

Abstract
Background
Nephrotic syndrome is traditionally classified on the basis of the response to standard steroid treatment. Mutations in more than 24 genes have been associated with nephrotic syndrome in children, although the great majority of steroid-resistant cases have been attributed to mutations in three main genes: NPHS1, NPHS2 and WT1. The aims of this study were to identify mutations in these genes more frequently reported as mutated and to characterize each variation using different in silico prediction algorithms in order to understand their biological functions.
Methods
We performed direct sequence analysis of exons 8 and 9 of WT1, 8 exons of NPHS2 and 29 exons of NPHS1, including NPHS2 and NPHS1 intron–exon boundary sequences, as well as 700 bp of the 5′ UTR from both genes in 27 steroid-resistant patients aged between 3 months and 18 years.
Results
Analysis of the NPHS2 gene revealed four missense mutations, one frameshift mutation and three variations in the 5′ UTR. Four patients presented compound heterozygosis, and four other patients presented one heterozygous alteration only. WT1 and NPHS1 gene analysis did not reveal any mutations.
Discussion
This is the first study focusing on genetics of SRNS in Brazilian children. Identification of mutations is important because it could influence physicians’ decision on patient treatment, as patients carrying mutations can be spared the side effects of immunosuppressive therapy and ultimately could be considered for kidney transplantation from a living donor.
Conclusions
After molecular analysis of the genes more frequently reported as mutated in 27 steroid-resistant nephrotic syndrome patients, we identified NPHS2 mutations confirming the hereditary character of the kidney disease in only 14.8 % of patients. Therefore, the next step is to perform a next generation sequencing based analysis of glomeluropathy-related panel of genes for the remaining patients in order to search for mutations in other genes related to steroid-resistant nephrotic syndrome.
Electronic supplementary material
The online version of this article (doi:10.1186/s12881-015-0231-9) contains supplementary material, which is available to authorized users.
Keywords: Steroid-resistant nephrotic syndrome, Children, Adolescents, Mutations, NPHS2, NPHS1, WT1
Background
Nephrotic syndrome (NS) is considered as one of the main kidney diseases in children and is characterized by proteinuria, edema, hypoalbuminemia and hyperlipidemia [1]. Depending on the age at diagnosis, NS is usually classified as congenital (CNS) when it manifests in utero or during the first 3 months of life; as infantile when it occurs during the first year; as childhood when symptoms occur between 1 and 12 years of age; and as juvenile with onset between 12 and 18 years of age [2]. Approximately 90 % of patients show a favorable outcome after steroid treatment of NS and are termed steroid-sensitive NS (SSNS) patients. Clinically, however, these SSNS patients are not a homogeneous group, as some continue to develop frequent relapses (FRNS) or even follow a steroid-dependent course (SDNS) of the disease. The remaining 10 % are steroid-resistant NS (SRNS) patients who do not respond to steroids or to any other immunosuppressive therapy; they show most commonly histopathological findings of focal segmental glomerulosclerosis (FSGS) and generally progress to end-stage renal disease [2].
A single-gene cause is responsible for approximately 30 % of SRNS cases, as reported in a recent paper by Sadowski et al. [3] on the largest international cohort studied so far for monogenic causes of SRNS. By using an in-house developed high-throughput approach, the investigators examined 1783 families and identified mutations in 21 out of 27 genes encoding proteins mainly expressed in the glomerular filtration barrier (GFB). The clinical course in affected children differs considerably with respect to the mode of inheritance, age, renal biopsy results, response to immunosuppressive treatment and extrarenal manifestations. One of the main genes, NPHS2 (OMIM *604766), encodes podocin, a member of the stomatin protein family that is localized at the slit membrane [4]. In the slit–diaphragm (SD) complex, podocin interacts with nephrin, a member of the immunoglobulin superfamily and the principal protein in the SD [5]. Podocin mediates the recruitment of nephrin into specialized microdomains of the cell plasma membrane where, in the so-called “lipid rafts”, both proteins play essential roles in the maintenance of the glomerular permeability barrier [6]. Moreover, podocin belongs to a family comprising more than 1800 proteins conserved throughout evolution that share approximately 150 amino acid domains similar to mitochondrial protein prohibitin (PHB). Many of the PHB domain proteins regulate membrane protein function by binding sterols and altering their local lipid environment [7]. Huber et al. [8] demonstrated that podocin binds to cholesterol and contributes to form a supercomplex of proteins and lipids that regulate ion channel complexes; the authors also showed that podocin interacts with the cytoplasmic tail of nephrin, thus potentiating the effect of nephrin cellular signaling.
Mutations in the NPHS2 gene were originally described in families with autosomal recessive SRNS (nephrotic syndrome type 2, OMIM #600995) [8] but later were found to be present also in sporadic cases [9], occurring in approximately 40 % of familial cases and 6–17 % of sporadic SRNS cases [9–11]. Familial and sporadic cases with compound heterozygous or homozygous mutations in NPHS2 typically have disease onset between 3 months and 5 years of age. Recently, Bouchireb et al. [12] pointed out that not all ethnic groups share the same frequency of mutations in this gene, following an extensive review of NPHS2 mutations in SRNS patients between 1999 and 2013. It was found that African-American and Asian patients show a low prevalence of NPHS2 mutations.
Regarding the NPHS1 gene (OMIM *602716) that encodes nephrin, two major mutations were originally identified in Finnish patients with CNS of the Finnish type (OMIM #256300) [13]. In non-Finnish patients, a large number of different mutations have been described [13–15]. In addition to the typical features of CNS, mutations in nephrin were also associated with childhood-onset SRNS in patients aged from 5 months to 8 years [16]. Furthermore, Koziell et al. [17] described the coexistence of mutations in NPHS2 and NPHS1 genes in patients with milder CNS.
