Gill's Formula (Vector, Cat. No. H-3401). Since all tumor cells showed positive staining, no quantification was performed other than intensity of staining (1 = weak, 2 = modest and 3 = strong) was analyzed.
Statistical analysis
Data present means ± SD (in vitro data) or SEM (in vivo data). Differences were analyzed using 2-tailed Student's t-test. Multiple comparisons were analyzed by 2-way ANOVA including Bonferroni correction. Survival curves were analyzed with Mantel-Cox test. Statistical analysis was performed using GraphPad Prism software (version 5.0a); P-values <0.05 were considered significant.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft SCHN 664/3–1 and SCHN 664/3–2 to MS, Elite Network of Bavaria (i-Target) to MS, SK and SE, GK 1202 to MS, SK and SE, GK 1202 student grants to TA and FöFoLe student grants to EB, SVK and HB.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work is part of the theses of EB, HB and SVK at the University of Munich. The authors thank Andrea Sendelhofert for conducting histopathology.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Siegel R, DeSantis C, Virgo K, Stein K, Mariotto A, Smith T, Cooper D, Gansler T, Lerro C, Fedewa S, et al.. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin 2012; 62:220-41; PMID:22700443 [DOI] [PubMed] [Google Scholar]
- 2.Sideras K, Braat H, Kwekkeboom J, van Eijck CH, Peppelenbosch MP, Sleijfer S, Bruno M. Role of the immune system in pancreatic cancer progression and immune modulating treatment strategies. Cancer Treat Rev 2014; 40:513-22; PMID:24315741; http://dx.doi.org/ 10.1016/j.ctrv.2013.11.005 [DOI] [PubMed] [Google Scholar]
- 3.Zheng L, Xue J, Jaffee EM, Habtezion A. Role of immune cells and immune-based therapies in pancreatitis and pancreatic ductal adenocarcinoma. Gastroenterology 2013; 144:1230-40; PMID:23622132; http://dx.doi.org/ 10.1053/j.gastro.2012.12.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lutz E, Yeo CJ, Lillemoe KD, Biedrzycki B, Kobrin B, Herman J, Sugar E, Piantadosi S, Cameron JL, Solt S, et al.. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor-secreting tumor vaccine for pancreatic adenocarcinoma. A Phase II trial of safety, efficacy, and immune activation. Ann Surg 2011; 253:328-35; PMID:21217520; http://dx.doi.org/ 10.1097/SLA.0b013e3181fd271c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lepisto AJ, Moser AJ, Zeh H, Lee K, Bartlett D, McKolanis JR, Geller BA, Schmotzer A, Potter DP, Whiteside T, et al.. A phase I/II study of a MUC1 peptide pulsed autologous dendritic cell vaccine as adjuvant therapy in patients with resected pancreatic and biliary tumors. Cancer Ther 2008; 6:955-64; PMID:19129927 [PMC free article] [PubMed] [Google Scholar]
- 6.Bauer C, Dauer M, Saraj S, Schnurr M, Bauernfeind F, Sterzik A, Junkmann J, Jakl V, Kiefl R, Oduncu F, et al.. Dendritic cell-based vaccination of patients with advanced pancreatic carcinoma: results of a pilot study. Cancer Immunol Immunother 2011; 60:1097-107; PMID:21547597; http://dx.doi.org/ 10.1007/s00262-011-1023-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacobs C, Duewell P, Heckelsmiller K, Wei J, Bauernfeind F, Ellermeier J, Kisser U, Bauer CA, Dauer M, Eigler A, et al.. An ISCOM vaccine combined with a TLR9 agonist breaks immune evasion mediated by regulatory T cells in an orthotopic model of pancreatic carcinoma. Int J Cancer 2011; 128:897-907; PMID:20473889; http://dx.doi.org/ 10.1002/ijc.25399 [DOI] [PubMed] [Google Scholar]
- 8.Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, Greenberg PD, Hingorani SR. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 2014; 63(11):1769-81; PMID:24555999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sandin LC, Eriksson F, Ellmark P, Loskog AS, Totterman TH, Mangsbo SM. Local CTLA4 blockade effectively restrains experimental pancreatic adenocarcinoma growth in vivo. Oncoimmunology 2014; 3:e27614; PMID:24701377; http://dx.doi.org/ 10.4161/onci.27614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, et al.. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011; 331:1612-6; PMID:21436454; http://dx.doi.org/ 10.1126/science.1198443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neesse A, Gress TM, Michl P. Therapeutic targeting of apoptotic pathways: novel aspects in pancreatic cancer. Curr Pharm Biotechnol 2012; 13:2273-82; PMID:21605073; http://dx.doi.org/ 10.2174/138920112802501953 [DOI] [PubMed] [Google Scholar]
