

Abstract
Obesity and alcohol consumption often coexist and work synergistically to promote steatohepatitis; however, the underlying mechanisms remain obscure. Here, we demonstrated that feeding mice a high-fat diet (HFD) for as little as 3 days markedly exacerbated acute ethanol binge-induced liver neutrophil infiltration and injury. Feeding mice with a HFD for 3 months plus a single binge of ethanol induced much more severe steatohepatitis. Moreover, 3-day or 3-month HFD-plus-ethanol binge (3d-HFD+ethanol or 3m-HFD+ethanol) treatment markedly upregulated the hepatic expression of several chemokines, including chemokine (C-X-C motif) ligand 1 (Cxcl1), which showed the highest fold (approximately 20-fold and 35-fold, respectively) induction. Serum CXCL1 protein levels were markedly elevated after the HFD+ethanol treatment. Blockade of CXCL1 with a CXCL1 neutralizing antibody or genetic deletion of the Cxcl1 gene reduced the HFD+ethanol-induced hepatic neutrophil infiltration and injury; whereas overexpression of the Cxcl1 exacerbated steatohepatitis in HFD-fed mice. Furthermore, the expression of Cxcl1 mRNA was upregulated in hepatocytes, hepatic stellate cells and endothelial cells isolated from HFD+ethanol-fed mice compared to mice that were only given a HFD, with the highest fold induction observed in hepatocytes. In vitro stimulation of hepatocytes with palmitic acid upregulated the expression of Cxcl1 mRNA, and this upregulation was attenuated after treatment with ERK1/2, JNK, or NF-κB inhibitors. In addition, hepatic or serum levels of free fatty acids (FFAs) were higher in HFD+ethanol-fed mice than in the control groups. In conclusion, a HFD combined with acute ethanol consumption synergistically induces acute liver inflammation and injury via the elevation of hepatic or serum FFAs and subsequent upregulation of hepatic CXCL1 expression and promotion of hepatic neutrophil infiltration.
Keywords: free fatty acid, palmitic acid, inflammation, alcoholic hepatitis
Introduction
It has been recognized for many years that being overweight or obese and consuming excess amounts of alcohol synergistically promote the development and progression of alcoholic liver injury, alcoholic hepatitis, cirrhosis, and hepatocellular carcinoma in patients.1-9 In a recent study, multivariate regression analysis of several factors indicated that abdominal girth is positively correlated with the liver fibrosis score in alcoholics, suggesting that visceral fat accumulation, independent of body mass index and metabolic syndrome criteria, plays an important role in promoting alcoholic liver injury.3
The synergistic effects of obesity and alcohol on steatohepatitis have also been observed in several animal models. For example, providing mice ad libitum with a high-fat diet (HFD) and alcohol in the drinking water synergistically increased the hepatic expression of profibrogenic genes, activation of hepatic stellate cells (HSCs) and extracellular matrix deposition.10 Moreover, feeding rats a HFD for one month followed by gavage with ethanol every 12 h for the last three days increased hepatic oxidative stress but did not increase serum ALT levels,11 and intragastric overfeeding of rats or mice with a HFD and co-administration of ethanol synergistically induced an elevation of serum ALT levels, hepatic steatosis, liver inflammation and fibrosis.12, 13 The data from mechanistic studies further indicate that alcohol and HFDs synergistically induce nitrosative, endoplasmic reticulum (ER), and mitochondrial stress and an upregulation of hepatic toll-like receptor 4 (TLR4), thereby contributing to steatohepatitis.10, 13 Additionally, moderate ethanol binges induce significant liver damage (hepatocyte apoptosis) in genetically obese (ob/ob) mice by increasing TNF-α and decreasing nuclear NF-κB activity.14 Moreover, a recent study reported that chronic ethanol feeding of genetically obese (ob/ob) mice exacerbated hepatic steatosis and injury via the impairment of hepatic lipid metabolism pathways mediated by the hepatic SIRT1-AMPK signaling system.15 In the present study, we developed a simple model of mixed steatohepatitis by feeding mice a HFD followed by gavage with a single dose of ethanol. The most striking finding was that feeding mice a HFD for as little as 3 days, which is known to impair glucose tolerance and hepatic insulin sensitivity,16-18 markedly exacerbated the acute ethanol binge-induced neutrophilia, hepatic neutrophil infiltration, and liver injury. Long-term (3 months) HFD feeding plus gavage of a single dose of ethanol caused much more severe steatohepatitis.
In response to tissue injury or infection, neutrophils are recruited from the blood into the affected tissue; this process is orchestrated by multiple adhesion molecules, chemokines and cytokines.19-21 These factors include endothelium-expressed molecules, such as E-selectin, P-selectin, vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule 1 (ICAM-1), neutrophil-expressed proteins, such as L-selectin (CD62L), E-selectin ligand 1 (ESL-1), P-selectin glycoprotein ligand 1 (PSGL-1) and CD44, and members of the CXC chemokine family including chemokine (C-X-C motif) ligand 1 (CXCL1), CXCL2 and CXCL8 (IL-8).19, 20 To examine the mechanisms underlying the synergistic effects of HFD-plus-binge ethanol feeding on neutrophil infiltration, we analyzed the expression of an array of adhesion molecules, chemokines and cytokines. Among these factors, CXCL1 showed the highest induction (up to 20-fold and 30-fold) in the liver after 3d-HFD+ethanol and 3m-HFD+ethanol feeding, respectively.
CXCL1 belongs to the CXC chemokine family and has several other names, including keratinocyte-derived chemokine (KC), cytokine-induced neutrophil chemoattractant-1 (CINC-1), growth-regulated protein oncogene-1 (Gro1), Gro-α, neutrophil-activating protein 3 (NAP-3) and melanoma growth-stimulating activity alpha (MSGA-α).22 In human, this protein is encoded by the CXCL1 gene. CXCL2 is also referred to as macrophage inflammatory protein 2-α (MIP2-α), Gro-β and Gro-2. CXCL1 and CXCL2 are 90% identical in their amino acid sequences and elicit their functions by signaling through the chemokine receptor CXCR2 (also known as IL-8 binding protein), which is a G protein-coupled chemokine receptor expressed on neutrophils.20, 22 Hepatic and serum levels of CXCL1 are markedly elevated, whereas hepatic CXCL2 expression is downregulated in patients with alcoholic hepatitis.23 Hepatic CXCL1 mRNA levels correlated positively with hepatic neutrophil infiltration in these patients.23 In addition, the CXCL1 rs4074 A allele is associated with the elevated levels of CXCL1 protein and is a genetic risk factor for alcoholic cirrhosis.24 In the current paper, we demonstrated that HFD-plus-binge ethanol synergistically upregulated hepatic and serum levels of CXCL1. Furthermore, we demonstrated that PA, oleic acid (OA) and linoleic acid (LA) upregulated the expression of Cxcl1 mRNA in mouse hepatocytes and that HFD+ethanol feeding resulted in elevated hepatic or serum free fatty acids (FFAs). Finally, we provided evidence suggesting that CXCL1 plays an important role in inducing liver neutrophil recruitment and liver injury after HFD+ethanol treatment.
Materials and Methods
Mice
Eight- to twelve-week-old male C57BL/6 mice were used in this study. Mice were housed in a temperature-controlled room with a 12-h dark/12-h light cycle. Wild-type, Tlr4-/- mice on a C57BL/6J background were purchased from the Jackson Laboratory (Bar Harbor, Maine). Cxcl1-/- mice on a C57BL/6J background were kindly provided by Dr. Sergio Lira (Mount Sinai, NY).25 All animals were cared for in accordance with the National Institutes of Health guidelines, and all animal experiments were approved by the National Institute on Alcohol Abuse and Alcoholism animal care and use committee.
HFD+ethanol binge feeding model
Mice were fed a HFD (60% kcal% fat; Cat# D12492, Research Diet, New Brunswick, NJ) for 3 days or 3 months. On the last day of HFD feeding, mice were given a single dose of ethanol (5 g/kg body weight as a 31.25% solution in water for 3d-HFD+ethanol model and 53% solution in water for 3m-HFD+ethanol model) or isocaloric dextrin-maltose by oral gavage and then euthanized at various time points following this treatment. Mice were also fed a control diet (10% kcal% fat; Cat# D12450, Research Diet) for 3 days or 3 months, followed by gavage of a single dose of ethanol or maltose.
Statistical analysis
Data are shown as the mean ± SD of 6-20 mice per group. Data from two groups were compared using an unpaired t-test. Data from three or more groups were analyzed by one-way ANOVA with Tukey's multiple comparisons test. Differences were considered significant when the P value was less than 0.05.
Other methods are described in the supporting materials
Results
Short-term or long-term HFD-plus-binge ethanol feeding synergistically induces liver injury
To examine the effects of HFD and acute ethanol binge on liver injury, mice were fed a HFD for 3 days (3d, short-term) or 3 months (3m, long-term), followed by gavage of a single dose of maltose or ethanol. As illustrated in Fig. 1A, serum ALT and AST levels were markedly elevated in 3m-HFD+ethanol mice, and also elevated in 3d-HFD+ethanol mice but to a lesser extent. Those levels returned to the basal levels 24 h post ethanol gavage in both groups (Supporting Fig. 1A-1B). In addition, feeding with a HFD alone for 3 months but not 3 days also elevated serum ALT and AST levels. Oral administration of ethanol alone did not elevate the serum ALT or AST levels in chow-fed mice.