A third important gene involved in the genetics of NS is WT1 (OMIM *607102), which encodes a zinc finger transcription factor. WT1-dominant mutations were originally identified to be responsible for Wilms’ tumor (WT), a childhood kidney cancer (OMIM #194070) [18, 19, 20]. WT1 mutations were also associated with Denys-Drash syndrome (OMIM #194080) [21, 22] and Frasier syndrome (OMIM #136680) [22]. However, dominant de novo mutations in exons 8 and 9 have been found as the cause of approximately 2.7–7 % of sporadic cases of isolated SRNS in infants and children. In a recent study, Lipska et al. [23] used the PodoNet cohort to describe the genotypic and phenotypic spectrum of WT1-associated kidney disease and found WT1 mutations in 6 % of sporadic SRNS patients, confirming the prevalence found by previous smaller studies.
Molecular data are clinically relevant to SRNS patients, because children who do not respond to corticosteroids and present with mutations in any of these genes can be spared the adverse effects of corticosteroid treatment. In addition, patients with mutations have a reduced risk of developing FSGS after renal transplantation, compared to patients without mutations, at a rate of 8 % versus 30 %, respectively [11].
In our study, we performed sequence analysis of the entire NPHS2 and NPHS1 genes and exons 8 and 9 of the WT1 gene in 27 SRNS Brazilian patients, including seven unrelated familial cases and 20 sporadic cases. We present nucleotide changes found after screening these 27 SRNS patients.
Methods
Patients
Between 2008 and 2013, 27 SRNS patients, comprising seven unrelated familial cases and 20 sporadic (isolated) cases, were included in this study. Prerequisites for enrollment included the presence of NS for at least 1 year and the age at disease onset being between 3 months and 18 years of age. The median age at disease onset was 4 years. From the 27 patients, 15 were males and 12 were females. Resistance to steroids was defined by nephrologists who followed up the patients, according to the International Study of Kidney Disease in Children definition [24]. The definition of chronic kidney disease (CKD) was classified according to the National Kidney Foundation for patients aged over 2 years [25]. Eight of 27 patients progressed to CKD and had already had a kidney transplant. Table 1 presents the age at disease onset, age at CKD diagnosis and at transplantation, and biopsy and molecular findings. The Ethics Committee from Faculdade de Ciências Médicas at Universidade Estadual de Campinas approved this study. Written informed consent for the study was obtained from all patients and their parents.
Table 1.
Clinical data of SRNS patients with NPHS2 and NPHS1 alterations
| Patient number | Gender | Sporadic/ familiala | Age at onset (years) | Renal Biopsyb | Age of onset to CKD - V (Yes/No)c | Age of transplant (Yes/No) | NPHS2 | ||
|---|---|---|---|---|---|---|---|---|---|
| Nucleotide change | Effect on coding | Ref. | |||||||
| (years) | (years) | ||||||||
| Steroid Resistant cases with two NPHS2 heterozygous alterations | |||||||||
| P6 | F | spo | 12 | FSGS | Yes/15 | Yes/15 | c.686G > A | p.Arg229Gln | [37] |
| c.851C > T | p.Ala284Val | [10] | |||||||
| P67 | F | spo | 2.2 | DMP | No | No | c.686G > A | p.Arg229Gln | [37] |
| c.928G > A | p.Glu310Lys | [38] | |||||||
| P103 | F | fam | 1.2 | FSGS | No | No | c.714delG | p.Lis239Argfs*13 | ps |
| c.779 T > A | p.Val260Glu | [12] | |||||||
| P154 | M | fam | 13 | FSGS | No | No | c.686G > A | p.Arg229Gln | [37] |
| c.851C > T | p.Ala284Val | [10] | |||||||
| Steroid Resistant cases with one NPHS2 heterozygous alteration | |||||||||
| P111 | F | spo | 3 | FSGS | No | No | c.-164C > T | - | ps |
| P68 | M | spo | 4 | CO | No | No | c.-196C > G | - | ps |
| P85 | M | spo | 6.11 | CO | Yes/8 | Yes | c.-537_-531delCTTTTTT | - | [40] |
| P72 | M | fam | 16 | FSGS | Yes/18 | Yes/18 | c.686G > A | p.Arg229Gln | [37] |
a Spo: sporadic; Fam: familial;b FSGS:focal segmental glomerular sclerosis; DMP: diffuse mesangial proliferation; CO: complex = minimal change disease or diffuse mesangial proliferation or focal segmental glomerular sclerosis; CKD: cchronic kidney disease; n.a.: not analyzed; nd: not determined
Molecular analysis of NPHS2, NPHS1 and WT1 genes
Genomic DNA was isolated from blood samples using standard methods [26]. Amplification of all coding exons, exon–intron boundaries and 700 bp of the 5′ UTR end from both NPHS2 and NPHS1 genes were performed by polymerase chain reaction (PCR). For WT1 gene we performed amplification of exons 8 and 9 only (for specific primers, see in Additional file 1: Tables S1, S2 and S3). After PCR reactions and direct sequencing of products (see Additional file 1), Chromas Pro v.1.5 and CLC Sequence Viewer v.6.6.2, were used to analyze and compare these sequences with the NPHS2, NPHS1 and WT1 reference sequences in the Ensembl database (ENSG00000116218, ENSG00000161270, ENSG00000184937, respectively; http://www.ensembl.org/index.html).