- 12.Xu C, Li H, Su C, Li Z. Viral therapy for pancreatic cancer: tackle the bad guys with poison. Cancer letters 2013; 333:1-8; PMID:23354590; http://dx.doi.org/ 10.1016/j.canlet.2013.01.035 [DOI] [PubMed] [Google Scholar]
- 13.van den Boorn JG, Hartmann G. Turning tumors into vaccines: co-opting the innate immune system. Immunity 2013; 39:27-37; PMID:23890061; http://dx.doi.org/ 10.1016/j.immuni.2013.07.011 [DOI] [PubMed] [Google Scholar]
- 14.Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 2006; 314:997-1001; PMID:17038589; http://dx.doi.org/ 10.1126/science.1132998 [DOI] [PubMed] [Google Scholar]
- 15.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, et al.. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006; 441:101-5; PMID:16625202; http://dx.doi.org/ 10.1038/nature04734 [DOI] [PubMed] [Google Scholar]
- 16.Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, et al.. 5′-Triphosphate RNA is the ligand for RIG-I. Science 2006; 314:994-7; PMID:17038590; http://dx.doi.org/ 10.1126/science.1132505 [DOI] [PubMed] [Google Scholar]
- 17.Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, Diamond MS, Colonna M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci U S A 2006; 103:8459-64; PMID:16714379; http://dx.doi.org/ 10.1073/pnas.0603082103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev 2009; 227:75-86; PMID:19120477; http://dx.doi.org/ 10.1111/j.1600-065X.2008.00737.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loo YM, Gale M Jr.. Immune signaling by RIG-I-like receptors. Immunity 2011; 34:680-92; PMID:21616437; http://dx.doi.org/ 10.1016/j.immuni.2011.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tormo D, Checinska A, Alonso-Curbelo D, Perez-Guijarro E, Canon E, Riveiro-Falkenbach E, Calvo TG, Larribere L, Megias D, Mulero F, et al.. Targeted activation of innate immunity for therapeutic induction of autophagy and apoptosis in melanoma cells. Cancer Cell 2009; 16:103-14; PMID:19647221; http://dx.doi.org/ 10.1016/j.ccr.2009.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poeck H, Besch R, Maihoefer C, Renn M, Tormo D, Morskaya SS, Kirschnek S, Gaffal E, Landsberg J, Hellmuth J, et al.. 5′-Triphosphate-siRNA: turning gene silencing and Rig-I activation against melanoma. Nature Med 2008; 14:1256-63; PMID:18978796; http://dx.doi.org/ 10.1038/nm.1887 [DOI] [PubMed] [Google Scholar]
- 22.Besch R, Poeck H, Hohenauer T, Senft D, Hacker G, Berking C, Hornung V, Endres S, Ruzicka T, Rothenfusser S, et al.. Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon-independent apoptosis in human melanoma cells. J Clin Invest 2009; 119:2399-411; PMID:19620789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meng G, Xia M, Xu C, Yuan D, Schnurr M, Wei J. Multifunctional antitumor molecule 5′-triphosphate siRNA combining glutaminase silencing and RIG-I activation. Int J Cancer 2014; 134:1958-71; PMID:23921958; http://dx.doi.org/ 10.1002/ijc.28416 [DOI] [PubMed] [Google Scholar]
- 24.Ellermeier J, Wei J, Duewell P, Hoves S, Stieg MR, Adunka T, Noerenberg D, Anders HJ, Mayr D, Poeck H, et al.. Therapeutic efficacy of bifunctional siRNA combining TGF-beta1 silencing with RIG-I activation in pancreatic cancer. Cancer Res 2013; 73:1709-20; PMID:23338611; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-3850 [DOI] [PubMed] [Google Scholar]
- 25.Dunn GP, Bruce AT, Sheehan KC, Shankaran V, Uppaluri R, Bui JD, Diamond MS, Koebel CM, Arthur C, White JM, et al.. A critical function for type I interferons in cancer immunoediting. Nat Immunol 2005; 6:722-9; PMID:15951814; http://dx.doi.org/ 10.1038/ni1213 [DOI] [PubMed] [Google Scholar]