Figure 1. Short-term or long-term HFD feeding exacerbates acute ethanol-induced liver injury.
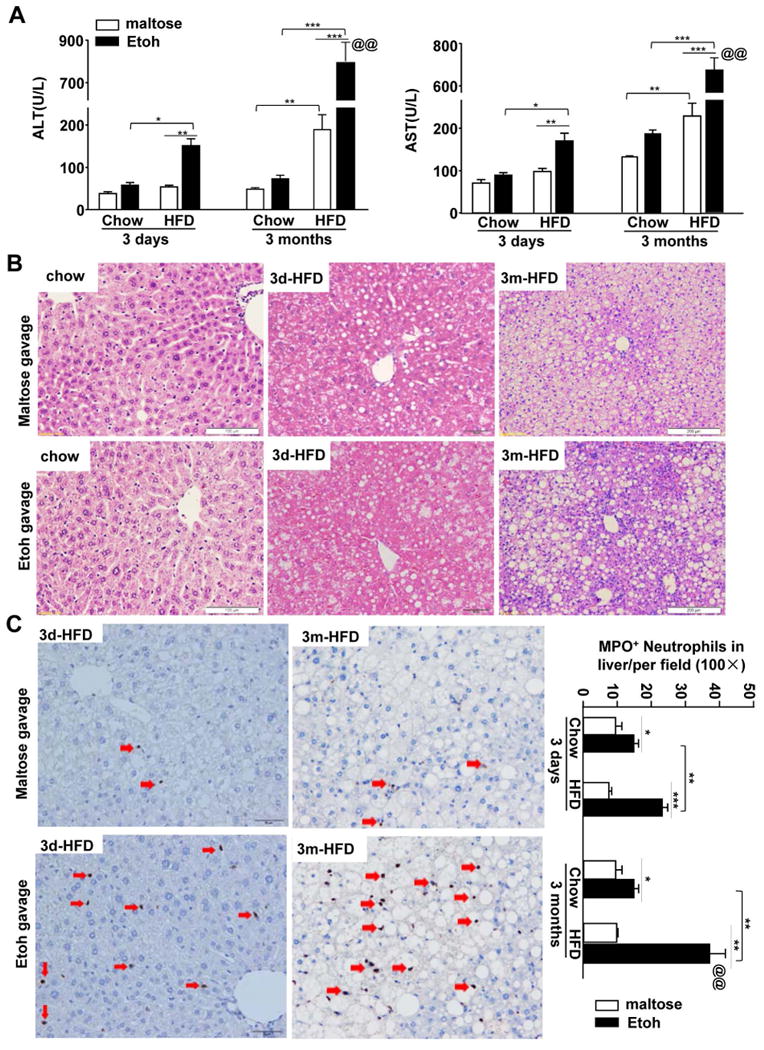
C57BL/6J mice were fed a HFD diet or chow for 3 days or 3 months, and then given maltose or ethanol by oral gavage 9 h prior to euthanization. (A) Serum ALT and AST levels. (B) Representative H&E staining of liver tissue sections. (C) Representative MPO staining is shown. The number of MPO+ cells/per field was determined and is shown on the right. Values represent the mean ± SD (n=8-20). **P<0.1, **P<0.01, ***P<0.001, as indicated. @@P<0.01 in comparison with 3d-HFD+Etoh group.
H&E staining (Fig. 1B) revealed that 3d-HFD feeding caused obvious hepatic steatosis, which is consistent with previous reports that short-term HFD feeding, i.e. for 3-4 days, is sufficient to induce hepatic steatosis and insulin resistance.16-18 Feeding mice a HFD for 3 months caused severe steatosis with obvious fat droplets in more than 90% hepatocytes. Acute ethanol binge did not further elevate steatosis in HFD-fed mice (Fig. 1B, supporting Fig. 1C)
HFD-plus-binge ethanol induces hepatic neutrophil infiltration and neutrophilia
Liver inflammation in HFD-plus-binge ethanol-fed mice was examined by immunostaining of the neutrophil marker myeloperoxidase (MPO) and real-time PCR analysis of inflammatory markers as well as flow cytometry analysis of hepatic leukocytes. As illustrated in Fig. 1C and Supporting Fig. 2A, the number of MPO+ neutrophils in the liver was markedly elevated in the 3d- and 3m-HFD-plus-binge ethanol groups with a higher number in the latter group than in the former group. Such neutrophil infiltration was observed as early as 3h post gavage in 3m-HFD+ethanol mice with peak effects occurring at 6h and 9h post gavage, and significant neutrophil infiltration was still observed 24h post gavage and declined 48h post gavage (Supporting Fig. 2B). Interestingly, MPO+ neutrophils were diffused in the parenchymal regions at 9 h post ethanol gavage in 3d-HFD- or 3m-HFD-fed mice (Fig. 1C). However, clusters of neutrophils were frequently observed around steatotic hepatocytes in 3m-HFD+ethanol group 24h post ethanol gavage (Supporting Fig. 2B).
Furthermore, as shown in Fig. 2A, gavage with a single dose of ethanol increased hepatic expression of Ly6G (a neutrophil marker) in chow- and HFD-fed mice, with higher levels in HFD-fed mice than in chow-fed mice. The hepatic Ly6G mRNA levels were much higher in 3m-HFD+ethanol-fed mice than in 3d-HFD+ethanol-fed mice (55-fold induction in 3m-HFD+ethanol verus 8-fold in 3d-HFD+ethanol). Interestingly, acute ethanol gavage downregulated hepatic expression of F4/80 (a marker of Kupffer cells and macrophages) in 3d-chow, 3d-HFD, and 3m-chow-fed mice compared to maltose gavage in these groups (Fig. 2B). Feeding mice with HFD for 3m increased hepatic expression of F4/80 about 2 folds compared to chow-fed mice, such expression was slightly increased after acute ethanol gavage (Fig. 2B).
Figure 2. HFD-plus-binge ethanol feeding synergistically induces liver neutrophil infiltration and neutrophilia.
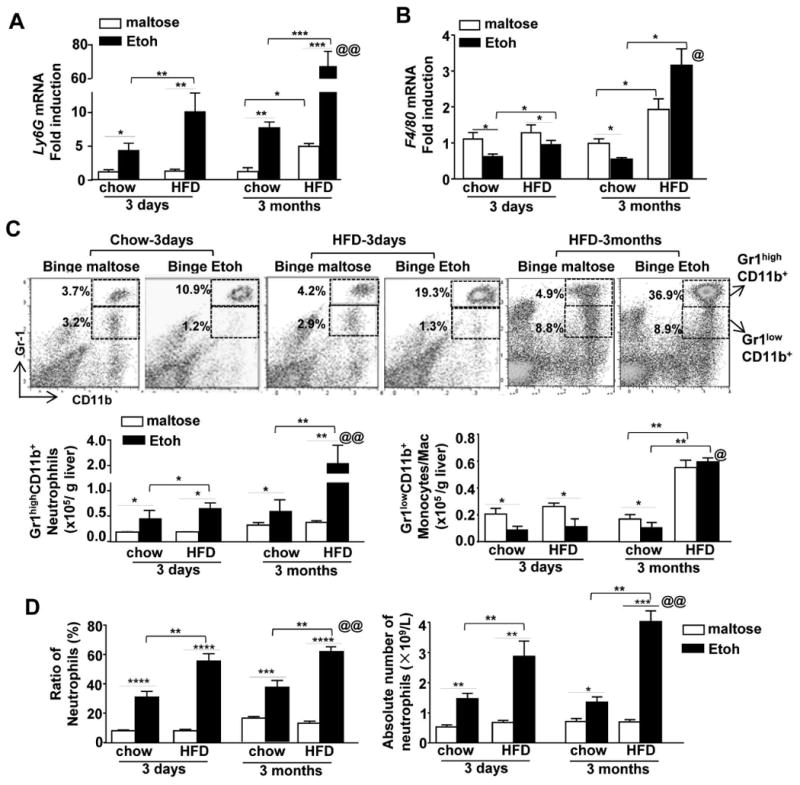
Mice were fed a HFD diet or chow for 3 days or 3 months, and then given maltose or ethanol by oral gavage 9 h prior to euthanization. (A, B) Hepatic expression of Ly6G or F4/80 mRNA levels was determined by real-time quantitative PCR. (C) Liver leukocytes from 3d-or 3m-HFD+ethanol-fed mice were isolated and subjected to flow cytometric analysis. The total numbers of neutrophils (Gr1highCD11b+) and monocytes/macrophages (Gr1lowCD11b+) were determined. (D) The total number and percentage of circulating neutrophils were determined. Values represent the mean ± SD (n=5-15). *P<0.05; **P<0.01; ***P<0.001, as indicated. @P<0.05; @@P<0.01 in comparison with 3d-HFD+Etoh group.
