

Abstract
Beginning in the 1980s, an alarming rise in the incidence of esophageal adenocarcinoma (EA) led to screening of patients with reflux to detect Barrett’s esophagus (BE) and surveillance of BE to detect early EA. This strategy, based on linear progression disease models, resulted in selective detection of BE that does not progress to EA over a lifetime (overdiagnosis) and missed BE that rapidly progresses to EA (underdiagnosis). Here we review the historical thought processes that resulted in this undesired outcome and the transformation in our understanding of genetic and evolutionary principles governing neoplastic progression that has come from application of modern genomic technologies to cancers and their precursors. This new synthesis provides improved strategies for prevention and early detection of EA by addressing the environmental and mutational processes that can determine “windows of opportunity” in time to detect rapidly progressing BE and distinguish it from slowly or non-progressing BE.
Keywords: genomics, chromosome instability, evolution, overdiagnosis
Overview
The challenges facing attempts to reduce mortality of EA by prevention and early detection include overdiagnosis and overtreatment of BE as well as failure to detect the vast majority of EAs when they are early and curable (“underdiagnosis”) 1–10. Overdiagnosis is defined as diagnosis of a disease, typically by screening, that will cause neither symptoms nor death during the lifetime of an individual 11, 12. A recent review found that 90% of individuals with BE die of causes unrelated to EA (overdiagnosis) and that 93% of EAs are not detected by current screening strategies and instead present as advanced, symptomatic EAs with high mortality (underdiagnosis) 13. Here, we explore the implications of incorporating advances in genome technology with genetic and evolutionary principles into a “new synthesis” of BE and EA. This synthesis will include genetic approaches to cancer and its precursors that can address the challenges of overdiagnosis and underdiagnosis. This review is focused on genetics (the classical science of heredity) and genomics (the comprehensive study of alterations in both the constitutive genome (the level of the individual) and the somatic genome (within cells of the body including neoplasms), and the evolution of neoplastic cell lineages during progression to cancer that can be evaluated by genetic and genomic approaches. These concepts are at the core of many current challenges in cancer control as seen by the clinical gastroenterologist. This review does not address alterations in DNA methylation, chromatin remodeling or proteomics.
Barrett’s esophagus, prevention and early detection of esophageal adenocarcinoma
In 1950, Norman Barrett proposed that chronic peptic ulcers of the esophagus arose in a columnar lining resulting from a congenital short esophagus 14. Three years later, Alison and Johnstone presented a series of case reports calling attention to the association between BE, gastroesophageal reflux and esophageal ulcers and offered the competing but not mutually exclusive hypothesis that the columnar lining resulted from gastric epithelial overgrowth of the ulcer during healing 15. In 1957, Barrett responded with an insightful manuscript that reviewed comparative anatomy studies indicating that the esophageal squamous lining extends into the anatomic stomach in many animals, including the horse, cow, rat, rabbit and platypus 16. He also reviewed embryology, citing the Johns manuscript that reported the embryonic human esophagus has a columnar lining at early stages of development before it is replaced by a squamous lining 17, similar to recent reports in mouse models 18. In 1975, Naef et al. reported a large series of 1225 patients with reflux esophagitis, 140 patients with BE and 12 EAs completing the triad that continues to dominate current clinical thought 19.
Beginning in the 1980s, the incidence of EA began to increase at an alarming rate in the United States and much of the western world 20–23. Given the associations between gastroesophageal reflux, BE and EA, it therefore seemed evident that the way to control the rising incidence of this deadly cancer would be to screen the population with reflux symptoms by endoscopy to detect BE and monitor those with BE by endoscopic biopsy surveillance for early detection of EA 24. This clinical response was not unreasonable given the prevailing paradigm of gradual linear progression to cancer that the investigators were taught during medical training 25. However, these well-intentioned efforts at early detection selectively identified patients in whom BE will cause neither EA, death nor symptoms during their lifetime while failing to detect the vast majority of EAs that continue to present at an advanced stage with poor prognoses 4, 5, 8, 9, 13, 26. Thus, seemingly paradoxically, these attempts to control EA based on accepted medical concepts resulted in overdiagnosis of benign BE and underdiagnosis of life-threatening EA 13. To understand the apparently paradoxical behavior of BE and EA as well as over- and underdiagnosis of many other cancers and “precancerous” conditions, long held clinical beliefs need to be re-evaluated in light of recent genetic and genomic advances.
These apparently paradoxical screening results are believed to be due to length-biased sampling, which postulates that screening tests selectively detect slowly or non-progressive conditions and miss rapidly progressing disease 25 (Figure 1). If this hypothesis is correct, then time and space become two of the most critical variables for early detection. To understand and overcome the challenges posed by length-biased sampling and evolution of resistance and relapse after therapy, recent genomic advances need to be reviewed as they pertain to somatic genomic evolution of cancer. We begin with a short history of medical and genetic theories of neoplastic progression.
Figure 1. Length-biased sampling.

Screening tests tend to be more effective at detecting slowly evolving neoplasms than those that progress rapidly. In some cases, neoplastic evolution occurs so quickly that the patient develops an advanced cancer that was not detected by screening. However, if the disease progresses sufficiently slowly or not at all, the patient can die of unrelated causes (“overdiagnosis”). This is believed to result from length-biased sampling. Research is needed to overcome length-biased sampling in BE screening including (1) identification of the duration of the window of opportunity in time so that screening intervals can be determined to detect rapidly evolving BE before it progresses to an incurable EA and (2) development of biomarkers that distinguish rapidly progressing BE from BE that evolves slowly or not at all.
Medical and genetic cancer progression theories
The National Cancer Act was passed while the first author of this manuscript was a graduate student in genetics. The department was a hotbed of discussion about a theory that cancer was a disease of branching evolution of somatic genomes in organs and tissues of the body 27. The author entered medical school to investigate this concept, where he encountered a competing theory. For more than a century, medical training has been based on the concept of gradual, linear progression of disease 25. This concept was reinforced by linear models that represented neoplastic progression as a gradual accumulation of molecular abnormalities leading to cancer 28, 29 (Figure 2). However, the authors of these papers and others have cautioned that progression would be more complex 30. For example, more recent studies indicate that somatic genome evolution in cancer may be branched and occur much more rapidly than anticipated 31–35 although there is also evidence that some cancers may evolve gradually 29, 36. The differences between these two progression models have profound implications for cancer prevention and early detection: Linear models predict that inhibiting a single step will interrupt progression to cancer whereas branched evolution provides avenues for resistance to therapies that target only one branch (Figure 2).
