

Abstract
Certain nucleoside/nucleotide reverse transcriptase (RT) inhibitors (NRTIs) are effective against HIV-1 and HBV. However, both viruses often acquire NRTI resistance, making it crucial to develop more potent agents that offer profound viral suppression. We report here that 4′-C-cyano-2-amino-2′-deoxyadenosine (CAdA) is a novel highly potent inhibitor of both HBV (IC50=0.4 nM) and HIV-1 (IC50=0.4 nM). In contrast, the approved anti-HBV NRTI entecavir (ETV) potently inhibits HBV (IC50=0.7 nM) but is much less active against HIV-1 (IC50=1,000 nM). Similarly, the highly potent HIV-1 inhibitor 4′-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) (IC50=0.3 nM) is less active against HBV (IC50=160 nM). Southern analysis using Huh-7 cells transfected with HBV-containing plasmids demonstrated that CAdA was potent against both wild-type (IC50=7.2 nM) and ETV-resistant HBV (IC50=69.6 nM for HBVETV-RL180M/S202G/M204V), whereas ETV failed to reduce HBVETV-RL180M/S202G/M204V DNA even at 1 μM. Once daily peroral administration of CAdA reduced HBVETV-RL180M/S202G/M204V viremia (p=0.0005) in human-liver-chimeric/HBVETV-RL180M/S202G/M204V-infected mice, while ETV completely failed to reduce HBVETV-RL180M/S202G/M204V viremia. None of the mice had significant drug-related body-weight or serum human-albumin concentration changes. Molecular modeling suggests that a shallower HBV-RT hydrophobic pocket at the polymerase active site can better accommodate the slightly shorter 4′-cyano of CAdA-triphosphate (TP), but not the longer 4′-ethynyl of EFdA-TP. In contrast, the deeper HIV-1-RT pocket can efficiently accommodate the 4′-substitutions of both NRTIs. The ETV-TP’s cyclopentyl ring can bind more efficiently at the shallow HBV-RT binding pocket. Conclusion: These data provide insights on the structural and functional associations of HBV- and HIV-1-RTs and show that CAdA may offer new therapeutic options for HBV patients.
The hepatitis B virus (HBV) is a double-stranded DNA virus, which belongs to the hepadnaviridae family, causes in humans acute and chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma1. In the United States, acute hepatitis B incidence has decreased by >80% from 1990 to 2007, a notable decline among children and adolescents predominantly due to the universal screening of pregnant women to prevent perinatal hepatitis B virus transmission with HBV vaccine2,3. However, the prevalence of HBV infection still remains essentially unchanged among persons over 50 years old3. It has especially been a serious threat among people who have multiple sexual exposures, men who have sex with men, and intravenous drug users2. Worse, such populations have a high risk for co-infection with human immunodeficiency virus-type 1 (HIV-1). Individuals co-infected with HIV-1 and HBV often have faster progression of liver fibrosis and an increased risk of end-stage liver diseases than patients with HBV infection alone4,5. To date, various strategies have been used to treat HBV infection. Interferon has some advantages, such as its limited treatment duration, the absence of the occurrence of resistant mutants, and durable responses, although inherent side effects of interferon are vexing in a number of patients6,7. In this regard, nucleoside/nucleotide reverse transcriptase (RT) inhibitors (NRTIs) have been used as a component of combined antiretroviral therapies (cART) for patients infected with HIV-1 and a few of such NRTIs are known to be also active against HBV. Those NRTIs that are currently being used to treat chronic HBV infections5 include lamivudine (3TC or LVD), adefovir (ADV), entecavir (ETV), telbivudine (LdT), and tenofovir (TDF). Such NRTIs act as HBV DNA polymerase [reverse transcriptase (RT)] inhibitors, and they have proven to be highly effective in reducing HBV viremia.
Currently, ETV is one of the most commonly used drugs against HBV because of its potent activity and low rate of HBV acquisition of drug resistance. However, certain HBV variants that acquired 3TC-associated amino acid substitutions such as M204V and L180M (HBVM204V and HBVL180M/M204V) are less susceptible to ETV by factors of 20 to 30 and tend to develop high levels of ETV resistance8. Those harboring HBVM204V, and/or HBVL180M/M204V are, therefore, recommended to receive TDF or ADV; however, a long-term treatment with TDF or ADV has been associated with certain adverse effects such as renal toxicity9,10. Since the cure of HBV infection or elimination of HBV in once HBV-infected individuals is as yet elusive, novel anti-HBV therapeutics that are potent against wild-type HBV (HBVWT) and have no or less long-term adverse effects are urgently needed.
We previously reported 4′-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA), which has a unique 4′-ethynyl moiety and retains the 3′-hydroxyl moiety in the ribose (Fig. 1), exerts highly potent anti-HIV-1 activity in vitro and in vivo11–16 and is currently undergoing clinical trials17. In the present study, we examined ~150 nucleoside analogs including EFdA and its congeners, 4′-C-cyano-2-amino-2′-deoxyadenosine (CAdA) and 4′-C-cyano-2′-deoxyguanosine (CdG) (Fig. 1)18 for their anti-HIV-1/HBV activity in vitro and in vivo.
Fig. 1.
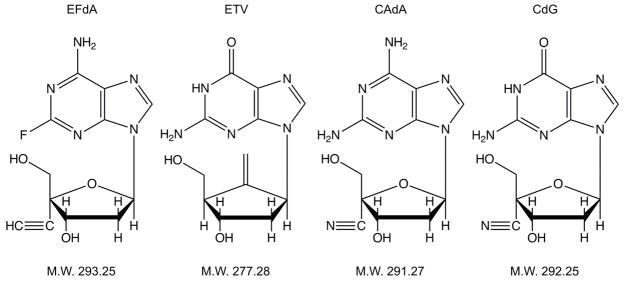
Structures and molecular weights of 4′-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA), entecavir (ETV), 4′-C-cyano-2-amino-2′-deoxyadenosine (CAdA), and 4′-C-cyano-2′-deoxyguanosine (CdG).
Materials and Methods
Cells, viruses, and antiviral agents
Various cell lines, HIV and HBV isolates, and nucleoside analogs employed in the present study are described in detail in the Supporting Information.
Anti-HIV and -HBV assays, cytotoxicity and mitochondrial DNA assays, and quantification of HBV and mitochondrial DNA
In vitro anti-HIV and -HBV assays, real-time HBV-PCR and Southern blot assays, methods for determining drug-associated cytotoxicity and mitochondrial toxicity and methods for quantification of HBV and mitochodrial DNA (mtNA) are described in detail in the Supporting Information.
HBV-DNA transfection and detection of core-associated HBV DNA from transfected cells using southern blot hybridization
Huh-7 cells were transfected with various HBV-containing plasmids and cultured in the presence of each drug and core-associated HBV DNA was extracted and subjected to the Southern blot assay. More detailed procedures are given in the Supporting Information.
HBV infection of human-liver-chimeric mice
Severe combined immunodeficiency mice transgenic for the urokinase-type plasminogen activator gene (uPA+/+/SCID+/+ mice) with their liver partly replaced with human hepatocytes (hu-liver-chimeric-uPA+/+/SCID+/+ mice) were infected with HBV and were treated with a test compound once a day over 14 days. More detailed procedures are given in the Supporting Information.