Flow cytometric analysis (Fig. 2C) of liver leukocytes demonstrated that acute ethanol gavage elevated the percentage and the total number of neutrophils (Gr1highCD11b+) in the livers of chow-fed mice and 3d- or 3m-HFD-fed mice with a higher number in the 3m-HFD+ethanol group than in the 3d-HFD-ethanol group. In contrast, acute ethanol binge reduced monocytes/macrophages (Gr1lowCD11b+) in chow or 3d-HFD-fed mice, and such reduction was comparable between these two groups. Interestingly, the number of monocytes/macrophages (Gr1lowCD11b+) was higher in 3m-HFD-fed mice than in chow or 3d-HFD-fed mice, and was not further decreased after acute ethanol binge (Fig. 2C), which is in line with the results in Fig. 2B. In addition, the reduction of monocytes/macrophages was further confirmed by flow cytometric analysis using F4/80 and CD11b markers (Supporting Fig. 2C); while the percentage of hepatic NK and NKT cells was not altered in 3m-HFD-fed mice after acute ethanol binge compared to maltose binge (Supporting Fig. 2D-E).
In addition to liver neutrophils, we also examined circulating neutrophils. As illustrated in Fig. 2D, the percentage and the number of circulating neutrophils were elevated after ethanol gavage in chow group, and were further increased in HFD+ethanol groups with higher levels in 3m-HFD+ethanol mice than in 3d-HFD+ethanol mice (Fig. 2D).
HFD-plus-binge ethanol synergistically upregulates hepatic and serum levels of CXCL1
To explore the mechanisms by which HFD+ethanol induces hepatic neutrophil accumulation, we examined hepatic expression of pro-inflammatory mediators and chemokines that promote hepatic neutrophil infiltration. As indicated in Supporting Fig. 3, acute ethanol gavage did not alter hepatic expression of several pro-inflammatory cytokines in 3d-HFD+ethanol mice, while hepatic expression of several cytokines was elevated in 3m-HFD-fed mice after acute ethanol gavage. Furthermore, as illustrated in Fig. 3, in the 3d-HFD group, acute ethanol binge upregulated hepatic expression of Cxcl1 mRNA by 20-fold, while other chemokines and adhesion molecules were either upregulated to a much lesser extent or not altered. In contrast, in the 3m-HFD group, hepatic expression of many chemokines and adhesion molecules was markedly elevated after acute ethanol binge, including approximately 30-fold induction of Cxcl1. Hepatic expression of several chemokine receptors was not upregulated and some of them (eg. Cxcr5 and Cxcr6) were even downregulated after acute ethanol binge in both 3d- and 3m-HFD-fed mice except Ccr1. Hepatic expression of Ccr1 was markedly upregulated in 3m-HFD+ethanol mice compared with 3m-HFD+maltose group (Fig. 3B).
Figure 3. HFD-plus-binge ethanol synergistically increases hepatic Cxcl1 mRNA and serum CXCL1 levels.
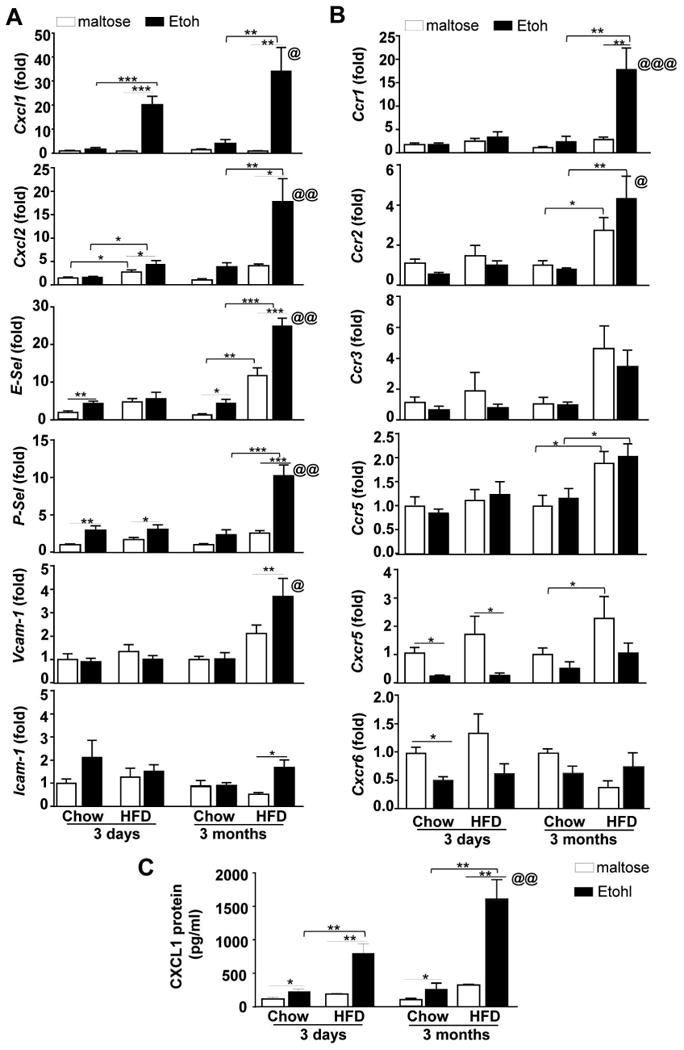
(A, B) Mice were fed chow or a HFD for 3 days or 3 months, followed by gavage with a single dose of maltose or ethanol. Mice were euthanatized 9 h post-gavage. (A, B) Liver tissues were collected, and total RNA was isolated and subjected to real-time PCR analysis. (B) Sera were collected to measure CXCL1 levels with an ELISA kit. Values represent the mean ± SD (n=5-10). *P<0.05; **P<0.01; ***P<0.001 as indicated. @P<0.05, @@P<0.01, @@@P<0.01 in comparison with 3d-HFD+Etoh group.
Becuase Cxcl1 mRNA showed the highest fold upregulation among other chemokines in both 3d- and 3m-HFD+ethanol-fed mice, we focused on the CXCL1 in the current paper, and further measured serum CXCL1 protein levels. As illustrated in Fig. 3C, the 3d- and 3m-HFD+ethanol treatment resulted in synergistically elevated serum CXCL1 levels to approximately 800 pg/ml and 1500 pg/ml, respectively, while 3d- or 3m-HFD+maltose feeding only slightly elevated the serum levels of CXCL1 protein.
TLR4 is not involved in the 3d- or 3m-HFD+ethanol-mediated elevation of serum CXCL1, hepatic neutrophil infiltration and injury
LPS, through binding to TLR4, has been shown to elevate serum CXCL1 levels during endotoxemic liver injury in mice.26 To determine whether the LPS/TLR4 axis was involved in the 3d- or 3m-HFD+ethanol-induced CXCL1 elevation and liver injury, WT and Tlr4KO mice were subjected to 3d- or 3m-HFD+ethanol feeding. As illustrated in supporting Fig. 4A and 4B, serum ALT and AST level, and serum CXCL1 protein levels were comparable between 3d- or 3m-HFD+ethanol-fed WT and Tlr4KO mice. As expected, hepatic Tlr4 mRNA was not detected in Tlr4KO mice, while hepatic expression of Ifng, IL-1b, IL-6, Tnfa, Ly6G was compabable between 3m-HFD+ethanol-fed WT and Tlr4KO mice (Supporting Figs. 4C, D). There was also no difference in the number of MPO+ neutrophils, hepatic triglyceride contents, hepatic and serum FFA levels between these two groups (Supporting Figs. 4D-G). Collectively, TLR4 does not contribute to 3d- or 3m-HFD+ethanol-induced elevation of CXCL1, liver neutrophil infiltration and injury.
Elevation of Cxcl1 expression in hepatocytes, and to a lesser extent in HSCs and sinusoidal ECs, following HFD+ethanol feeding
To identify which organ is mainly responsible for the elevated serum CXCL1 levels, we measured Cxcl1 mRNA in different organs. As illustrated in Figure 4A, acute ethanol gavage elevated Cxcl1 mRNA by 30-fold and 5-fold in the liver and epididymal fat, respectively, in 3d-HFD-fed mice, while its expression was not elevated in the heart, spleen, kidney and subcutaneous fat.
Figure 4. Upregulation of Cxcl1 mRNA expression in the liver and liver cells from HFD-plus-binge ethanol-fed mice.
