

Abstract
Prohibitin 1 (PHB1) is an evolutionary conserved pleiotropic protein that participates in diverse processes depending on its subcellular localization and interactome. Recent data have indicated a diverse role for PHB1 in the pathogenesis of obesity, cancer and inflammatory bowel disease, among others. Data presented here suggest that PHB1 is also linked to cholestatic liver disease.
PHB1 expression is markedly reduced in patients with primary billiary cirrhosis and biliary atresia and Alagille syndrome, two major pediatric cholestatic conditions. In the experimental model of bile duct ligation, silencing of PHB1 induced liver fibrosis, reduced animal survival and induced bile duct proliferation. Importantly, the modulatory effect of PHB1 is not dependent on its known mitochondrial function. Importantly, d PHB1 interacts with Histone Deacetylase 4 (HDAC4) in the presence of bile acids. Hence, PHB1 depletion leads to increased nuclear HDAC4 content and its associated epigenetic changes. Remarkably, HDAC4 silencing and the administration of the HDAC inhibitor parthenolide during obstructive cholestasis in vivo promote genomic reprogramming leading to the regression of the fibrotic phenotype in the liver-specific Phb1 KO mice.
Conclusion
Our data identify PHB1 as an important mediator of cholestatic liver injury regulating the activity of HDAC4, which controls specific epigenetic marks. These results identify potential novel strategies to treat liver injury and fibrosis, particularly as a consequence of chronic cholestasis.
Keywords: Prohibitin1, HDAC4, cholestasis, fibrosis
INTRODUCTION
Prohibitins (PHBs) comprise an evolutionary conserved and ubiquitously expressed family of proteins with a variety of suggested activities in different cellular compartments1. Two homologous PHB proteins, PHB1 and PHB2, form a large multimeric complex predominantly found in the inner mitochondrial membrane where it exerts a chaperone-like function to stabilize newly synthesized mitochondrial proteins2 and maintains the organization and stability of mitochondrial nucleoids3. PHBs are essential for mitochondrial biogenesis in yeast4 and mammals5, in part by regulating the stability of optic atrophy 1 (OPA1), a mitochondrial fusion regulating protein6. Although PHBs are best known for their role in mitochondria, recent reports indicate that they are involved in other multiple cellular pathways due to their subcellular localization. PHB1 is found in the nucleus, where it interacts with Rb and p53 among other proteins to induce a change in the transcriptional activity of E2F7 and p538,9. Moreover, its transcriptional repression requires histone-deacetylase (HDAC) activity. Indeed, it has been previously described that PHB1 inhibits E2F activity through a mechanism that involves HDAC1 and co-repressors such as NCoR17. These nuclear effects have been associated with the inhibition of cell cycle progression9 and the induction of apoptosis7.
Given the implication of PHB1 in many vital functions, we generated liver-specific Phb1 knockout (Phb1 KO) mice10, which developed liver fibrosis spontaneously. In humans, reduced levels of PHB1 have been observed in obese people at risk of developing non-alcoholic steatohepatitis11. Here we describe that PHB1 expression is markedly reduced in common human cholestatic liver diseases and provide the molecular basis underlying the PHB1’s protective role in the liver.
Our present studies reveal that a mitochondrial-independent mechanism contributes to the fibrotic phenotype associated with the absence of PHB1. The apoptotic response and the alteration of the gene profile associated to bile acid metabolism in the absence of PHB1 were dependent on an over-stimulation/representation of HDAC4. Notably, we provide evidence that dynamic regulation of acetylation/deacetylation by HDAC4 plays a key role in determining the PHB1-dependent liver injury. Parthenolide treatment leads to a switch in the balance of ubiquitin-dependent protein homeostasis, which consistently reduces the levels of HDAC4 resulting in a regression of the fibrotic phenotype in Phb1 KO mice. Importantly, similar effects are detected when silencing HDAC4 during obstructive cholestasis in bile duct ligated Phb1 KO mice.
These results determine that in PHB1 absent cholestatic liver disease, HDAC4 emerges as a new druggable target for liver injury.
MATERIALS AND METHODS
Human samples
PBC liver samples were obtained from the Biodonostia Health Research Institute-Donostia University Hospital and from the Hospital Clinic in Barcelona. Each patient signed an informed consent document. PBC liver samples were obtained by percutaneous biopsy in 7 patients. The diagnosis of PBC was established by liver biopsy with characteristic features of the disease and presence of anti-mitochondrial antibodies. Liver samples from children with biliary atresia (n=9), Alagille syndrome (n=9), and liver disease associated with total parenteral nutrition (n=9) were obtained at the time of transplantation. The diagnosis of biliary atresia was made on clinical, laboratory, radiological, and histopathological findings. Heterozygous mutations in JAG1 gene were demonstrated in all children with Alagille syndrome. The age of the patients ranged from 22 months to 5 years at the time of transplantation. For the PHB1 mRNA expression analysis in alcoholic and viral cirrhosis we used samples provided by Dr. Erica Villa, from 47 patients with liver cirrhosis detected during surveillance They had preserved liver function and corresponded to BCLC stage A (n=34) and B (n=13)12. Healthy human liver was used as control for immunostaining and qPCR analyses. Patients or patient parents gave informed consent to all clinical investigations, in according to the principles embodied in the Declaration of Helsinki.