Molecular modeling
Structural models of ternary complexes of HBV- and HIV-1-RTs in complex with primer-templates (pt) and NRTI-triphosphates(TP) were built. Structural analysis, visualization, and figures were generated using SYBYL-X (Tripos Associates, St. Louis, MO) and Maestro version 9.3 (Schrödinger, LLC, New York). More detailed experimental procedures are given in the Supporting Information.
Results
Determination of the activity of nucleoside analogs against HIV-1 using MT4 cells
We tested approximately 150 mostly newly designed and synthesized nucleoside analogs for antiviral activity against HIV-1. We also examined 13 NRTIs, which are or were in clinical use or under clinical trials as anti-HIV-1 and/or anti-HBV NRTIs for anti-HIV-1 activity as reference drugs. When examined with the MTT assay using MT-4 cells, 12 of the 13 reference drugs exerted moderate to highly potent activity against a wild-type strain HIV-1NL4-3. As shown in Table 1, while ETV and ddI were moderately potent against HIV-1NL4-3 (IC50 values of ~1 μM), 3TC, FTC, TDF, ADV, ABC, d4T, Ed4T, and ddC were substantially active with an IC50 values ranging from 0.23 to 0.65 μM. AZT proved to be more active with an IC50 value of 0.03 μM. EFdA showed the most potent activity with an IC50 value of 0.0003 μM, as previously reported16. LdT, an FDA-approved anti-HBV therapeutic, showed no detectable anti-HIV-1 activity when examined at concentrations of up to 100 μM. Of note, two novel compounds, CAdA and CdG (Fig. 1), were identified among the ~150 nucleoside analogs to be very active against HIV-1NL4-3 (Tables 1 and S1). It is of note that CAdA and CdG exerted potent anti-HIV-1 activity against the wild-type HIV-1 isolate (HIV-1NL4-3) with IC50 values of 0.00016 and 0.000042 μM, respectively (Table S1). Both analogs had reduced activity by 8–97 fold against 3 multi-drug-resistant HIV-1 clinical isolates examined. Yet, if the absolute concentrations are considered, the activity of both CAdA and CdG was significantly potent against the 3 variants, and they also showed potent activity against HIV-2 strains (Table S1). In contrast, the activity of all the four FDA-approved anti-HIV-1 drugs examined was significantly decreased against the variants with the IC50 values of 0.22 to >10 μM compared to that of CAdA and CdG (Table S1).
Table 1.
Anti-HIV-1 activity, anti-HBV activity, and cytotoxicity of NRTIs
| Drug | IC50 (μM) against
|
CC50 (μM) in
|
S.I. with
|
|||
|---|---|---|---|---|---|---|
| Anti-HIV-1 | Anti-HBV | MT-4 | HepG2 2.2.15 | Anti-HIV-1 | Anti-HBV | |
| ETV | 1.0 ± 0.4 | 0.0007 ± 0.0002 | 39.1 ± 8.7 | > 100 | 38 | > 140,000 |
| ddI | 1.1 ± 0.04 | 3.7 ± 0.7 | > 100 | > 100 | > 89 | > 27 |
| 3TC | 0.29 ± 0.08 | 0.03 ± 0.01 | > 100 | > 100 | > 340 | > 3,300 |
| FTC | 0.23 ± 0.10 | 0.02 ± 0.01 | > 100 | > 100 | > 430 | > 5,000 |
| TDF | 0.58 ± 0.39 | 0.04 ± 0.01 | 40.4 ± 11.3 | > 100 | 70 | > 2,500 |
| ADV | 0.24 ± 0.12 | 0.20 ± 0.08 | 5.1 ± 1.0 | > 100 | 21 | > 500 |
| ABC | 0.58 ± 0.03 | 3.9 ± 1.0 | > 100 | > 100 | > 170 | > 26 |
| d4T | 0.65 ± 0.03 | > 100 | > 100 | > 100 | > 150 | N.D. |
| Ed4T | 0.26 ± 0.01 | > 100 | > 100 | > 100 | > 380 | N.D. |
| ddC | 0.33 ± 0.13 | 0.75 ± 0.12 | > 100 | > 100 | > 300 | > 130 |
| AZT | 0.03 ± 0.01 | > 100 | 30.5 ± 3.1 | > 100 | 1,020 | N.D. |
| EFdA | 0.0003 ± 0.0001 | 0.16 ± 0.03 | 19.2 ± 5.9 | > 100 | 64,000 | > 630 |
| LdT | > 100 | 0.007 ± 0.001 | > 100 | > 100 | N.D. | > 14,000 |
| CAdA | 0.0004 ± 0.0001 | 0.0004 ± 0.0002 | 3.5 ± 1.2 | 3.8 ± 1.0 | 8,750 | 9,500 |
| CdG | 0.0004 ± 0.0001 | 0.0004 ± 0.0001 | 2.8 ± 0.9 | 3.2 ± 0.7 | 7,000 | 8,000 |
Abbreviations: IC50: 50% inhibitory concentration, CC50: 50% cytotoxic concentration, S.I.: selectivity index. (CC50/IC50), N.D.: not determined
Anti-HIV-1 activity was determined using MT-4 cells, while anti-HBV activity was determined using HepG2 2.2.15 cells. Each assay was conducted in duplicate or triplicate. The values represent mean values (± 1 S.D.) out of two or three independent experiments.
Determination of the activity of nucleoside analogs against HBV using HepG2 2.2.15 cells
We then attempted to evaluate the anti-HBV activity of all the compounds described above. In general, no in vitro inhibition assay of de novo HBV infection using hepatocyte-derived cells and infectious HBV particles has been established. In the present study, in order to evaluate whether a given test compound reduced the amount of intracellular HBV DNA in HepG2 2.2.15 cells, DNA isolated from HepG2 2.2.15 cells, which were cultured in the presence of each test compound over 12 days, was subjected to real-time HBV-PCR as described in the Materials & Methods section. A standard curve for HBV quantification, in which the CT (cycle threshold) values for each reaction were obtained, was generated with serial dilutions of a plasmid (pHBVWT) containing 1.24-fold HBV genome of a wild-type HBV (genotype C2/Ce:AB246345, HBVWT)19, and the CT value of each test sample was converted to an HBV copy number. A representative data set of the HBV DNA decrease caused by ETV is illustrated in Supplementary Fig. 1. The HepG2 2.2.15 cells intracellularly contain HBV DNA in the forms of both integrated HBV DNA and episomal DNA. The amount of integrated HBV DNA does not change with the addition of anti-HBV agents (such as ETV) but episomal DNA amounts decrease if reverse transcription of HBV RNA is blocked as seen in Fig. S1. When cultured alone, HepG2 2.2.15 cells proved to contain 9.0 × 105 copies/well (each well contains approximately 6.0 × 105 cells at the conclusion of the 12-day period of culture); however, in the presence of ETV, the number of HBV copies continued to decrease down to ~0.9 × 105 copies/well in a dose-response fashion at up to 100 nM and no further decrease in the copy number was seen at and beyond 100 nM of ETV. The S.D. values were mostly within 5% of the mean values as determined in triplicate (Fig. S1). These data indicate that the assay system used in the present study is appropriately reproducible and quantitative and that each cell contains ~2 copies of HBV, in line with the initial report that a HepG2 2.2.15 cell has two copies integrated in cellular DNA in a head-to-head tandem fashion20.