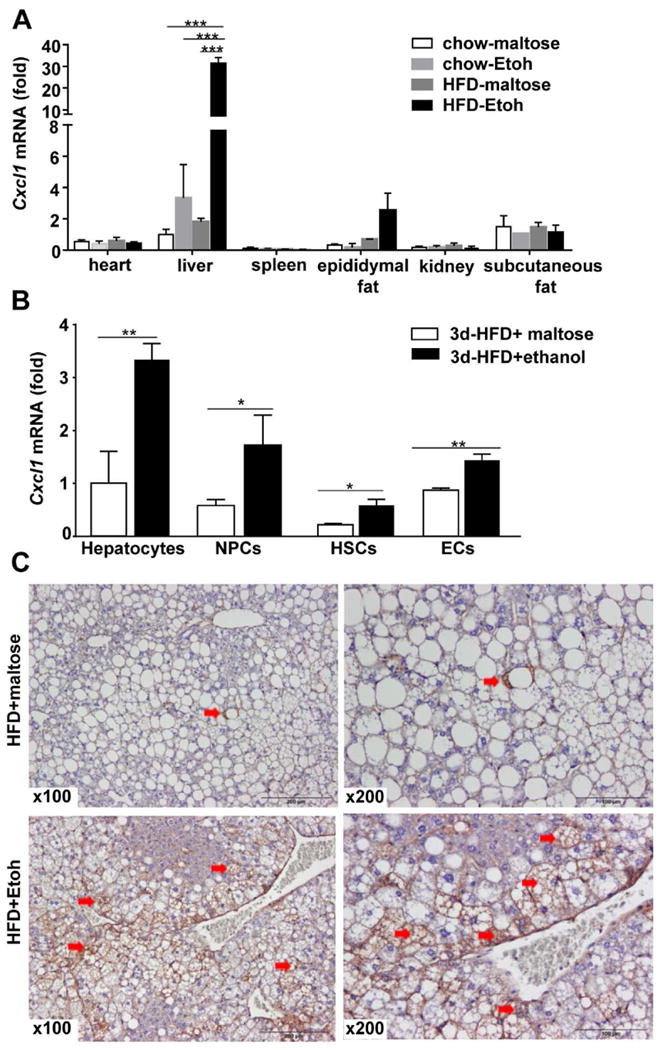
(A, B), Mice were fed a HFD for 3 days followed by a single dose of ethanol or maltose; 9 h after gavage, tissues and cells were collected. Various organs were collected and subjected to real-time PCR analysis of Cxcl1 mRNA (panel A). Hepatocytes, NPCs, HSCs and ECs were isolated and subjected to real-time PCR analysis to determine the levels ofCxcl1 mRNA (panel B). (C) Mice were fed a HFD for 3 months, followed by a single dose of ethanol or maltose; 9 h after gavage, liver tissues were collected for immunohistochemistry analyses of CXCL1 protein. Values represent the mean ± SD (n=5-10). *P<0.05; **P<0.01; ***P<0.001 as indicated. Arrows in panel C indicate positive CXCL1 staining in hepatocytes.
It has been reported that hepatocytes and liver non parenchymal cells (NPCs) can produce CXCL1 after stimulation with a variety of inflammatory mediators.26-29 To further understand the mechanisms by which 3d-HFD-ethanol feeding upregulates hepatic Cxcl1 mRNA expression, we investigated which cell types express Cxcl1 in the liver. We first isolated hepatocytes and NPCs and examined Cxcl1 mRNA in these cells. As illustrated in Fig. 4B, Cxcl1 mRNA levels were upregulated in both hepatocytes and NPCs from 3d-HFD+ethanol-fed mice compared to 3d-HFD+maltose-fed mice. Next, hepatic stellate cells (HSCs) and sinusoidal endothelial cells (ECs) were isolated from the total NPCs of mice in 3d-HFD+maltose or 3d-HFD+ethanol groups, and Cxcl1 mRNA was measured. As illustrated in Fig. 4B, the levels of Cxcl1 mRNA were increased in HSCs and sinusoidal ECs by approximately 2-fold. Collectively, Cxcl1 mRNA expression was increased in hepatocytes, HSCs and ECs from 3d-HFD+ethanol-fed mice compared to 3d-HFD+maltose-fed mice, with the highest induction observed in hepatocytes.
Immunostaining analysis revealed that a few steatotic hepatocytes expressed CXCL1 in 3m-HFD+maltose mice, whereas a much greater number of hepatocytes strongly expressed CXCL1 in 3m-HFD+ethanol mice (Fig. 4C).
HFD+ethanol feeding elevates hepatic or serum FFAs, which upregulate Cxcl1 expression in hepatocytes
Previous studies revealed that treatment with PA induced the production of IL-8 (CXCL8) in human hepatocytes via NF-κB- and JNK-dependent mechanisms.29 This notion led us to hypothesize that PA may contribute to the elevated serum CXCL1 levels observed after HFD+ethanol feeding. To test this hypothesis, we first measured serum FFA levels. As illustrated in Fig. 5A, serum FFA levels were significantly elevated in 3m-HFD+ethanol-fed mice but not in 3d-HFD+ethanol-fed mice compared to other groups. Next, we measured hepatic FFA levels, and the results revealed that hepatic FFA levels were markedly elevated in 3d-HFD+ethanol mice compared to HFD+maltose mice (Fig. 5B). Interestingly, hepatic FFA levels were higher in 3m-HFD-fed mice than in 3m-chow-fed mice. Acute ethanol binge did no further elevated hepatic FFA levels (Fig. 5B).
Figure 5. HFD-plus-binge ethanol feeding elevates hepatic or serum levels of FFAs, and FFAs upregulate Cxcl1 mRNA expression in mouse hepatocytes.
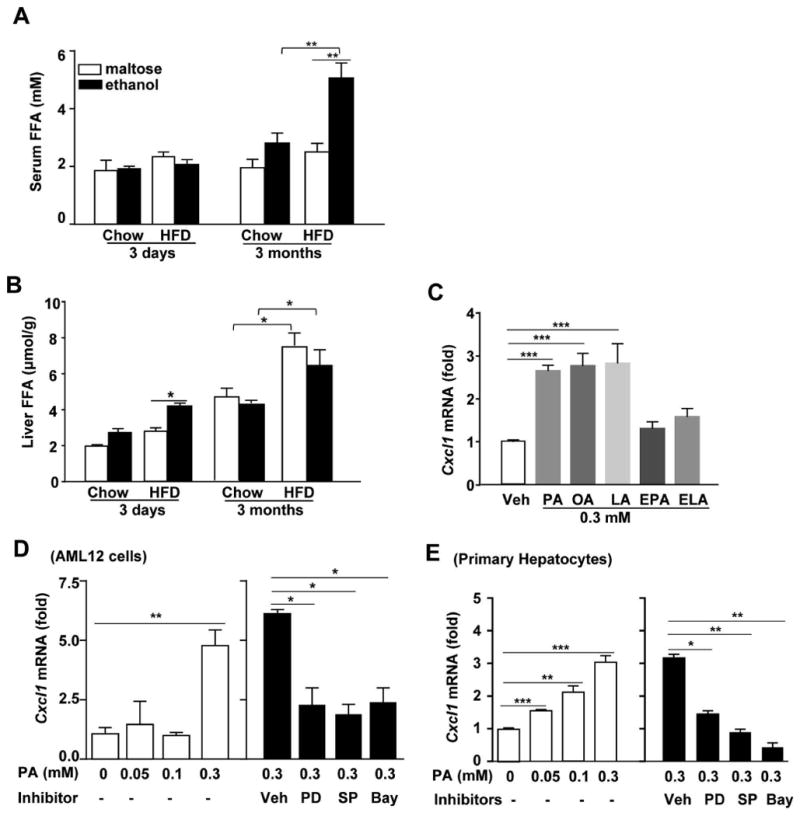
(A, B) Mice were fed a HFD for 3 days or 3 months, followed by gavage of a single dose of ethanol or maltose. Serum and hepatic FFAs levels were determined 9 h post-gavage. (C) Primary mouse hepatocytes were stimulated with 0.3 mM palmitic acid (PA), oleic acid (OA), linoleic acid (LA), ethyl palmitic acid (EPA) or ethyl linoleic (ELA) for 3 h, and Cxcl1 mRNA expression was determined by real-time PCR. (D, E) AML12 cells and primary mouse hepatocytes were treated with various doses of PA with or without pre-treatment with an ERK1/2 inhibitor (PD98059, 100μM), JNK inhibitor (SP600125, 50μM) or NF-κB inhibitor (Bay-7085, 10μM). Cxcl1 mRNA expression was determined by real-time PCR. Values represent the mean ± SD (n=4-6). *P<0.05; **P<0.01; ***P<0.001 as indicated.
In vitro treatment with PA, OA or LA but not EPA or ELA upregulated the expression of Cxcl1 mRNA in primary mouse hepatocytes (Fig. 5C). Fig. 5D-E shows that PA treatment upregulated Cxcl1 mRNA expression in the mouse hepatocyte cell line AML12 or primary mouse hepatocytes. Such Cxcl1 mRNA induction was diminished by the treatment with ERK1/2, JNK and NF-κB specific inhibitors. TLR4 was shown to contribute FFA-mediated effects;30 but these studies were not reproduced by others.31 Here we demonstrated that treatment with PA induced comparable elevation of Cxcl1 mRNA expression in wild-type and TLR4KO hepatocytes (Supporting Fig. 5), suggesting PA-mediated upregulation of Cxcl1 mRNA is TLR4-independent.