Figure 2. Linear and branched evolution of cancer.

Panel A. The linear model of disease has dominated medical thinking about early detection for more than a century. In its recent versions, it has postulated that a slow, gradual linear occurrence of molecular abnormalities (1, 2, 3, 4) cause changes in tissues (A, B, C, D) before the onset of cancer. This model predicts that interrupting any event (e.g., B) in the linear pathway will prevent progression. Panel B. Recent advances in genome technologies have reported that cancers arise by “branched evolution”. In some cases, such as in BE, an early branch leads to a state in which the esophageal metaplasia can remain stable for prolong periods of time even though it has some genomic alterations (B). However, in other BE, progression is branched. In this case inhibiting one step (e.g., E) will not necessarily block progression, which can proceed through C→D.
In 1859 Charles Darwin proposed that organisms evolved by natural selection 37. Shortly thereafter in 1865, Mendel published his work on inheritance in peas, but his genetic studies were lost and only rediscovered in 1900 38. In subsequent decades, the “modern synthesis” reconciled Darwin’s theory of evolution with Mendelian genetics as source of hereditary variation on which natural selection acted to promote evolution. The theory of neoplastic evolution predicted that genomic instability would promote branched evolution of cancers, each with a unique somatic genome that might require individualized therapy and carry the potential for evolution of resistance to preventive and therapeutic interventions was proposed by Nowell in 1976 27. Yet evolutionary principles have been poorly integrated into medical thought, training and practice. For example, a recent review evaluated 6,228 abstracts on therapeutic resistance and/or relapse and found evolutionary terms were used in only about 1%. Detailed coding of 22 recent papers revealed a higher proportion of use of evolutionary methods or theory, but this was less than 10% 39.
Modern genome sequencing and SNP array technologies have provided the breakthroughs to confirm and extend Nowell’s theory of branched evolution that predicts emergence of resistant populations of cells after therapy in contrast to linear models that predict inhibition of any step in progression will block downstream events and cancer 32, 40 (Figure 2). These advances will revolutionize the care of patients with BE and EA by facilitating a “new synthesis” incorporating genetics, genomics and somatic genome evolution that can (1) track neoplastic cell lineages regardless of morphology including both BE and neosquamous epithelium, (2) determine which BE will remain in relative genomic stasis (nonprogressing) while others evolve rapidly to EA, (3) identify environmental carcinogens that cause specific mutation signatures providing a new approach to prevention, and (4) give early warning of resistance or relapse after therapy so the patient’s disease may be appropriately treated. Modern genome technologies have also provided evidence that neoplastic evolution may be accelerated by increased mutation rates and even more rapidly by punctuated or catastrophic chromosomal events that may occur in one or a few cell divisions (Figure 3) 35, 41. This rapid evolution generates genetic and genomic variants (diversity) on which selection can act to promote progression to EA 40, 42. It is imperative that our approaches to prevention and early detection define “windows of opportunity” in time to detect these rapidly evolving genomes 43, 44.
Figure 3. Punctuated and catastrophic evolution can decrease the window of opportunity for early detection.

Panel A. Progression of a neoplasm over time has historically been thought to occur by a slow, gradual mutation rate that would typically take decades to accumulate the genetic changes needed to produce a cancer (“gradualism”). However, recent results from genome analyses of advanced cancers have provided evidence that genomic alterations may occur at vastly different rates. For example, point mutations may occur slowly resulting in relatively gradual rates of progression (black line), but exposure to environmental mutagens such as tobacco smoke (lung cancer) and sunlight (melanoma) as well as inherited conditions, such as mutations that cause microsatellite instability, can increase the mutation rate leading to rapid evolution of cancer (green line). A recent exome sequencing study of a large number of EAs found that MIN was uncommon with only 4/149 that had such high mutation frequencies (2.7%) 47. Panel B. Chromosome instability, which has been historically defined as an increased rate of gain or loss of whole chromosomes or large regions of chromosomes, causes “punctuated” evolutionary jumps (blue line). Some genomic errors involving chromosomes can lead to catastrophic evolution. These include chromothripsis (chromosome shattering) and whole genome doublings, which may occur in a single cell division (red line). A series of events may accelerate progression from punctuated to catastrophic evolution and cancer. For example, BE develops chromosome instability (“punctuated evolution” blue line) within four years of the diagnosis of EA and genome doublings (“catastrophic evolution” red line) that can be detected by SNP arrays within two years of EA diagnosis (red line). When evaluated by SNP arrays, non-progressing BE typically has a limited number of chromosomal alterations that developed before the patient was seen clinically and tend to remain stable for prolonged periods up to more than two decades (“stasis” yellow).
Three genomes may contribute to cancer evolution: (1) somatic genomes that evolve to cancer in nuclei of cells in organs and tissues of the body, (2) mitochondrial genomes and (3) inherited, constitutive genomes propagated in the germline. Advancing genomic technologies including exome and whole genome sequencing and high density SNP arrays have revealed that EA genomes have very high mutation and chromosome aberration frequencies 45–52. The inherited constitutive genome may have rare highly penetrant mutations that can be detected in family studies 53–58. Alternatively, risk may be detected by genome-wide association studies (GWAS) 59, 60, 61. Below, known contributions of each of these genomes to risk of cancer are reviewed, beginning with the somatic nuclear genome, which is the genome that evolves to cancer.
The ability to study genetic and genomic alterations in BE as it does or does not evolve to EA in space and time provides an opportunity to develop studies that meet best practice standards for biomarker research as well as genetic studies (Table 1).
Table 1. Genetic and evolutionary principles for early detection and prevention biomarkers using genomic data.
