

Abstract
Recent studies have shown that arsenic trioxide can induce a clinical remission in patients with acute promyelocytic leukemia. However, the molecular mechanisms of action remain to be elucidated. In this research, we performed the MTT assay to evaluate the cytotoxic effects of arsenic trioxide (ATO) to HL-60 cells and to compare their relative sensitivity to that of HepG2, and Jurkat T cells. We also performed the thiobarbituric acid test to determine the levels of malondialdehyde (MDA) plus 4-hydroxy-2 (E)-nonenal (4-HAE) production in these three cell lines following exposure to arsenic trioxide. The result of MTT assay clearly demonstrated that ATO has a significant cytotoxic effect on HL-60, Jurkat, and HepG2 cells; showing 24 hrs LD50 values of 6.4 ± 0.6 μg/mL, 15 ± 3.84 μg/mL, and 23.2 ± 6.03 μg/mL, respectively. These data indicated that HL-60 cells are about twice as sensitive to arsenic toxicity compared to Jurkat T cells and about 3 times more sensitive to arsenic trioxide compared to HepG2 cells. The result of the thiobarbituric acid test demonstrated that arsenic trioxide treatment resulted in a significant increase (p <0.05) of MDA and HAE production, indicating that oxidative stress plays a key role in arsenic induced toxicity and cell injury. MDA and HAE levels were significantly higher in arsenic trioxide-treated HL-60 cells, indicating that these cells appear to be more sensitive to oxidative stress than HepG2 and Jurkat T- cells. In summary, these results indicate that the pharmacology of ATO as an effective anti-cancer drug is associated with its cytotoxic effects in human promyelocytic leukemic cells. This cytotoxicity is found to be mediated by oxidative stress, a biomarker of cellular injury.
Keywords: Arsenic trioxide, HL-60, Jurkat, HepG2, MDA, HAE, lipid peroxidation
I. INTRODUCTION
Arsenic trixiode has been shown to inhibit both proliferation and viability when tested against a panel of lymphoma cell lines (1), and arsenicals have activity in vitro against myeloma cell lines and primary myeloma cells (2). Recently, arsenic trioxide (Trisenox) has been used as an anticancer agent in the treatment of acute promyelocytic leukemia. One of the major mechanisms by which arsenic exerts its toxic effect is through impairment of cellular respiration by the inhibition of various mitochondrial enzymes, and the uncoupling of oxidative phosphorylation. In addition, arsenic toxicity results from its ability to interact with sulphydryl groups of proteins and enzymes, and to substitute phosphorous group in a variety of biochemical reactions (3).
Although arsenic trioxide (ATO) has cytotoxic effects on several cancer cell, its molecular mechanisms of action remain to be elucidated. Hence, the aim of the present study was to assess the relative sensitivity of HL-60, Jurkat, and HepG2 cells to arsenic trioxide toxicity and to determine the role of oxidative stress in ATO induced cytotoxicity to these cells.
II. MATERIALS AND METHODS
II.1. Chemicals and Test Media
Arsenic trioxide (As2O3), CASRN 1327-53-3, MW 197.84, with an active ingredient of 100% (w/v) arsenic in 10% nitric acid was purchased from Fisher Scientific in Houston, Texas. Growth medium RMPI 1640 containing 1 mmol/L L-glutamine was purchased from Gibco BRL products (Grand Island, NY). Ninety six-well plates were purchased from Costar (Cambridge, MA). Fetal bovine serum (FBS), antibiotics (penicillin G and streptomycin), phosphate buffered saline (PBS), and MTT assay kit were obtained from Sigma Chemical Company (St. Louis, MO).
II.2. Tissue Culture
In the laboratory, cells were stored in the liquid nitrogen until use. They were thawed by gentle agitation of their containers (vials) for 2 minutes in a bath at 37°C. After thawing, the content of each vial of cell was transferred to a 25 cm2 tissue culture flask, diluted with up to 10 mL of RMPI 1640 (HL-60 and Jurkat T-cells) or DMEM (HepG2 cells) containing 1 mmol/L L-glutamine (GIBCO/BRL, Gaithersburg, MD) and supplemented with 10% (v/v) fetal bovine serum (FBS), 1% (w/v) penicillin/streptomycin. The 25 cm2 culture flasks (2 × 106 viable cells) were observed under the microscope, following by incubation in a humidified 5 % CO2 incubator at 37°C. Three times a week, they were diluted to maintain under the same conditions at a density of 5 ° 105/mL and harvested in the exponential phase of growth. The cell viability was assessed by the trypan blue exclusion test (Life Technologies) and manually counted using a hemocytometer.