Blockade of CXCL1 ameliorates HFD+ethanol-induced neutrophilia, hepatic neutrophil infiltration and liver injury
To define the functions of CXCL1 in HFD+ethanol-induced hepatic neutrophil infiltration and injury, HFD+ethanol-fed mice were treated with an anti-CXCL1 antibody or a control IgG antibody. As illustrated in Fig. 6A, compared to IgG treatment, administration of the anti-CXCL1 antibody markedly reduced the percentage and the total number of circulating neutrophils. In contrast, treatment with the anti-CXCL1 antibody did not affect the total number of circulating monocytes and lymphocytes in these HFD+ethanol-fed mice (Supporting Fig. 6). Real-time PCR and immunostaining analyses demonstrated that hepatic neutrophil infiltration, as measured by hepatic gene expression of the neutrophil marker Ly6G and the total number of MPO+ neutrophils, was markedly attenuated in anti-CXCL1 antibody-treated mice compared to IgG-treated mice (Fig. 6B, and Supporting Fig. 7). Finally, serum ALT and AST levels were significantly lower after anti-CXCL1 treatment compared to IgG treatment in 3d- or 3m-HFD-ethanol-fed mice (Figs. 6C), whereas hepatic triglyceride levels were not altered by anti-CXCL1 antibody treatment (Supporting Fig. 8).
Figure 6. Blockade of CXCL1 ameliorates HFD-plus-binge ethanol-induced neutrophilia, hepatic neutrophil infiltration, and liver injury.
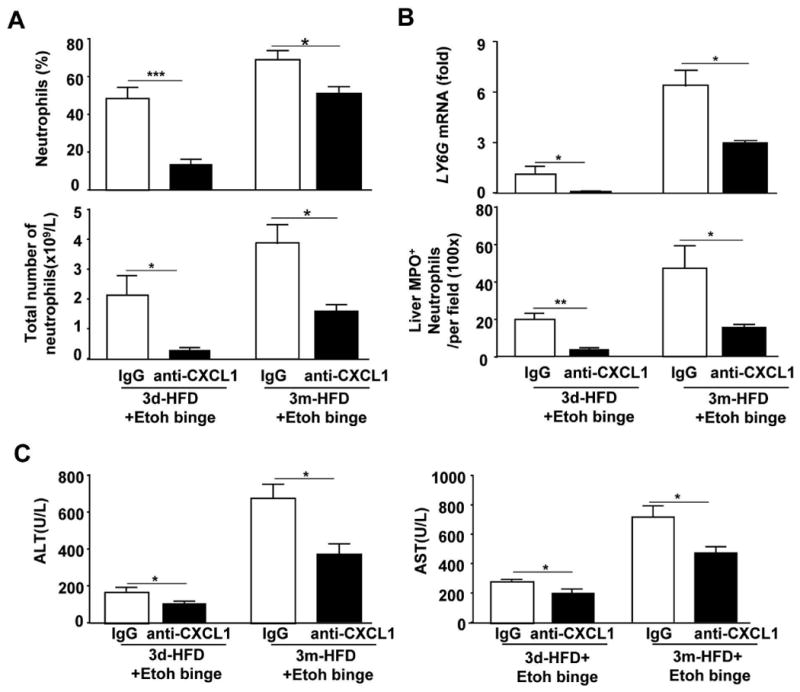
Mice were fed a HFD for 3 days or 3 months, followed by gavage with a single dose of ethanol. An anti-CXCL1 antibody or IgG antibody was administered to the mice 15 min before gavage. Mice were euthanatized 9 h post-gavage. (A) Blood leukocytes were collected, analyzed and counted. (B) Liver tissues were collected for real-time PCR analysis of Ly6G mRNA or subjected to immunostaining with an anti-MPO antibody, and the number of MPO+ cells/per field was determined. (C) Serum levels of ALT and AST were measured. Values represent the mean ± SD (n=5-8). *P<0.05, ***P<0.001 as indicated.
Disruption of the Cxcl1 gene reduces HFD+ethanol-induced liver inflammation and injury, while overexpression of Cxcl1 exacerbates steatohepatitis in 3m-HFD-fed mice
The functions of CXCL1 in HFD+ethanol induced liver inflammation and injury were further determined by using Cxcl1KO mice. As illustrated in Fig. 7A, the total number and the percentage of neutrophils in the blood, and the number of MPO+ neutrophils in the liver were markedly lower in Cxcl1KO mice compared to those in WT mice post 3d- or 3m-HFD+ethanol feeding. In line with this, Cxcl1KO mice had lower levels of serum ALT and AST compared to WT mice (Fig. 7B).
Figure 7. Genetic deletion of the Cxcl1 gene ameliorates HFD-plus-binge ethanol-induced hepatic neutrophil infiltration and injury, while overexpression of the Cxcl1 exacerbates liver inflammation and injury.
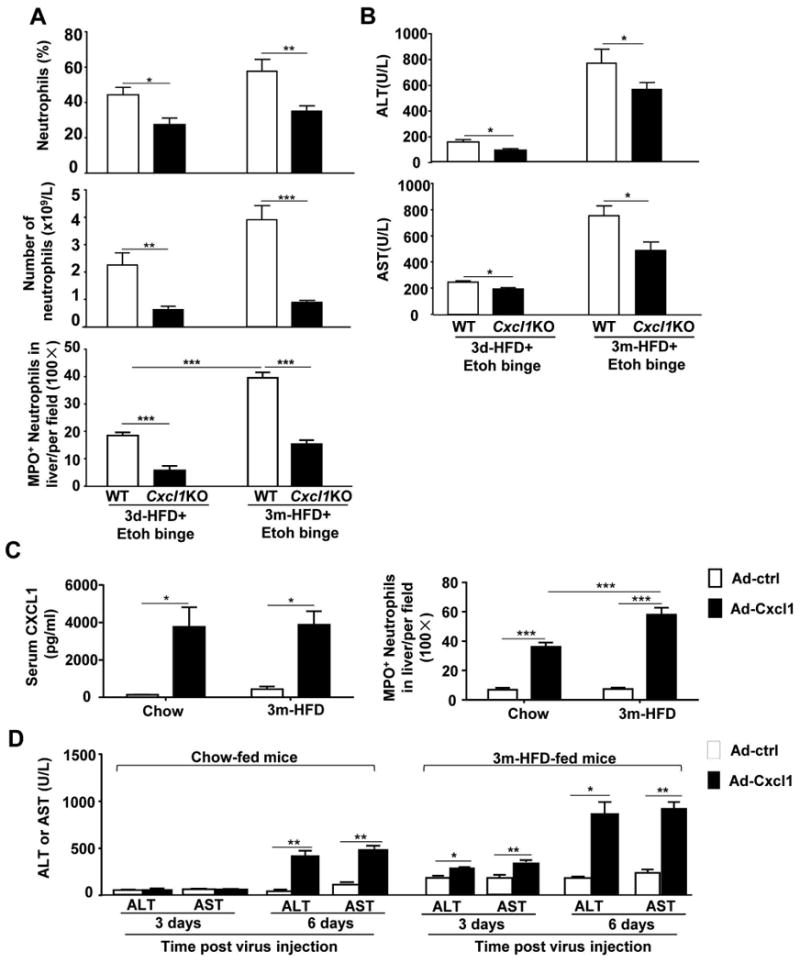
(A, B) Mice were fed a HFD for 3 days or 3 months, followed by gavage with a single dose of ethanol. Mice were euthanatized 9 h post-gavage. Circulating neutrophils and hepatic neutrophils were determined and are shown in panel A. Serum levels of ALT and AST are shown in panel B. (C, D) Chow- and 3m-HFD-fed mice were injected with Ad-control or Ad-Cxcl1 viruses, serum CXCl1 protein levels and hepatic neutrophil infiltration were determined 6 days post virus injection (panel C); serum ALT and AST levels were determined (panel D). Values represent the mean ± SD (n=6-10). *P<0.05; **P<0.01; ***P<0.001 as indicated.
To further determine whether acute ethanol binge-induced elevation of CXCL1 contributes to liver inflammation and injury in HFD-fed mice, a gain-of-function approach was used via the administration of Ad-Cxcl1 into HFD-fed mice to induce elevation of CXCL1. As illustrated in Fig. 7C, administration of Ad-Cxcl1 elevated serum CXCL1 protein levels to approximately 4000 pg/ml in chow or HFD-fed mice. The number of MPO+ neutrophils was markedly elevated in Ad-Cxcl1-treated mice with much higher levels in HFD-fed mice than in chow-fed mice (Fig. 7C, supporting Fig. 9). In addition, serum levels of ALT and AST were elevated in Ad-Cxcl1-treated chow-fed and HFD-fed mice compared to Ad-control virus-treated mice, and those levels were much higher in HFD-fed mice than in chow-fed mice (Fig. 7D).
CXCL1 has no direct cytotoxicity on primary mouse hepatocytes in vitro
A previous paper suggests that CXCL1 may directly induce hepatocyte death without inducing neutrophil recruitment in a model of CCl4-induced liver injury, but there was no direct evidence.32 To confirm whether CXCL1 had the direct hepatotoxicity, we examined Cxcr2 (a receptor for CXCL1) mRNA expression and the cytotoxicity of CXCL1 in primary mouse hepatocytes. As shown in Fig. 8A, compared to the spleen, the liver expressed very low levels of Cxcr2 mRNA. The expression of Cxcr2 mRNA was upregulated about 2-fold in the livers from HFD+ethanol-fed mice. Furthermore, expression of Cxcr2 mRNA in hepatocytes was 40- to 50-fold lower than that in splenocytes and neutrophils (Fig. 8B).