Best practices for translating genomic and evolutionary alterations. A few cautionary comments are in order about the development and use of somatic genetic and genomic alterations as biomarkers for cancer risk management. Multiple studies have embraced some aspects of the approach outlined here, such as using normal constitutive DNA as a control to be certain that changes are due to genomic alterations in BE, whereas other aspects, such as examining multiple samples obtained at multiple time points, are rarely used. For example, prominent studies from TCGA (in EA and in other cancer types), while accomplishing their goals of generating a valuable catalog of mutations that develop in within tumors, are not well suited for identifying biomarkers of risk progression since they did not examine patients who don’t progress to cancer or examine samples at multiple positions in space within the esophagus and/or multiple time points 136, 137. Incorporating multiple measures of genomic alterations as they evolve in space and time within BE is one of the two greatest and easiest advances that can be made in current BE research. The second greatest need is use of an EA outcome because many BE studies rely upon surrogate dysplasia endpoints. Formal criteria for using surrogate markers in clinical studies have been well described 138, 139. Surrogate endpoints must be reproducible, accurately represent the true endpoint (EA), and have strong predictive ability to distinguish patients who will progress to cancer from those who will not 138. The current dysplasia classification system does not meet these criteria because it is not reproducible 140–142, does not accurately represent the true endpoint EA 69, 143, 144, and has highly variable outcomes in predicting future progression to EA 3, 69, 143, 144. The current practice of normalizing genetic biomarkers to dysplasia grade guarantees that the genetic markers will be just as irreproducible as histopathology when they are used in other centers. Changing this practice is a second advance that, combined with the ability to study genetic and genomic alterations in BE as it does or does not evolve to EA in space and time, can provide a more robust analysis of how the cancer develops and evolves as well as providing more effective use of limited numbers of cancer outcomes 31, 44. Some problems can begin to be overcome by determining the spatial distribution/evolution of genetic alterations surrounding EAs at the time they are diagnosed 44, 145. This practice will likely increase as diversity within EAs becomes increasingly recognized as essential for planning therapeutic strategies. Other challenges will likely be overcome as technologies advance to allow robust analysis to be performed on archived FFPE material. Biorepositories of fresh frozen material obtained prospectively are very rare, but biopsies taken for histologic assessment may be repurposed to allow analysis of the evolution of EA over space and time in a larger set of patients. Where applicable, we have included in Table 1 examples of studies that illustrate use of these principles.
|
Genomic Evolution of EA and BE
Overview
Recent studies indicate that the concept of gradual, linear progression with long time intervals for early detection may not apply to many EAs and other cancers that appear to arise by chromosome instability, which has historically been defined as an increased rate of gain or loss of whole chromosomes or large regions of chromosomes 62. Studies using modern genomic advances in sequencing and SNP arrays have reported rapid “punctuated” and/or “catastrophic” chromosome evolutionary events that can develop in one or a few cell divisions 34, 35, 41, 44, 48 (Figure 3).
All somatic genomic mechanisms leading to increased chromosome or nucleotide evolution may result in rapid generation of cellular diversity within the BE segment on which selection can act to promote rapid somatic genomic evolution 34, 35, 41, 44, 48. This rapid evolution can lead to shorter “windows of opportunity” for early detection by decreasing time intervals required for progression to EA (Figure 3). In this regard, longitudinal studies of BE in space and time using EA endpoints may provide unique insights that are directly applicable to early detection of other cancers, including breast, lung, colon and ovarian among others that have been reported to evolve through stages of punctuated chromosome instability and catastrophic whole genome doublings in TCGA (The Cancer Genome Atlas) 41. In fact, nearly 40% of all cancers have been reported to have undergone at least one whole genome doubling63.
Changes in selective pressures including medical treatments may also lead to rapid evolution when selection favors a minority cell population in the BE segment and the majority population is at a selective disadvantage in the new environment 40, 64. Reported crypt to crypt variation in BE could provide such a source of variants for resistance to therapeutic interventions 65–67.
Genetic and genomic studies of BE and EA
Although this review focuses on recent evidence available as a result of technology advances, it should never be forgotten that this knowledge base has been built on pioneering studies performed by a large number of investigators prior to the spectacular technological advances that make the current studies possible. Historically, genetic and genomic studies of BE and EA have been focused more on chromosome alterations than mutations because of technology availability. DNA content flow (or image) cytometry 68–73, FISH (fluorescent in situ hybridization) 74–80, array comparative genomic hybridization 51, 52, 81–86, and TP53 analyses 31, 87–96 have all contributed to increasing knowledge of the complexity of chromosome instability in the EA genome and the BE genome as it progresses towards cancer. In general, these founding studies support conclusions of more recent genomic investigations providing a broad base on which to develop new approaches to early detection, prevention and therapy.
In the early days of BE genetic research, mutation studies focused on known genes, such as TP53 and CDKN2A because DNA sequencing technology was limited 97–99. In a study evaluating a panel of tumor suppressor genes and DNA content abnormalities (tetraploidy, aneuploidy), only the chromosome instability markers, loss of heterozygosity (LOH), tetraploidy and aneuploidy, provided independent cancer risk assessment in multivariate analysis 100. Interestingly, use of aspirin or other NSAIDs was associated with reduced risk of progression to EA in patients with 17pLOH and DNA content tetraploidy and aneuploidy in this study.
Genomic studies of EA
At the time of writing this manuscript, TCGA study of EA has not been published. The interested reader should certainly review the TCGA study when its results become available because it is likely to be the standard reference for some time. Much has already been learned from several sequencing and high density SNP array studies that have shown that EAs typically have massive genomic alterations, including high frequencies of mutations and chromosome alterations (see Supplementary Data in 45–49, 52, 101 for comprehensive listing of mutations and copy number alterations detected in EA). However, caution is urged in interpreting cancer-only study designs because, as shown below, common early events that are frequently detected in this type of design can also be found at equal frequency in BE that does not progress to cancer 44. Basing risk assessment on these frequent, non-progressing alterations can exacerbate overdiagnosis and overtreatment in BE.
DNA sequencing studies of EAs
EAs arise in a highly genotoxic environment in which the distal esophagus is exposed to high levels of local and systemic injury from reflux of acid, bile and other gastric contents, tobacco products, and inflammatory responses to the injury, all of which are mutagenic 2. EAs have very high mutation frequencies exceeded only by bladder, melanoma, and lung cancer 45, 47, 102. In the largest study, Dulak et al evaluated 149 normal/tumor pairs by exome sequencing, with 15 also evaluated by whole genome sequencing47. The median mutation frequency across the genome per cancer was 26,161 with whole genome sequencing (range 18,881–66,225 mutations per cancer).