Experimental procedures in animals
Phb1 KO animals were generated as described10. Males from 8–12 weeks of age were treated in accordance with the Spanish Guide for the Care and Use of Laboratory Animals. Parthenolide was intraperitoneally injected at a dose of 3mg/kg 24h and 1h before bile duct ligation (BDL) or twice a week during two weeks. Liver specimens were snap-frozen for subsequent analysis.
Isolation of biliary trees
C57BL6 WT and Phb1 KO mice were subjected to bile duct ligation and after 7 days biliary trees were isolated via collagenase perfusion of the liver13.
In vivo silencing
For PHB1 and OPA1 silencing BDL was performed in 12-week-old WT mice and 3 days later they received via tail vein injection, either 200 μl of a 0.75 μg/μl solution of PHB1, OPA1 specific shRNA or control shRNA (pSM2c Open Biosystems). Animals were sacrificed 14 days after BDL. For the HDAC4 silencing, 12-week old Phb1 KO mice received via tail vein injection, 1 day before and 2 days after the BDL, 100 μl of a 25 μM solution of HDAC4 or control siRNA.
RESULTS
Alteration of PHB1 levels is associated with cholestatic liver disease
We have previously reported that PHB1 protein is frequently down-regulated in obese people at risk of developing non-alcoholic steatohepatitis11. Indeed, liver-specific PHB1 knockout (Phb1 KO) mice develop fibrosis spontaneously, with abnormal ductular proliferation10. However, it is unknown if PHB1 levels confer sensitivity to liver cholestatic injury. We assessed the levels of PHB1 in livers from patients with primary biliary cirrhosis (PBC), one of the most common cholestatic liver diseases in the adult population and in two representative severe pediatric cholestatic disorders as biliary atresia and Alagille syndrome14. Remarkably, the levels of PHB1 were significantly reduced in these pathologies in comparison to normal healthy livers (Fig. 1A, B). We also measured the levels of PHB1 in samples from children with end-stage liver disease associated with total parenteral nutrition and from a prospective study of patients with liver cirrhosis at mRNA level. In all these conditions, PHB1 gene expression was comparable to that of healthy livers (Fig. 1B, C). Interestingly, liver biopsy specimens from patients with alcohol and viral cirrhosis displayed higher PHB1 protein levels than normal liver (Fig. 1D). Finally, we observed that PHB1 expression decreased at protein and mRNA levels in wild type (WT) mice subjected to bile duct ligation (BDL), a well-established experimental model of obstructive cholestasis leading to fibrosis (Fig. 1E, F). Overall, these data strongly support a role of PHB1 during cholestatic liver injury.
Figure 1. Reduced PHB1 levels are associated with cholestatic liver disease.

(A) PHB1 levels in normal healthy liver (NL) and PBC human samples. PHB1 mRNA levels in human (B) NL, biliary atresia (BA), Alagille syndrome (ALS) and parenteral nutrition-associated liver disease (TPN) and (C) NL and BCLC A and B stage viral and alcoholic liver cirrhosis. (D) PHB1 levels in human NL, viral and alcoholic cirrhosis. PHB1 (E) protein and (F) gene expression in WT mice at different time points after BDL. (Values are mean ± Stdev. *p<0.05, **p<0.01, ***p<0.001 [PBC, BA, ALS, Virus or Alcohol vs NL, BDL vs Control).
PHB1 deficiency predisposes to liver injury
Cholestasis is the read out of a bile duct damage caused by the accumulation of toxic hydrophobic bile acids. In response to repeated injury, both biliary epithelial cells (BECs) and hepatocytes loose their balance to evoke inflammation and collagen deposition, which further damage the liver and lead to fibrosis. To examine the potential role of PHB1 in a cholestatic liver we adopted the BDL animal model, an experimental model of human obstructive cholestatic liver disease14. We have previously reported that Phb1 KO mice develop spontaneously liver injury10. As expected, the response of Phb1 KO mice to BDL was more deleterious than WT animals. After 3 days, the survival of Phb1 KO mice was reduced to 45% (Suppl Fig. 1A). Moreover, features such as F4/80 immunostaining, necrosis and collagen-1 liver expression 3 and 7 days post-BDL and the activation of JNK in Phb1 KO mice revealed more severe liver damage (Suppl Fig. 1B, C). Indeed, Phb1 KO mice 3 and 7 days post-BDL had a strong regulation in the gene profile associated to an inflammatory response as showed by TNFα, TNFR2 and IL-6 levels, while markers related to proliferation like HGF decreased, reflecting an impairment also in the regenerative response (Suppl. Fig. 1D). Furthermore, the liver injury associated to the absence of PHB1 implied a dysregulation in bile acid metabolism with the down-regulation of FXR, as well as increased expression of CYP7A1, the rate-limiting enzyme of bile acids synthesis and in proapototic markers such as Bax (Suppl. Fig. 1D).