Figure 8. Hepatocytes express low levels of Cxcr2 mRNA, and CXCL1 does not induce hepatocyte death in vitro.
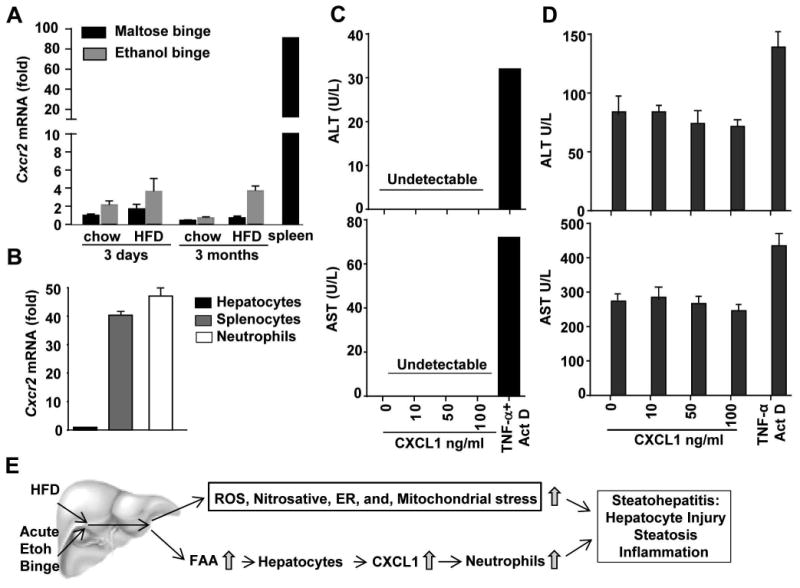
(A) Mice were fed chow or a HFD for 3 days or 3 months, followed by gavage with a single dose of maltose or ethanol. Mice were euthanatized 9 h post-gavage. Liver tissues were collected for real-time PCR analysis of Cxcr2 mRNA. Spleen tissues from normal C57BL/6 mice were used as positive controls for Cxcr2 mRNA expression. (B) Hepatocytes, splenocytes, and blood neutrophils were isolated from C57BL/6 mice, and subjected to real-time PCR for Cxcr2 mRNA. (C, D) Hepatocytes were isolated from chow-fed (panel C) or 3m-HFD-fed mice (panel D), and were incubated with various concentrations of CXCL1 or TNF-α+Actinomycin D. Supernatant was collected 24 h post incubation and used for measurement of ALT and AST. (E) The mechanisms underlying the synergistic effect of HFD and acute ethanol binge on steatohepatitis.
Incubation of primary mouse hepatocytes with CXCL1 protein even at high concentrations (up to 100 ng/ml) did not induce cell death detected by TUNEL staining (Supporting Fig. 10) and ALT or AST release into the cell culture supernatant (Fig. 8C), whereas positive controls from incubation with TNF-α plus actinomycin D demonstrated marked hepatocyte death (Fig. 8C, and Supporting Fig. 10). In addition, incubation of steatotic hepatocytes isolated from HFD-fed mice with CXCL1 protein did not enhance ALT or AST release into the cell culture supernatant (Fig. 8D).
Discussion
Alcohol and HFD consumption are known to synergistically induce nitrosative, ER, and mitochondrial stress and an upregulation of hepatic TLR4, which are important mechanisms contributing to the synergistic effect of alcohol and HFD on the induction of steatohepatitis.10, 13 In the present study, we have identified another important mechanism by which HFD-plus-binge ethanol synergistically exacerbates acute steatohepatitis via the induction of CXCL1 and subsequent hepatic neutrophil infiltration (Fig. 8E).
The most striking finding from this study was that feeding mice a HFD for as little as 3 days markedly exacerbated the acute ethanol-induced hepatic neutrophil infiltration and liver injury. This may not be very surprising because short-term HFD feeding, i.e. for 3-4 days, is sufficient to induce hepatic steatosis and hepatic insulin resistance.16-18 Although feeding mice a HFD alone for 3 days induced steatosis, hepatic neutrophil infiltration and serum ALT levels were not elevated. Acute gavage with a single dose of ethanol resulted in slight elevation of serum ALT and AST, increased hepatic neutrophil infiltration and steatosis in chow-fed mice, and all of these effects (except steatosis) were markedly aggravated in 3d-HFD-fed mice. Furthermore, feeding mice a HFD for 3 months plus a single binge of ethanol caused much more severe steatohepatitis than 3d-HFD+binge ethanol feeding.
One of the most fascinating features in this HFD+binge ethanol feeding model is dramatic hepatic neutrophil infiltration. Neutrophil infiltration has been implicated in promoting liver damage in various types of liver diseases 33 including drug-induced liver injury 34-37 and alcoholic liver injury.38, 39 The significant increase in hepatic neutrophil infiltration likely also participates in HFD+ethanol-induced liver damage because blockade of CXCL1 by an anti-CXCL1 neutralizing antibody or disruption of the Cxcl1 gene prevented hepatic neutrophil infiltration and hepatocellular damage. In contrast to marked hepatic neutrophil infiltration, hepatic macrophages were reduced in 3d-HFD or slightly elevated in 3m-HFD fed mice post ethanol binge. It has been shown that short-term ethanol feeding or acute ethanol gavage induces apoptosis of Kupffer cells or macrophages in vivo,40, 41 and that in vitro incubation with ethanol directly induced apoptosis of mouse peritoneal macrophages or macrophage cell line J774.41 Thus, the reduction of hepatic macrophages after acute ethanol binge in 3d-HFD-fed mice was probably due to the pro-apoptotic effects of ethanol on Kupffer cells/macrophages. These apoptotic Kupffer cells/macrophages likely produce damage-associated molecular pattern molecules that subsequently contribute to neutrophil activation in this HFD-plus-binge ethanol model.
Another important finding from this study is the identification of CXCL1 as an important chemokine that was highly elevated in the liver and serum from HFD+ethanol-fed mice. Among several chemokines and adhesive molecules that we examined, hepatic Cxcl1 mRNA was upregulated by more than 20-fold and 35-fold in the 3d- and 3m-HFD-plus-binge ethanol groups, respectively, compared to their corresponding HFD+maltose groups. Serum CXCL1 protein levels were also significantly elevated in 3d- and 3m-HFD+ethanol-fed mice, reaching approximately 800 pg/ml and 1500 pg/ml, respectively. Moreover, the fact that HFD+ethanol feeding highly upregulated Cxcl1 mRNA in the liver but not in the other organs suggests that the liver is the major source of the increased serum CXCL1 levels in this model. The next obvious question was which cell type in the liver was mainly responsible for CXCL1 production. Previous studies have reported that hepatocytes and NPCs can produce CXCL1.27, 28 Here, we demonstrated that Cxcl1 mRNA expression was upregulated in hepatocytes, HSCs and ECs after 3d-HFD+ethanol binge feeding, with the highest induction observed in hepatocytes. Given that hepatocytes constitute 80% of the liver volume and 60% of the total liver cells, we concluded that hepatocytes are largely responsible for the upregulation of Cxcl1 in the liver after HFD-plus-binge ethanol feeding; NPCs also contribute to this CXCL1 production but to a lesser extent.
The expression of CXCL1 is upregulated by LPS 26 and the LPS receptor TLR4 has been shown to promote HFD-induced nonalcoholic steatohepatitis.42, 43 However, we demonstrated that genetic deletion of the LPS receptor TLR4 had no effect on 3d- or 3m-HFD-plus-binge ethanol-induced hepatic CXCL1 expression, neutrophilia, and liver injury, suggesting that TLR4 does not contribute to HFD+ethanol binge-induced elevation of CXCL1, acute liver injury and inflammation. Why TLR4 contributes to HFD-induced steatohepatitis but is not involved in HFD-plus-binge ethanol binge-induced liver injury is not clear. One reason may be that 3m-HFD+ethanol feeding caused much greater elevation of serum CXCL1 than 3m-HFD alone does (1500 pg/ml circulating CXCL1 protein in 3m-HFD+ethanol vs 300 pg/ml in 3m-HFD alone), and this high elevation of CXCL1 in HFD+ethanol is independent TLR4. A recent study reported that serum CXCL1 levels were elevated in chronic-plus-binge ethanol feeding model, but those levels were much lower than in HFD+ethanol-fed mice (200 pg/ml in chronic-plus-binge ethanol model 44 versus 600-1500 pg/ml in HFD+ethanol model). Both TLR2 and TLR9 contribute to chronic ethanol-plus-binge ethanol-mediated elevation of serum CXCL1 protein.44 However, we found that HFD+ethanol treatment induced similar levels of CXCL1 elevation and liver injury between WT and TLR9KO mice (unpublished data), suggesting that the TLR9 pathway does not contribute to HFD+ethanol-induced elevation of CXCL1. Instead, we identified a different mechanism underlying hepatic CXCL1 upregulation in the HFD+ethanol model. First, acute ethanol binge markedly elevated hepatic and serum FFA levels in 3d- and 3m-HFD-fed mice, respectively. Second, in vitro experiments demonstrated that several FFAs were able to directly stimulate Cxcl1 mRNA expression in hepatocytes, which was dependent on proper ERK, JNK and NF-κB activation. Collectively, our data suggest that acute ethanol binge induces elevation of hepatic or serum FFAs in HFD-fed mice, which directly upregulate hepatic CXCL1 production and induce acute liver injury in those mice.