Bioinformatics analysis
The impact of missense and 5′ UTR variations on podocin or nephrin structure and function was evaluated using selected bioinformatics tools. SIFT (SortingIntolerantFromTolerant; http://sift.jcvi.org/) [27], Polyphen (PolymorphismPhenotyping; http://genetics.bwh.harvard.edu/pph2/) [28] and Align GVGD (http://agvgd.iarc.fr/agvgd_input.php/) [29] were used to assess potentially damaging effects of non-synonymous amino acid substitutions (see in Additional file 1: Table S4). In order to verify conserved amino acid residues, we performed multiple alignment analysis using ClustalW program [30] that lines up orthologs from different species to reveal the identities, similarities and differences. Human nephrin and podocin sequences were compared with orthologous sequences available in UniProt data bank (http://www.uniprot.org/). Moreover, in order to better understand if missense mutations affected exonic splicing enhancers or silencers (ESEs and ESS, respectively) leading to splicing defects, we further performed prediction analysis using two premRNA splicing prediction tools: Human Splicing Finder (HSF) [31] and SpliceAid2 [32]. The three 5′ UTR heterozygous alterations were analyzed using Alibaba2 gene regulation program (http://www.gene-regulation.com/pub/programs/alibaba2/index.html) that predicts transcription factor binding sites in the DNA query using data from TRANSFAC® Public eukaryotic transcription factors [33].
Results and discussion
Since the discovery that inherited structural defects in the GFB are responsible for a large proportion of SRNS cases, major breakthrough studies on the discovery of genes and their implication in the disease have been carried out mainly in European and North American countries [3, 4, 14, 34, 35, 36]. In Brazil, genetic screening of patients with NS in childhood is still incipient. This is the first study focusing on genetics of SRNS in Brazilian children. Identification of mutations is important because it could influence physicians’ decision on patient treatment, as patients carrying mutations can be spared the side effects of immunosuppressive therapy and ultimately could be considered for kidney transplantation from a living donor.
Here we present results for a cohort of 27 SRNS patients from a public hospital that is a reference center in the South East region of Brazil where approximately 200 patients with NS are routinely followed up. Of note, the 27 SRNS patients represented a frequency of 13.5 % (27/200), which is in line with the reported worldwide frequency of 10–20 % for this form of the disease [2, 11]. However, the cohort was not representative of the entire country, since the Brazilian population consists of about 205 million of highly miscegenated people with diverse and regional features. However, there is no other genetic study on NS patients in Brazil for comparison.
Only four patients (two familial and two sporadic cases) representing a total of 14.8 % showed two heterozygous alterations that confirmed the hereditary character of their kidney disease. Mutations in WT1 exons 8 and 9 and NPHS1 gene were not found. NPHS2 analysis, however, showed eight alterations: four missense mutations, one frameshift mutation and three variations in the 5’ UTR (Table 1).
Sporadic cases
Among the 20 sporadic cases, the c.686G > A transition in exon 5 leading to p.Arg229Gln [36] was identified in association with two different pathogenic missense mutations (Table 1). Patient P6 was compound heterozygous [p.Arg229Gln];[p.Ala284Val], as indicated by the c.851C > T nucleotide transition [9] observed from NPHS2 sequencing. Compound heterozygosis for the two mutations was demonstrated by the analysis of patient P6’s father carrying p.Ala284Val and mother carrying p.Arg229Gln (Fig. 1). Patient P67 was compound heterozygous [p.Arg229Gln];[p.Glu310Lys] which resulted from c.928G > A transition in exon 8 [37]. Individual mutations were also inherited from each parent, indicating an autosomal recessive inheritance for SRNS in both cases (Fig. 1). The p.Arg229Gln variant is the most frequently reported non-synonymous NPHS2 variant in Caucasians; there are reports on its allele frequency showing that it is more frequent among European descendants than African descendants in the United States [10, 11, 37]. Tsukaguchi et al. demonstrated that trans-association of p.Arg229Gln with pathogenic mutations may result in a less severe phenotype such as late-onset SRNS. The most commonly associated mutation is p.Ala284Val, and notably this combination is predominant among South American populations from Chile and Argentina with adult-onset SRNS [37]. Recently, Tory et al., observed that p.Arg229Gln podocin presented a subcellular mislocalization only when co-expressed with podocins carrying amino acid substitutions encoded on exons 7–8, but not with substitutions encoded on exons 1–6 [38]. The authors suggested that disease-associated 3′ mutations exert a dominant-negative effect on p.Arg229Gln podocin, on the basis of a structural model of dimerization; heterodimers of p.Ala284Val podocin with p.Arg229Gln podocin showed an altered three-dimensional structural arrangement, and the authors therefore proposed this different structure contributed to the retention of p.Arg229Gln podocin within cytoplasmic compartments. However, when pathogenic mutations were associated with wild-type podocin, they behaved as recessive alleles. In summary, Tory et al. [39] described a phenomenon in an autosomal recessive disorder, in which the pathogenicity of one allele depended on that of the other allele, and they proposed a pattern of mutation-dependent recessive inheritance in NPHS2-associated SRNS that is beyond Mendel’s laws. Based on these criteria, the pathogenic association [p.Arg229Gln]; [mutEx7–8] was identified in both patients P6 and P67. Patient P6 was diagnosed with SRNS when proteinuria occurred at the age of 12 years, thus characterizing a late-onset form of the disease. Renal biopsy revealed FSGS, and the patient progressed to CKD in 3 years when she had a kidney transplant; after approximately 1 year post-transplant, her renal function has now returned to normal. In contrast, patient P67 developed proteinuria at 2 years and 2 months of age, and renal biopsy demonstrated diffuse mesangial proliferation (Table 1). The same [p.Arg229Gln];[p.Glu310Lys] association was identified by Machuca et al. [37] in three patients, but all three presented with FSGS. In silico evaluation using SpliceAid2 and HSF splicing prediction tools for c.686G > A (p.Arg229Gln), c.851C > T (p.Ala284Val) and c.928G > A (p.Glu310Lys) gave the following results: SpliceAid2 prediction indicated that c.686A did not show significant modifications in splicing motifs; c.851 T did not modify protein-binding recognition motifs; however, c.928A variant disrupted a hnRNP A1 protein-binding motif in the mutated sequence (Fig. 2). HSF prediction indicated that c.686A did not show modification in sequence motifs (data not shown); c.851 T created an exonic ESS site; for c.928A substitution, there was an ESE site broken after HSF prediction (Table 2).