In order to sort out whether the liver damage observed in Phb1 KO mice was due to the chronic absence of the gene, we transiently down-regulated PHB1 by i.v injection of shRNAs in WT mice during BDL (Suppl Fig 1E). Lowering PHB1 expression resulted in decreased survival and higher levels of caspase 3 and JNK activity (Fig. 2A–C). We also observed more necrotic areas and collagen deposition, higher CK19, αSMA, and F4/80 immunostaining compared to control animals (Fig. 2D) and increased ALT and AST activities (Fig. 2E). Therefore, the decrease of PHB1 levels after BDL induced a more aggressive fibrotic phenotype linked to an inflammatory and ductular response. PHB1 is important for mitochondrial function predominantly through the regulation of OPA1 stability6. We therefore assessed whether the deleterious effect observed in Phb1 KO mice during BDL was due to the regulation of OPA1. Specific reduction of OPA1 by i.v injection of shRNAs in WT mice did not alter mortality rates and did not show worsening of BDL-induced liver injury (Suppl Fig 1E, Fig. 1A). Indeed, caspase 3 activity (Fig. 2B), JNK activation (Fig. 2C), the presence of necrotic areas, collagen, the staining for αSMA, CK19 and F4/80 (Fig. 2D) and the activities of ALT and AST (Fig. 2E) were equal or even lower in OPA1-silenced animals as compared to controls.
Figure 2. The absence of PHB1 predisposes to liver injury.

(A) Kaplan Meier curve, (B) Caspase 3 activity, (C) WB analysis with the indicated antibodies, (D) H&E, CK19, αSMA, Sirius red and F4/80 staining in livers and (E) AST, ALT and bilirubin levels in serum from WT, shPHB1 and shOPA1 livers 14 days after BDL. (F) qPCR analysis of the indicated genes in the biliary trees (n=2) isolated from WT and Phb1 KO mice 7 days after BDL. (Values are mean ± SEM. *p<0.05, **p<0.01, ***p<0.001 [shPHB1 and shOPA1 vs Control, Phb1 KO vs WT).
Consistent with the in vivo results, increased JNK activity (Suppl. Fig. 1F) correlated with a major apoptotic response in Phb1 KO and silenced PHB1 hepatocytes after deoxycholic acid (DCA) treatment (Suppl Fig 2A left panel), while OPA1 silencing had the opposite effect (Suppl Fig 2A right panel).
Thus, the effect of PHB1 knockdown in the liver was not mediated through the down-regulation of OPA1 suggesting an independent role of its well-known chaperone activity. Although other functions of PHB1 related to mitochondrial dysfunction cannot be excluded.
PHB1 deficiency modulates cholangiocytes activation after BDL
The reduction of Phb1 levels in cholestatic liver injury and the increased sensitivity of Phb1 KO to BDL, prompted us to evaluate the behavior of their biliary epithelial cells (BECs). For that purposed we harvested livers in Phb1 KO mice and in WT animals at day 7 post-BDL, which corresponds to the peak of ductular proliferation15. The RNA was extracted from the ‘biliary tree’ fraction, in which the bile ducts are separated from the hepatocytes by perfusion digestion13. First, we found no contamination of the purified biliary trees with hepatocytes through the evaluation of albumin levels. Importantly, PHB1 levels in BECS from AlbCre Phb1 KO and WT mice remained unaltered. We detected the enrichment of markers related to a fibrotic phenotype and activation of cholangiocytes as represented by higher levels of CK19, Col1A1 and αSMA in those BECs derived from AlbCre Phb1 KO mice in comparison to the control mice. Indeed, biliary trees derived from Phb1 KO showed significantly higher levels of TNFα, an indicator of inflammation and genes related to regenerative and proliferative activities such as cyclin D1, as well as Epcam and VEGF (Fig 2F). These data underscore that the absence of Phb1 in the hepatocytes result in a ductular proliferation leading to a fibrotic phenotype.
PHB1 regulates the expression of HDAC4
It has been shown that regulatory changes in histone acetylation rates occur in the absence of PHB116. Moreover, a strong implication has been identified between HDAC activity and liver fibrosis17. Therefore, we analyzed the regulatory pattern of HDACs associated to PHB1 levels. As we mentioned before, the levels of PHB1 decrease significantly after 7 days of BDL in WT mice and in human cholestatic patients (Fig 1A and E). We evaluated the levels of HDAC 1 to 6 and importantly (data not shown), we found that only HDAC4 protein is overexpressed basally in Phb1 KO livers and hepatocytes, and in livers after 3 and 7 days of BDL (Fig. 3A). This upregulation was also detected at mRNA levels after BDL (Fig. 3B). Moreover, we observed a significant increase of HDAC4 at mRNA level in the i.v PHB1 silenced WT mice 14 days after BDL (Suppl. Fig. 2B).
Figure 3. HDAC4 expression in Phb1 KO mice.

HDAC4 expression in (A) liver sections from WT and Phb1 KO mice at basal conditions and after BDL (left) and WT and Phb1 KO livers and hepatocytes by WB (right), (B) WT and Phb1 KO livers at basal conditions and after BDL at mRNA level and (C) nuclear and cytosolic fractions from WT and Phb1 KO livers and hepatocytes. (D) IP of PHB1 and WB against HDAC4 in WT hepatocytes stimulated with DCA. (E) HDAC4 in human samples from NL and PBC. (Values are mean ± SEM. *p<0.05 [Phb1 KO vs WT; PBC vs NL).
Interestingly, we observe that the downregulation of PHB1 after 7 days of BDL correlates with higher nuclear HDAC4 localization in WT mice. Also, specifically a nuclear HDAC4 localization was increased in Phb1 KO (Fig. 3A, 3C, Suppl. Fig. 2C). Altogether, our data suggest that PHB1 could play a role in HDAC4 subcellular localization. No changes in global H3 acetylation levels were detected in Phb1 KO livers compared to WT animals (Suppl. Fig. 2D). To examine if this shift in HDAC4 compartmentalization and therefore its activity could be mediated by PHB1, we performed coimmunoprecipitation studies. We found a slightly interaction between PHB1 and HDAC4 in WT hepatocytes (Fig. 3D). Importantly, this interaction was enhanced by DCA. These data support the hypothesis that in the livers where PHB1 is absent, an aberrant transcriptional regulation could be mediated through the hyperactivation of HDAC4. Interestingly, immunohistochemical analysis revealed a significant increase of HDAC4 expression in patients with initial stages of PBC where PHB1 is also found down-regulated (Fig. 3E).