We consequently examined the ~150 nucleoside analogs plus the 13 reference NRTIs for activity against HBV in the real-time HBV-PCR assay described above. Anti-HBV activity of 5 selected reference nucleoside/nucleotide analogs is shown in Fig. 2A. All these 5 analogs significantly reduced the number of HBV copies in similar dose-response fashions. The order of the potency of the five analogs was: ETV > LdT > 3TC > TDF > ddC and their IC50 values were 0.0007, 0.007, 0.03, 0.04, and 0.75 μM, respectively (Fig. 2A & Table 1). CAdA and CdG were found to be highly active against HBV both with an IC50 value of 0.0004 μM.
Fig. 2.

(A) Reduction of HBV DNA levels in HepG2 2.2.15 cells with ETV, LdT, 3TC, TDF, ddC, CAdA, and CdG. Intracellular HBV-DNA levels in each well were quantified using real-time HBV-PCR and shown as percent controls compared to that without drug (control=100%). All data are shown as the average values obtained from at least 3 independent experiments. (B) Comparative analysis of anti-HBV and anti-HIV-1 activity of various NRTIs. The X-axis shows the potency (IC50 values) of anti-HBV activity, while the Y-axis shows the potency of anti-HIV-1 activity. Of note, bis-pivaloyloxymethyl derivatives of acyclic nucleosides have 9–23 fold greater antiviral potency than their corresponding unmodified compounds such as PMPA. Bis-pivaloyloxymethyl-PMEA, known as adefovir dipivoxil, is also more potent than its corresponding unmodified compound (PMEA). This is due to their greater cellular uptake and intracellular formation of the active diphosphorylated metabolites38. ADV and TDF have now been commercially used, and we thus used these prodrugs instead of their active forms.
In order to corroborate the anti-HBV activity observed using the real-time HBV-PCR assay, we also conducted a Southern blot assay and determined whether ETV, CAdA, and CdG would block HBV replication (Fig. S2A). Huh-7 cells were first transfected with pHBVWT. The results revealed the presence of a single-stranded (SS) DNA signal as cultured in the absence of agents (Fig. S2A). The signal was clearly seen in the cells treated with a low concentration of ETV (1 nM; Fig. S2A). However, the signal weakened in a dose-dependent manner and was virtually lost in the cells exposed to 100 nM ETV. The IC50 value calculated by measuring the changes in the signal density was 14.1 nM. As examined under the same conditions, the IC50 values of CAdA and CdG against HBVWT were 7.2 nM and 9.8 nM, respectively. Thus, the potency determined with the Southern blot assay was overall comparable among the three compounds, consistent with the data obtained with the real-time HBV-PCR assay, while the absolute IC50 values in the Southern blot assay were greater by 16 to 24-fold than in the real-time HBV-PCR assay.
Relationship of anti-HIV-1 & anti-HBV activity of nucleoside analogs examined
Approximately 65% of the ~150 nucleoside analogs examined in the present study showed no activity against HBV or HIV-1; ~20% were active against both HBV and HIV-1; ~10% active against HBV only; while ~5% active against HIV-1 only. Among the ~20% nucleoside analogs active against both viruses, CAdA and CdG were most potent against both viruses and were further examined in the present study. Fig. 2B illustrates the activity of selected NRTIs against HBV and HIV-1. The X-axis shows the potency of anti-HBV activity, while the Y-axis the potency of anti-HIV-1 activity. AZT, Ed4T, and d4T were all substantially potent against HIV-1, while these three drugs show virtually no activity against HBV. ABC, ddI, and ddC were substantially potent against HIV-1 and HBV, while ADV, 3TC, TDF, and FTC were substantially potent against HIV-1 but more potent against HBV. LdT was substantially potent against HBV but was virtually inert against HIV-1. EFdA was extremely potent against HIV-1; however, the compound was not that potent against HBV. ETV proved to be highly potent against HBV but it was not that potent against HIV-1. Of note, both CAdA and CdG were highly potent against both HIV-1 and HBV (Fig. 2B).
CAdA and CdG block HBVETV-RL180M/S202G/M204V and HBVADV-RA181T/N236T replication
We subsequently asked whether CAdA and CdG blocked the replication of ETV-resistant HBV (HBVETV-RL180M/S202G/M204V) and ADV-resistant HBV (HBVADV-RA181T/N236T). As expected, ETV at 1 to 1,000 nM concentrations failed to block HBVETV-RL180M/S202G/M204V replication; even the highest concentration (1,000 nM) of ETV failed to achieve 50% signal reduction as shown in Fig. S2B & Table 2. By contrast, both CAdA and CdG significantly blocked HBVETV-RL180M/S202G/M204V replication in a dose-response fashion (Fig. S2B); the IC50 values of CAdA and CdG were 70 ± 14 and 42 ± 34 nM, respectively (Table 2). The average fold-reductions of the potency of CAdA and CdG against HBVETV-RL180M/S202G/M204V were only 9.7 and 4.3, respectively, compared with their IC50 values against HBVWT (Table 2). The data clearly indicate that HBVETV-RL180M/S202G/M204V is highly susceptible to both CAdA and CdG. We also determined the activity of CAdA and CdG against HBVADV-RA181T/N236T. As shown in Table 2 and Fig. S2, both CAdA and CdG were active against HBVADV-RA181T/N236T with IC50s of 0.09 and 0.04 μM, respectively, although ADV had a significantly greater IC50 value of 39.5 μM.
Table 2.
In vitro susceptibility of the HBVWT, HBVETV-RL180M/S202G/M204V, and HBVADV-RA181T/N236T to ETV, ADV, CAdA, and CdG.
| Drug | IC50 (μM) against
|
||
|---|---|---|---|
| HBVWT | HBVETV-RL180M/S202G/M204V | HBVADV-RA181T/N236T | |
| ETV | 0.014 ± 0.008 [n=2] | >1 (>71.4) [n=2] | 0.18 (13.2) [n=1] |
| ADV | 7.7 [n=1]* | 36.4 ± 23.5 (4.7) [n=2] | 39.5 ± 13.3 (5.1) [n=2] |
| CAdA | 0.007 ± 0.01 [n=2] | 0.07 ± 0.01 (9.7) [n=2] | 0.09 ± 0.07 (12.9) [n=2] |
| CdG | 0.01 ± 0.002 [n=2] | 0.04 ± 0.03 (4.3) [n=2] | 0.04 ± 0.02 (4.0) [n=2] |
Referenced from Ref 37
The IC50 numbers were obtained using Southern blotting assay. The IC50 values represent average values (±1 S.D.) out of two experiments except for the IC50 value of ETV against HBVADV-RA181T/N236T and that of ADV against HBVWT. The numbers in parentheses represent the fold changes of IC50 values compared to those against HBVWT. The numbers in brackets represent the replicate numbers.
CAdA and CdG Block HBV Replication in Hu-liver-chimeric SCID Mice
We then asked whether CAdA and CdG blocked the replication of HBV in hu-liver-chimeric-uPA+/+/SCID+/+ mice. In 8 weeks following inoculation of these mice with HBVWT, they were orally gavaged with ETV, CAdA, or CdG (prepared at a concentration of 0.002 mg/mL in saline) using oral sondes so that the dose administered resulted in 0.02 mg/kg/day. Just before the administration of ETV, the HBV copy numbers in their plasma were as high as ~5 × 109; however, the viremia levels significantly went down on day 7 of ETV administration and the viremia reduction further went down even after the termination of ETV administration to the lowest (day 21) (Fig. 3A, upper-left panel). Around day 21 and beyond, the viremia went up slowly, but the viremia level on day 42 was still lower than the day 1 viremia level. Both CAdA and CdG also significantly blocked the replication of HBV under the same conditions (upper panel in Fig. 3A). The greatest magnitudes of serum HBV-DNA level reduction with CAdA and CdG were by -2.9 or -2.7 log10 copies/ml, respectively. Over days 3–14, the decrease of HBV DNA copy numbers was significantly greater in CAdA-treated mice than in ETV-treated mice (p=0.013: repeated measures analysis of variance with two-tailed p values corrected for multiple comparisons). Over days 17–41, the decrease of HBV DNA copy numbers in CAdA-treated mice and CdG-treated mice were significantly greater than in ETV-treated mice (p=0.0026 and p=0.0015, respectively). This was thought to be partly due to CAdA- and CdG-treated mice starting from lower levels due to the effects during days 3–14.
