

Abstract
Chaperone-mediated autophagy (CMA) and the ubiquitin-proteasomal system (UPS) are two major protein degradation systems responsible for maintaining cellular homeostasis, but how these two systems are regulated after traumatic brain injury (TBI) remains unknown. TBI produces primary mechanical damage that must be repaired to maintain neuronal homeostasis. The level of lysosomal-associated membrane protein type 2A (LAMP2A) is the hallmark of CMA activity. The level of polyubiquitinated proteins (ubi-proteins) reflects UPS activity. This study utilized a moderate fluid percussion injury model in rats to investigate the changes in CMA and the UPS after TBI. Induction of CMA was manifested by significant upregulation of LAMP2A and secondary lysosomes during the periods of 1–15 days of recovery after TBI. In comparison, the levels of ubi-proteins were increased only moderately after TBI. The increases in the levels of LAMP2A and 70 kDa heat-shock protein for CMA after TBI were seen mainly in the secondary lysosome-containing fractions. Confocal and electron microscopy further showed that increased LAMP2A or lysosomes were found mainly in neurons and proliferated microglia. Because CMA and the UPS are two major routes for elimination of different types of cellular aberrant proteins, the consecutive activation of these two pathways may serve as a protective mechanism for maintaining cellular homeostasis after TBI.
Key words: : chaperone-mediated autophagy, HSC70, HSP70, Iba-1, LAMP2, lysosome, ubiquitin, traumatic brain injury
Introduction
Traumatic brain injury (TBI) initiates a cascade of pathophysiological processes including protein degradation pathways such as autophagy and the ubiquitin-proteasomal system (UPS). Autophagy is the process of lysosomal degradation of unnecessary or dysfunctional cellular components. There are three basic types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). Macroautophagy, commonly referred to as autophagy, is the process in which cellular contents are sequestrated by a double-layer membrane to form autophagosomes, and then autophagosomes fuse with lysosomes to degrade the cellular contents. Unlike macroautophagy that requires autophagosome formation, microautophagy is mediated by direct lysosomal engulfment of the cytoplasmic content. In comparison, chaperone-mediated autophagy or CMA refers to the chaperone-dependent selection of soluble cytosolic proteins that are then delivered to lysosomes for degradation. These three types of autophagy work either together or sequentially to maintain cellular homeostasis, particularly under stress conditions.1
Primary lysosomes are derived from the endoplasmic reticulum and Golgi apparatus pathway.2 After loading with degradation materials, primary lysosomes transform into secondary lysosomes and thus are deposited into heavier subcellular fractions.3 Secondary lysosome in tissue sections can be recognized by electron microscopy (EM) as membrane structure containing electron-dense materials.2,3 Lysosome-associated membrane protein-2 (LAMP2) is a major lysosomal membrane protein with a single transmembrane (TM) domain, a large heavily glycosylated N-terminal intraluminal domain and a short cytosolic C-terminal tail.4 There are three splicing isoforms (LAMP2-A, -B and -C), which differ in their TM domain and cytosolic tail fragments.4 Therefore, LAMP2-A can be differentiated from LAMP2-B and -C by using an antibody recognizing the cytosolic tail fragment specific to LAMP2A. Among these isoforms, only LAMP2A is involved in CMA.1
Cellular aberrant proteins refer to those that are unfolded, misfolded, or damaged in cells. They expose their hydrophobic segments and thus are prone to toxic aggregation in cells.5 The UPS and CMA are independent cellular systems for degradation of cellular aberrant proteins. Both the UPS and CMA are active at all times and maximally activated in response to stress.6,7 Usually, after onset of a stress, macroautophagy and the UPS react first as early as 0.5 h and remain at high activity for about 4–8 h. If the stress persists for more than 10 h, the cells further initiate CMA for the next 36–72 h period to remove aberrant proteins.8
During the CMA process, HSC70 or its inducible form HSP70 recognizes aberrant cytosolic proteins and delivers them into the lysosome via a lysosomal membrane receptor channel in an ATP dependent manner. The lysosomal receptor channel is made up of the multimers of the LAMP2A isoform.1 Previous studies show that the binding of the substrate proteins to LAMP2A is the rate-limiting step for CMA, and thus the cellular LAMP2A level correlates directly with CMA activity.
The objective of this study is to investigate the role of CMA and the UPS after TBI. This study shows that CMA and the UPS are consecutively upregulated for maintaining cellular homeostasis after TBI.
Methods
Materials
Leupeptin, pepstain, aprotinin, phenylmethylsulfonyl fluoride (PMSF), dithiothreitol (DTT), Triton X-100 (TX100), sodium dodecyl sulfate (SDS), propidium iodide, and other chemicals were purchased from Sigma (St. Louis, MO). The following antibodies were used: anti-LAMP2 (Invitrogen, Camarillo, CA), anti-ubiquitin (1:200, Millipore, Billerica, MA), anti-Iba-1 (Millipore), and anti-beta-actin (Cell Signaling Tech, Danvers, MA, #4970). The anti-mouse or anti-rabbit secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PA).
TBI model
The parasagittal fluid percussion injury (FPI) model was produced using male Sprague–Dawley rats weighing 270 to 320 g (Charles River Laboratories, Raleigh, NC). All experimental procedures were in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Animal Care and Use Committee in the University of Miami Miller School of Medicine. All feasible measures were taken to reduce animal suffering and the numbers of animals used for these experiments.
The basic surgical preparation for brain injury was performed according to the previously described methods.10,11 Animals were maintained for at least 7 days before the experiment in a temperature-regulated room (23–25°C) on a 12 h light/dark cycle. The rats were fasted, but allowed free access to water overnight before the procedure. Moderate TBI was produced with fluid-percussion pressure levels of 2.0±0.2 atmospheres (atm). Rats were anesthetized with 3.0% isoflurane in a gas mixture of 70% N2O and 30% O2. The femoral artery was cannulated to deliver pancuronium bromide (0.5 mg/kg, intravenously) every 1 h during the surgical procedure to immobilize the rats.
An endotracheal tube was inserted, and the rats were mechanically ventilated with 70% N2O, 0.5% - 1.5% isoflurane, and a balance of O2. The animals were then placed in a stereotaxic frame, and a 4.8 mm craniotomy was made over the right parietal cortex (3.8 mm posterior to bregma, 2.5 mm lateral to the midline). A plastic injury tube (18 gauge modified Precision Glide needle hub, Becton Dickinson, Franklin Lakes, NJ) was placed over the exposed dura and fixed with dental acrylic.