The authors also reported a high frequency of AA>AC transversions at AA nucleotides, a mutation signature that has been confirmed by other studies 48. This signature has been reported only in esophageal and gastric adenocarcinomas 103, 104. Other more common mutation signatures, such as the APOBEC cytidine deaminase signature and an aging signature, have also been identified 102. In the Dulak study, 8,331 genes had mutations in at least one EA, but only TP53 was mutated at high frequency (72%). This ground breaking manuscript reported many mutated genes that had not been previously detected in EAs that were potential targets for therapy, but only TP53 was mutated at sufficiently high frequency to have a major impact on early detection or prevention. Other, smaller exome sequencing studies and one whole genome sequencing study of 22 patients have also reported that TP53 is the only commonly mutated gene in EAs 45, 48, 101, 105. Localized regions of hypermutation (“kataegis”), a BRCA-deficiency signature and a previously unknown signature have also been reported in subsets of EA 48. These different mutation signatures presumably represent the highly genotoxic environment in which EA arises, but no direct causality has yet been demonstrated for many of them 48.
Chromosome alterations in EAs
EAs have high frequencies of somatic chromosome evolution, including classical chromosome instability, which can occur in a series of “punctuated” events, followed by catastrophic chromosome evolutionary events, including whole genome doublings, which can develop in a single cell division 41, 46–50, 52, 83, 106, 107. Chromothripsis (“chromosome shattering”) can also develop in one or a few cell divisions 35, 48. These modern sequencing and SNP array data provide additional support for the concept that cancer evolves through a genome doubling (“tetraploidization”) followed by additional chromosome instability, which has been well recognized in the cytogenetic and flow cytometric literature for several decades 108. Evidence of chromothripsis was found in 36% of EAs in a recent combined study of whole genome sequencing (22 EAs) and SNP arrays (101 EAs) 48. Interestingly, the same study also reported evidence of breakage-fusion-bridge cycles that can develop as a result of telomere attrition 48. The breakage-fusion-bridge cycle findings are consistent with findings of other investigators that short telomeres in (1) BE are associated with chromosome instability 109, (2) in the blood are a risk factor for progression from BE to EA 110 and (3) are found in EAs themselves 111.
DNA sequencing studies in BE
Less is known about mutations in BE, and much of the knowledge that exists comes from patients who progressed to EA and had co-existing BE that was sequenced 45, 101. In one study, exome sequencing was performed to evaluate 11 EAs, two of which had matching samples from BE 45. In one patient, 65 of 78 mutations detected in EA were also found in BE. In the second patient, 31 of 39 mutations detected in EA were also found in adjacent BE 45. In a second paper, biopsies from a single EA and adjacent BE were evaluated by whole genome sequencing, and the investigators found that the mutational profiles of EA and BE were remarkably similar 101. However, these case-report studies did not include nonprogressing control populations.
These observations were recently extended in a cross-sectional study that included several components 105. This study performed whole genome sequencing in a discovery set of 22 EAs. Mutations detected above background rate in the discovery set and in pathways of interest were then validated in a larger set of 90 EAs. Combining mutations found in discovery and validation resulted in only 15 genes that were mutated in four or more samples. Consistent with the results of Dulak et al., the only gene that was mutated at high frequency was TP53 (69%). Twenty-six genes were then evaluated in a cross-sectional analysis of 66 biopsies from 40 patients who were always negative for dysplasia during follow-up and 43 biopsies from 39 patients who had coexisting high-grade dysplasia. Twenty-one of 40 patients whose biopsies were consistently non-dysplastic (53%) had mutations in the BE segment. The mutational frequency was not significantly different between non-dysplastic BE, high-grade dysplasia and EA; only TP53 and SMAD4 were associated with advanced stages of progression, and SMAD4 was mutated at low frequency in only 13% of EAs. Initial validation of a non-endoscopic screening device (“Cytosponge”) to detect TP53 mutations in this study is a major step forward toward developing more effective screening strategies for high-risk BE and early EA 105.
Chromosome studies in BE
Genomic studies in BE have consistently reported the presence of chromosome alterations. As the density of markers has increased, small localized regions of copy number alterations and LOH appear to be frequently found in fragile sites some of which contain genes such as CDKN2A, WWOX and FHIT that are deleted frequently 86, 112. It was initially hypothesized that these findings might be due to chronic reflux exposure and genotoxic stress due to oxidative damage, and stalling at DNA replication forks that merited evaluation as a biomarker of EA risk in patients with BE 112. There is also evidence that massive and small chromosome alterations can be detected in BE before EA 44, 50, 113. Early studies using low density STR polymorphisms implicated loss of chromosome arm 9p and CDKN2A in progression to EAs 100. However, large, well-designed studies have reported that the frequencies of small homozygous deletions in fragile sites involving CDKN2A, FHIT, and WWOX as well as chromosome 9p loss or LOH are not significantly different between progressors to EA and non-progressors 44, 113. This represents an important principle of early detection and prevention research: well-designed studies with non-progressing control populations are required to distinguish those lesions whose increased risk warrant therapy from benign changes that do not progress. Further research is necessary to characterize the roles of these changes in BE.
The largest longitudinal study of BE using SNP arrays was a case-cohort study of 248 patients with BE of whom 79 progressed to EA while 169 did not 44. Chromosome alterations, including homozygous deletions, losses, gains, and balanced gains, were assessed in a defined protocol evaluating one endoscopic biopsy by SNP arrays every two centimeters in the Barrett’s segment using a constitutive genome control. The patients were evaluated at the baseline endoscopy in the study and the penultimate endoscopy (next to last endoscopy in patients who did not progress to EA or endoscopy before cancer in patients who progressed). Non-progressors largely maintained stable genomes with only minor changes involving fragile sites and small genetic regions including 9p LOH and small deletions and homozygous deletion on 3p, 9p and 16q, the sites of FHIT, CDKN2A, and WWOX. These abnormalities have been also observed at high frequency in non-progressors in previous studies 113. It has recently been proposed that everyone may develop similar genetic alterations during their lifetimes without progressing to clinically evident cancer 114.
In contrast, massive genomic alterations including widespread evidence of chromosome instability were detected beginning 48 months before the diagnosis of cancer, followed by catastrophic genome doublings and widespread aneuploidization in the 24 months before cancer 44. Strikingly, this pattern of chromosome instability followed by whole genome doublings has been observed in many types of cancer including esophageal, breast, colon, lung, and ovarian 41, 115. This sequence is also very similar to a previous report in which 17p LOH was strongly associated with development of increased 4N (G2/tetraploid) populations that were followed by development of aneuploid cell populations in BE approximately 17 months later 116.