Fig. 3.

Antiviral activity of NRTIs against HBV in hu-liver-chimeric-uPA+/+/SCID+/+ mice. (A) In 8 weeks following inoculation of the mice with HBVWT (upper panels) HBVETV-RL180M/S202G/M204V (lower panels), ETV, CAdA, or CdG was administered (0.02 mg/kg/day). Serum HBV-DNA levels following indicated treatments were determined by real-time PCR (n=2–3 per group). Each line illustrates the changes in DNA copy numbers in an individual mouse. CAdA brought about significantly greater levels of HBVWT viremia reduction over days 3–14 days (p=0.013) compared to ETV. CdG also brought about HBVWT viremia reduction, which however was not significant compared to ETV (p=0.062). CAdA and CdG also elicited significantly greater levels of HBVETV-RL180M/S202G/M204V viremia reduction by day 7 (p=0.0005 in both groups) and the reduction was seen even after the conclusion of the administration. Thick bars denote the administration period (14 days) with a dose of 0.02 mg/kg/day. Comparison was made between ETV and CAdA or CdG. (B) Human albumin levels in serum and body weights of the treated mice were monitored. Each line illustrates the changes in albumin levels and body weights in an individual mouse. There are no significant differences among ETV, CAdA, and CdG. Thick bars denote the administration period (14 days) with a dose of 0.02 mg/kg/day.
In 8 weeks following inoculation of the hu-liver-chimeric-uPA+/+/SCID+/+ mice with an HBVETV-RL180M/S202G/M204V, the viremia levels had reached 8.6 × 105 – 7.7 × 106 (lower panels in Fig. 3A). ETV, at a dose of 0.02 mg/kg/day, showed essentially no reduction in HBVETV-RL180M/S202G/M204V viremia levels (lower panel, Fig. 3A). By contrast, CAdA and CdG at the same dose of 0.02 mg/kg/day, brought about a significant level of viremia reduction by day 7 of administration. The greatest magnitudes of viremia reduction with CAdA and CdG were -1.4 log10 copies/ml (average of -1.6 and -1.1) and -1.2 log10 copies/ml (average of −0.9 and −1.5), respectively (Fig. 3A). Over the 14-day period of administration, the viremia reduction in both CAdA- and CdG-receiving mice was significantly lower compared to that in ETV-receiving mice (p=0.0005).
ETV, CAdA, and CdG caused no significant changes in body-weights and serum human albumin levels in mice
Human albumin (h-albumin) levels in serum and body weights of the treated mice were also monitored during the treatment (Fig. 3B). HBVWT-infected mice treated with CAdA or CdG had moderately decreased h-albumin levels; but mice treated with ETV also had a comparable decrease of h-albumin levels over the period of observation (p=0.4~0.5). HBVETV-RL180M/S202G/M204V-infected mice treated with ETV, CAdA, or CdG also had a gradual decrease in their h-albumin levels; however, there was no significant difference observed between the three groups. There was no significant difference in body weights among ETV-treated, CAdA-treated, and CdG-treated groups (Fig. 3B).
CAdA and CdG appear not to have significant hepatotoxicity
Certain NRTIs are known to inhibit the activity of human DNA polymerase γ, an enzyme essential for mitochondrial DNA (mtDNA) replication and repair, and to cause a variety of adverse effects including peripheral neuropathy, myopathy, pancreatitis, and lactic acidosis with hepatic steatosis. Thus, we examined whether CAdA had caused any hepatotoxicity in the CAdA-receiving hu-liver-chimeric-uPA+/+/SCID+/+ mice described above. The mice were sacrificed upon the conclusion of CAdA administration over 14 days and their liver tissues were histologically examined. Liver chimerism harboring human hepatocytes (pink area) and mouse hepatocytes (red area) was evident (Fig. S3A). No necrotic or steatotic changes were identified in the CAdA-receiving mice. In the portal region, bile ductule cell hyperplasia (open arrowheads) with a few mononuclear cell infiltration was noted, and single cell necrosis (closed arrowheads) and ceroid-laden macrophages (arrows) were sporadically observed in parenchyma (Fig. S3B); however, these minor changes were also seen in the mice with HBV infection but receiving no agents (vehicle only; Fig. S3C & S3D). Thus, it was concluded that CAdA did not cause any identifiable liver toxicities over the 14-day period of its administration.
We further examined the effects of CAdA and CdG on cellular mtDNA, L-lactate synthesis, and viable cell numbers using HepG2 cells at concentrations much higher than the circulation of the hu-liver-chimeric-uPA+/+/SCID+/+ mice receiving CAdA or CdG. After 12 days of culture, cells and supernatants were harvested and subjected to DNA extraction (for real-time mtDNA-PCR) and determination of L-lactate levels in the culture medium. As shown in Fig. 4A, no reduction of mtDNA synthesis was observed and no L-lactate level changes were identified in the cells exposed to ETV (up to 100 μM), although slight and moderate decreases in the cell viability were seen as determined in MTT assay at 10 and 100 μM, respectively. Another NRTI, ddC, which is known to inhibit the activity of DNA polymerase γ in humans21, significantly decreased mtDNA synthesis at 1 μM and beyond without causing any significant decrease in L-lactate amounts or viable cell numbers (Fig. 4B). CAdA and CdG at concentrations of 1 μM and beyond significantly lowered the levels of mtDNA (Fig. 4C & 4D). However, CAdA and CdG severely lowered the levels of L-lactate and viable cell numbers, in particular, at 10 and 100 μM, which was thought to be responsible for the lowered mtDNA levels.
Fig. 4.

Effects of ETV, ddC, CAdA, and CdG on mitochondrial DNA (mtDNA) synthesis, L-lactate levels, and viable cell numbers using HepG2 cells. HepG2 cells were cultured in the presence of various concentrations of a test agent. After 12 days of culture, cells were harvested and subjected to DNA extraction for mtDNA determination (real-time mtDNA-PCR) and supernatants harvested at the same time were subjected to the determination of L-lactate levels. The number of viable cells was determined using the MTT assay on day 5 of culture under the same conditions described above and the average numbers are shown as reference. Note that no reduction of mtDNA synthesis was observed with ETV (up to 100 μM), while ddC significantly decreased mtDNA synthesis at 1 μM and beyond. Arrows indicate the IC90 concentration for each agent against HBVWT determined from the assays conducted for and illustrated in Table 1. No significant changes in L-lactate levels in the medium were seen with ETV or ddC (panels A and B). CAdA and CdG showed significant reduction in the amounts of mtDNA and L-lactate at 1 μM and beyond as assessed on day 12; however, a significant reduction of viable cell numbers had been noted as assessed on day 5 of culture (panels C and D).