HDAC inhibition modulates the apoptotic response in Phb1 KO hepatocytes
To evaluate if the overactivation of HDACs is part of the mechanism that mediates liver injury in the absence of PHB1 the pan inhibitor of HDAC activity, trichostatin A (TSA) was tested in the presence of bile acids. Interestingly, TSA reduced Phb1 KO primary hepatocytes sensibility to DCA apoptotic stimuli analyzed by caspase 3 activity (Suppl Fig. 3A). These data support that a hyper-activation of HDAC could participate in the pathological phenotype associated to PHB1 ablation.
In order to discriminate the impact that HDAC class I and II could play in the Phb1 KO hepatocytes injury, we employed different inhibitors (rocilinostat, PCI34051, mocetinostat, parthenolide and apicidin) and measured the apoptotic response in the presence of DCA. Only parthenolide, the HDAC inhibitor with anti-inflammatory features18, displayed a potent anti-apoptotic effect in Phb1 KO hepatocytes (Suppl Fig. 3A). We reasoned that the effect of TSA and parthenolide in Phb1 KO hepatocytes might result, among other effects, in a proper regulation of the gene expression profile associated with bile acid metabolism and inflammatory response that is essential to preserve liver homeostasis. Indeed, TSA and parthenolide-treated hepatocytes showed increased levels of FXR, and reduced levels of CYP7A1, HDAC4, TNFα, TRAIL and Bax suggesting a less toxic effect of bile acids as a results of specific HDAC inhibition, resulting in the attenuation of the Phb1 KO hepatocytes apoptotic response (Suppl Fig. 3B).
Parthenolide reduces liver damage after BDL in Phb1 KO mice
Importantly, parthenolide exerted a protective effect from the liver injury after BDL in Phb1 KO mice. Indeed, parthenolide treatment resulted in a reduction of the mortality rate of this mice after BDL (Fig. 4A) associated with a lower apoptotic response as revealed by a reduction of necrotic areas, Tunel-staining (Fig. 4B), as well as decreased ALT (8431±957 vs. 4225±210 U/L) and AST (4805±300 vs. 2242±438 U/L) activities compared to control Phb1 KO mice. Additionally, markers of fibrosis like αSMA (Fig. 4B) and the proinflammatory cytokines TNFα, and IL-6 as well as TNFR2 (Fig. 4C) were reduced in the presence of the drug in Phb1 KO mice treated with parthenolide. Importantly, low levels of HDAC4 mRNA were identified under the drug treatment (Fig 4C). Indeed, the protein levels of TNFα in the livers were reduced in the presence of the HDAC inhibitor (Fig. 4D). Interestingly, parthenolide restored FXR and CYP7A1 levels. (Fig. 4C).
Figure 4. Parthenolide reduces liver damage after BDL in Phb1 KO mice.

(A) Kaplan Meier curve, (B) H&E, αSMA and TUNEL staining, (C) indicated genes expression, (D) TNFα levels measured by ELISA and (E) HDAC4 levels in control and parthenolide treated Phb1 KO animals after BDL. (F) Caspase 3 activity after DCA (100μM) (upper panel) and WB against HDAC4 (lower panel) in WT, Phb1 KO, Phb1 KO treated with parthenolide and Phb1 KO overexpressing HDAC4 treated with parthenolide primary hepatocytes. (Values are mean ± SEM. *p>0.05, **p>0.01 [Parthenolide vs Phb1 KO, Phb1 KO vs WT and Parthenolide + HDAC4 vs Parthenolide]).
Parthenolide has been reported as an inductor of HDAC1 degradation by proteasome activity through the action of Mdm219. While no changes were detected in HDAC1 (data not shown), parthenolide treatment resulted in the decrease of HDAC4 protein levels (Fig. 4E and Suppl. Fig. 3C), which we speculated could be due to proteasomal degradation. Indeed, we found that 26S proteasome activity decreased after BDL in Phb1 KO mice, and that this activity was restored with parthenolide treatment (Suppl Fig. 3D). Moreover, the amount of ubiquitinated proteins was significantly higher after BDL in Phb1 KO mice, and, in contrast, parthenolide prevented this accumulation (Suppl Fig. 3E). Certainly, the use of tandem ubiquitin-binding entities (TUBEs)20 showed lower levels of ubiquitinated hepatic HDAC4 protein after BDL in the presence of parthenolide (Suppl. Fig. 3F). Overall, these data suggest that parthenolide restored protein homeostasis of HDAC4, which significantly attenuated liver injury in Phb1 KO mice. Importantly, keeping the levels of HDAC4 elevated through its overexpression in Phb1 KO hepatocytes under DCA treatment counteracts the reduction in the apoptotic response detected in the presence of parthenolide (Fig. 4F). Therefore, this result highlighted the importance of HDAC4 levels in the effect mediated by parthenolide treatment.