Mitochondrial DNA mutations have been reported in EA, BE and cell cultures derived from BE 117–120. No large scale studies have been reported in either BE or EA, and their role in progression is currently unknown. Mitochondrial mutations have been used innovatively to characterize genetic lineages that assess clonal relationships between Barrett’s metaplasia and esophageal squamous cells 67. Using a combination of DNA sequencing to detect mutations in cytochrome c oxidase and immunohistochemistry to detect the enzymatic activity, the authors were able to demonstrate that Barrett’s glands were clonal. They also showed that the clonal glands were able to develop all the differentiated cell lineages found in BE. The glands were able to spread forming patches within the epithelium, and in one patient regenerating squamous epithelium and the underlying glandular epithelium shared a clonal mutation establishing that squamous and metaplastic epithelium were derived from a common precursor.
The constitutive genome has been evaluated in family studies and two genome-wide association studies (GWAS). A genetic component to developing reflux, BE and EA has been suspected based on GWAS studies 121, analysis of familial clusters 53–57, 122 and twin studies 123, 124. A complete family history is recommended for patients being seen for BE or EA 125. The impact of identification of genetic risk factors on patients and families who are at risk for inherited BE and EA is profound for both patients who inherit the risk and for relatives who do not. Although they represent a small portion of the population and the number of people who develop EA, the benefits of early diagnosis and prevention are great in these patients. Research to identify the genetic loci that account for the increased risk in family studies of BE and EA is ongoing.
Two GWAS studies have been published. One reported that variants at the MHC locus and at chromosome 16q24.1 predispose to development of BE 61. The closest protein-coding gene to the 16q24.1 locus is FOXF1, which has been implicated in esophageal development and structure. The other GWAS identified four associations, including 19p13 in CRTC1, which has also been implicated in esophageal development 59. Other loci identified include one at 9q22 in BARX1, which codes for a transcription factor for esophageal specification. A third was at 3p14 near FOXP1, which is known to regulate esophageal development. This study also confirmed the previously reported association with BE near FOXF1 at 16q24, which was also associated with EA. One study reported that some CDKN2A polymorphisms were associated with reduced risk of EA 126. This protective association is reminiscent of reports that CDKN2A abnormalities are associated with clonal expansions in BE but the chromosome instability leading to 17p LOH, tetraploidy and aneuploidy is required for progression to EA 64.
Integrative team science, a path forward: “The future ain’t what it used to be.” 127
It would be a mistake to think of genetics and genomics as simply “biomarkers” because the modern synthesis presaged an era in which genetic and genomic approaches can permit analyses of evolving cell lineages that can track clones over space and time in individual BE segments as well as in human population studies. Team science approaches that incorporate advances in genome technologies combined with application of evolutionary principles of selection of genetic variants will revolutionize the care of patients with BE and EA (Figure 4).
Figure 4. Integrative team science for genetic/genomic studies of BE.
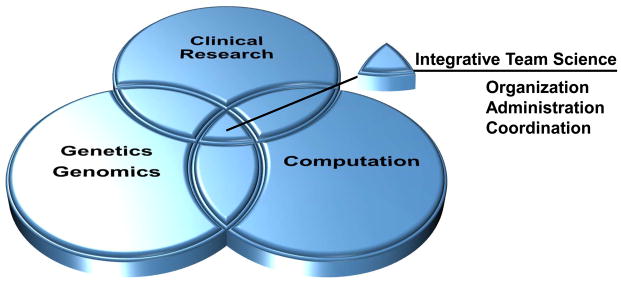
BE is a complex adaptive system that can evolve into a stable state that persists for the lifetime of 90–95% of individuals or enter a process of dynamic, stochastic somatic genomic evolution in space and time that leads to EA. The path forward will require multidisciplinary studies that include: (1) Genetics, genomics, and evolutionary biology, (2) advanced computational approaches, (3) clinical and epidemiological research with well annotated biospecimens and (4) integration in an organizational structure with smoothly functioning translational research units. The size of the teams can be variable ranging from two or three collaborators with appropriate expertise to large consortiums. It is likely that success will also require specialized computational support to answer specific clinical research questions. For example, studies of the altered developmental program that leads to BE could be studied at the genomic, expression and/or proteomic levels in BE biopsies and genetic model systems 18, 146. Alternatively, the program could be investigated in comparative genomic studies, which will become increasingly available as more species are sequenced. For example, genes required for acid secretion have been recently found to be mutated or deleted in the platypus147, which Barrett evaluated in his comparative anatomy studies 16. The probability of success can be enhanced by increased education and training in genetics, genomics and evolutionary biology as part of GI programs and GI national meetings. The path forward is through collaboration and team science, with each team building the structure required for their specific research hypotheses and questions around institutional strengths.
The power of genetic lineage analysis is also illustrated by the seemingly simple experiment described above using DNA sequencing of the mitochondrial genome that found that regenerating squamous epithelium and underlying glandular epithelium were derived from a common precursor 67. A subsequent somatic genetic study evaluated pre- and post-ablation epithelia in patients with BE 128. In this study, somatic mutations involving TP53 and/or CDKN2A were found in post-ablation neosquamous epithelium and deep esophageal glands. Non-dysplastic BE epithelium was also found to contain mutant clones that were subsequently found in EA demonstrating a lineage that evolved and progressed over time. There have been at least four case reports of esophageal squamous cell carcinomas arising after various forms of ablation of BE 129–131. Genomic evaluation of resection margins after endoscopic therapy might therefore be used for detection of residual disease much like pathology margins are currently used after surgery. Somatic genomic assessment can also be used to monitor squamous and columnar epithelium after ablation therapy.
Although it is not covered in detail in this review of genetics and genomics in BE and EA, it is worth mentioning that recent research by a number of investigators has shown BE intestinal metaplasia has a number of properties that appear to be selective protective adaptations to the harsh, genotoxic environment in which BE arises 2, 132. Some of these adaptations, such as crypt architecture have long been thought to have evolved as a mechanism to prevent cancer by decreasing clonal expansions of mutations 133. Observations of specific mutation signatures in EAs may indicate the presence of environmental mutagens against which the protective adaptation might have been lost or never evolved. This could guide prevention strategies for EA by eliminating the environmental mutagen or other risk factors such as obesity 2, 134.