Molecular Modeling of HIV-1-RT and HBV- RT Complexed with ETV, EFdA, and CAdA
We finally conducted molecular modeling of HIV-1-RT and HBV-RT complexed with ETV-TP, EFdA-TP, or CAdA-TP and primer/template (p/t). The sugar moiety of all three agents in the triphosphate form was found close to a hydrophobic pocket in both HIV-1-RT and HBV-RT. The pocket in HIV-1-RT is constructed by residues A114, Y115, P157, F160, M184, and D185 (Fig. 5A, C, E, 6A, C & E) and that in HBV-RT by residues A87, F88, P177, L180, M204, and D205 (Fig. 5B, D, F, 6B, D & F).
Fig. 5.

Molecular models of HIV-1-reverse transcriptase (RT)/template-primer/NRTI- triphosphates (TP)s and HBV-polymerase (RT)/template-primer/NRTI-TPs. Stereoviews of the ternary complexes of HIV-1-RT/template-primer with ETV-TP (panel A), EFdA-TP (panel C), and CAdA-TP (panel E), are shown in surface representations: fingers, palm, and thumb of RT are colored in blue, red, and green, respectively. Stereoviews of HBV-RT/template-primer with ETV-TP (panel B), EFdA-TP (panel D), and CAdA-TP (panel F) are also shown. The template (dark gray), primer (yellow), and NRTI-TP are rendered as balls-and-sticks. Individual atoms in NRTI-TP are colored as carbon in white, nitrogen blue, oxygen red, phosphorus orange, and fluorine aquamarine. The arrows denote the positions of 4′-ethynyl (panels C and D) and 4′-cyano (panels E and F) moieties. The hydrophobic pocket comprises residues A114, Y115, P157, F160, M184, and D185 in HIV-1-RT and is shown in light pink. In HBV-RT, the pocket is formed by residues A87, F88, P177, L180, M204, and D205 and is shown in light pink. The hydrophobic pocket that accommodates the 4′-susbtituent is deeper in HIV-1-RT than in HBV-RT.
Fig. 6.

Interactions of ETV-TP, EFdA-TP, and CAdA-TP with the hydrophobic pocket of HIV-1-RT and HBV-RT in stereoview. The cyclopentyl ring of ETV-TP does not have favorable interactions with the slightly polar Y115 of HIV-1-RT (panel A) but has strong hydrophobic interactions with the corresponding residue F88 of HBV-RT (panel B). The sugar ring of EFdA-TP has strong hydrophobic interactions with HIV-1-RT (panel C). The sugar ring of EFdA-TP seems to have favorable interactions with HBV-RT (panel D) but to a lesser extent compared to EFdA-TP with HIV-1-RT. The ethynyl group of EFdA-TP has good hydrophobic contact with F160 of HIV-1-RT (panel C); however, it is not accommodated well in the shallow cavity of HBV-RT (panel D) and is probably responsible for EFdA’s lesser activity against HBV. The shorter 4′-cyano of CAdA-TP is accommodated equally well in the cavities of HIV-1-RT (panel E) and HBV-RT (panel F).
In the ternary complex of HIV-1-RT and ETV-TP, the cyclopentyl ring (pseudo-sugar) and exocyclic alkene (=CH2) moieties of ETV-TP have some hydrophobic interactions with A114, Y115, and the aliphatic chain of D185 of HIV-1-RT (Fig. 6A). By contrast, in HBV-RT complexed with ETV-TP, the equivalent residues (A87, F88, and D205)(Fig. 6B) engage in tight hydrophobic interactions with ETV-TP. There are additional hydrophobic contacts involving L180 of HBV-RT and the ETV-TP’s cyclopentyl ring, which are not present in HIV-1-RT since the amino acid residue equivalent to L180 of HBV-RT is F160, which does not appear to interact with ETV-TP in the HIV-1-RT/ETV-TP complex, explaining why ETV is highly potent against HBV but not against HIV-1. While EFdA-TP’s 4′-ethynyl interacts very well with the hydrophobic pocket of HIV-1-RT as previously described15, we found that the 4′-ethynyl also interacts with the pocket of HBV-RT and that CAdA-TP’s 4′-cyano interacts with the hydrophobic pockets of both HIV-1-RT and HBV-RT (Fig. 5C–F, shown as pink surface representation)(Fig. 6C–F). However, there is a clear and noticeable difference between the architectures of the hydrophobic pocket of the two enzymes. The HIV-1-RT’s pocket is deep (Fig. 5C & Fig. 6C), in contrast to HBV-RT’s pocket, which is considerably shallower, thus potentially limiting the position of certain NRTI-TPs (Fig. 5D & Fig. 6D). Indeed, the slightly longer 4′-ethynyl of EFdA-TP has reduced binding or misalignment of EFdA-TP in the HBV-RT pocket, although the same 4′-ethynyl attains strong binding to the HIV-1-RT pocket (Fig. 5C, 5D, 6C & 6D). On the other hand, the 4′-cyano of CAdA-TP is accommodated very well in HIV-1-RT as well as HBV-RT’s shallower hydrophobic pocket (Fig. 5E, 5F, 6E & 6F). These findings should at least in part explain why EFdA is highly potent against HIV-1 but not against HBV, while CAdA is highly potent against both HIV-1 and HBV. Since 2,6-diaminopurine nucleoside analogs are prodrugs to 2′-deoxyguanosine nucleoside analogs22, we hypothesize that CAdA may get converted to CdG. Thus, the modeling insights gained from the complexes of HIV-1-RT and HBV-1-RT with CAdA-TP would apply to CdG as well.
Discussion
At the beginning of the present study, we found that EFdA, a highly potent anti-HIV-1 agent, was much less potent against HBVWT in cell-based assays. On the other hand, ETV proved to be highly active against HBVWT, while it was much less active against HIV-1. Furthermore, AZT, the first known nucleoside analog active against HIV-123,24 was virtually inert against HBVWT, while LdT that has been used for the treatment of HBV infection was essentially inert against HIV-1 (Fig. 2B). In this regard, HBV-RT, in terms of amino acid sequence homology, is close to HIV-1-RT and murine leukemia virus (MuLV)-RT, but with less than 25% amino acid sequence identity. Nevertheless, the study of sequence alignment performed by Das et al.25 has demonstrated that the functionally important amino acid residues are highly conserved among the RTs of HBV, HIV-1, and MuLV, especially in the catalytic region of those RTs that include the conserved domains A-G26,27. It was thought from the observed differential activity of EFdA, ETV, AZT, and LdT against HIV-1 and HBV that the catalytic regions of HIV-1 and HBV should sufficiently differ so that HIV-1-RT and HBV-RT differentially recognize those nucleosides as good or poor substrates. Thus, we hypothesized that we could successfully identify lead compounds active against HBV among modified nucleosides that we examined and found essentially inert against HIV-1 or inappropriate as anti-HIV-1 therapeutics in our initial in vitro screening assays conducted in the 1980s and 1990s. Indeed, both CAdA and CdG were among the modified nucleoside analogs we previously reported to be active against HIV-118.