Of particular importance was the successful attenuation of the spontaneously developed fibrosis, characteristic of Phb1 KO mice, after 15 days of parthenolide treatment. We detected lower AST activity (268±68 vs. 63±16 U/L) and marked reduction in Smad2/3 and F4/80 protein levels in parthenolide-treated Phb1 KO livers (Suppl. Fig. 4A). Moreover, mRNA levels of profibrogenic markers like TGFβ and αSMA were decreased in the presence of this drug. Additionally, the levels of FXR increased while those of CYP7A1 diminished (Fig. 5A). Importantly, the protein levels of the inflammatory cytokine TNFα were also reduced (Fig. 5B).
Figure 5. Parthenolide delays liver fibrosis in Phb1 KO mice.
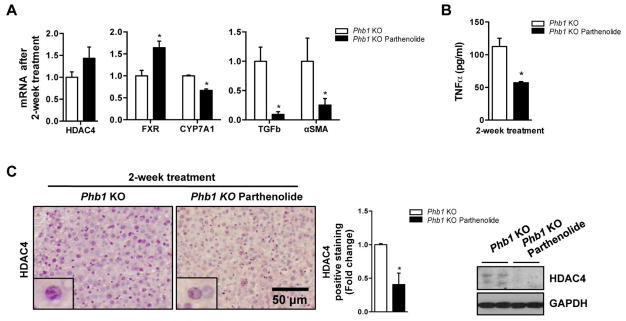
(A) Indicated genes expression, (B) TNFα levels measured by ELISA and (C) HDAC4 levels in control and parthenolide treated Phb1 KO animals. Values are mean ± SEM. *p > 0.05, **p<0.01 [Parthenolide vs Phb1 KO]).
Alternatively, parthenolide induced the hepatic 26S proteasome activity (Suppl. Fig. 4B). Indeed, this trigger of the proteasome degradation was correlated with a remarkable reduction of HDAC4 protein levels by IHC and Western blot analysis in the liver animals under treatment, with no changes at mRNA level (Fig 5A, C). Additionally, global levels of ubiquitin were also lower (Suppl. Fig. 4C). The use of ubiquitin-traps revealed the formation of ubiquitin chains on HDAC4 in the Phb1 KO mice that demonstrated the defective proteasome-mediated degradation in these animals, which was corrected upon parthenolide treatment (Suppl Fig 4D). Our results propose a direct effect of parthenolide on protein ubiquitination and therefore protein homeostasis in liver fibrosis. Importantly HDAC4 was identified as a specific target of this drug.
HDAC4 silencing attenuates liver damage after BDL in Phb1 KO mice
Finally, to confirm that the increase of HDAC4 expression contributed to the liver fibrosis observed in the liver-specific Phb1 KO, we silenced HDAC4 with siRNAs in Phb1 KO hepatocytes and evaluated the response to bile acids exposure. Notably, HDAC4 silencing reduced significantly the apoptotic response induced by DCA analyzed by caspase 3 activity (Suppl. Fig. 5A). We further validated this in vivo by i.v injection of siHDAC4 in Phb1 KO mice after BDL, which revealed a decreased in the liver damage as shown by H&E, Sirius red, CK19 and F4/80 staining (Fig. 6A) and by the significant reduction of bilirubin levels from 24±2 to 12±5 mg/dl. Importantly, we observed a decrease in JNK activation both in vitro and in vivo (Fig. 6B and Suppl. Fig. 5B), together with a deeply down-regulation of genes related to the inflammatory response like TNFα, TNFR2, CXCL1 and CCL2, apoptotic genes like Bax and those linked to bile acids metabolism, with the exception of FXR, which was markedly induced (Fig. 6C and Suppl. Fig. 5C). These data support the implication of HDAC4 during cholestatic liver damage in the context of low levels of PHB1.
Figure 6. HDAC4 silencing protects Phb1 KO mice from bile acid injury.

(A) HDAC4, H&E, Sirius red, CK19 and F4/80 staining, (B) WB with the indicated antiboides and (C) expression of the indicated genes on livers from Phb1 KO siControl and siHDAC4 animals 7 days after BDL. (Values are mean ± SEM. *p> 0.05, **p<0.01 [siHDAC4 vs siControl]).
DISCUSSION
Recent data have indicated a diverse role for PHB1 in the pathogenesis of diseases such as obesity, cancer and inflammatory bowel disease, among others11. Data presented here suggest that PHB1 is also linked to cholestatic liver disease. Thus, reduced PHB1 expression is observed in representative cholestatic disorders that manifest in adulthood or infancy, such as PBC, biliary atresia and Alagille syndrome. This effect can hardly be attributed to the cirrhotic state of the liver, as no changes in PHB1 mRNA levels were detected in patients with alcoholic or viral cirrhosis. Reinforcing these data, we have identified a down-regulation of PHB1 at mRNA and protein levels after the model of obstructive cholestasis such as BDL. Importantly, the transient silencing of PHB1 in mice has also a harmful effect in the liver after BDL, as well as in hepatocytes treated with bile acids. Altogether, these data identify a new link between PHB1 and cholestatic liver injury paving the way for new therapeutic approaches.