Before and after TBI, blood gases and mean arterial blood pressure were monitored and maintained at physiologic levels. Brain temperature was monitored with a thermistor probe placed in the left temporalis muscle, whereas core temperature was determined with a rectal thermometer. Brain temperature was maintained at 37°C with self-adjusting feedback heating lamps. Blood gases, blood glucose, and hematocrit values were monitored 15 min before TBI, 15 min after TBI, and then once every hour for up to 4 h after TBI. All animals were maintained within physiologic ranges for mean arterial pressure (120–140 mm Hg), pO2 blood gas levels (105–170 mm Hg), pCO2 blood gas levels (35–45 mm Hg), and blood pH (7.38–7.41).
Three sets of tissue samples from a total of 56 rats were prepared for biochemical, confocal, and EM studies, respectively. The biochemical samples were prepared from a series of sham-operated control rats and rats subjected to TBI followed by 4 h and 1, 3, 5, and 15 days of recovery. The confocal microscopic sections were prepared from another series of sham-operated control rats and rats subjected to TBI followed by 1, 3, 5, and 15 days of recovery. The EM sections were prepared from the third series of sham-operated control rats and rats subjected to TBI followed by 3 and 15 days of recovery.
For the biochemical analysis, we performed a sample size estimate using SigmaStat with a power of 0.80, indicating that there was an 80% chance of detecting a statistical difference between groups. Based on this analysis, four animals in every sham-operated control or post-TBI experimental group were used for preparing all three sets of tissue samples. Sham-operated rats were subjected to identical surgical procedures, but without the fluid-percussion injury pulse.
After TBI, anesthesia was discontinued, and the animals were returned to their cages. At the recovery time point to collect brain samples, the animals were anesthetized, tracheotomized, and artificially ventilated with 70% N2O, 0.5–1.5% isoflurane, and a balance of O2. A 50 mL centrifuge tube with an open bottom was embedded into a skin incision on the top of the skull and then filled with liquid nitrogen while the respiration was maintained with the ventilator. Brains were carefully removed from the liquid nitrogen-frozen heads with a saw, a hammer, and a chisel. The right, injured ipsilateral parietal cortex was dissected in a glove box freezer (−12°C) as described previously.12 This brain dissection method prevents brain biochemical changes during the processes of decapitation and isolation of brains from the skull.12
For confocal microscopy, animals were anesthetized and ventilated with a respirator. They were then perfused via ascending aorta with ice-cold 4% paraformaldehyde in phosphate-buffered saline (PBS), sectioned with a vibratome, and stored in an anti-freeze solution at −20°C until use. For EM, rats were perfused with ice-cold 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M cacodylate buffer.
Confocal and electron microscopy
For light microscopic examination, 50 μm vibratome sections were stained with acid fuchsin and celestine blue. Brain sections at bregma −3.60 mm of the hippocampal level were examined by light microscopy.
For confocal microscopy, double-labeled fluorescence immunocytochemistry was performed on coronal brain sections (50 μm) according to the method described in our previous studies.6 Because FPI damages the lateral neocortical and hippocampal CA3 areas,11 brain sections at the hippocampal level (bregma−3.6 mm) were used for confocal microscopic studies. Briefly, after washing and blocking, brain sections were incubated overnight at 4°C with a primary antibody (e.g., Invitrogen LAMP2A antibody or a cell marker antibody, 1:250 dilution in TBS, 0.1% Triton X-100, and 3% bovine serum albumin). After washing, the sections were incubated either with fluorescein-labeled anti-rabbit secondary antibody (1:500, Life technologies, Carlsbad, CA) or anti-mouse secondary antibody (1:500, Life technologies) for 1 h at room temperature.
For propidium iodide (PI) staining of brain sections, 10 μL of PI stock solution (1 μg/mL in PBS) was added to 1 mL of the secondary antibody solution before incubation with secondary antibody. PI can penetrate into all cells after paraformaldehyde fixation, and thus stains all nuclei in paraformaldehyde-fixed brain sections. Dead neurons can often be recognized with PI staining because they have shrunken and polygonal nuclei, and the lack of immunostaining in the cytoplasm.6
Sections were mounted with Vectashield mounting medium (Vector Laboratories Inc., Burlingame, CA). At the hippocampal level (bregma −3.6 mm), the center area of the lateral neocortex, middle segments of hippocampal CA1 and CA3, and middle segment of the DG upper blade were examined with a Zeiss 510 laser-scanning confocal microscope (Zeiss Inc., Thornwood, NY).
For EM, coronal brain sections at the same hippocampal level (bregma −3.6 mm) were prepared according to the method described previously.6,13 Briefly, brains after perfusion-fixation were sectioned with a vibratome at 100 μm. For the conventional EM, brain sections were postfixed for 2 h with 1% osmium tetroxide in 0.1 M cacodylate buffer immediately, rinsed in distilled water, stained with 1% aqueous uranyl acetate overnight, dehydrated, and embedded between two glass slides with Durcopan ACM. A small piece of embedded tissue section about 2 square millimeters either from the center of the lateral neocortical area or the middle segment of hippocampal CA3 was dissected out from embedded brain sections and mounted onto a resin block. The ultrathin sections were cut, counterstained with lead citrate, and evaluated with a ZEISS 10A Transmission Electron Microscope (Zeiss Inc.).
For the ethanolic phosphotungstic acid (EPTA) EM, brain sections were dehydrated in an ascending series of ethanol to 100% and stained for 45 min with 1% phosphotungstic acid (PTA) prepared by dissolving 0.1g of PTA in 10 mL of 100 ethanol and adding 100 μL of 95% ethanol. Sections were then embedded in Durcupan ACM. Ultrathin sections (0.1 □m) were prepared and examined with an electron microscope without additional staining.
Preparation of subcellular fractions
Brain tissue homogenate and subcellular fractions were prepared from sham-operated rats and rats subjected to moderate (2.0 atm) TBI followed by 4 h and 1, 3, 5, and 15 days of recovery according to the method described previously.14 The TBI ipsilateral neocortical tissues were dissected and chopped into small pieces in a −15°C glove box freezer, and then homogenized on ice with a Dounce homogenizer (35 strokes, 4°C) in 10 volumes of homogenization buffer: 15 mmol/L Tris pH 7.6, 0.25 mol/L sucrose, 1 mmol/L MgCl2, 2.5 mmol/L EDTA, 1 mmol/L EGTA (ethylene glycol-bis [b-amino ethyl ether] tetraacetic acid), 1 mmol/L dithiothreitol, 1.25 μg/mL pepstatin A, 10 μg/mL leupeptin, 2.5 μg/mL aprotinin, 0.5 mmol/L phenylmethylsulfonyl fluoride, 0.1 mmol/L Na3VO4, 50 mmol/L NaF, and 2 mmol/L Na4P2O7.