Various 4′-modified nucleoside analogs were reported to be active against HIV-1 in the 1990s28–30, although most of those analogs were overly cytotoxic and none of them entered clinical trials. However, the addition of a fluorine atom at the 2-position in the purine moiety of potent but cytotoxic 4′-ethynyl-2′-deoxyadenosine31, generating EFdA, successfully conferred highly potent anti-HIV-1 activity on EFdA and reduced the potential toxicity, especially the inhibition of human DNA polymerase γ activity12. Moreover, the half-life of EFdA-TP, the active form of EFdA, proved to be ~17 hours or longer12. EFdA is currently under clinical trials as a potential therapeutic for treating HIV-1 infection. The mechanism, by which EFdA exerts potent activity against a wide spectrum of multi-NRTI-resistant HIV-1 variants and the emergence of HIV-1 variants resistant to EFdA is significantly delayed and the multitude of EFdA resistance is highly limited32, is thought to be derived from multiple mechanisms involved in its activity against HIV-1. EFdA-TP has been shown to block HIV-1-RT as a translocation defective NRTI, which significantly slows proviral DNA synthesis and acts as a de facto immediate chain terminator. Although non-translocated EFdA-monophosphate (MP)-terminated primers are unblocked, they are efficiently converted back to the EFdA-MP-terminated form15. Moreover, EFdA-TP functions as a delayed chain terminator, allowing incorporation of an additional physiological dNTP before blocking DNA extension, thereby probably enabling EFdA-MP-terminated to be free from excision. Moreover, EFdA-MP is highly efficiently misincorporated by HIV-1-RT, making the mismatched primers extremely difficult to be extended and efficiently disallowing the excision of EFdA-TP from the mismatched primers33.
Considering that the multiple antiviral mechanisms involved represent a unique feature of the 4′-modified NRTIs, we hypothesized that CAdA and CdG would also act as efficient HBV DNA chain terminators through the mechanism(s) mentioned above, which should differ from the mechanism(s) through which ETV exerts its activity against HBV and that CAdA and CdG would also be active against HBVETV-RL180M/S202G/M204V. As expected, both CAdA and CdG proved to significantly reduce the production of both HBVWT and HBVETV-RL180M/S202G/M204V in cell-based assays and productively HBV-infected hu-liver-chimeric-uPA+/+/SCID+/+ mice (Fig. 2 & Fig. 3). The IC50 value difference in the HBV signal reduction in the Southern blot between HBVWT and HBVETV-RL180M/S202G/M204V was 9.7-fold for CAdA treatment and 4.3-fold for CdG treatment (Table 2). In the mouse model, the difference in the viremia reduction magnitude between HBVWT and HBVETV-RL180M/S202G/M204V was 30-fold (−2.9 vs −1.4 log10 copies/ml) for CAdA treatment and 30-fold (−2.7 vs −1.2 log10 copies/ml) for CdG treatment. However, it should be noted that the potential efficacy against HBVETV-RL180M/S202G/M204V of CAdA and CdG in the clinical settings is to be determined only in carefully managed clinical trials.
Certain NRTIs such as stavudine (d4T) and zalcitabine (ddC) serve as substrates of mitochondrial polymerase γ, resulting in mtDNA depletion, which leads to decreased synthesis of mitochondrial proteins that maintain oxidative phosphorylation pathways. In the early stages of mitochondrial toxicity, glycolysis is inhibited and the metabolism of pyruvate is shifted to L-lactate, causing a decrease of energy production and bringing about myopathy, peripheral neuropathy, and life-threatening hepatic steatosis with lactic acidosis. In the present study, the short-term administration of either of CAdA or CdG showed no significant liver toxicity or general toxicity as examined with body-weight and serum human albumin level changes as indicators in the productively HBV-infected hu-liver-chimeric-uPA+/+/SCID+/+ mice. In terms of the potential mitochondrial toxicity and hepatotoxicity of nucleoside analogs including CAdA and CdG, it is noteworthy that clevudine, one of the recently developed anti-HBV NRTIs, reportedly attained sustained suppression of HBV-DNA for several months after cessation of therapy34, was associated with myopathy characterized by the depletion of mtDNA35. Potential toxicities such as mitochondrial toxicity of CAdA or CdG have to be carefully examined in controlled clinical trials in humans.
It is noteworthy that we have previously determined the intracellular metabolism of EFdA, which is a nucleoside analog, is structurally related to CAdA and CdG, and is highly potent against HIV-112,14. In the case of EFdA, the intraperitoneal administration (20 mg/kg) resulted in a maximal concentration of 4 μg/ml in 30 min, more than 1,000-fold higher than its in vitro anti-HIV-1 IC90 concentration (~5 nM)14. Since both CAdA and CdG exerted highly potent activity in vitro and in vivo as shown in the present study, it is strongly suggested that both analogs well penetrate their target cells and are well triphosphorylated intracellularly, although detailed pharmacokinetics and pharmacodynamics of CAdA and CdG remain to be determined.
In our structural modeling of HIV-1-RT and HBV-RT in complex with ETV-TP, EFdA-TP, or CAdA-TP, there appear to be three major factors contributing to greater potency of ETV against HBV compared to HIV-1: (i) the shallower pocket in HBV-RT compared to HIV-1-RT, (ii) the presence of a phenylalanine (F88) in HBV-RT in place of the slightly polar tyrosine (Y115) in HIV-1-RT (Fig. 6A & 6B) that appears to contribute to a stronger hydrophobic interaction with the exocyclic alkene-containing hydrophobic ring of ETV-TP and (iii) favorable interactions of L180 in HBV-RT with ethene-cyclopentyl ring in ETV-TP, which is absent in HIV-1-RT due to the slightly distal location of F160 at equivalent position. Therefore, the stronger hydrophobic interactions of the pseudo-sugar (exocyclic alkene containing cyclopentyl ring) of ETV-TP in HBV-RT compared to those in HIV-1-RT may contribute to the enhanced binding of ETV-TP leading to a greater inhibition of HBV compared to HIV-1 (Table 1)36, although it is possible that other factors also contribute to the observed potency differences. In the models of EFdA-TP, the 4′-ethynyl binding pocket in HIV-1-RT (Fig. 5C & 6C) is deeper compared to that in HBV-RT (Fig. 5D & 6D), which may increase the interactions and affinity of EFdA-TP with HIV-1-RT compared to that in HBV-RT. The sugar ring of CAdA-TP has a 4′-cyano instead of the 4′-ethynyl in EFdA. The 4′-cyano group is slightly shorter than the 4′-ethynyl (by ~0.1 Å), and both moieties can be accommodated in the deeper hydrophobic pocket of HIV-1-RT (Fig. 6C, E & G), accounting for the similar antiviral potencies of EFdA and CAdA against HIV-1 (IC50 values: 0.4 vs. 0.3 nM, Table 1). Moreover, the 4′-cyano-containing NRTI-TPs can be accommodated in the shallow pocket of HBV-RT, but the slightly longer 4′-ethynyl may lead to reduced binding or misalignment of EFdA in the same pocket. These subtle differences between lengths of 4′-cyano and 4′-ethynyl may explain to a certain extent the difference in the potency of EFdA-TP compared to that of CAdA in HBV-RT (Fig. 5D, F, 6D & F). However, differences in the catabolism of CAdA and EFdA to respective triphosphates in different cell types may also affect the potency of these NRTIs.
In conclusion, the data presented in this study suggest that CAdA and CdG represent promising candidates as novel therapeutics for HBVETV-RL180M/S202G/M204V infection and that further structural optimization of 4′-modified nucleosides should help develop more novel NRTIs that have unique and better antiviral profiles against HBV.