Fig. 1.
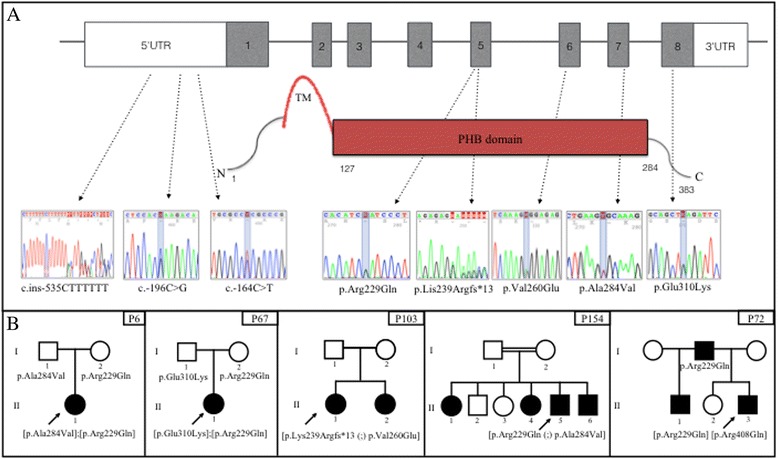
Schematic structure of NPHS2/podocin showing the position of alterations and patient’s heredograms. a Eight exons of NPHS2 gene and podocin illustration showing electropherograms of the three 5’ UTR variants, four missense mutations and the frameshift mutation in the corresponding exons/domains identified in five SRNS patients. TM = transmembrane domain; PHB = prohibitin domain. b Sporadic cases P6, P67 (first two boxes on the left) and family cases P103, P154 and P72 pedigrees. Black squares and circles represent patients with kidney disease. Arrow indicates the index case. Mutations are annotated below the individuals that had their NPHS2 or NPHS1 genes screened. P103 (II1) and her sister (II2) presented [p.Lys239Argfs*13 (;) p.Val260Glu] association. P154 (II4) and her two brothers (II5 and II6) presented the [p.Arg229Gln (;) p.Ala284Val] association
Fig. 2.

Splice Aid2 and Alibaba2 gene regulation in silico predictions. a Splice Aid2 graphic outcomes from c.928G > A (p.Glu310Lys) and c.779 T > A, (p.Val260Glu) substituions. Top: For c.928G > A the ESS recognition site for hnRNP A1 is broken after G > A substituion. Below: after c.779 T > A substitution (right) the histogram result shows that, besides the two hnRNPs (H1 and H2) proteins that putatively binds the normal ESS recognition site, a new motif is created, with the potential of being recognized by the hnRNP A1 and by other four SR proteins. Arrows on the left of histograms indicate proteins that bind ESEs and ESSs, respectively. The bars have variable width and height respectively related to the number of nucleotides of the binding site and to its score (binding affinity). Next to each bar there is the label showing the name of the protein predicted to bind to the recognition motif. Below the histogram there are two lines: one with red bars, representing the recognition motifs and with a bracket representing a putative splicing site; the other line represents the DNA sequence analyzed. A green circle highlights the nucleotide substitution. b Alibaba2 gene regulation prediction analysis showing above: wild c.-164C and mutant c.-164 T sequences (highlighted). Wild sequence was recognized by putative transcription factors ETF-1 and GLI3 and, after substitution, these sites were abolished and a new recognition site for putative WT1 transcription factor was created. Below: no change after c.-196C > G substitution
Table 2.
Exonic mutations in silico prediction
| Enhancer motifs analysis through ESE finder matrices for SRp40, SC35, SF2/ASF and SRp55 proteins | |||||||
|---|---|---|---|---|---|---|---|
| Sequence Variant | Sequence positiona | Linked SRb protein | Reference motif | Linked SRb protein | Mutant motif | Variation | |
| (value 0–100)c | (value 0–100)c | ||||||
| c.779 T > A | 776 | - | - | SF2/ASF | AAGAGGA (80.37) | New ESS site | |
| c.851C > T | 845 | - | - | SF2/ASF | CTGAAGT (75.38) | New ESS site | |
| c.928G > A | 923 | SF2/ASF | CAGCTGA (78.10) | - | - | Site broken | |
| Potencial splice sites prediction through HSF matrices | |||||||
| Sequence Variant | Sequence position | Splice site type | Motif | New splice site | Wild typed | Mutantd | Variaton |
| c.779 T > A | 769 | Acceptor | GGAATCAAAGTGGA | ggaatcaaagagGA | 38.61 | 67.56 | New site +74.98 |
aPosition in cDNA from ATG codon; bSR = serine/arginine rich proteins; c = Consensus values go from 0 to 100 and the threshold is defined differently for each algorithm. Threshold for SF2/ASF from ESE finder matrices is 72.98. Every signal with a score above the defined threshold is considered to be a potential ESE/ESS. dFor HSF matrices consensus values go from 0 to 100 and threshold is 65. Every signal with a score above the threshold is considered to be a splice site (donor or acceptor). When a mutation occurs if the WT score is under the threshold and the score variation is above +10 % for HSF it is considered that the mutation creates a new splice site
Considering compound heterozygosis for the two mutations, the percentage of patients with sporadic SRNS was 10 % (2/20). This result is in accordance with data published by others that indicated frequencies for patients bearing two heterozygous mutations varying from 6 to 17 % among sporadic SRNS cases [9–11].