Homogenates were centrifuged at 800g at 4°C for 10 min to obtain P1 pellets (containing the heaviest cellular components, including the nuclei and secondary lysosomes) and supernatants (S1). The S1 was further centrifuged at 10,000g at 4°C for 10 min to obtain crude cell membrane/synaptosomal/mitochondrial pellet (P2) and its supernatant (S2). The S2 were centrifuged again at 165,000g at 4°C for 1 h to obtain the cytosol S3 and the microsomal pellet P3 that contains intracellular membrane structures such as the endoplasmic reticulum (ER), lysosome, and Golgi. All pellet fractions were suspended in homogenization buffer containing 0.1% Triton X-100. Each subcellular fraction was assayed for total protein concentration using the Coomassie Plus assay kit (Bio-Rad Laboratories, Hercules, CA).
Western blot analysis
Equal protein amounts in subcellular fractions were electrophoresed on 10% sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE) and then transferred to Immobilon-P membranes (Millipore). The membranes were incubated with 3% BSA in TBS for 30 min, and then overnight at 4°C with a primary antibody. After washing, the membranes were further incubated with horseradish peroxidase-conjugated anti-rabbit or anti-mouse secondary antibodies for 1 h at room temperature (1:2,000; Cell Signaling Technology, Billerica, MA).
In some experiments, the β-actin levels on the immunoblots were determined as endogenous protein loading controls. The immunoblots were developed using enhanced chemiluminescence (ECL, Pierce Biosciences, Rockford, IL) and developed on Kodak X-omat LS film (Eastman Kodak Company, New Haven, CT). Densitometry was performed with Kodak ID image analyses software (Eastman Kodak Company). All Western blot data were expressed as the ratio of the levels of the protein of interest and β-actin.
Deglycosylation assay
Each sample solution (10 μL) containing 10 μg of P1 fraction, distilled water, and 1 μL of 10 times glycoprotein denaturing buffer (5% SDS, 0.4M DTT), was denatured by heating at 100°C for 10 min. After denaturation, the samples were incubated at 37°C for 1 h with a cocktail of enzymes: 2 μL of 10x G7 Reaction Buffer (0.5M sodium phosphate, pH 7.5 at 25°C), 2 μL of 10% NP-40, 2 μL PNGase F (New England Biolabs, Ipswich, MA), and 4 μL distilled H2O. After incubation, SDS sample buffer (five times stock solution) was added to each reaction mixture and then subjected to Western blot analysis.
Statistical analysis
Data are expressed as mean±standard deviation (SD) as a percentage of sham-operated control levels. One-way analysis of variance followed by the Tukey post hoc test was used for statistical analysis. For comparison of two groups of experimental data, unpaired Student t test was used. *p<0.05 between sham and post-TBI groups.
Results
Histopathology
The TBI model used for this study is well established and has been used in many previous studies.11,15 To confirm tissue damage after TBI, the vibratome brain sections from sham-operated control rats and rats subjected to 2.0 atm FPI followed by 1, 3, 5, and 15 days of recovery were histologically stained and examined by light microscopy. Brain damage was not seen in sham-operated control sections, and observed in the cortical contusion area and CA3 of the hippocampus in all post-TBI brain sections examined (data not shown, but see Fig. 3). These results are consistent with previous studies.16,17
FIG. 3.

Confocal microscopy of lysosomal-associated membrane protein type 2A (LAMP2A). (A) Confocal microscopic images of hippocampal CA1, CA3, DG, and neocortical (Cx) regions. Brain sections were obtained from sham-operated rats and rats subjected to traumatic brain injury (TBI) followed by 1 day of recovery and were double-labeled with anti-LAMP2A antibody (green) and PI (red). At the hippocampal level (bregma−3.6 mm), the center area of the lateral neocortex, middle segments of hippocampal CA1 and CA3, and middle segment of the DG upper blade were examined with a Zeiss 510 laser-scanning confocal microscope. Arrows point to LAMP2A in neurons. Double-arrows indicate TBI dead neurons. Size bars=20 μm. (B) Confocal microscopy of neocortical regions of brain sections double-labeled with anti-LAMP2A antibody (green) and anti-Iba-1 antibody (red). Brain sections were from a sham-operated rat and rats subjected to TBI followed by 3 and 15 days of recovery. Arrows point to LAMP2A immunostained neurons, and arrowheads indicate LAMP2A and Iba-1 stained microglia. Size bars=20 μm. Color image is available online at www.liebertpub.com/neu
Upregulation of LAMP2A after TBI
The biochemical hallmark of CMA is the LAMP2A level that correlates directly with CMA activity.9 It is well established that lysosomes are morphologically and biochemically heterogeneous.18 To study lysosomal protein redistribution, tissue homogenate and P1, P2, and P3 subcellular fractions from neocortical brain tissues were analyzed with Western blotting. The LAMP2A antibody (Invitrogen 51-2200) recognized two major protein bands on Western blots (Fig. 1A, arrows).19 The upper band was not markedly altered, while the lower band was significantly increased in P1, P2, and P3 fractions after TBI (Fig. 1A, arrows). Quantitative analysis of the lower band indicated that it was significantly increased at 1, 3, 5, and 15 days of recovery after TBI (Fig. 1A).
FIG. 1.

Western blot analysis of lysosomal-associated membrane protein type 2A (LAMP2A). (A) Western blots (upper panels) and quantitative analysis (lower panels) of LAMP2A in homogenate (H), P1, P2, and P3 subcellular fractions after traumatic brain injury (TBI). Samples were prepared from the neocortical tissues of sham-operated control rats and rats subjected to TBI followed by 4 h and 1, 3, 5, and 15 days recovery. Data are normalized with β-actin and are expressed as mean±standard deviation (SD) of fold of control (n=4). *p<0.05 between sham and TBI animals, one-way analysis of variance followed by the Tukey post hoc test. (B) Western blots (upper panel) and quantitative analysis (lower panels) of deglycosylated (deglyc.) LAMP2A in P1 fraction. Data are normalized with β-actin and are expressed as mean±SD of fold of control (n=3). *p<0.05 between sham and TBI animals, unpaired t test.
LAMP2A is extensively glycosylated. To further verify the LAMP2A changes after TBI, P1 samples were treated with a mixture of deglycosylation enzymes (Fig. 1B). After deglycosylation treatment, the upper band was not moved, but the lower band was shifted from about 80 kDa to 45 kDa after electrophoresis. The 45 kDa deglycosylated band was significantly increased after TBI. Because glycosylated LAMP2A is about 80 kDa, and nonglycosylated is 44,961 Dalton, this study confirmed that the lower band is LAMP2A. The data are consistent with a previous study.19
LAMP2A is not neuron-specific. To study whether upregulation of LAMP2A is indicative of the lysosomal activity in neurons after TBI, brain sections were examined with transmission EM. Neocortical neurons from sham-operated control rats possessed Golgi apparatus (G), ribosomal rosettes (black arrows), nucleus (N), rough endoplasmic reticulum (ER), and mitochondria (M), and relatively lightly stained primary lysosome (black double arrow) (Fig. 2A). Darkly stained secondary lysosomes were rarely seen in sham-operated control neurons but easily identified in neurons at 3 and 15 days of recovery after TBI (Fig. 2A, white arrows).