Cholestatic liver injury is characterized by mitochondrial dysfunction and oxidative stress. Although disturbances in the bile acid balance induces apoptosis in hepatocytes through the generation of reactive oxygen species and mitochondrial impairment with the release of cytochrome C, we propose that this is not the main function exerted by PHB1 during cholestatic liver injury. Among other findings, we show that OPA1 silencing upon BDL has the opposite outcome than the profibrotic effect exerted in the absence of PHB1. The dynamin-like GTPase OPA1 is a major organizer of the mitochondrial inner membrane and a repressor of cellular apoptosis through the sequestration of cytochrome C and the regulation of the cell cycle9. These data suggest that liver injury linked to the absence of PHB1 is not totally dependent of its well-known chaperone role of OPA1 in the mitochondria and other functions of PHB1 related to mitochondrial dysfunction cannot be excluded.
Exogenous expression of PHB1 in intestinal epithelial cells reduced the inflammatory responses and regulated the transcription of multiple cytokines21. Consistently, we have shown that the absence of PHB1 in the liver after BDL resulted in the up-regulation of inflammatory markers, while genes related with bile acid metabolism were deeply regulated in the Phb1 KO versus WT animals as shown by the down-regulation of FXR a central regulator of bile acid synthesis and CYP7A1. It has been previously reported that FXR-mediated gene expression is suppressed during hepatic inflammation22. TNFα is a key cytokine that plays a central role during liver fibrosis orchestrating an inflammatory crosstalk between hepatocytes and hepatic immune cells23. Indeed, TNFα levels were also increased not only in the liver but also in Phb1 KO biliary tree after BDL versus control mice. Proliferating cholangiocytes, typical of obstructive cholestasis, are more sensitive to TNFα-mediated cell injury24. Likewise, we have identified an increase in the expression of pro-fibrogenic markers in the biliary tree derived from Phb1 KO mice and a profuse ductular reaction in those animals compared to control mice.
The different roles proposed for prohibitin proteins are attributed to their subcellular localization and interactome1. At the subcellular level, PHB1 has been reported in mitochondrial membranes as well as the nucleus and cytoplasm. PHB1 can interact with Nrf2 and behave as a co-activator of ARE25. Additionally, PHB1 could inhibit E2F activity through a mechanism that involves HDAC1 and co-repressors such as NCoR1. HDAC4 expression is regulated by multiple mechanisms including transcriptional regulation. TNFα has been identified as one of the cytokines that regulate HDAC4 levels and interestingly, the levels of TNFα and HDAC4 are elevated in Phb1 KO in comparison to WT hepatocytes. The treatment with HDAC inhibitors counteracts this effect decreasing the amount of HDAC4 available in the liver. Importantly, we have also revealed that TNFα levels were reduced upon HDAC inhibitors treatment, so we cannot exclude a negative feedback mechanism to control HDAC4 expression mediated by this cytokine.
Regarding HDAC4 activity, this is modulated by multiple mechanisms encompass its interaction with HDAC3 that can associate in a complex with the nuclear receptor corepressors NCoR and SMRT to mediate transcriptional repression by nuclear receptors26 and postranslational modifications that determine its nucleocytoplasmic shuttling. These include modification by multiple kinases and phosphatases and its interaction with different proteins such as 14-3-327. However, the complete interactome of HDAC4 is still not defined. We now demonstrate that lower levels of PHB1 are linked to a more nuclear localization of HDAC4 as shown in control mice after BDL, in Phb1 KO mice and very importantly, in PBC patients. These data suggest that a shift in the cellular compartimentalization of HDAC4-PHB1 could be associated with cholestatic liver damage. Indeed, we have found that PHB1 interacts with HDAC4 mainly throughout the response to bile acids avoiding its shuttling to the nucleus. These findings support a role of PHB1 as a negative regulator of HDAC4 activity, sequestering it in the cytoplasm. Although, the nuclear interactome of HDAC4 in the absence of PHB1 needs to be further explored, we can suggest that the absence of PHB1 leads to epigenetic changes sensitizing the liver to the apoptotic response in a HDAC4 dependent manner.
HDAC activity is generally linked to transcriptional repression28. The misbalance in HDAC activity is associated with cancer and several disorders including fibrosis, with aberrations in inflammatory and chemokine related genes29. Specifically, HDAC4 has been related to fibrogenesis in vivo30 and as we mentioned before, in PBC patients with low expression of PHB1.
Parthenolide is a sesquiterpene lactone derived from the plant feverfew that is actively investigated as a potential therapeutic drug for several human cancers31. Notably, parthenolide induces ubiquitination and proteasomal degradation of HDAC1 in treated cells19. It has been previously described that PHB1 deficiency induces a down-regulation of proteasome activity in human hepatoma cells32. Accordingly, we have found that parthenolide increases hepatic 26S proteasome activity in Phb1 KO mice resulting in the degradation of HDAC4 and the regression of the liver injury. Indeed, HDAC4 has been reported before as a target of ubiquitination33. No changes in HDAC1 were detected under these conditions. This loss of HDAC4 resulted in epigenetic activation of key genes related to bile acid metabolism like FXR and down-regulation of profibrogenic and inflammatory markers.
Finally, stressing the importance of HDAC4 in cholestatic liver injury, its specific knockdown both in vitro and in vivo was sufficient to diminish the damage detected in the absence of PHB1, restoring the expression of important genes related to inflammation, bile acid metabolism and apoptosis.
Overall, our data identify PHB1 as a critical modulator in cholestatic liver injury regulating HDAC4 activity and specific epigenetic marks. These results also confirm that HDAC4 activity is central during hepatic damage caused by PHB1 deficiency. This mechanism might be particularly important for the identification of novel therapeutic targets, including HDAC4, to treat liver injury and chronic cholestatic diseases.