Moreover, among the sporadic cases, there were also three cases in which we identified only one heterozygous alteration, the promoter variants c.-164C > T, c.-196C > G and c.-537_-531delCTTTTTT in patients P111, P68 and P85, respectively. Di Duca et al. [39], who studied regulatory elements in the NPHS2 promoter, described c.-537_-531delCTTTTTT (rs146791300, MAF = 0.069; http://www.ncbi.nlm.nih.gov/) variant as a “functional polymorphism” that downregulated the gene expression of podocin by 85 % when transfected in podocytes. As there are no records of c.-164C > T and c.-196C > G variants in the database (http://www.ensembl.org), we screened the 700 bp of the 5′ UTR in 278 control subjects. We identified the c.-196G in heterozygosis in two controls (2/278, <1 %), but neither c.-537_-531delCTTTTTT nor c.-164 T were found, suggesting a possible role as disease-causing mutations. Alibaba2 gene regulation prediction algorithm showed that the C > G transversion in position c.-196 did not disrupt the SP1 transcription binding site located in this region (data not shown). However, this was not the case for C > T transition in −164 position that created a new WT1 transcription factor binding site (Fig. 2). It is well known that WT1 plays a key role during kidney and genital development [19, 20]. However, after birth, WT1 expression persists in the glomerular visceral epithelial cells where the protein probably contributes to the functional maintenance of differentiated podocytes [40]. In fact, it was previously demonstrated that WT1 binds and activates a glomerular-specific NPHS1 enhancer in vitro [41]. Although there are no reports of WT1 acting as a trans-regulator element of the NPHS2 promoter, we cannot discard a possible role for WT1 in this process, since mechanisms by which transcription factors regulate the expression of their target genes in podocytes are not yet fully understood. Recently, He et al. [42] identified a putative enhancer at 743 bp in the human NPHS2 promoter that is combinatorially regulated by Lmx1b and FoxC transcription factors in podocytes. Therefore, with respect to c.-164C > T identified in our study, further expression and electrophoretic mobility shift assays are essential to better understand its role in podocin production. What is already known is that, in proteinuric renal disease, podocin expression is a dynamic process in which dramatic decreases or abnormal distributions of podocin expression in glomeruli of children with NS have already been demonstrated [43].
Familial cases
When considering only familial cases, two out of seven unrelated cases presented two heterozygous alterations. The new frameshift p.Lys239Argfs*13 was identified in association with the already described p.Val260Glu missense in patient P103 and her sister (Fig. 1). We considered compound heterozygosis as the most probable genotype characterizing an autosomal recessive inheritance in this family, although we could not analyze their parent’s NPHS2 gene. The frameshift p.Lys239Argfs*13 results from the c.714delG deletion in exon 5, and putatively produces amino acid changes from residues 239 to 251 and creates a premature stop codon at residue 252. Therefore, this mutation might affect podocin interaction with nephrin via the PHB domain, since podocin PHB domain comprises amino acids 127 to 284 [44]. The missense p.Val260Glu is the result of c.779 T > A transversion in exon 6 [11]. The protein resulting from this substitution is retained in the endoplasmic reticulum and loses its ability to recruit nephrin in lipid rafts [45]. After in silico evaluation of c.779 T > A (p.Val260Glu) by HSF and SpliceAid2 splicing prediction tools, this substitution was classified with the potential of interfering with splicing in different ways: by activating a new exonic cryptic acceptor splice site, as predicted by HSF matrices; and by creating an ESS site, as predicted by exonic regulatory sequences (ESRs) algorithm [46] as well as HSF matrices (Table 2). SpliceAid2 prediction indicated that, after T > A substitution, new ESE and ESS putative binding motifs were created that might be recognized by different serine and arginine-rich (SR) proteins and heterogeneous nuclear ribonucleoproteins (hnRNP), respectively (Fig. 2). In patient P154, we also identified the [p.Arg229Gln (;) p.Ala284Val] association, as described for patient P6. Patient P154 is a 13-year-old boy with FSGS. He has five siblings, and we analyzed the NPHS2 gene in two of the siblings, a sister and a brother with FSGS who also presented the [p.Arg229Gln (;) p.Ala284Val] association. Analysis of the mother revealed p.Arg229Gln; however, a DNA sample from the father was not available for analysis. Our findings also included an SRNS patient with only one heterozygous alteration in the NPHS2 gene, which diverged from the concept that SRNS is an autosomal recessive disease. The p.Arg229Gln substitution was identified in patient P72 whose father also showed the disease-associated polymorphism. Patient P72 had the disease onset at 16 years of age when he was diagnosed with FSGS upon renal biopsy. He progressed to CKD and had kidney transplant at the age of 18 years. His father was diagnosed with FSGS and had kidney transplant about 30 years ago, with no FSGS recurrence thereafter. The family also reported that the patient’s brother died of renal failure. Therefore, this family history suggests a possible genetic origin for the disease. Since NPHS2 mutations are expected to act in a recessive mode in SRNS patients, our hypothesis is that mutations in other genes might be contributing to the disease in this family. NPHS1 analysis revealed the c.1223G > A substitution in exon 10, leading to the p.Arg408Gln variant [14] (Fig. 1). There is some evidence, however, that this non-synonymous substitution is probably neutral, since it has been described as a polymorphism frequently found in homozygosis and heterozygosis in control individuals [15, 47]. In order to verify its frequency among Brazilians, we analyzed 100 controls and found 2 % were heterozygous for this variant (data not shown). In addition, patient P72’s father did not show this NPHS1 variant, which led to the conclusion that there might be a mutation in one of the many other genes implicated in NS that were not screened in the present work. Besides these three cases with mutations, the other four cases with a family history of SRNS did not show any mutations in the NPHS2 gene. Therefore, the percentage of familial cases carrying two NPHS2 mutations in our study was 28.57 % (2/7), whereas other groups reported percentages of around 40 % [9–11]. It is likely the small study sample size here could explain this difference in our frequency result.