FIG. 2.

Electron micrographs of neocortical neurons after traumatic brain injury (TBI). (A) Osmium-uranyl-lead stained brain sections of sham-operated control rats and rats subjected to TBI followed by 3 and 15 days recovery after TBI. Black arrows point to ribosomal rosettes, a black double arrow points to a primary lysosome, larger white arrows indicate secondary lysosomes, and arrowheads denote electron-dense vesicles. G=Golgi apparatus; N=nucleus; M=mitochondria; ER=rough endoplasmic reticulum; scale bars=0.5 μm. (B) Ethanolic phosphotungstic acid (EPTA)-stained neocortical neurons. EPTA stained basic proteins in the nuclei (N), ER-associated polyribosomes (double-arrows) and the secondary lysosomes (larger arrows); scale bars=1 μm.
Furthermore, clusters of electron-dense vesicles with electron-dense inclusions deriving from the trans face of the proliferated Golgi stacks were often seen at 15 days after TBI (Fig. 2A, TBI 15 days, arrowheads). The identity of these vesicles is unknown, but the ultrastructure may be similar to either transporting vesicles or newly formed lysosomes in the trans-Golgi network according to previous studies.20 These ultrastructural features suggest that the lysosomal activity is markedly upregulated after TBI.
In a previous study, we found that EPTA selectively stained secondary lysosomes in post-ischemic neurons.6 To further identify the secondary lysosomes after TBI, we performed an EPTA EM. In this method, the lipid component of membranes was not directly visible, because the material was dehydrated through absolute ethanol and acetone without osmication, leading to the extraction of most lipids in the EPTA stained material (Fig. 2B).
EPTA normally stains basic proteins in the nuclei (Fig. 2B, N) and ribosomes (Fig. 2B, double arrows) as well as denatured proteins located in the secondary lysosomes under EM (Fig. 2B, TBI-3d and TBI-15d, larger arrows), while leaving other subcellular structures relatively unstained, thus making identification of the secondary lysosomes easy.6 Similar to the conventional osmium-uramium-lead EM, EPTA EM showed that the secondary lysosomes were rarely found in neurons of sham-operated control rats, but easily seen in neurons at 3 and 15 days of recovery after TBI (Fig. 2B, larger arrows).
To study further the LAMP2A protein cellular distribution, we performed confocal microscopy using brain sections from sham-operated control rats and rats subjected to TBI followed by 24 h of recovery. LAMP2A immunoreactivity was distributed in CA1, CA3, DG, and neocortical (Cx) neurons of both sham-operated control (Fig. 3A, sham) and post-TBI brains (Fig. 3A, TBI-1 day), but appeared as a more intense punctuated pattern in the CA3 and Cx areas at the ipsilateral side of brain sections after TBI (Fig. 3A, TBI-1 day). Some dead neurons in CA3 and Cx after TBI were seen based on the PI-stained shrunken nuclei and the lost of the LAMP2A immunoreactivity (Fig. 3A, TBI-1 day, double arrows).
Microglia proliferation was observed in the contusion neocortical area after TBI particularly during the later periods of recovery as demonstrated by microglial marker Iba-1 immunolabeling (Fig. 3, TBI-3 days and TBI-15 days, red color, arrowheads). Double staining confocal microscopy of LAMP2A (Invitrogen 51-2200) and Iba-1 further demonstrated that LAMP2A immunoreactivity was increased both in neurons (Fig. 3B, green color, arrows) and microglia (Fig. 3B, red color, arrowheads) at 3 and 15 days of recovery after TBI.
The UPS is a parallel route with CMA for the clearance of cellular aberrant proteins. To study whether the UPS was also upregulated after TBI, the same sets of subcellular fraction samples as used in Figure 1 were used to study the levels of polyubiquitinated protein conjugates (ubi-proteins) using Western blotting (Fig. 4). Ubi-proteins on Western blots typically appeared as a high molecular-weight smear caused by heterogeneity of the modified proteins (Fig. 4).
FIG. 4.

Western blot analysis of ubiquitinated proteins (ubi-proteins). Western blots (upper panels) and quantitative analysis (lower panels) of ubi-proteins in P1, P2, P3, and free ubiquitin in S3 subcellular fractions after traumatic brain injury (TBI). Brain samples were from the same sets of tissue samples of Figure 1. Data are expressed as mean±standard deviation of fold of control (n=4) *p<0.05 between sham and TBI animals; one-way analysis of variance followed by the Tukey post hoc test.
Quantitative analysis showed that ubi-proteins were significantly and transiently increased and then returned to or below the control level in P1 fraction after TBI (Fig. 4). In comparison, the increases in ubi-proteins continued in P2 and P3 fractions during the recovery periods of 4 h–15 days after TBI (Fig. 4). Polyubiquitinated proteins or ubi-protein smear bands were mostly above 90 kDa on Western blots. There was no pattern change on Western blots between smaller and larger molecular size ubi-protein bands after TBI (Fig. 4). The cytosolic pool of 7.6 kDa free ubiquitin in S3 fraction was not significantly altered after TBI (Fig. 4). Double staining confocal microscopy further demonstrated that immunostaining of ubiquitin (Fig. 5, red color) and LAMP2A (Fig. 5, green color) were colocalized in neurons at 3 days of recovery after TBI.
FIG. 5.
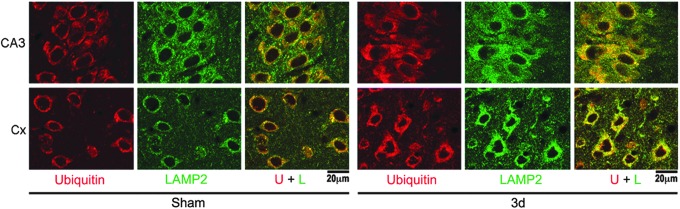
Confocal microscopy of lysosomal-associated membrane protein type 2A (LAMP2A) and ubiquitin immunoreactivity. Confocal microscopic images of hippocampal CA3 and neocortical (Cx) regions of brain sections double-labeled with anti-LAMP2A (green) and anti-ubiquitin antibodies (red). Brain sections were from sham-operated rats and rats subjected to traumatic brain injury followed by 3 days of recovery. Size bars=20 μm. Color image is available online at www.liebertpub.com/neu
HSC70 and its inducible form HSP70 are molecular chaperones for delivery of aberrant proteins to lysosomes during CMA. Western blot analysis showed that the level of HSC70 was unchanged, but its inducible form HSP70 was significantly upregulated starting at 1 day, peaking at 3 days, and remaining elevated for as long as 15 days of recovery after TBI in P1, P2, and P3 subcellular fractions after TBI (Fig. 6, A, B). HSP70 was also significantly upregulated, although to a much less degree, in cytosolic S3 fraction during 3–15 days after TBI. These changes occurred virtually in the same time course as those of LAMP2A after TBI.