Supplementary Material
Acknowledgments
Financial Support: This work is supported by grants from NIH CA172086 (to S.C.L.; J.M.M and M.L.M-C.), ETORTEK-2011 (to M.L.M.-C), Sanidad Gobierno Vasco 2013 (to M.L.M.-C), FIS PI11/01588 (to M.L.M.-C), Asociación Española contra el Cáncer (P.F-T, JMB and M.L.M-C), SAF 2011-29851 and Educación Gobierno Vasco (to J.M.M.), FIS PI12/00402 and Program Ramón y Cajal (to N.B), Sanidad Gobierno Vasco 2012 (to M.V.R.), Ciberehd is funded by the Instituto de Salud Carlos III.
Abbreviations
- BDL
bile duct ligation
- DCA
deoxycholic acid
- HDAC
histone deacetylase
- OPA1
Optic Atrophy 1
- PBC
primary biliary cirrhosis
- PHB1
Prohibitin 1
- TSA
trichostatin A
- TUBE
tandem ubiquitin-binding entity
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Author contribution:
Lucía Barbier-Torres: Acquisition of data; analysis and interpretation of data. Critical revision of the manuscript.
Naiara Beraza: Acquisition of data; analysis and interpretation of data. Critical revision of the manuscript.
Pablo Fernandez-Tussy: Acquisition of data; analysis and interpretation of data. Critical revision of the manuscript.
Fernando Lopitz-Otsoa: Acquisition of data; analysis and interpretation of data. Critical revision of the manuscript.
David Fernández-Ramos: Acquisition of data; analysis and interpretation of data. Critical revision of the manuscript.
Imanol Zubiete-Franco: Acquisition of data; analysis and interpretation of data. Critical revision of the manuscript.
Marta Varela-Rey: Critical revision of the manuscript
Teresa C Delgado: Acquisition of data; analysis and interpretation of data. Critical revision of the manuscript.
Virginia Gutiérrez: Acquisition of data; analysis and interpretation of data; statistical analysis.
Juan Anguita: Analysis and interpretation of data. Critical revision of the manuscript.
Albert Pares: Material support. Critical revision of the manuscript.
Jesus M. Banales: Material support. Critical revision of the manuscript.
Erica Villa: Material support: Critical revision of the manuscript.
Juan Caballería: Material support: Critical revision of the manuscript.
Luis Alvarez: Material support. Critical revision of the manuscript.
Shelly C Lu: Critical revision of the manuscript; obtained funding.
José M Mato: Critical revision of the manuscript; obtained funding.
María L Martínez-Chantar: Study concept and design; analysis and interpretation of data; study supervision; drafting of the manuscript; obtained funding.
References
- 1.Mishra S, Murphy LC, Murphy LJ. The Prohibitins: emerging roles in diverse functions. J Cell Mol Med. 2006;10:353–363. doi: 10.1111/j.1582-4934.2006.tb00404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nijtmans LG, et al. Prohibitins act as a membrane-bound chaperone for the stabilization of mitochondrial proteins. EMBO J. 2000;19:2444–2451. doi: 10.1093/emboj/19.11.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kasashima K, Sumitani M, Satoh M, Endo H. Human prohibitin 1 maintains the organization and stability of the mitochondrial nucleoids. Exp Cell Res. 2008;314:988–996. doi: 10.1016/j.yexcr.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 4.Steglich G, Neupert W, Langer T. Prohibitins regulate membrane protein degradation by the m-AAA protease in mitochondria. Mol Cell Biol. 1999;19:3435–3442. doi: 10.1128/mcb.19.5.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Merkwirth C, Langer T. Prohibitin function within mitochondria: essential roles for cell proliferation and cristae morphogenesis. Biochim Biophys Acta. 2009;1793:27–32. doi: 10.1016/j.bbamcr.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 6.Merkwirth C, et al. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 2008;22:476–488. doi: 10.1101/gad.460708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang S, Fusaro G, Padmanabhan J, Chellappan SP. Prohibitin co-localizes with Rb in the nucleus and recruits NCoR and HDAC1 for transcriptional repression. Oncogene. 2002;21:8388–8396. doi: 10.1038/sj.onc.1205944. [DOI] [PubMed] [Google Scholar]
- 8.Fusaro G, Dasgupta P, Rastogi S, Joshi B, Chellappan S. Prohibitin induces the transcriptional activity of p53 and is exported from the nucleus upon apoptotic signaling. J Biol Chem. 2003;278:47853–47861. doi: 10.1074/jbc.M305171200. [DOI] [PubMed] [Google Scholar]
- 9.Kathiria AS, et al. Prohibitin attenuates colitis-associated tumorigenesis in mice by modulating p53 and STAT3 apoptotic responses. Cancer Res. 2012;72:5778–5789. doi: 10.1158/0008-5472.CAN-12-0603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ko KS, et al. Liver-specific deletion of prohibitin 1 results in spontaneous liver injury, fibrosis, and hepatocellular carcinoma in mice. Hepatol Baltim Md. 2010;52:2096–2108. doi: 10.1002/hep.23919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Theiss AL, Sitaraman SV. The role and therapeutic potential of prohibitin in disease. Biochim Biophys Acta. 2011;1813:1137–1143. doi: 10.1016/j.bbamcr.2011.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forner A, Reig ME, de Lope CR, Bruix J. Current strategy for staging and treatment: the BCLC update and future prospects. Semin Liver Dis. 2010;30:61–74. doi: 10.1055/s-0030-1247133. [DOI] [PubMed] [Google Scholar]