Furthermore, we also found that, among four patients bearing two heterozygous mutations, two (2/4, 50 %) presented the [p.Arg229Gln];[p.Ala284Val] association already described as predominant in South American populations [37]; apparently, this is also a common association in South Eastern Brazil. Patient P6’s grandparents are from Brazil, and the patient herself does not have a “hispanic” background. She was the only patient who had available information about her grandparents’ origin. The data reported here also demonstrated a considerable incidence of single heterozygous NPHS2 alterations, but we cannot correlate them with a favorable outcome, since two patients (P85 and P72) progressed to CKD Class V (Table 1). Of note, one of these two patients presented a promoter variation, and, in fact, we identified three promoter variations that might downregulate podocin in the GFB of these patients. Therefore, further functional studies are necessary, in order to evaluate if these promoter variants alter the regulatory mechanisms and influence the maintenance of the barrier’s functional integrity.
Conclusions
Our study reports the identification of NPHS2 mutations in only 14.8 % of both sporadic and familial SRNS cases in the Brazilian patients analyzed, after screening for mutations in the NPHS2, NPHS1 and WT1 genes. The study did not reveal mutations in NPHS1 and WT1. Therefore, target-oriented next generation sequencing analysis of glomerulopathy-related panel of genes is recommended to search for mutations in other genes related to SRNS.
Acknowledgments
We are grateful to the bioestatistic Rodrigo Secolin from “Laboratório de Biologia Molecular da Faculdade de Ciências Médicas (FCM, UNICAMP)” and to the following nephrologists for having sent the blood of some patients for molecular diagnosis, always trusting in our research: Dr. Diana Régia Bezerra Feitosa, Dr. Jovânia Heringer Sobrinho and Dr. Andréia Ribeiro from “Hospital Municipal José de Carvalho Florence São José dos Campos”; Dr. Flavia Vanesca Felix Leão, Dr. Maria Aparecida de Paula Cançado, Dr. Paulo Koch Nogueira and Luciana Feltran from “Setor Nefrologia Departamento de Pediatria da Universidade Federal de São Paulo”; Dr. Alberto Zagury from Rio de Janeiro - RJ and Dr. Liliane Prates (patients from Piracicaba – SP).
Funding
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP - 2012/51109-9 to MPdeM) and the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq - 478444/08-7 to G.G-J and 141072/2010 to MSG).and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP - 2012/51109-0 to MPdeM).
Additional file
Supplemental Information. Table S1. NPHS2 specific primers. Table S2. WT1 exons 8 and 9 especific primers. Table S3. NPHS1 specific primers. Methods (PCR reactions and sequencing). Table S4. NPHS2 and NPHS1 missenses in silico predictions. (DOC 84 kb)
Footnotes
Vera MS Belangero and Gil Guerra-Junior contributed equally to this work.
Competing interests
The author(s) declare that they have no competing interests.
Authors’ contributions
MSG carried out the molecular genetic studies of NPHS2, NPHS1 and WT1 genes, sequences alignments, in silico predictions and drafted the manuscript. ACGBL was responsible for the clinical evaluation of the patients and contributed with writing clinical description of cases for the manuscript draft. CSCP participated in genomic DNA isolation from blood samples. MLS participated on molecular genetic studies of NPHS2, NPHS1 and WT1 genes. SRS participated on molecular genetic studies of NPHS1 gene. TBH participated on molecular genetic studies of NPHS2, NPHS1 and WT1 genes. ATM-G participated in the design of the study. VMSB participated in the design of the study and was responsible for the clinical evaluation of the patients and contributed with writing of clinical description of cases for the manuscript draft. GG-J participated in the design of the study and helped to draft the manuscript. MP-de-M conceived the study, participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Authors’ information
Not applicable.
Availability of data and materials
Not applicable.
Contributor Information
Mara Sanches Guaragna, Phone: + 55 19 3521 1091, Email: mara.guaragna@gmail.com.
Anna Cristina GB Lutaif, Email: aclutaif@uol.com.br.
Cristiane SC Piveta, Email: cristianepiveta@yahoo.com.br.
Marcela L. Souza, Email: malopes.souza@gmail.com
Suéllen R. de Souza, Email: suellen.souza05@gmail.com
Taciane B. Henriques, Email: tacianebh@gmail.com
Andréa T. Maciel-Guerra, Email: atmg@uol.com.br
Vera MS Belangero, Email: vmsbelangero@gmail.com.
Gil Guerra-Junior, Email: gileandrea@uol.com.br.