FIG. 6.
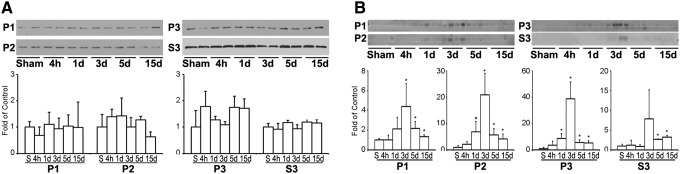
Western blot analysis of HSC70 and HSP70. Western blots (upper panels) and quantitative analysis (lower panels) of HSC70 (A) and HSP70 (B) in P1, P2, P3, and S3 subcellular fractions after traumatic brain injury (TBI). Brain samples were obtained from the same sets of tissue samples as in Figure 1. Data were normalized with β-actin and are expressed as mean±standard deviation of fold of control (n=4). *p<0.05 between sham and TBI animals; one-way analysis of variance followed by the Tukey post hoc test.
Discussion
There are several cellular degradation pathways including macroautophagy (commonly referring to as autophagy), microautophagy, and CMA, as well as the UPS. Evidence for changes in macroautophagy has been reported in several models of TBI and is a target for therapeutic interventions. For instance, Cordaro and associates21 report that co-treatment with palmitoylethanolamide and flavonoid luteolin reduces brain damage and the mactoautophagy activity after TBI. The study of Bao and colleagues22 shows that Apelin-13 attenuates brain damage and suppresses autophagy after TBI. Sun and associates23 demonstrate that administration of bone marrow stromal cells is able to significantly suppress TBI-induced autophagy activity. Sarkar and colleagues24 concluded that the accumulation of autophagosomes after severe TBI was because of lysosomal dysfunction. Taken together these studies emphasize the importance of macroautophagy in the pathophysiology and management of TBI.
In comparison with changes in macroautophagy, this study shows that the UPS and CMA are consecutively upregulated after TBI. The upregulation of the UPS activity occurs earlier starting from 4 h of recovery and continues for as long as 15 days after TBI. In comparison, the upregulation of LAMP2A and HSP70 takes place between 24 and 72 h and continues also for as long as 15 days of recovery after TBI. The increases in LAMP2A and HSP70 were seen mainly in the secondary lysosome-containing fractions.
Electron or confocal microscopic examinations further show that the increase in LAMP2A or secondary lysosomes occurs in living neurons and proliferated microglia, whereas TBI-damaged neurons lose their LAMP2A immunoreactivity. We did not see marked changes in LAMP2A immunostaining in other cell types of brain in the TBI-affected neocortical, CA1, CA3, and DG areas during the post-TBI phase. Because CMA and the UPS are two major routes for elimination of cellular aberrant proteins, the consecutive upregulation of these two pathways may serve as protective mechanisms for maintaining cellular homeostasis after TBI.
Upregulation of the UPS after TBI
The biochemical hallmark of the UPS activity is the upregulation of ubi-proteins. It was reported previously that free ubiquitin protein levels were significantly reduced in both ipsilateral cerebral cortex and hippocampus during the periods of 1–7 days after TBI, while ubi-proteins were not increased until 3 days after TBI.25,26 In comparison, the present study observed no significant change in the free ubiquitin and only moderate increases in ubi-proteins during the periods of 4 h to 15 days after TBI. The discrepancy is likely because more severe TBI (about 2.5 atm) was used in the previous studies than that in the present report (about 2.0 atm).
Previous studies demonstrate that FPI especially at a higher degree of severity may leads to post-TBI hypoxia, ischemia, edema, and blood–brain barrier breakdown.11,27,28 Reductions of ATP after moderate FPI, however, are generally transient or mild, and rapidly recover to baseline.29–33 ATP decreases significantly after 2.5 atm, but not after mild 1.5 atm mild FPI.31 Because protein misfolding and ubiquitination is directly related to the depletion of cellular ATP,5–7,34,35 the moderate change in protein ubiquitination after moderate FPI shown in this study may be directly related to the degree of ATP reduction and injury severity.
Upregulation of CMA after TBI
In comparison with more moderate upregulation of the UPS, this study further demonstrates that CMA is significantly upregulated in a delayed fashion between 1–15 days after TBI. Currently, there are no reports of CMA after TBI in the literature. CMA is constitutively active but maximally upregulated and accounts for the degradation of 30% of cytosolic proteins under stress conditions.36 Levels of LAMP2A can be increased via transcriptional upregulation as in the case of oxidative stress, or through changes in the degradation rate of LAMP-2A at the lysosomal membrane as occurs when CMA is upregulated during prolonged starvation.36 As in the case of this study, increased transcription of LAMP2A may also be plausible as oxidative stress occurs after TBI.37
There are three splicing isoforms of LAMP2 (A, B and C). These isoforms have different cytosolic segments.4,38 The LAMP2A antibody from Invitrogen (#51-2200) was made specifically against the LAMP2A cytosolic peptide segment (GLKRHHTGYEQF). This peptide differs significantly in aminoacid composition from the cytosolic regions of LAMP2B (GRRKSRTGYQSV) and LAMP2C (GRRKTYAGYQTL). The significant different composition makes the LAMP2A antibody very unlikely to recognize LAMP2B and LAMP2C, as shown experimentally.38
This LAMP2A (Invitrogen #51-2200) has been used for both Western blotting and immunocytochemistry in several previous publications.8,19,39–41 As shown in Figure 1, this LAMP2A antibody cross-reacts with a non-LAMP2A protein in brain tissue samples on Western blots. For that reason, it may not be ideal to use this antibody for immunostaining of tissue sections. The results are consistent with a previous study.19 There are situations that an antibody recognizes antigen in its native structure and location, but recognize additional bands when protein are denatured and solubilized in Western blot analysis. It is also possible the top band recognized by this Invitrogen LAMP2A antibody does not change, thus serving as a background staining, while the lower LAMP2A band is significantly altered, thus reflecting the LAMP2A immunostaining pattern changes in brain sections after TBI.