Supplementary Material
Acknowledgments
Financial Support: This work was supported in part by the Intramural Research Program of Center for Cancer Research, National Cancer Institute, National Institutes of Health (H.M.), in part by a Health and Labor Sciences Research Grant [Practical Research on Hepatitis (Research on the innovative development and the practical application of new drugs for hepatitis B)] (H.M., Ya.T., K.H.), and in part by a Grant from International Research Center Aiming at the Control of AIDS, Kumamoto University (H.M.). S.G.S was supported by R01AI112417 and R01AI076119.
Abbreviations
- HBV
hepatitis B virus
- HIV-1
human immunodeficiency virus-type 1
- RT
reverse transcriptase
- NRTI
nucleoside/nucleotide reverse transcriptase inhibitor
- cART
combined antiretroviral therapies
- mtDNA
mitochondrial DNA
- SCID
severe combined immunodeficiency
- uPA
urokinase-type plasminogen activator
- TP
triphosphates
- 3TC
lamivudine
- ADV
adefovir dipivoxil
- ETV
entecavir
- LdT
telbivudine
- TDF
tenofovir disoproxil fumarate
- HBVWT
wild-type HBV
- EFdA
4′-ethynyl-2-fluoro-2′-deoxyadenosine
- CAdA
4′-C-cyano-2-amino-2′-deoxyadenosine
- CdG
4′-C-cyano-2′-deoxyguanosine
- Ed4T
2′,3′-didehydro-3′-deoxy-4′-ethynylthymidine
- FTC
emtricitabine
- AZT
3′-azido-3′-deoxythymidine
- ABC
abacavir
- d4T
2′,3′-didehydro-3′-deoxythymidine
- ddI
2′,3′-dideoxyinosine
- ddC
2′,3′-dideoxycytidine
- CT
cycle threshold
- SS
single stranded
- ETV-R
entecavir resistance
- ADV-R
adefovir resistance
Footnotes
Potential conflict of interest; S.K. and H.M. are among co-inventors on a patent for 4′-ethynyl-2-fluoro-2′-deoxyadenosine; all the rights, title, and interest to the patent have been assigned to Yamasa Corporation, Chiba, Japan. The other authors declare no competing financial interests.
Additional Supporting Information may be found in the online version of this article.
References
- 1.Shepard CW, Simard EP, Finelli L, Fiore AE, Bell BP. Hepatitis B virus infection: epidemiology and vaccination. Epidemiol Rev. 2006;28:112–125. doi: 10.1093/epirev/mxj009. [DOI] [PubMed] [Google Scholar]
- 2.Daniels D, Grytdal S, Wasley A. Surveillance for acute viral hepatitis - United States, 2007. MMWR Surveill Summ. 2009;58:1–27. [PubMed] [Google Scholar]
- 3.Wasley A, Kruszon-Moran D, Kuhnert W, Simard EP, Finelli L, McQuillan G, et al. The prevalence of hepatitis B virus infection in the United States in the era of vaccination. J Infect Dis. 2010;202:192–201. doi: 10.1086/653622. [DOI] [PubMed] [Google Scholar]
- 4.Thio CL, Seaberg EC, Skolasky R, Jr, Phair J, Visscher B, Munoz A, et al. HIV-1, hepatitis B virus, and risk of liver-related mortality in the Multicenter Cohort Study (MACS) Lancet. 2002;360:1921–1926. doi: 10.1016/s0140-6736(02)11913-1. [DOI] [PubMed] [Google Scholar]
- 5.Levy V, Grant RM. Antiretroviral therapy for hepatitis B virus-HIV-coinfected patients: promises and pitfalls. Clin Infect Dis. 2006;43:904–910. doi: 10.1086/507532. [DOI] [PubMed] [Google Scholar]
- 6.Lau GK, Piratvisuth T, Luo KX, Marcellin P, Thongsawat S, Cooksley G, et al. Peginterferon Alfa-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis B. N Engl J Med. 2005;352:2682–2695. doi: 10.1056/NEJMoa043470. [DOI] [PubMed] [Google Scholar]
- 7.Buster EH, Flink HJ, Cakaloglu Y, Simon K, Trojan J, Tabak F, et al. Sustained HBeAg and HBsAg loss after long-term follow-up of HBeAg-positive patients treated with peginterferon alpha-2b. Gastroenterology. 2008;135:459–467. doi: 10.1053/j.gastro.2008.05.031. [DOI] [PubMed] [Google Scholar]
- 8.Levine S, Hernandez D, Yamanaka G, Zhang S, Rose R, Weinheimer S, et al. Efficacies of entecavir against lamivudine-resistant hepatitis B virus replication and recombinant polymerases in vitro. Antimicrob Agents Chemother. 2002;46:2525–2532. doi: 10.1128/AAC.46.8.2525-2532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fisher EJ, Chaloner K, Cohn DL, Grant LB, Alston B, Brosgart CL, et al. The safety and efficacy of adefovir dipivoxil in patients with advanced HIV disease: a randomized, placebo-controlled trial. AIDS. 2001;15:1695–1700. doi: 10.1097/00002030-200109070-00013. [DOI] [PubMed] [Google Scholar]
- 10.Karras A, Lafaurie M, Furco A, Bourgarit A, Droz D, Sereni D, et al. Tenofovir-related nephrotoxicity in human immunodeficiency virus-infected patients: three cases of renal failure, Fanconi syndrome, and nephrogenic diabetes insipidus. Clin Infect Dis. 2003;36:1070–1073. doi: 10.1086/368314. [DOI] [PubMed] [Google Scholar]
- 11.Ohrui H. 2′-Deoxy-4′-C-ethynyl-2-fluoroadenosine, a nucleoside reverse transcriptase inhibitor, is highly potent against all human immunodeficiency viruses type 1 and has low toxicity. Chem Rec. 2006;6:133–143. doi: 10.1002/tcr.20078. [DOI] [PubMed] [Google Scholar]
- 12.Nakata H, Amano M, Koh Y, Kodama E, Yang G, Bailey CM, et al. Activity against human immunodeficiency virus type 1, intracellular metabolism, and effects on human DNA polymerases of 4′-ethynyl-2-fluoro-2′-deoxyadenosine. Antimicrob Agents Chemother. 2007;51:2701–2708. doi: 10.1128/AAC.00277-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawamoto A, Kodama E, Sarafianos SG, Sakagami Y, Kohgo S, Kitano K, et al. 2′-Deoxy-4′-C-ethynyl-2-halo-adenosines active against drug-resistant human immunodeficiency virus type 1 variants. Int J Biochem Cell Biol. 2008;40:2410–2420. doi: 10.1016/j.biocel.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 14.Hattori S, Ide K, Nakata H, Harada H, Suzu S, Ashida N, et al. Potent activity of a nucleoside reverse transcriptase inhibitor, 4′-ethynyl-2-fluoro-2′-deoxyadenosine, against human immunodeficiency virus type 1 infection in a model using human peripheral blood mononuclear cell-transplanted NOD/SCID Janus kinase 3 knockout mice. Antimicrob Agents Chemother. 2009;53:3887–3893. doi: 10.1128/AAC.00270-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Michailidis E, Marchand B, Kodama EN, Singh K, Matsuoka M, Kirby KA, et al. Mechanism of inhibition of HIV-1 reverse transcriptase by 4′-Ethynyl-2-fluoro-2′-deoxyadenosine triphosphate, a translocation-defective reverse transcriptase inhibitor. J Biol Chem. 2009;284:35681–35691. doi: 10.1074/jbc.M109.036616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murphey-Corb M, Rajakumar P, Michael H, Nyaundi J, Didier PJ, Reeve AB, et al. Response of simian immunodeficiency virus to the novel nucleoside reverse transcriptase inhibitor 4′-ethynyl-2-fluoro-2′-deoxyadenosine in vitro and in vivo. Antimicrob Agents Chemother. 2012;56:4707–4712. doi: 10.1128/AAC.00723-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Merck Signs Two Deals for Novel HIV Drug Candidates and Initiates Phase II Clinical Trial of MK-1439 for HIV. MERCK; [Last accessed 9 December 2014]. Available from: http://www.merck.com/licensing/our-partnership/Merck-chimerix-partnership.html. [Google Scholar]