II.3. Cytotoxicity/ MTT Assay
Human leukemia HL-60 cells and human Jurkat-T cells were maintained in RMPI 1640. Human liver carcinoma (HepG2) cells were maintained in DMEM. Both RPMI 1640 and DMEM containing 1 mmol/L L-glutamine were supplemented with 10% (v/v) fetal bovine serum (FBS), 1% (w/v) penicillin/streptomycin, and incubated at 37°C in humidified 5% CO2 incubator. To 180 μL aliquots in six replicates of the cell suspension (5 × 105/mL) seeded to 96 well polystyrene tissue culture plates, 20 μL aliquots of ATO solutions (1.25, 2.5, 5, 10, 20, and 40μg/mL) were added to each well using distilled water as solvent. Cells incubated in culture medium alone served as a control for cell viability (untreated wells). All chemical exposures were carried in 96 well tissue culture plates for the purpose of chemical dilutions. Cells were placed in the humidified 5% CO2 incubator for 24 hrs at 37°C. After incubation, 20 μL aliquots of MTT solution (5 mg/mL in PBS) were added to each well and re-incubated for 4 hours at 37°C following by low centrifugation at 800 rpm for 5 minutes for HL-60 and Jurkat T-cells. Then, the 200 μL of supernatant culture medium were carefully aspirated and 200 μL aliquots of dimethylsulfoxide (DMSO) were added to each well to dissolve the formazan crystals, following by incubation for 10 minutes to dissolve air bubbles. The culture plate was placed on a Biotex Model micro-plate reader and the absorbance was measured at 550 nm. The amount of color produced is directly proportional to the number of viable cell. All assays were performed in six replicates for each concentration. Cell viability rate was calculated as the percentage of MTT absorption as follows: % survival = (mean experimental absorbance/mean control absorbance×100).
II. 4. Assay of Lipid Peroxidation
Statistical Analysis
Data were presented as means ± SDs. Statistical analysis was done using one way analysis of variance (ANOVA) for multiple samples and Student's t-test for comparing paired sample sets. P-values less than 0.05 were considered statistically significant. The percentages of cell viability and MDA plus HAE levels were presented graphically in the form of histograms, using Microsoft Excel computer program.
III. RESULTS
III.1. Cytotoxicity/MTT Assay
Data presented in (Fig 2) clearly demonstrate that arsenic trioxide has a significant cytotoxic effect on HL-60, Jurkat, and HepG2 cells. LD50 values of 6.4 ± 0.57 μg/mL, 15 ± 3.84 μg/mL, and 23.2 ± 6.03 μg/mL were computed for HL-60, Jurkat, and HepG2 cells, respectively. These data indicate that HL-60 cells are about twice as sensitive to ATO compared to Jurkat T-cells and about 3 times more sensitive to ATO compared to HepG2 cells (Fig. 2, 3). Taken together, our results demonstrated a strong dose-response relationship with regard to ATO cytotoxicity. At low doses of exposure, arsenic trioxide produces a slight linear increase in cell viability. On the other hand, it produces a non-linear gradual decrease in cell viability at higher doses.
Figure 2.
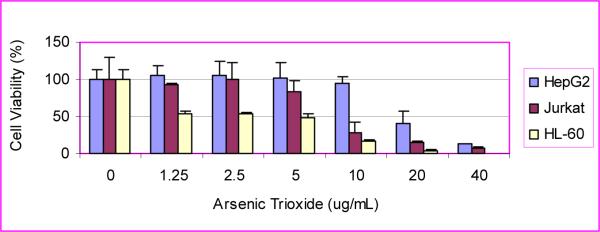
Cytotoxicity of ATO to HepG2, Jurkat, and HL-60 cells.
Figure 3.
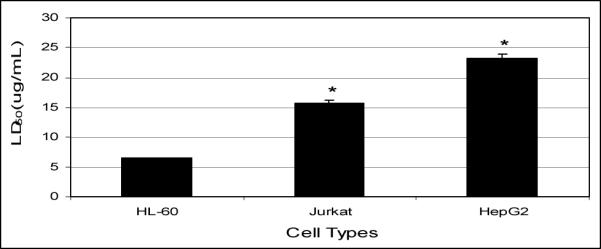
Mean values of 24 h-LD50 of ATO to HL-60, Jurkat, and HepG2 cells.
III. 2. Lipid Peroxidation Assay
The standard curve generated from lipid peroxidation assay is presented in (Fig 4), and the effect of arsenic trioxide on lipid peroxidation is presented in (Fig 5). Results of this assay indicated an elevated production of MDA plus HAE in ATO-treated cells compared to the control cells. At low doses of exposure, the release of MDA plus HAE was higher in HL-60 cells compared to Jurkat and HepG2 cells, indicating that HL-60 cells is more sensitive to ATO than other cell lines.