Darkly stained secondary lysosomal structures under the conventional EM are rarely seen in control neurons, but easily identified in neurons after TBI. These lysosomal structures can also be stained with EPTA under EM as shown in Figure 2B. Because EPTA stains only proteins,42 these lysosomal structures are likely secondary lysosomes, rather than lysosomes with lipofuscin vesicles. In lysosomal storage diseases, lysosomal enzyme deficiencies lead to accumulation of dysfunctional cellular lysosomes.
Could the increase in the LAMP2A level or the EM secondary lysosomes after TBI be because of lysosomal dysfunction? This may not be the case, because several previous studies show that mechanical stress generally activates, rather than inhibits lysosomal turnover.43–47 As shown in the present study, LAMP2A is increased in both neurons and proliferated microglia (Fig. 3B). Therefore, as discussed above, the upregulation of LAMP2A after TBI may be because of not only the degradation rate,36 but also the formation of new lysosomes after TBI.
EM is an excellent tool for analysis of subcellular fine structures. Using EM to estimate the lysosomal number alterations after TBI must be with caution, however. The reason is that there are significant amounts of unforeseen challenges when using EM as a quantitative tool for estimating three-dimensional subcellular structures even using a stereological method.48 This is also because of the natures of the diffuse axonal mechanical damage and multiple foci pathology after TBI.49 To overcome these obstacles, the present study employed a combination analysis of using both more quantitative Western blot analysis of lysosomal marker protein LAMP2A, as well as EM and confocal microscopic cellular distribution of the lysosomes to show the upregulation of CMA after TBI.
CMA substrate proteins contain a common consensus pentapeptide known as “KFERQ-like motif.” In this process, the motif-containing substrate proteins are recognized and then delivered by HSC70 or HSP70 to lysosomes where they are bound to the receptor channel composed of multimer LAMP2A.36 The present study shows that HSC70 is unchanged, but HSP70 is significantly upregulated in the lysosome-containing subcellular fractions during the period of 3–15 days after TBI. The upregulation of LAMP2A also occurs during the period of 3–15 days after TBI. The results are in line with previous observations that CMA is enhanced after heat shock.50
Significance of CMA and the UPS pathway after moderate TBI
This study shows that the UPS and CMA are consecutively upregulated after TBI. Is upregulation of UPS and CMA helpful or harmful in neurons after TBI or simply another marker of potential neuronal stress? A common pathology of neurodegeneration is the toxic protein misfolding and deposition, which may be because of the failure of the proteolytic systems to adequately dispose of deleterious proteins.5 Protein deposition can be seen in TBI-affected brain regions.51
The increases in the activities of CMA and the UPS may be cellular attempts to eliminate pathogenic proteins.45 Overactivation of these cellular degradation processes might also be detrimental, however. For instance, overactivated lysosomes might leak destructive enzymes after brain injury.5,52 Overactivation of microglia may induce collateral damage to salvageable neurons after TBI.53
On the other hand, activation of microglia may also play a beneficial role in removing damaged cellular structures and remodeling synaptic networks in the late recovery phase after TBI.54 Therefore, moderate inhibition of CMA might reduce microglial overactivation-induced collateral damage in the acute phase, whereas promoting CMA in the late recovery phase might facilitate brain repair and networking.
TBI is an important risk factor for the onset of Parkinson and Alzheimer diseases.55–59 Alpha-synuclein is significantly increased in TBI patient cerebrospinal fluid.60 Deposition of aberrant proteins is seen in TBI animal models and patients.55,57 CMA is a major cellular system for removal of α-synuclein, Tau, huntingtin, ubiquitin carboxyl-terminal esterase L1, calcineurin 1, and the cytoplasmic tail of amyloid precursor protein.58,61 Many of these neurodegenerative disease-related proteins can act as CMA inhibitors and thus compromise CMA.62 Overexpression of LAMP2A can effectively decrease the levels of neurodegenerative disease-related aberrant proteins and reduce neurodegeneration.63 Therefore, upregulation of CMA may be beneficial to remove toxic aberrant proteins during the late period of recovery after TBI.
Acknowledgment
This work was supported by National Institutes of Health grants NS040407 and NS030291, AHA EIA grant 0940042N and Veteran Affair Merit Award I01BX001696-01.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Kaushik S., and Cuervo A.M. (2012). Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 22, 407–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Winchester B.G. (2001). Lysosomal membrane proteins. Eur. J. Paediatr. Neurol. 5 Suppl A, 11–19 [DOI] [PubMed] [Google Scholar]
- 3.De Matteis M.A., and Luini A. (2008). Exiting the Golgi complex. Nat. Rev. Mol. Cell Biol. 9, 273–284 [DOI] [PubMed] [Google Scholar]
- 4.Treusch S., Knuth S., Slaugenhaupt S.A., Goldin E., Grant B.D., and Fares H. (2004). Caenorhabditis elegans functional orthologue of human protein h-mucolipin-1 is required for lysosome biogenesis. Proc. Natl. Acad. Sci. U. S. A., 101, 4483–4488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luo T., Park Y., Sun X., Liu C., and Hu B. (2013). Protein misfolding, aggregation, and autophagy after brain ischemia. Transl. Stroke Res. 4, 581–588 [DOI] [PubMed] [Google Scholar]
- 6.Hu B.R., Martone M.E., Jones Y.Z., and Liu C.L. (2000). Protein aggregation after transient cerebral ischemia. J. Neurosci. 20, 3191–3199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu B.R., Janelidze S., Ginsberg M.D., Busto R., Perez-Pinzon M., Sick T.J., Siesjö BK, and Liu CL. (2001). Protein aggregation after focal brain ischemia and reperfusion. J. Cereb. Blood Flow Metab. 21, 865–875 [DOI] [PubMed] [Google Scholar]
- 8.Cuervo A.M., and Dice J.F. (1996). A receptor for the selective uptake and degradation of proteins by lysosomes. Science 273, 501–503 [DOI] [PubMed] [Google Scholar]