Maricilda P. De Mello, Email: mpmello869@gmail.com
References
- 1.Gbadegesin R, Smoyer W. Nephrotic Syndrome. In: Mosby Elsevier, editor. Comprehensive Pediatric Nephrology, First. 2008. pp. 205–18. [Google Scholar]
- 2.Benoit G, Machuca E, Antignac C. Hereditary nephrotic syndrome: a systematic approach for genetic testing and a review of associated podocyte gene mutations. Pediatr Nephrol. 2010;25:1621–32. doi: 10.1007/s00467-010-1495-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, et al. A Single-Gene Cause in 29.5 % of Cases of Steroid-Resistant Nephrotic Syndrome. J Am Soc Nephrol. 2014;26:1279–89. doi: 10.1681/ASN.2014050489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet. 2000;24:349–54. doi: 10.1038/74166. [DOI] [PubMed] [Google Scholar]
- 5.Tryggvason K. Unraveling the mechanisms of glomerular ultrafiltration: nephrin, a key component of the slit diaphragm. J Am Soc Nephrol. 1999;10:2440–5. doi: 10.1681/ASN.V10112440. [DOI] [PubMed] [Google Scholar]
- 6.Huber TB, Simons M, Hartleben B, Sernetz L, Schmidts M, Gundlach E, et al. Molecular basis of the functional podocin-nephrin complex: mutations in the NPHS2 gene disrupt nephrin targeting to lipid raft microdomains. Hum Mol Genet. 2003;12:3397–405. doi: 10.1093/hmg/ddg360. [DOI] [PubMed] [Google Scholar]
- 7.Morrow IC, Parton RG. Flotillins and the PHB domain protein family: rafts, worms and anaesthetics. Traffic. 2005;6:725–40. doi: 10.1111/j.1600-0854.2005.00318.x. [DOI] [PubMed] [Google Scholar]
- 8.Fuchshuber A, Jean G, Gribouval O, Gubler MC, Broyer M, Beckmann JS, et al. Mapping a gene (SRN1) to chromosome 1q25-q31 in idiopathic nephrotic syndrome confirms a distinct entity of autosomal recessive nephrosis. Hum Mol Genet. 1995;4:2155–8. doi: 10.1093/hmg/4.11.2155. [DOI] [PubMed] [Google Scholar]
- 9.Karle SM, Uetz B, Ronner V, Glaeser L, Hildebrandt F, Fuchshuber A. Novel mutations in NPHS2 detected in both familial and sporadic steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2002;13:388–93. doi: 10.1681/ASN.V132388. [DOI] [PubMed] [Google Scholar]
- 10.Ruf RG, Lichtenberger A, Karle SM, Haas JP, Anacleto FE, Schultheiss M, et al. Patients with mutations in NPHS2 (podocin) do not respond to standard steroid treatment of nephrotic syndrome. J Am Soc Nephrol. 2004;15:722–32. doi: 10.1097/01.ASN.0000113552.59155.72. [DOI] [PubMed] [Google Scholar]
- 11.Weber S, Gribouval O, Esquivel EL, Morinière V, Tête M-J, Legendre C, et al. NPHS2 mutation analysis shows genetic heterogeneity of steroid-resistant nephrotic syndrome and low post-transplant recurrence. Kidney Int. 2004;66:571–9. doi: 10.1111/j.1523-1755.2004.00776.x. [DOI] [PubMed] [Google Scholar]
- 12.Bouchireb K, Boyer O, Gribouval O, Nevo F, Huynh-Cong E, Morinière V, et al. NPHS2 mutations in steroid-resistant nephrotic syndrome: a mutation update and the associated phenotypic spectrum. Hum Mutat. 2014;35:178–86. doi: 10.1002/humu.22485. [DOI] [PubMed] [Google Scholar]
- 13.Kestilä M, Lenkkeri U, Männikkö M, Lamerdin J, McCready P, Putaala H, et al. Positionally cloned gene for a novel glomerular protein--nephrin--is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1:575–82. doi: 10.1016/S1097-2765(00)80057-X. [DOI] [PubMed] [Google Scholar]
- 14.Lenkkeri U, Männikkö M, McCready P, Lamerdin J, Gribouval O, Niaudet PM, et al. Structure of the gene for congenital nephrotic syndrome of the finnish type (NPHS1) and characterization of mutations. Am J Hum Genet. 1999;64:51–61. doi: 10.1086/302182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schoeb DS, Chernin G, Heeringa SF, Matejas V, Held S, Vega-Warner V, et al. Nineteen novel NPHS1 mutations in a worldwide cohort of patients with congenital nephrotic syndrome (CNS) Nephrol Dial Transplant. 2010;25:2970–6. doi: 10.1093/ndt/gfq088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Philippe A, Nevo F, Esquivel EL, Reklaityte D, Gribouval O, Tête M-J, et al. Nephrin mutations can cause childhood-onset steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2008;19:1871–8. doi: 10.1681/ASN.2008010059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koziell A, Grech V, Hussain S, Lee G, Lenkkeri U, Tryggvason K, et al. Genotype/phenotype correlations of NPHS1 and NPHS2 mutations in nephrotic syndrome advocate a functional inter-relationship in glomerular filtration. Hum Mol Genet. 2002;11:379–88. doi: 10.1093/hmg/11.4.379. [DOI] [PubMed] [Google Scholar]
- 18.Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell. 1990;60:509–20. doi: 10.1016/0092-8674(90)90601-A. [DOI] [PubMed] [Google Scholar]
- 19.Gessler M, Poustka A, Cavenee W, Neve RL, Orkin SH, Bruns GA. Homozygous deletion in Wilms tumours of a zinc-finger gene identified by chromosome jumping. Nature. 1990;343:774–8. doi: 10.1038/343774a0. [DOI] [PubMed] [Google Scholar]
- 20.Denys P, Malvaux P, Van Den Berghe H, Tanghe W, Proesmans W. Association of an anatomo-pathological syndrome of male pseudohermaphroditism, Wilms’ tumor, parenchymatous nephropathy and XX/XY mosaicism. Arch françaises pédiatrie. 1967;24:729–39. [PubMed] [Google Scholar]
- 21.Drash A, Sherman F, Hartmann WH, Blizzard RM. A syndrome of pseudohermaphroditism, Wilms’ tumor, hypertension, and degenerative renal disease. J Pediatr. 1970;76:585–93. doi: 10.1016/S0022-3476(70)80409-7. [DOI] [PubMed] [Google Scholar]
- 22.Frasier SD, Bashore RA, Mosier HD. Gonadoblastoma associated with pure gonadal dysgenesis in monozygous twins. J Pediatr. 1964;64:740–5. doi: 10.1016/S0022-3476(64)80622-3. [DOI] [PubMed] [Google Scholar]