Figure 4.
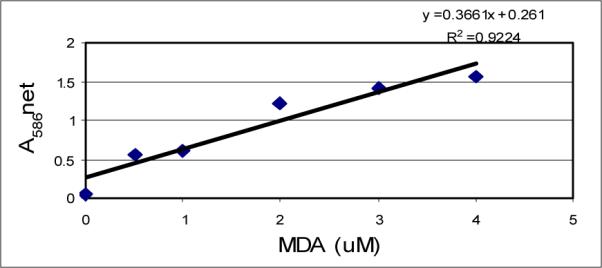
MDA standard curve showing the net absorbance at 586 nm as a function of MDA concentration.
Figure 5.
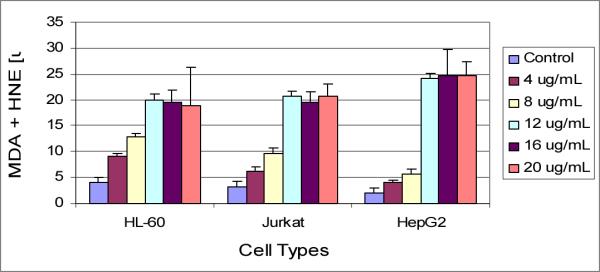
Effects of different concentrations of ATO on MDA plus HAE production in HL-60, Jurkat, and HepG2 cells.
IV. DISCUSSION
IV.1. Cytotoxicity/MTT Assay
Cytotoxicity can be defined as the cell killing property of a chemical compound independent from the mechanism of death. We examined the cytotoxic effect of arsenic trioxide on the HL-60, Jurkat, and HepG2 cells. Findings from our study clearly showed that arsenic trioxide is highly cytotoxic to these three cell lines. Several studies have addressed the cytotoxicity of arsenic to various cells. A study has shown that when using 0.5 to 1 μM/L of ATO, apoptosis was induced in the monocytic cells line NB4 (4). Cytotoxicity studies of two multiple myeloma (MM) derived cell lines, RPMI 8226 and U266, found that 1.0 μM/L ATO inhibited cell proliferation resulting in a weak degree of apoptosis induction, and 2.0 μM/L ATO- induced cell apoptosis. These results showed that ATO exerts apoptosis inducing and growth-inhibiting effects on MM derived cells (5). Recently, we reported that ATO is cytotoxic to human liver carcinoma (HepG2) cells, showing a LD50 of 8.55 ± 0.58 μg/ml after 48 hrs of exposure (6, 7). Findings from other studies suggest that low doses of arsenic are effective in APL and show considerable promise in preclinical models of other tumor types (8). Taken together, these results indicated that HL-60 cells are more sensitive to ATO than HepG2 cells and many other cell lines.
IV. 2. Lipid Peroxidation Assay
Because heavy metals such as arsenic are powerful catalysts of lipid peroxidation processes (9), the induction of lipid peroxidation in arsenic trioxide - treated cells was determined by estimating the levels of malondialdehyde (MDA) plus 4-hydroxy-2 (E)-nonenal (4-HAE). Our results demonstrated that ATO treatment resulted in a significant increase (p <0.05) of MDA plus HAE, indicating that oxidative stress plays an important role in arsenic induced toxicity and cell injury.
These studies suggest that the pharmacology of ATO as an effective anti-cancer drug is associated with its cytotoxic effects in human promyelocytic leukemic cells, which is found to be mediated through oxidative stress, a biomarker of cellular injury. Damage to cell organelles produced by lipid peroxidation has been demonstrated by many investigations. However, there is not a firm evidence about the mechanism of lipid peroxidation. One of the primary effects of ATO in the cell is induction of oxidative stress, which is followed by the development of apoptosis (10, 11). The metal-induced lipid peroxidation is mostly attributed to increased production of free radicals (9, 12). Our results indicate that excess ATO increased oxidative stress, as is evident from increased lipid peroxidation. Several authors (13, 14) have reported similar increase in lipid peroxidation products (MDA) when plant cells were treated with copper.
V. CONCLUSIONS
Arsenic trioxide exerts a potent cytotoxic effect on human leukemia (HL-60) cells and other cell lines by inhibiting cell proliferation and inducing cell death. Such effects have been observed in cultured cell lines and animal models, as well as clinical studies. Findings from these studies indicate that the pharmacology of ATO as an effective anti-cancer drug is associated with its cytotoxic effects in human promyelocytic leukemic cells. This cytotoxicity is found to be mediated by oxidative stress, a biomarker of cellular injury.