- 9.Bandyopadhyay U., Kaushik S., Varticovski L., and Cuervo A.M. (2008). The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell Biol. 28,5747–5763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tomura S., de Rivero Vaccari J.P., Keane R.W., Bramlett H.M., and Dietrich W.D. (2012). Effects of therapeutic hypothermia on inflammasome signaling after traumatic brain injury. J. Cereb. Blood Flow Metab. 32, 1939–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blaya M.O., Bramlett H.M., Naidoo J., Pieper A.A., and Dietrich W.D. (2014). Neuroprotective efficacy of a proneurogenic compound after traumatic brain injury. J. Neurotrauma 31, 476–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ponten U., Ratcheson R.A., and Siesjo B.K. (1973). Metabolic changes in the brains of mice frozen in liquid nitrogen. J. Neurochem. 21, 1211–1216 [PubMed] [Google Scholar]
- 13.Martone M.E., Jones Y.Z., Young S.J., Ellisman M.H., Zivin J.A., and Hu B.R. (1999). Modification of postsynaptic densities after transient cerebral ischemia: a quantitative and three-dimensional ultrastructural study. J. Neurosci. 19, 1988–1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu B., Liu C., Bramlett H., Sick T.J., Alonso O.F., Chen S., and Dietrich W.D. (2004). Changes in trkB-ERK1/2-CREB/Elk-1 pathways in hippocampal mossy fiber organization after traumatic brain injury. J. Cereb. Blood Flow Metab. 24, 934–943 [DOI] [PubMed] [Google Scholar]
- 15.Lotocki G., de Rivero Vaccari J.P., Perez E.R., Sanchez-Molano J., Furones-Alonso O., Bramlett H.M., and Dietrich W.D. (2009). Alterations in blood-brain barrier permeability to large and small molecules and leukocyte accumulation after traumatic brain injury: effects of post-traumatic hypothermia. J. Neurotrauma 26, 1123–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McIntosh T.K., Vink R., Noble L., Yamakami I., Fernyak S., Soares H., and Faden A.L. (1989). Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience 28, 233–244 [DOI] [PubMed] [Google Scholar]
- 17.Dietrich W.D., Alonso O., Halley M., and Busto R. (1996). Delayed posttraumatic brain hyperthermia worsens outcome after fluid percussion brain injury: a light and electron microscopic study in rats. Neurosurgery 38, 533–541 [DOI] [PubMed] [Google Scholar]
- 18.Cuervo A.M., Dice J.F., and Knecht E. (1997). A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J. Biol. Chem. 272, 5606–5615 [DOI] [PubMed] [Google Scholar]
- 19.Malkus K.A., and Ischiropoulos H. (2012). Regional deficiencies in chaperone-mediated autophagy underlie α-synuclein aggregation and neurodegeneration. Neurobiol. Dis. 46, 732–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanai M., Soji T., and Herbert D.C. (1997). Biogenesis and function of lipolysosomes in developing chick hepatocytes. Microsc. Res. Tech. 39, 444–452 [DOI] [PubMed] [Google Scholar]
- 21.Cordaro M., Impellizzeri D., Paterniti I., Bruschetta G., Siracusa R., De Stefano D., Cuzzocrea S., and Esposito E. (2014). Neuroprotective effects of Co-ultraPEALut on secondary inflammatory process and autophagy involved in traumatic brain injury. J. Neurotrauma 2015. Epub ahead of print [DOI] [PubMed] [Google Scholar]
- 22.Bao H.J., Zhang L., Han W.C., and Dai D.K. (2015). Apelin-13 attenuates traumatic brain injury-induced damage by suppressing autophagy. Neurochem. Res. 40, 89–97 [DOI] [PubMed] [Google Scholar]
- 23.Sun L., Gao J, Zhao M., Jing X., Cui Y., Xu X., Wang K., Zhang W., and Cui J. (2014). The effects of BMSCs transplanation on autophagy by CX43 in the hippocampus following traumatic brain injury in rats. Neurol. Sci. 35, 677–682 [DOI] [PubMed] [Google Scholar]
- 24.Sarkar C., Zhao Z., Aungst S., Sabirzhanov B., Faden A.I., and Lipinski M.M. (2014). Impaired autophagy flux is associated with neuronal cell death after traumatic brain injury. Autophagy 10, 2208–2222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yao X., Liu J., and McCabe J.T. (2007). Ubiquitin and ubiquitin-conjugated protein expression in the rat cerebral cortex and hippocampus following traumatic brain injury (TBI). Brain Res. 1182, 116–122 [DOI] [PubMed] [Google Scholar]
- 26.Sakai K., Fukuda T., and Iwadate K. (2014). Immunohistochemical analysis of the ubiquitin proteasome system and autophagy lysosome system induced after traumatic intracranial injury: association with time between the injury and death. Am. J. Forensic Med. Pathol. 35, 38–44 [DOI] [PubMed] [Google Scholar]
- 27.McIntosh T.K., Soares H., Thomas M., and Cloherty K. (1990). Development of regional cerebral oedema after lateral fluid-percussion brain injury in the rat. Acta Neurochir. Suppl. (Wien). 51, 263–264 [DOI] [PubMed] [Google Scholar]
- 28.Bramlett H.M., and Dietrich W.D. (2004). Pathophysiology of cerebral ischemia and brain trauma: similarities and differences. J. Cereb. Blood Flow Metab. 24, 133–150 [DOI] [PubMed] [Google Scholar]
- 29.Sullivan P.G., Keller J.N., Mattson M.P., and Scheff S.W. (1998). Traumatic brain injury alters synaptic homeostasis: implications for impaired mitochondrial and transport function. J. Neurotrauma 15, 789–798 [DOI] [PubMed] [Google Scholar]
- 30.Lee S.M., Wong M.D., Samii A., and Hovda D.A. (1999). Evidence for energy failure following irreversible traumatic brain injury. Ann. N. Y. Acad. Sci. 893, 337–340 [DOI] [PubMed] [Google Scholar]
- 31.Buczek M., Alvarez J., Azhar J., Zhou Y., Lust W.D., Selman W.R., and Ratcheson R.A. (2002). Delayed changes in regional brain energy metabolism following cerebral concussion in rats. Metab. Brain Dis. 17, 153–167 [DOI] [PubMed] [Google Scholar]
- 32.Marklund N., Salci K., Ronquist G., and Hillered L. (2006). Energy metabolic changes in the early post-injury period following traumatic brain injury in rats. Neurochem. Res. 31, 1085–1093 [DOI] [PubMed] [Google Scholar]
- 33.Aoyama N., Lee S.M., Moro N., Hovda D.A., and Sutton R.L. (2008). Duration of ATP reduction affects extent of CA1 cell death in rat models of fluid percussion injury combined with secondary ischemia. Brain Res. 1230, 310–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartl F.U., and Hayer-Hartl M. (2002). Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 295, 1852–1858 [DOI] [PubMed] [Google Scholar]