- 26.Chen ZH, Zhang H, Savarese TM. Gene deletion chemoselectivity: codeletion of the genes for p16(INK4), methylthioadenosine phosphorylase, and the α- and β-interferons in human pancreatic cell carcinoma lines and its implications for chemotherapy. Cancer Res 1996; 56:1083-90; PMID:8640765 [PubMed] [Google Scholar]
- 27.Megjugorac NJ, Young HA, Amrute SB, Olshalsky SL, Fitzgerald-Bocarsly P. Virally stimulated plasmacytoid dendritic cells produce chemokines and induce migration of T and NK cells. J Leukoc Biol 2004; 75:504-14; PMID:14742635; http://dx.doi.org/ 10.1189/jlb.0603291 [DOI] [PubMed] [Google Scholar]
- 28.Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD. IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J Immunol 2002; 168:3195-204; http://dx.doi.org/ 10.4049/jimmunol.168.7.3195 [DOI] [PubMed] [Google Scholar]
- 29.Duewell P, Steger A, Lohr H, Bourhis H, Hoelz H, Kirchleitner SV, Stieg MR, Grassmann S, Kobold S, Siveke JT, et al.. RIG-I-like helicases induce immunogenic cell death of pancreatic cancer cells and sensitize tumors toward killing by CD8 T cells. Cell Death Differ 2014; 21(12):1825-37; PMID:25012502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005; 7:469-83; PMID:15894267; http://dx.doi.org/ 10.1016/j.ccr.2005.04.023 [DOI] [PubMed] [Google Scholar]
- 31.Bauer C, Bauernfeind F, Sterzik A, Orban M, Schnurr M, Lehr HA, Endres S, Eigler A, Dauer M. Dendritic cell-based vaccination combined with gemcitabine increases survival in a murine pancreatic carcinoma model. Gut 2007; 56:1275-82; PMID:17395611; http://dx.doi.org/ 10.1136/gut.2006.108621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhoopathi P, Quinn BA, Gui Q, Shen XN, Grossman SR, Das SK, Sarkar D, Fisher PB, Emdad L. Pancreatic cancer-specific cell death induced in vivo by cytoplasmic-delivered polyinosine-polycytidylic acid. Cancer Res 2014; 74:6224-35; PMID:25205107; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-0819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zoglmeier C, Bauer H, Norenberg D, Wedekind G, Bittner P, Sandholzer N, Rapp M, Anz D, Endres S, Bourquin C. CpG blocks immunosuppression by myeloid-derived suppressor cells in tumor-bearing mice. Clin Cancer Res 2011; 17:1765-75; PMID:21233400; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-2672 [DOI] [PubMed] [Google Scholar]
- 34.Pace L, Vitale S, Dettori B, Palombi C, La Sorsa V, Belardelli F, Proietti E, Doria G. APC activation by IFN-α decreases regulatory T cell and enhances Th cell functions. J Immunol 2010; 184:5969-79; http://dx.doi.org/ 10.4049/jimmunol.0900526 [DOI] [PubMed] [Google Scholar]
- 35.Courtin A, Richards FM, Bapiro TE, Bramhall JL, Neesse A, Cook N, Krippendorff BF, Tuveson DA, Jodrell DI. Anti-tumour efficacy of capecitabine in a genetically engineered mouse model of pancreatic cancer. PloS one 2013; 8:e67330; PMID:23840665; http://dx.doi.org/ 10.1371/journal.pone.0067330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, et al.. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009; 324:1457-61; PMID:19460966; http://dx.doi.org/ 10.1126/science.1171362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol 2013; 31:51-72; PMID:23157435; http://dx.doi.org/ 10.1146/annurev-immunol-032712-100008 [DOI] [PubMed] [Google Scholar]
- 38.Ali HM, Urbinati G, Raouane M, Massaad-Massade L. Significance and applications of nanoparticles in siRNA delivery for cancer therapy. Expert Rev Clin Pharmacol 2012; 5:403-12; PMID:22943120; http://dx.doi.org/ 10.1586/ecp.12.33 [DOI] [PubMed] [Google Scholar]
- 39.Corbett TH, Roberts BJ, Leopold WR, Peckham JC, Wilkoff LJ, Griswold DP Jr., Schabel FM Jr.. Induction and chemotherapeutic response of two transplantable ductal adenocarcinomas of the pancreas in C57BL/6 mice. Cancer Res 1984; 44:717-26; PMID:6692374 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.