Figure 1.
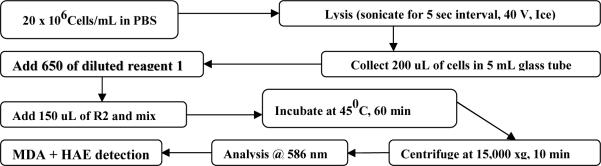
Schematic representation of the steps in Lipid Peroxidation assay
ACKNOWLEDGEMENTS
This research was financially supported by a grant from the National Institutes of Health (Grant No. 1G12RR13459), through the RCMI-Center for Environmental Health at Jackson State University. The authors thank Dr. Abdul Mohamed; Dean of College of Science, Engineering, and Technology for his technical support in this research.
REFERENCES
- 1.Zhang K, Ohnishi K, Shigeno K, Fujisawa S, Naito K, Nakamura S, Takeshita K, Takeshita A, Ohno R. The induction of apoptosis and cell cycle arrest by arsenic trioxide in lymphoid neoplasms. Leukemia (Baltimore) 1998;12:1383–1391. doi: 10.1038/sj.leu.2401112. [DOI] [PubMed] [Google Scholar]
- 2.Rousselot P, Labaume S, Marolleau JP, Larghero J, Noguera MK, Brouet JC, Fermand JP. Arsenic trioxide and melarsoprol induce apoptosis in plasma cell lines and in plasma cells from myeloma patients. Cancer Res. 1999;59:1041–1048. [PubMed] [Google Scholar]
- 3.Li JH, Rossman TC. Inhibition of DNA ligase activity by arsenite: A possible mechanism of its comutagenesis. Mol. Toxicol. 1989;2:1–9. [PubMed] [Google Scholar]
- 4.Chen C-S, Siegel DM. Arsenical keratosis. eMedicine J. 2001;2(6) [Google Scholar]
- 5.Jai P, Chen G, Huang X, Cai X, Yang J, Wang L, Zhou Y, Shen Y, Zhou L, Yu Y, Chen S, Zhang X, Wang Z. Arsenic trioxide induces multiple myeloma cell apoptosis via disruption of mitochondrial transmembrane potentials and activation of caspace-3. Chin. Med. J. (Engl) 1999;(114):19–24. [PubMed] [Google Scholar]
- 6.Tchounwou PB, Wilson BA, Abdelgnani AA, Ishaque AB, Patlolla AK. Differential cytotoxicity and gene expression in human liver carcinoma (HepG2) cells exposed to arsenic trioxide and monosodium acid methanearsonate (MSMA) Int J Mol Sci. 2002;3:1117–1132. [Google Scholar]
- 7.Tchounwou PB, Yedjou CG, Dorsey WC. Arsenic Trioxide induced transcriptional activation and expression of stress genes in human liver carcinoma cells (HepG2) Cellular and Molecular Biology™. 2003;49(7):1071–1079. [PubMed] [Google Scholar]
- 8.Soignet SL, Frankel SR, Douer D, Tallman MS, Kantarjian H, Calleja E, Stone RM, Kalaycio M, Scheinberg DA, Steinherz P, Sievers EL, Coutré S, Dahlberg S, Ellison R, Warrell RP., Jr. United States multicenter study of arsenic trioxide in relapsed acute promyelocytic leukemia. J. Clin. Oncol. 2001;19:3852–3860. doi: 10.1200/JCO.2001.19.18.3852. [DOI] [PubMed] [Google Scholar]
- 9.Halliwell B, Gutteridge JMC. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J. 1984;219:1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith KR, Klei LR, Barchowsky A. Arsenite stimulates plasma membrane NADPH oxidase in vascular endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2001;280:L442–9. doi: 10.1152/ajplung.2001.280.3.L442. [DOI] [PubMed] [Google Scholar]
- 11.Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- 12.Aust SD, Marehouse LA, Thomas CE. Role of metals in oxygen radical reactions. J. Free Radic. Biol. Med. 1985;1:3–25. doi: 10.1016/0748-5514(85)90025-x. [DOI] [PubMed] [Google Scholar]
- 13.Rama Devi S, Prasad MNV. Copper toxicity in Ceratophyllum demersum L. (Coontail), a free floating macrophyte: Response of antioxidant enzymes and antioxidants. Plant Sci. 1998;138:157–165. [Google Scholar]
- 14.Mazhoudi S, Chaoui A, Ghorbal MH, El Ferjani E. Response of antioxidative enzymes to excess copper in tomato (Lycopersicm esculentum, Mill) Plant Sci. 1997;127:129–137. [Google Scholar]