- 35.Tsigelny I.F., and Nigam S.K. (2004). Complex dynamics of chaperone-protein interactions under cellular stress. Cell Biochem. Biophys. 40, 263–276 [DOI] [PubMed] [Google Scholar]
- 36.Cuervo A.M. (2011). Chaperone-mediated autophagy: Dice's ‘wild’ idea about lysosomal selectivity. Nat. Rev. Mol. Cell Biol. 12, 535–541 [DOI] [PubMed] [Google Scholar]
- 37.Hall E.D., Vaishnav R.A., and Mustafa A.G. (2010). Antioxidant therapies for traumatic brain injury. Neurotherapeutics 7, 51–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cuervo A.M., and Dice J.F. (2000). Unique properties of lamp2a compared to other lamp2 isoforms. J. Cell Sci. 113, 4441–4450 [DOI] [PubMed] [Google Scholar]
- 39.Natarajan R., and Linstedt A.D. (2004). A cycling cis-Golgi protein mediates endosome-to-Golgi traffic. Mol. Biol. Cell 15, 4798–4806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roscoe W., Veitch G.I., Gong X.Q., Pellegrino E., Bai D., McLachlan E., Shao Q., Kidder G.M., and Laird D.W. (2005). Oculodentodigital dysplasia-causing connexin43 mutants are non-functional and exhibit dominant effects on wild-type connexin43. J. Biol. Chem. 280, 11458–11466 [DOI] [PubMed] [Google Scholar]
- 41.Wei J., Fujita M., Nakai M., Waragai M., Sekigawa A., Sugama S., Takenouchi T., Masliah E., and Hashimoto M. (2009). Protective role of endogenous gangliosides for lysosomal pathology in a cellular model of synucleinopathies. Am. J. Pathol. 174, 1891–1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu B.R., Park M., Martone M.E., Fischer W.H., Ellisman M.H., and Zivin J.A. (1998). Assembly of proteins to postsynaptic densities after transient cerebral ischemia. J. Neurosci. 18, 625–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clark R.S., Bayir H., Chu C.T., Alber S.M., Kochanek P.M., and Watkins S.C. (2008). Autophagy is increased in mice after traumatic brain injury and is detectable in human brain after trauma and critical illness. Autophagy 4, 88–90 [DOI] [PubMed] [Google Scholar]
- 44.Sadasivan S., Dunn W.A., Jr., Hayes R.L., and Wang K.K. (2008). Changes in autophagy proteins in a rat model of controlled cortical impact induced brain injury. Biochem. Biophys. Res. Commun. 373, 478–481 [DOI] [PubMed] [Google Scholar]
- 45.Liu C.L., Chen S., Dietrich D., and Hu B.R. (2008). Changes in autophagy after traumatic brain injury. J. Cereb. Blood Flow Metab. 28, 674–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.King J.S. (2012). Mechanical stress meets autophagy: potential implications for physiology and pathology. Trends Mol. Med. 18, 583–588 [DOI] [PubMed] [Google Scholar]
- 47.Lin C.J., Chen T.H., Yang L.Y., and Shih C.M. (2014). Resveratrol protects astrocytes against traumatic brain injury through inhibiting apoptotic and autophagic cell death. Cell Death Dis. 5, e1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mühlfeld C., Nyengaard J.R., and Mayhew T.M. (2010). A review of state-of-the-art stereology for better quantitative 3D morphology in cardiac research. Cardiovasc. Pathol. 19, 65–82 [DOI] [PubMed] [Google Scholar]
- 49.Farkas O., and Povlishock J.T. (2007). Cellular and subcellular change evoked by diffuse traumatic brain injury: a complex web of change extending far beyond focal damage. Prog Brain Res. 161, 43–59 [DOI] [PubMed] [Google Scholar]
- 50.Yabu T., Imamura S., Mohammed M.S., Touhata K., Minami T., Terayama M., and Yamashita M. (2011). Differential gene expression of HSC70/HSP70 in yellowtail cells in response to chaperone-mediated autophagy. FEBS J. 278, 673–685 [DOI] [PubMed] [Google Scholar]
- 51.Sharp D.J., Scott G., and Leech R. (2014). Network dysfunction after traumatic brain injury. Nat. Rev. Neurol. 10, 156–166 [DOI] [PubMed] [Google Scholar]
- 52.Hayashi T., Shoji M., and Abe K. (2006). Molecular mechanisms of ischemic neuronal cell death—with relevance to Alzheimer's disease. Curr. Alzheimer Res. 3, 351–358 [DOI] [PubMed] [Google Scholar]
- 53.de Rivero Vaccari J.P., Dietrich W.D., and Keane R.W. (2014). Activation and regulation of cellular inflammasomes: gaps in our knowledge for central nervous system injury. J. Cereb. Blood Flow Metab. 34, 369–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beynon S.B., and Walker F.R. (2012). Microglial activation in the injured and healthy brain: what are we really talking about? Practical and theoretical issues associated with the measurement of changes in microglial morphology. Neuroscience 225, 162–171 [DOI] [PubMed] [Google Scholar]
- 55.Uryu K., Laurer H., McIntosh T., Praticò D., Martinez D., Leight S., Lee V.M., and Trojanowski J.Q. (2002). Repetitive mild brain trauma accelerates Abeta deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of Alzheimer amyloidosis. J. Neurosci. 22, 446–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Van Den Heuvel C., Thornton E., and Vink R. (2007). Traumatic brain injury and Alzheimer's disease: a review. Prog. Brain Res. 161, 303–316 [DOI] [PubMed] [Google Scholar]
- 57.Kawai N., Kawanishi M., Kudomi N., Maeda Y., Yamamoto Y., Nishiyama Y., and Tamiya T. (2013). Detection of brain amyloid β deposition in patients with neuropsychological impairment after traumatic brain injury: PET evaluation using Pittsburgh Compound-B. Brain Inj. 27, 1026–1031 [DOI] [PubMed] [Google Scholar]
- 58.Shahaduzzaman M., Acosta S., Bickford P.C., and Borlongan C.V. (2013). α-Synuclein is a pathological link and therapeutic target for Parkinson's disease and traumatic brain injury. Med. Hypotheses 81, 675–680 [DOI] [PubMed] [Google Scholar]
- 59.Sivanandam T.M., and Thakur M.K. (2012). Traumatic brain injury: a risk factor for Alzheimer's disease. Neurosci. Biobehav. Rev. 36, 1376–1381 [DOI] [PubMed] [Google Scholar]
- 60.Mondello S., Buki A., Italiano D., and Jeromin A. (2013). α-Synuclein in CSF of patients with severe traumatic brain injury. Neurology 80, 1662–1668 [DOI] [PubMed] [Google Scholar]
- 61.Koga H., and Cuervo A.M. (2011). Chaperone-mediated autophagy dysfunction in the pathogenesis of neurodegeneration. Neurobiol. Dis. 43, 29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cuervo A.M., and Wong E. (2014). Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 24, 92–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xilouri M., Brekk O.R., Kirik D., and Stefanis L. (2013). LAMP2A as a therapeutic target in Parkinson disease. Autophagy 9, 2166–2168 [DOI] [PubMed] [Google Scholar]