GWAS studies have identified a number of highly intriguing loci that are involved in esophageal development 59, 61. Recent studies of p63 knockout mice have shown that the embryonic mouse esophagus is lined by a columnar epithelium that is remarkably similar to BE 18. These studies, combined with rediscovery of the Johns manuscript, will fuel the debate and drive scientific inquiries about the origin of BE 17. This research suggests that the ability to generate Barrett’s metaplasia in response to a reflux environment is the result of a developmental program that could be the target of additional research into the origin of the BE. A better understanding of how the Barrett’s epithelium originates could allow development of better screening strategies to identify patients at risk for developing BE in the population in general and potentially screening for high risk BE.
Researchers are searching for inherited highly penetrant mutations that predispose to BE and EA in familial clusters. Identification of the inherited genes will have a profound effect on the families: One sibling may inherit the mutation and a second will be unaffected. They will live different lives, and their parents, physicians and counselors will need to be sensitive to their different needs. Once, long ago when the first author of this paper was in training, a senior attending physician seeing a patient with an inherited susceptibility to a disease said to the patient “You’re a mutant”. We are entering a world of genomic medicine, and our training programs need to include genome biologists, geneticists and genetic counselors so that future gastroenterologists, who will be involved in the care of these patients, will be attentive to their needs.
Summary
A recent perspective on BE and EA appropriately commented that “The current strategy can be construed as representing not a ‘war’ on oesophageal adenocarcinoma, but rather a war on Barrett oesophagus” 13 with the unintended consequence of overdiagnosis. Assuming the goal is to reduce mortality of EA, this “war on BE” will fail since the current strategies for early detection result in 90% overdiagnosis of benign BE that causes neither death nor symptoms over a lifetime and 93% underdiagnosis of early EA. This strategy can become even more deeply flawed if overdiagnosis leads to overtreatment.
As Ruth Sager said, “Cancer is a disease of the genome.”135 Multidisciplinary research teams using proper application of genetic and evolutionary principles combined with modern genomic advances can markedly improve our ability to diagnose risk, define windows of opportunity for early detection and guide interventions to prevent EA (Figure 4). Successful incorporation of the genetic, genomic and evolutionary principles described here will lead to a “new synthesis” of BE and EA based on genetic and genomic advances applied in a context of evolutionary dynamics over time and space that reduce both over- and underdiagnosis. Successful research will lead to more accurate identification of patients in whom interventions can prolong functional life by preventing EA.
Acknowledgments
The authors thank the Barrett’s esophagus/esophageal adenocarcinoma research community for their contributions in identifying critical genetic and genomic components involved in progression from BE to EA. This review has attempted to reference these founding contributions, and the authors apologize to anyone they have missed.
Financial Support: National Cancer Institute (NCI) P01CA091955 and NCI RC1 CA 146973 supported Brian J. Reid, Thomas G. Paulson and Xiaohong Li.
Abbreviations
- BE
Barrett’s esophagus
- EA
esophageal adenocarcinoma
- TCGA
The Cancer Genome Atlas
Footnotes
Disclosures: No conflicts of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rustgi AK, El-Serag HB. Esophageal carcinoma. N Engl J Med. 2014;371:2499–509. doi: 10.1056/NEJMra1314530. [DOI] [PubMed] [Google Scholar]
- 2.Reid BJ, Li X, Galipeau PC, et al. Barrett’s oesophagus and oesophageal adenocarcinoma: time for a new synthesis. Nat Rev Cancer. 2010;10:87–101. doi: 10.1038/nrc2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma P, Falk GW, Weston AP, et al. Dysplasia and cancer in a large multicenter cohort of patients with Barrett’s esophagus. Clin Gastroenterol Hepatol. 2006;4:566–72. doi: 10.1016/j.cgh.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Anderson LA, Murray LJ, Murphy SJ, et al. Mortality in Barrett’s oesophagus: results from a population based study. Gut. 2003;52:1081–4. doi: 10.1136/gut.52.8.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conio M, Cameron AJ, Romero Y, et al. Secular trends in the epidemiology and outcome of Barrett’s oesophagus in Olmsted County, Minnesota. Gut. 2001;48:304–9. doi: 10.1136/gut.48.3.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hage M, Siersema PD, van Dekken H, et al. Oesophageal cancer incidence and mortality in patients with long-segment Barrett’s oesophagus after a mean follow-up of 12. 7 years. Scand J Gastroenterol. 2004;39:1175–9. doi: 10.1080/00365520410003524. [DOI] [PubMed] [Google Scholar]
- 7.Macdonald CE, Wicks AC, Playford RJ. Final results from 10 year cohort of patients undergoing surveillance for Barrett’s oesophagus: observational study. Bmj. 2000;321:1252–5. doi: 10.1136/bmj.321.7271.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moayyedi P, Burch N, Akhtar-Danesh N, et al. Mortality rates in patients with Barrett’s oesophagus. Aliment Pharmacol Ther. 2008;27:316–20. doi: 10.1111/j.1365-2036.2007.03582.x. [DOI] [PubMed] [Google Scholar]
- 9.Solaymani-Dodaran M, Logan RF, West J, et al. Mortality associated with Barrett’s esophagus and gastroesophageal reflux disease diagnoses-a population-based cohort study. Am J Gastroenterol. 2005;100:2616–21. doi: 10.1111/j.1572-0241.2005.00340.x. [DOI] [PubMed] [Google Scholar]
- 10.Hvid-Jensen F, Pedersen L, Drewes AM, et al. Incidence of adenocarcinoma among patients with Barrett’s esophagus. N Engl J Med. 2011;365:1375–83. doi: 10.1056/NEJMoa1103042. [DOI] [PubMed] [Google Scholar]
- 11.Welch HG, Black WC. Overdiagnosis in cancer. J Natl Cancer Inst. 2010;102:605–13. doi: 10.1093/jnci/djq099. [DOI] [PubMed] [Google Scholar]