- 13.Liu Z, et al. Interleukin-6, hepatocyte growth factor, and their receptors in biliary epithelial cells during a type I ductular reaction in mice: interactions between the periductal inflammatory and stromal cells and the biliary epithelium. Hepatol Baltim Md. 1998;28:1260–1268. doi: 10.1002/hep.510280514. [DOI] [PubMed] [Google Scholar]
- 14.Glaser SS, et al. Recent advances in the regulation of cholangiocyte proliferation and function during extrahepatic cholestasis. Dig Liver Dis Off J Ital Soc Gastroenterol Ital Assoc Study Liver. 2010;42:245–252. doi: 10.1016/j.dld.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bai H, et al. Yes-associated protein regulates the hepatic response after bile duct ligation. Hepatol Baltim Md. 2012;56:1097–1107. doi: 10.1002/hep.25769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dart DA, Brooke GN, Sita-Lumsden A, Waxman J, Bevan CL. Reducing prohibitin increases histone acetylation, and promotes androgen independence in prostate tumours by increasing androgen receptor activation by adrenal androgens. Oncogene. 2012;31:4588–4598. doi: 10.1038/onc.2011.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joanna F, et al. Histone deacetylase inhibition and the regulation of cell growth with particular reference to liver pathobiology. J Cell Mol Med. 2009;13:2990–3005. doi: 10.1111/j.1582-4934.2009.00831.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwok BH, Koh B, Ndubuisi MI, Elofsson M, Crews CM. The anti-inflammatory natural product parthenolide from the medicinal herb Feverfew directly binds to and inhibits IkappaB kinase. Chem Biol. 2001;8:759–766. doi: 10.1016/s1074-5521(01)00049-7. [DOI] [PubMed] [Google Scholar]
- 19.Gopal YNV, Arora TS, Van Dyke MW. Parthenolide specifically depletes histone deacetylase 1 protein and induces cell death through ataxia telangiectasia mutated. Chem Biol. 2007;14:813–823. doi: 10.1016/j.chembiol.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 20.Hjerpe R, et al. Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep. 2009;10:1250–1258. doi: 10.1038/embor.2009.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Theiss AL, et al. Prohibitin protects against oxidative stress in intestinal epithelial cells. FASEB J Off Publ Fed Am Soc Exp Biol. 2007;21:197–206. doi: 10.1096/fj.06-6801com. [DOI] [PubMed] [Google Scholar]
- 22.Huang X, Zhao W, Huang W. FXR and liver carcinogenesis. Acta Pharmacol Sin. 2015;36:37–43. doi: 10.1038/aps.2014.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tacke F, Luedde T, Trautwein C. Inflammatory pathways in liver homeostasis and liver injury. Clin Rev Allergy Immunol. 2009;36:4–12. doi: 10.1007/s12016-008-8091-0. [DOI] [PubMed] [Google Scholar]
- 24.Carrion AF, Bhamidimarri KR. Liver transplant for cholestatic liver diseases. Clin Liver Dis. 2013;17:345–359. doi: 10.1016/j.cld.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 25.Yang H, et al. Activation of a Novel c-Myc-miR27-Prohibitin 1 Circuitry in Cholestatic Liver Injury Inhibits Glutathione Synthesis in Mice. Antioxid Redox Signal. 2015;22:259–274. doi: 10.1089/ars.2014.6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goodwin MM, et al. Histone deacetylases and the nuclear receptor corepressor regulate lytic-latent switch gene 50 in murine gammaherpesvirus 68-infected macrophages. J Virol. 2010;84:12039–12047. doi: 10.1128/JVI.00396-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang AH, et al. Regulation of Histone Deacetylase 4 by Binding of 14-3-3 Proteins. Mol Cell Biol. 2000;20:6904–6912. doi: 10.1128/mcb.20.18.6904-6912.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delcuve GP, Khan DH, Davie JR. Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin Epigenetics. 2012;4:5. doi: 10.1186/1868-7083-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cohen AL, et al. Genomic pathway analysis reveals that EZH2 and HDAC4 represent mutually exclusive epigenetic pathways across human cancers. BMC Med Genomics. 2013;6:35. doi: 10.1186/1755-8794-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mannaerts I, et al. Class II HDAC inhibition hampers hepatic stellate cell activation by induction of microRNA-29. PloS One. 2013;8:e55786. doi: 10.1371/journal.pone.0055786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghantous A, Sinjab A, Herceg Z, Darwiche N. Parthenolide: from plant shoots to cancer roots. Drug Discov Today. 2013;18:894–905. doi: 10.1016/j.drudis.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 32.Sánchez-Quiles V, et al. Prohibitin deficiency blocks proliferation and induces apoptosis in human hepatoma cells: molecular mechanisms and functional implications. Proteomics. 2010;10:1609–1620. doi: 10.1002/pmic.200900757. [DOI] [PubMed] [Google Scholar]
- 33.Cernotta N, Clocchiatti A, Florean C, Brancolini C. Ubiquitin-dependent degradation of HDAC4, a new regulator of random cell motility. Mol Biol Cell. 2011;22:278–289. doi: 10.1091/mbc.E10-07-0616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.