- 18.Kohgo S, Yamada K, Kitano K, Iwai Y, Sakata S, Ashida N, et al. Design, efficient synthesis, and anti-HIV activity of 4′-C-cyano- and 4′-C-ethynyl-2′-deoxy purine nucleosides. Nucleosides Nucleotides Nucleic Acids. 2004;23:671–690. doi: 10.1081/NCN-120037508. [DOI] [PubMed] [Google Scholar]
- 19.Sugiyama M, Tanaka Y, Kato T, Orito E, Ito K, Acharya SK, et al. Influence of hepatitis B virus genotypes on the intra- and extracellular expression of viral DNA and antigens. HEPATOLOGY. 2006;44:915–924. doi: 10.1002/hep.21345. [DOI] [PubMed] [Google Scholar]
- 20.Sells MA, Chen ML, Acs G. Production of hepatitis B virus particles in HepG2 cells transfected with cloned hepatitis B virus DNA. Proc Natl Acad Sci USA. 1987;84:1005–1009. doi: 10.1073/pnas.84.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Birkus G, Hitchcock MJ, Cihlar T. Assessment of mitochondrial toxicity in human cells treated with tenofovir: comparison with other nucleoside reverse transcriptase inhibitors. Antimicrob Agents Chemother. 2002;46:716–723. doi: 10.1128/AAC.46.3.716-723.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weckbecker G, Cory JG. 2,6-Diaminopurinedeoxyriboside as a prodrug of deoxyguanosine in L1210 cells. Cancer Res. 1987;47:2218–2223. [PubMed] [Google Scholar]
- 23.Mitsuya H, Weinhold KJ, Furman PA, St Clair MH, Lehrman SN, Gallo RC, et al. 3′-Azido-3′-deoxythymidine (BW A509U): an antiviral agent that inhibits the infectivity and cytopathic effect of human T-lymphotropic virus type III/lymphadenopathy-associated virus in vitro. Proc Natl Acad Sci USA. 1985;82:7096–7100. doi: 10.1073/pnas.82.20.7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitsuya H, Broder S. Strategies for antiviral therapy in AIDS. Nature. 1987;325:773–778. doi: 10.1038/325773a0. [DOI] [PubMed] [Google Scholar]
- 25.Das K, Xiong X, Yang H, Westland CE, Gibbs CS, Sarafianos SG, et al. Molecular modeling and biochemical characterization reveal the mechanism of hepatitis B virus polymerase resistance to lamivudine (3TC) and emtricitabine (FTC) J Virol. 2001;75:4771–4779. doi: 10.1128/JVI.75.10.4771-4779.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Allen MI, Deslauriers M, Andrews CW, Tipples GA, Walters KA, Tyrrell DL, et al. Identification and characterization of mutations in hepatitis B virus resistant to lamivudine. Lamivudine Clinical Investigation Group. HEPATOLOGY. 1998;27:1670–1677. doi: 10.1002/hep.510270628. [DOI] [PubMed] [Google Scholar]
- 27.Bartholomeusz A, Tehan BG, Chalmers DK. Comparisons of the HBV and HIV polymerase, and antiviral resistance mutations. Antivir Ther. 2004;9:149–160. [PubMed] [Google Scholar]
- 28.Maag H, Rydzewski RM, McRoberts MJ, Crawford-Ruth D, Verheyden JP, Prisbe EJ. Synthesis and anti-HIV activity of 4′-azido- and 4′-methoxynucleosides. J Med Chem. 1992;35:1440–1451. doi: 10.1021/jm00086a013. [DOI] [PubMed] [Google Scholar]
- 29.O-Yang C, Wu HY, Fraser-Smith EB, Walker KAM. Synthesis of 4′-cyanothymidine and analogs as potent inhibitors of HIV. Tetrahedron Lett. 1992;33:37–40. [Google Scholar]
- 30.Kohgo S, Kodama E, Shigeta S, Saneyoshi M, Machida H, Ohrui H. Synthesis of 4′-substituted nucleosides and their biological evaluation. Nucleic Acids Symp Ser. 1999;42:127–128. doi: 10.1093/nass/42.1.127. [DOI] [PubMed] [Google Scholar]
- 31.Kodama EI, Kohgo S, Kitano K, Machida H, Gatanaga H, Shigeta S, et al. 4′-Ethynyl nucleoside analogs: potent inhibitors of multidrug-resistant human immunodeficiency virus variants in vitro. Antimicrob Agents Chemother. 2001;45:1539–1546. doi: 10.1128/AAC.45.5.1539-1546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maeda K, Desai DV, Aoki M, Nakata H, Kodama EN, Mitsuya H. Delayed emergence of HIV-1 variants resistant to 4′-ethynyl-2-fluoro-2′-deoxyadenosine: comparative sequential passage study with lamivudine, tenofovir, emtricitabine and BMS-986001. Antivir Ther. 2014;19:179–189. doi: 10.3851/IMP2697. [DOI] [PubMed] [Google Scholar]
- 33.Michailidis E, Huber AD, Ryan EM, Ong YT, Leslie MD, Matzek KB, et al. 4′-Ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) Inhibits HIV-1 Reverse Transcriptase with Multiple Mechanisms. J Biol Chem. 2014;289:24533–24548. doi: 10.1074/jbc.M114.562694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dienstag JL. Hepatitis B virus infection. N Engl J Med. 2008;359:1486–1500. doi: 10.1056/NEJMra0801644. [DOI] [PubMed] [Google Scholar]
- 35.Seok JI, Lee DK, Lee CH, Park MS, Kim SY, Kim HS, et al. Long-term therapy with clevudine for chronic hepatitis B can be associated with myopathy characterized by depletion of mitochondrial DNA. HEPATOLOGY. 2009;49:32080–2086. doi: 10.1002/hep.22959. [DOI] [PubMed] [Google Scholar]
- 36.Seifer M, Hamatake RK, Colonno RJ, Standring DN. In vitro inhibition of hepadnavirus polymerases by the triphosphates of BMS-200475 and lobucavir. Antimicrob Agents Chemother. 1998;42:3200–3208. doi: 10.1128/aac.42.12.3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hayashi S, Murakami S, Omagari K, Matsui T, Iio E, Isogawa M, et al. Characterization of Novel Entecavir Resistance Mutations. J Hepatol. 2015 Mar 24; doi: 10.1016/j.jhep.2015.03.020. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 38.Srinivas RV, Robbins BL, Connelly MC, Gong YF, Bischofberger N, Fridland A. Metabolism and in vitro antiretroviral activities of bis(pivaloyloxymethyl) prodrugs of acyclic nucleoside phosphonates. Antimicrob Agents Chemother. 1993;37:2247–2250. doi: 10.1128/aac.37.10.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.