- 23.Lipska BS, Ranchin B, Iatropoulos P, Gellermann J, Melk A, Ozaltin F, et al. Genotype-phenotype associations in WT1 glomerulopathy. Kidney Int. 2014;85:1169–78. doi: 10.1038/ki.2013.519. [DOI] [PubMed] [Google Scholar]
- 24.ISKDC The primary nephrotic syndrome in children. Identification of patients with minimal change nephrotic syndrome from initial response to prednisone. A report of the International Study of Kidney Disease in Children. J Pediatr. 1981;98:561–4. doi: 10.1016/S0022-3476(81)80760-3. [DOI] [PubMed] [Google Scholar]
- 25.Hogg RJ, Furth S, Lemley KV, Portman R, Schwartz GJ, Coresh J, et al. National Kidney Foundation’s Kidney Disease Outcomes Quality Initiative clinical practice guidelines for chronic kidney disease in children and adolescents: evaluation, classification, and stratification. Pediatrics. 2003;111:1416–21. doi: 10.1542/peds.111.6.1416. [DOI] [PubMed] [Google Scholar]
- 26.Sambrook J, Fritsh EF, Maniatis T. Molecular cloning: A laboratory manual. New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 27.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–74. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tavtigian SV, Deffenbaugh AM, Yin L, Judkins T, Scholl T, Samollow PB, et al. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet. 2006;43:295–305. doi: 10.1136/jmg.2005.033878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Desmet F-O, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37 doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piva F, Giulietti M, Nocchi L, Principato G. SpliceAid: a database of experimental RNA target motifs bound by splicing proteins in humans. Bioinformatics. 2009;25:1211–3. doi: 10.1093/bioinformatics/btp124. [DOI] [PubMed] [Google Scholar]
- 33.Grabe N. AliBaba2: context specific identification of transcription factor binding sites. In Silico Biol. 2002;2:S1–15. [PubMed] [Google Scholar]
- 34.Sanna-Cherchi S, Burgess KE, Nees SN, Caridi G, Weng PL, Dagnino M, et al. Exome sequencing identified MYO1E and NEIL1 as candidate genes for human autosomal recessive steroid-resistant nephrotic syndrome. Kidney Int. 2011;80:389–96. doi: 10.1038/ki.2011.148. [DOI] [PubMed] [Google Scholar]
- 35.Gee HY, Zhang F, Ashraf S, Kohl S, Sadowski CE, Vega-Warner V, et al. KANK deficiency leads to podocyte dysfunction and nephrotic syndrome. J Clin Invest. 2015;125:2375–84. doi: 10.1172/JCI79504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsukaguchi H, Sudhakar A, Le TC, Nguyen T, Yao J, Schwimmer JA, et al. NPHS2 mutations in late-onset focal segmental glomerulosclerosis: R229Q is a common disease-associated allele. J Clin Invest. 2002;110:1659–66. doi: 10.1172/JCI0216242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Machuca E, Hummel A, Nevo F, Dantal J, Martinez F, Al-Sabban E, et al. Clinical and epidemiological assessment of steroid-resistant nephrotic syndrome associated with the NPHS2 R229Q variant. Kidney Int. 2009;75:727–35. doi: 10.1038/ki.2008.650. [DOI] [PubMed] [Google Scholar]
- 38.Tory K, Menyhárd DK, Nevo F, Gribouval O, Kerti A, Stráner P, et al. Mutation-dependent recessive inheritance in NPHS2-associated steroid-resistant nephrotic syndrome. Beyond Mendel’s laws. Eur J Hum Genet. 2013;21, Supple:50. doi: 10.1038/ng.2898. [DOI] [PubMed] [Google Scholar]
- 39.Di Duca M, Oleggini R, Sanna-Cherchi S, Pasquali L, Di Donato A, Parodi S, et al. Cis and trans regulatory elements in NPHS2 promoter: implications in proteinuria and progression of renal diseases. Kidney Int. 2006;70:1332–41. doi: 10.1038/sj.ki.5001767. [DOI] [PubMed] [Google Scholar]
- 40.Hammes A, Guo JK, Lutsch G, Leheste JR, Landrock D, Ziegler U, et al. Two splice variants of the Wilms’ tumor 1 gene have distinct functions during sex determination and nephron formation. Cell. 2001;106:319–29. doi: 10.1016/S0092-8674(01)00453-6. [DOI] [PubMed] [Google Scholar]
- 41.Guo G, Morrison DJ, Licht JD, Quaggin SE. WT1 activates a glomerular-specific enhancer identified from the human nephrin gene. J Am Soc Nephrol. 2004;15:2851–6. doi: 10.1097/01.ASN.0000143474.91362.C4. [DOI] [PubMed] [Google Scholar]
- 42.He B, Ebarasi L, Zhao Z, Guo J, Ojala JRM, Hultenby K, et al.: Lmx1b and FoxC Combinatorially Regulate Podocin Expression in Podocytes. J Am Soc Nephrol 2014; DOI: 10.1681/ASN.2012080823 [DOI] [PMC free article] [PubMed]
- 43.Guan N, Ding J, Zhang J, Yang J. Expression of nephrin, podocin, alpha-actinin, and WT1 in children with nephrotic syndrome. Pediatr Nephrol. 2003;18:1122–7. doi: 10.1007/s00467-003-1240-z. [DOI] [PubMed] [Google Scholar]
- 44.Relle M, Cash H, Brochhausen C, Strand D, Menke J, Galle PR, et al. New perspectives on the renal slit diaphragm protein podocin. Mod Pathol. 2011;24:1101–10. doi: 10.1038/modpathol.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roselli S, Moutkine I, Gribouval O, Benmerah A, Antignac C. Plasma membrane targeting of podocin through the classical exocytic pathway: effect of NPHS2 mutations. Traffic. 2004;5:37–44. doi: 10.1046/j.1600-0854.2003.00148.x. [DOI] [PubMed] [Google Scholar]
- 46.Goren A, Ram O, Amit M, Keren H, Lev-Maor G, Vig I, et al. Comparative analysis identifies exonic splicing regulatory sequences--The complex definition of enhancers and silencers. Mol Cell. 2006;22:769–81. doi: 10.1016/j.molcel.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 47.Lahdenkari A-T, Kestilä M, Holmberg C, Koskimies O, Jalanko H. Nephrin gene (NPHS1) in patients with minimal change nephrotic syndrome (MCNS) Kidney Int. 2004;65:1856–63. doi: 10.1111/j.1523-1755.2004.00583.x. [DOI] [PubMed] [Google Scholar]