- 12.Etzioni R, Urban N, Ramsey S, et al. The case for early detection. Nat Rev Cancer. 2003;3:243–52. doi: 10.1038/nrc1041. [DOI] [PubMed] [Google Scholar]
- 13.Vaughan TL, Fitzgerald RC. Precision prevention of oesophageal adenocarcinoma. Nat Rev Gastroenterol Hepatol. 2015;12:243–248. doi: 10.1038/nrgastro.2015.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barrett NR. Chronic peptic ulcer of the oesophagus and “oesophagitis”. British Journal of Surgery. 1950;38:175–182. doi: 10.1002/bjs.18003815005. [DOI] [PubMed] [Google Scholar]
- 15.Allison PR, Johnstone AS. The oesophagus lined with gastric mucous membrane. Thorax. 1953;8:87–101. doi: 10.1136/thx.8.2.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barrett NR. The lower esophagus lined by columnar epithelium. Surg. 1957;41:881–894. [PubMed] [Google Scholar]
- 17.Johns BAE. Developmental changes in the oesophageal epithelium in man. J Anat. 1952;86:431–439. [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Ouyang H, Yamamoto Y, et al. Residual embryonic cells as precursors of a Barrett’s-like metaplasia. Cell. 2011;145:1023–35. doi: 10.1016/j.cell.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naef AP, Savary M, Ozzello L. Columnar-lined lower esophagus: an acquired lesion with malignant predisposition. Report on 140 cases of Barrett’s esophagus with 12 adenocarcinomas. Journal of Thoracic and Cardiovascular Surgery. 1975;70:826–35. [PubMed] [Google Scholar]
- 20.Blot WJ, Devesa SS, Kneller RW, et al. Rising incidence of adenocarcinoma of the esophagus and gastric cardia. JAMA. 1991;265:1287–9. [PubMed] [Google Scholar]
- 21.Cook MB, Chow WH, Devesa SS. Oesophageal cancer incidence in the United States by race, sex, and histologic type, 1977–2005. Br J Cancer. 2009;101:855–9. doi: 10.1038/sj.bjc.6605246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castro C, Bosetti C, Malvezzi M, et al. Patterns and trends in esophageal cancer mortality and incidence in Europe (1980–2011) and predictions to 2015. Ann Oncol. 2014;25:283–90. doi: 10.1093/annonc/mdt486. [DOI] [PubMed] [Google Scholar]
- 23.Kroep S, Lansdorp-Vogelaar I, Rubenstein JH, et al. Comparing trends in esophageal adenocarcinoma incidence and lifestyle factors between the United States, Spain, and the Netherlands. Am J Gastroenterol. 2014;109:336–43. doi: 10.1038/ajg.2013.420. quiz 335, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sampliner RE. Practice guidelines on the diagnosis, surveillance, and therapy of Barrett’s esophagus. The Practice Parameters Committee of the American College of Gastroenterology. Am J Gastroenterol. 1998;93:1028–32. doi: 10.1111/j.1572-0241.1998.00362.x. [DOI] [PubMed] [Google Scholar]
- 25.Croswell JM, Ransohoff DF, Kramer BS. Principles of cancer screening: lessons from history and study design issues. Semin Oncol. 2010;37:202–15. doi: 10.1053/j.seminoncol.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bytzer P, Christensen PB, Damkier P, et al. Adenocarcinoma of the esophagus and Barrett’s esophagus: a population- based study. Am J Gastroenterol. 1999;94:86–91. doi: 10.1111/j.1572-0241.1999.00776.x. [DOI] [PubMed] [Google Scholar]
- 27.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–8. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 28.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 29.Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science. 2013;339:1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sprouffske K, Pepper JW, Maley CC. Accurate reconstruction of the temporal order of mutations in neoplastic progression. Cancer Prev Res (Phila) 2011;4:1135–44. doi: 10.1158/1940-6207.CAPR-10-0374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barrett MT, Sanchez CA, Prevo LJ, et al. Evolution of neoplastic cell lineages in Barrett oesophagus. Nat Gen. 1999;22:106–9. doi: 10.1038/8816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Bruin EC, McGranahan N, Mitter R, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014;346:251–6. doi: 10.1126/science.1253462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baca SC, Prandi D, Lawrence MS, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153:666–77. doi: 10.1016/j.cell.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stephens PJ, Greenman CD, Fu B, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones S, Chen WD, Parmigiani G, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A. 2008;105:4283–8. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Darwin C. On the Origin of Species. John Murray; 1859. [Google Scholar]
- 38.Mendel G. Versuche über Plflanzen-hybriden. Verhandlungen des naturforschenden Ver-eines in Brünn. 1865:3–47. [Google Scholar]
- 39.Aktipis CA, Kwan VS, Johnson KA, et al. Overlooking evolution: a systematic analysis of cancer relapse and therapeutic resistance research. PLoS One. 2011;6:e26100. doi: 10.1371/journal.pone.0026100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–13. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carter SL, Cibulskis K, Helman E, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–21. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maley CC, Galipeau PC, Finley JC, et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet. 2006;38:468–73. doi: 10.1038/ng1768. [DOI] [PubMed] [Google Scholar]
- 43.Li X, Blount PL, Vaughan TL, et al. Application of biomarkers in cancer risk management: evaluation from stochastic clonal evolutionary and dynamic system optimization points of view. PLoS Comput Biol. 2011;7:e1001087. doi: 10.1371/journal.pcbi.1001087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li X, Galipeau PC, Paulson TG, et al. Temporal and spatial evolution of somatic chromosomal alterations: a case-cohort study of Barrett’s esophagus. Cancer Prev Res (Phila) 2014;7:114–27. doi: 10.1158/1940-6207.CAPR-13-0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Agrawal N, Jiao Y, Bettegowda C, et al. Comparative Genomic Analysis of Esophageal Adenocarcinoma and Squamous Cell Carcinoma. Cancer Discov. 