Roh et al. recently reported that a selective CXCR2 blockade inhibits hepatic neutrophil infiltration and injury in chronic-plus-binge ethanol feeding model.44 Because CXCR2 is a receptor for CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL7, and CXCL8,45 therefore, all of those chemokines may contribute to chronic-plus-binge ethanol-induced liver inflammation and injury. In the current study, by using loss-of-function and gain-of-function approaches, we clearly demonstrated that CXCL1 plays an important role in promoting HFD+ethanol-induced acute steatohepatitis. First, HFD-plus-binge ethanol feeding markedly upregulated hepatic Cxcl1 mRNA expression and elevated serum CXCL1 levels. Second, blockade of CXCL1 with a neutralizing antibody or genetic deletion ameliorated HFD+ethanol-induced neutrophilia, hepatic neutrophil infiltration and injury but did not affect hepatic steatosis. Previous studies have implicated CXCL1 in promoting neutrophil infiltration and liver injury induced by D-galactosamine/endotoxin (D-Gal/ET),26 ischemia/reperfusion46 and focal brain damage.47 However, Dorman et al. 48 reported that treatment with anti-KC and anti-MIP-2 antibodies did not attenuate D-Gal/ET-induced liver injury in C3Heb/FeJ mice, and no differences in liver injury were observed between wild-type BALB/cJ and CXCR2-/- mice. The reason behind the discrepancy between these experiments is not clear but may be related to the different backgrounds of the mice, liver injury models and the severity of liver injury used in these studies. Furthermore, overexpression of CINC (a rat homolog of CXCL1) via adenovirus-mediated gene delivery markedly induced neutrophilia and liver injury in rats.49 In the current paper, we demonstrated that administration of adenovirus-Cxcl1 resulted in hepatic Cxcl1 overexpression and elevated hepatic neutrophil infiltration and injury in HFD-fed mice, providing direct evidence that CXCL1 promotes steatohepatitis. All of these data support the notion that CXCL1 promotes hepatic neutrophil infiltration that subsequently contributes to liver injury. Surprisingly, Stefanovic et al. reported that treatment of mice with CXCL1 protein exacerbated liver damage without inducing hepatic neutrophil infiltration in a model of CCl4-induced liver injury, 32 Thus, authors concluded that CXCL1 directly induced hepatocyte death. However, Stefanovic et al. did not test whether CXCL1 had direct cytotoxicity against hepatocytes in vitro. In the present study, we observed that treatment with CXCL1 protein did not induce hepatocyte death in vitro, which is consistent with extremely low levels of CXCR2 expression on hepatocytes. Therefore, in this HFD-plus-binge ethanol model, the hepatotoxicity of CXCL1 is mediated via the induction of hepatic neutrophil infiltration and is not due to the direct cytotoxicity on hepatocytes.
Clinical implications of this study
The finding that a single oral binge exposure to ethanol can trigger acute liver injury in the presence of short-term or long-term HFD feeding highlights the risk of even episodic binge drinking in obese/overweight individuals. Although this model does not represent a model of chronic ASH, it can trigger acute liver injury. The obvious clinical implication of this finding is that obese individuals should avoid binge drinking large amounts of alcohol, as this would likely cause significant acute liver damage. These findings should also promote clinical studies on the effect of the interaction between obesity and binge drinking on neutrophilia, liver inflammation and hepatic injury. For example, the hepatic expression of CXC chemokines (e.g., CXCL1/Gro-α, IL-8, CXCL5, and CXCL6) was found to be upregulated and correlated with neutrophil infiltration and the severity of disease in patients with alcoholic hepatitis.23 Hepatic expression of CXCL1, IL-8 and their receptors is also upregulated in the liver of obese patients with NASH.50 Thus, it would be interesting to examine whether obesity, as measured by BMI, and binge alcohol drinking synergistically promote increased hepatic levels of CXC chemokines, neutrophilia, and the severity of liver disease in these patients. Another important feature of the present model is the rapidity of the development of neutrophil-mediated steatohepatitis, which would make it uniquely suitable for future mechanistic studies.
Supplementary Material
Acknowledgments
This work was supported by the intramural program of NIAAA, NIH. Dr. Binxia Chang was supported partially by a fund from Beijing 302 Hospital.
Abbreviations
- ALT
alanine aminotransferase
- CINC-1
cytokine-induced neutrophil chemoattractant-1
- CXCL1
(C-X-C motif) ligand 1
- d
day(s)
- ECs
endothelial cells
- FFAs
free fatty acids
- h
hour(s)
- HFD-binge
high-fat diet feeding plus binge ethanol
- HSC
hepatic stellate cell
- KC
keratinocyte-derived chemokine
- LA
linoleic acid
- m
months
- NPCs
non parenchymal cells
- MPO
Myeloperoxidase
- OA
oleic acid
- PA
palmitic acid
- TLR4
toll-like receptor 4
Footnotes
No conflicts of interest exist for any of the authors.
References
- 1.Naveau S, Giraud V, Borotto E, et al. Excess weight risk factor for alcoholic liver disease. Hepatology. 1997;25:108–11. doi: 10.1002/hep.510250120. [DOI] [PubMed] [Google Scholar]
- 2.Iturriaga H, Bunout D, Hirsch S, et al. Overweight as a risk factor or a predictive sign of histological liver damage in alcoholics. Am J Clin Nutr. 1988;47:235–8. doi: 10.1093/ajcn/47.2.235. [DOI] [PubMed] [Google Scholar]
- 3.Naveau S, Dobrin AS, Balian A, et al. Body fat distribution and risk factors for fibrosis in patients with alcoholic liver disease. Alcohol Clin Exp Res. 2013;37:332–8. doi: 10.1111/j.1530-0277.2012.01927.x. [DOI] [PubMed] [Google Scholar]
- 4.Lu XL, Luo JY, Tao M, et al. Risk factors for alcoholic liver disease in China. World J Gastroenterol. 2004;10:2423–6. doi: 10.3748/wjg.v10.i16.2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruhl CE, Everhart JE. Joint effects of body weight and alcohol on elevated serum alanine aminotransferase in the United States population. Clin Gastroenterol Hepatol. 2005;3:1260–8. doi: 10.1016/s1542-3565(05)00743-3. [DOI] [PubMed] [Google Scholar]
- 6.Alatalo PI, Koivisto HM, Hietala JP, et al. Effect of moderate alcohol consumption on liver enzymes increases with increasing body mass index. Am J Clin Nutr. 2008;88:1097–103. doi: 10.1093/ajcn/88.4.1097. [DOI] [PubMed] [Google Scholar]
- 7.Shen Z, Li Y, Yu C, et al. A cohort study of the effect of alcohol consumption and obesity on serum liver enzyme levels. Eur J Gastroenterol Hepatol. 2010;22:820–5. doi: 10.1097/MEG.0b013e3283328b86. [DOI] [PubMed] [Google Scholar]
- 8.Loomba R, Bettencourt R, Barrett-Connor E. Synergistic association between alcohol intake and body mass index with serum alanine and aspartate aminotransferase levels in older adults: the Rancho Bernardo Study. Aliment Pharmacol Ther. 2009;30:1137–49. doi: 10.1111/j.1365-2036.2009.04141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hart CL, Morrison DS, Batty GD, et al. Effect of body mass index and alcohol consumption on liver disease: analysis of data from two prospective cohort studies. BMJ. 2010;340:c1240. doi: 10.1136/bmj.c1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gabele E, Dostert K, Dorn C, et al. A new model of interactive effects of alcohol and high-fat diet on hepatic fibrosis. Alcohol Clin Exp Res. 2011;35:1361–7. doi: 10.1111/j.1530-0277.2011.01472.x. [DOI] [PubMed] [Google Scholar]
- 11.Demori I, Voci A, Fugassa E, et al. Combined effects of high-fat diet and ethanol induce oxidative stress in rat liver. Alcohol. 2006;40:185–91. doi: 10.1016/j.alcohol.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 12.Tsukamoto H, Towner SJ, Ciofalo LM, et al. Ethanol-induced liver fibrosis in rats fed high fat diet. Hepatology. 1986;6:814–22. doi: 10.1002/hep.1840060503. [DOI] [PubMed] [Google Scholar]
- 13.Xu J, Lai KK, Verlinsky A, et al. Synergistic steatohepatitis by moderate obesity and alcohol in mice despite increased adiponectin and p-AMPK. J Hepatol. 2011;55:673–82. doi: 10.1016/j.jhep.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robin MA, Demeilliers C, Sutton A, et al. Alcohol increases tumor necrosis factor alpha and decreases nuclear factor-kappab to activate hepatic apoptosis in genetically obese mice. Hepatology. 2005;42:1280–90. doi: 10.1002/hep.20949. [DOI] [PubMed] [Google Scholar]
- 15.Everitt H, Hu M, Ajmo JM, et al. Ethanol administration exacerbates the abnormalities in hepatic lipid oxidation in genetically obese mice. Am J Physiol Gastrointest Liver Physiol. 2013;304:G38–47. doi: 10.1152/ajpgi.00309.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ji Y, Sun S, Xia S, et al. Short term high fat diet challenge promotes alternative macrophage polarization in adipose tissue via natural killer T cells and interleukin-4. J Biol Chem. 2012;287:24378–86. doi: 10.1074/jbc.M112.371807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee YS, Li P, Huh JY, et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes. 2011;60:2474–83. doi: 10.2337/db11-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wiedemann MS, Wueest S, Item F, et al. Adipose tissue inflammation contributes to short-term high-fat diet-induced hepatic insulin resistance. Am J Physiol Endocrinol Metab. 2013;305:E388–95. doi: 10.1152/ajpendo.00179.2013. [DOI] [PubMed] [Google Scholar]
- 19.Wong CH, Heit B, Kubes P. Molecular regulators of leucocyte chemotaxis during inflammation. Cardiovasc Res. 2010;86:183–91. doi: 10.1093/cvr/cvq040. [DOI] [PubMed] [Google Scholar]
- 20.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–75. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 21.Marra F, Tacke F. Roles for Chemokines in Liver Disease. Gastroenterology. 2014;147:577–594. e1. doi: 10.1053/j.gastro.2014.06.043. [DOI] [PubMed] [Google Scholar]
- 22.Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol. 2000;18:217–42. doi: 10.1146/annurev.immunol.18.1.217. [DOI] [PubMed] [Google Scholar]
- 23.Dominguez M, Miquel R, Colmenero J, et al. Hepatic expression of CXC chemokines predicts portal hypertension and survival in patients with alcoholic hepatitis. Gastroenterology. 2009;136:1639–50. doi: 10.1053/j.gastro.2009.01.056. [DOI] [PubMed] [Google Scholar]
- 24.Nischalke HD, Berger C, Lutz P, et al. Influence of the CXCL1 rs4074 A allele on alcohol induced cirrhosis and HCC in patients of European descent. PLoS One. 2013;8:e80848. doi: 10.1371/journal.pone.0080848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shea-Donohue T, Thomas K, Cody MJ, et al. Mice deficient in the CXCR2 ligand, CXCL1 (KC/GRO-alpha), exhibit increased susceptibility to dextran sodium sulfate (DSS)-induced colitis. Innate Immun. 2008;14:117–24. doi: 10.1177/1753425908088724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X, Klintman D, Liu Q, et al. Critical role of CXC chemokines in endotoxemic liver injury in mice. J Leukoc Biol. 2004;75:443–52. doi: 10.1189/jlb.0603297. [DOI] [PubMed] [Google Scholar]
- 27.Maher JJ, Lozier JS, Scott MK. Rat hepatic stellate cells produce cytokine-induced neutrophil chemoattractant in culture and in vivo. Am J Physiol. 1998;275:G847–53. doi: 10.1152/ajpgi.1998.275.4.G847. [DOI] [PubMed] [Google Scholar]
- 28.Maher JJ. Rat hepatocytes and Kupffer cells interact to produce interleukin-8 (CINC) in the setting of ethanol. Am J Physiol. 1995;269:G518–23. doi: 10.1152/ajpgi.1995.269.4.G518. [DOI] [PubMed] [Google Scholar]
- 29.Joshi-Barve S, Barve SS, Amancherla K, et al. Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology. 2007;46:823–30. doi: 10.1002/hep.21752. [DOI] [PubMed] [Google Scholar]
- 30.Shi H, Kokoeva MV, Inouye K, et al. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–25. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Erridge C, Samani NJ. Saturated fatty acids do not directly stimulate Toll-like receptor signaling. Arterioscler Thromb Vasc Biol. 2009;29:1944–9. doi: 10.1161/ATVBAHA.109.194050. [DOI] [PubMed] [Google Scholar]
- 32.Stefanovic L, Brenner DA, Stefanovic B. Direct hepatotoxic effect of KC chemokine in the liver without infiltration of neutrophils. Exp Biol Med (Maywood) 2005;230:573–86. doi: 10.1177/153537020523000809. [DOI] [PubMed] [Google Scholar]
- 33.Xu R, Huang H, Zhang Z, et al. The role of neutrophils in the development of liver diseases. Cell Mol Immunol. 2014 doi: 10.1038/cmi.2014.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Williams CD, Bajt ML, Sharpe MR, et al. Neutrophil activation during acetaminophen hepatotoxicity and repair in mice and humans. Toxicol Appl Pharmacol. 2014;275:122–33. doi: 10.1016/j.taap.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marques PE, Amaral SS, Pires DA, et al. Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology. 2012;56:1971–82. doi: 10.1002/hep.25801. [DOI] [PubMed] [Google Scholar]
- 36.Ju C, Reilly T. Role of immune reactions in drug-induced liver injury (DILI) Drug Metab Rev. 2012;44:107–15. doi: 10.3109/03602532.2011.645579. [DOI] [PubMed] [Google Scholar]
- 37.Liu ZX, Han D, Gunawan B, et al. Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology. 2006;43:1220–30. doi: 10.1002/hep.21175. [DOI] [PubMed] [Google Scholar]
- 38.Jaeschke H. Neutrophil-mediated tissue injury in alcoholic hepatitis. Alcohol. 2002;27:23–7. doi: 10.1016/s0741-8329(02)00200-8. [DOI] [PubMed] [Google Scholar]
- 39.Bertola A, Park O, Gao B. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: a critical role for E-selectin. Hepatology. 2013;58:1814–23. doi: 10.1002/hep.26419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cohen JI, Roychowdhury S, McMullen MR, et al. Complement and alcoholic liver disease: role of C1q in the pathogenesis of ethanol-induced liver injury in mice. Gastroenterology. 2010;139:664–74. 674, e1. doi: 10.1053/j.gastro.2010.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singhal PC, Reddy K, Ding G, et al. Ethanol-induced macrophage apoptosis: the role of TGF-beta. J Immunol. 1999;162:3031–6. [PubMed] [Google Scholar]
- 42.Li L, Chen L, Hu L, et al. Nuclear factor high-mobility group box1 mediating the activation of Toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology. 2011;54:1620–30. doi: 10.1002/hep.24552. [DOI] [PubMed] [Google Scholar]
- 43.Ye D, Li FY, Lam KS, et al. Toll-like receptor-4 mediates obesity-induced non-alcoholic steatohepatitis through activation of X-box binding protein-1 in mice. Gut. 2012;61:1058–67. doi: 10.1136/gutjnl-2011-300269. [DOI] [PubMed] [Google Scholar]
- 44.Roh YS, Zhang B, Loomba R, et al. TLR2 and TLR9 contribute to alcohol-mediated liver injury through induction of CXCL1 and neutrophil infiltration. Am J Physiol Gastrointest Liver Physiol. 2015 doi: 10.1152/ajpgi.00031.2015. ajpgi 00031 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Allen SJ, Crown SE, Handel TM. Chemokine: receptor structure, interactions, and antagonism. Annu Rev Immunol. 2007;25:787–820. doi: 10.1146/annurev.immunol.24.021605.090529. [DOI] [PubMed] [Google Scholar]
- 46.Lentsch AB, Yoshidome H, Cheadle WG, et al. Chemokine involvement in hepatic ischemia/reperfusion injury in mice: roles for macrophage inflammatory protein-2 and KC. Hepatology. 1998;27:1172–7. doi: 10.1002/hep.510270440. [DOI] [PubMed] [Google Scholar]
- 47.Campbell SJ, Hughes PM, Iredale JP, et al. CINC-1 is an acute-phase protein induced by focal brain injury causing leukocyte mobilization and liver injury. FASEB J. 2003;17:1168–70. doi: 10.1096/fj.02-0757fje. [DOI] [PubMed] [Google Scholar]
- 48.Dorman RB, Gujral JS, Bajt ML, et al. Generation and functional significance of CXC chemokines for neutrophil-induced liver injury during endotoxemia. Am J Physiol Gastrointest Liver Physiol. 2005;288:G880–6. doi: 10.1152/ajpgi.00317.2004. [DOI] [PubMed] [Google Scholar]
- 49.Maher JJ, Scott MK, Saito JM, et al. Adenovirus-mediated expression of cytokine-induced neutrophil chemoattractant in rat liver induces a neutrophilic hepatitis. Hepatology. 1997;25:624–30. doi: 10.1002/hep.510250322. [DOI] [PubMed] [Google Scholar]
- 50.Bertola A, Bonnafous S, Anty R, et al. Hepatic expression patterns of inflammatory and immune response genes associated with obesity and NASH in morbidly obese patients. PLoS One. 2010;5:e13577. doi: 10.1371/journal.pone.0013577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.