2012;2:899–905. doi: 10.1158/2159-8290.CD-12-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dulak AM, Schumacher SE, van Lieshout J, et al. Gastrointestinal adenocarcinomas of the esophagus, stomach, and colon exhibit distinct patterns of genome instability and oncogenesis. Cancer Res. 2012;72:4383–93. doi: 10.1158/0008-5472.CAN-11-3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dulak AM, Stojanov P, Peng S, et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet. 2013;45:478–86. doi: 10.1038/ng.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nones K, Waddell N, Wayte N, et al. Genomic catastrophes frequently arise in esophageal adenocarcinoma and drive tumorigenesis. Nat Commun. 2014;5:5224. doi: 10.1038/ncomms6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bandla S, Pennathur A, Luketich JD, et al. Comparative genomics of esophageal adenocarcinoma and squamous cell carcinoma. Ann Thorac Surg. 2012;93:1101–6. doi: 10.1016/j.athoracsur.2012.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gu J, Ajani JA, Hawk ET, et al. Genome-wide catalogue of chromosomal aberrations in barrett’s esophagus and esophageal adenocarcinoma: a high-density single nucleotide polymorphism array analysis. Cancer Prev Res (Phila) 2010;3:1176–86. doi: 10.1158/1940-6207.CAPR-09-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paulson TG, Maley CC, Li X, et al. Chromosomal instability and copy number alterations in Barrett’s esophagus and esophageal adenocarcinoma. Clin Cancer Res. 2009;15:3305–14. doi: 10.1158/1078-0432.CCR-08-2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nancarrow DJ, Handoko HY, Smithers BM, et al. Genome-wide copy number analysis in esophageal adenocarcinoma using high-density single-nucleotide polymorphism arrays. Cancer Res. 2008;68:4163–72. doi: 10.1158/0008-5472.CAN-07-6710. [DOI] [PubMed] [Google Scholar]
- 53.Chak A, Ochs-Balcom H, Falk G, et al. Familiality in Barrett’s esophagus, adenocarcinoma of the esophagus, and adenocarcinoma of the gastroesophageal junction. Cancer Epidemiol Biomarkers Prev. 2006;15:1668–73. doi: 10.1158/1055-9965.EPI-06-0293. [DOI] [PubMed] [Google Scholar]
- 54.Groves C, Jankowski J, Barker F, et al. A family history of Barrett’s oesophagus: another risk factor? Scand J Gastroenterol. 2005;40:1127–8. doi: 10.1080/00365520510023189. [DOI] [PubMed] [Google Scholar]
- 55.Munitiz V, Parrilla P, Ortiz A, et al. High risk of malignancy in familial Barrett’s esophagus: presentation of one family. J Clin Gastroenterol. 2008;42:806–9. doi: 10.1097/MCG.0b013e3180329015. [DOI] [PubMed] [Google Scholar]
- 56.Poynton AR, Walsh TN, O’Sullivan G, et al. Carcinoma arising in familial Barrett’s esophagus. Am J Gastroenterol. 1996;91:1855–6. [PubMed] [Google Scholar]
- 57.Romero Y, Cameron AJ, Locke GR, 3rd, et al. Familial aggregation of gastroesophageal reflux in patients with Barrett’s esophagus and esophageal adenocarcinoma. Gastroenterology. 1997;113:1449–56. doi: 10.1053/gast.1997.v113.pm9352846. [DOI] [PubMed] [Google Scholar]
- 58.Orloff M, Peterson C, He X, et al. Germline mutations in MSR1, ASCC1, and CTHRC1 in patients with Barrett esophagus and esophageal adenocarcinoma. JAMA. 2011;306:410–9. doi: 10.1001/jama.2011.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Levine DM, Ek WE, Zhang R, et al. A genome-wide association study identifies new susceptibility loci for esophageal adenocarcinoma and Barrett’s esophagus. Nat Genet. 2013;45:1487–93. doi: 10.1038/ng.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Palles C, Chegwidden L, Li X, et al. Polymorphisms near TBX5 and GDF7 are associated with increased risk for Barrett’s esophagus. Gastroenterology. 2015;148:367–78. doi: 10.1053/j.gastro.2014.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Su Z, Gay LJ, Strange A, et al. Common variants at the MHC locus and at chromosome 16q24. 1 predispose to Barrett’s esophagus. Nat Genet. 2012;44:1131–6. doi: 10.1038/ng.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rajagopalan H, Nowak MA, Vogelstein B, et al. The significance of unstable chromosomes in colorectal cancer. Nature Reviews Cancer. 2003;3:695–701. doi: 10.1038/nrc1165. [DOI] [PubMed] [Google Scholar]
- 63.Zack TI, Schumacher SE, Carter SL, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45:1134–1140. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reid BJ, Kostadinov R, Maley CC. New strategies in Barrett’s esophagus: integrating clonal evolutionary theory with clinical management. Clin Cancer Res. 2011;17:3512–9. doi: 10.1158/1078-0432.CCR-09-2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leedham SJ, Preston SL, McDonald SA, et al. Individual crypt genetic heterogeneity and the origin of metaplastic glandular epithelium in human Barrett’s oesophagus. Gut. 2008;57:1041–8. doi: 10.1136/gut.2007.143339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Graham TA, McDonald SA. Genetic diversity during the development of Barrett’s oesophagus-associated adenocarcinoma: how, when and why? Biochem Soc Trans. 2010;38:374–9. doi: 10.1042/BST0380374. [DOI] [PubMed] [Google Scholar]
- 67.Nicholson AM, Graham TA, Simpson A, et al. Barrett’s metaplasia glands are clonal, contain multiple stem cells and share a common squamous progenitor. Gut. 2012;61:1380–9. doi: 10.1136/gutjnl-2011-301174. [DOI] [PubMed] [Google Scholar]
- 68.Rabinovitch PS, Longton G, Blount PL, et al. Predictors of progression in Barrett’s esophagus III: baseline flow cytometric variables. American Journal of Gastroenterology. 2001;96:3071–83. doi: 10.1111/j.1572-0241.2001.05261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reid BJ, Levine DS, Longton G, et al. Predictors of progression to cancer in Barrett’s esophagus: baseline histology and flow cytometry identify low- and high-risk patient subsets. American Journal of Gastroenterology. 2000;95:1669–76. doi: 10.1111/j.1572-0241.2000.02196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Robaszkiewicz M, Hardy E, Volant A, et al. Flow cytometric analysis of cellular DNA content in Barret’s esophagus. A study of 66 cases. Gastroenterol Clin Biol. 1991;15:703–10. [PubMed] [Google Scholar]
- 71.Teodori L, Gohde W, Persiani M, et al. DNA/protein flow cytometry as a predictive marker of malignancy in dysplasia-free Barrett’s esophagus: thirteen-year follow-up study on a cohort of patients. Cytometry. 1998;34:257–63. doi: 10.1002/(sici)1097-0320(19981215)34:6<257::aid-cyto3>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 72.Dunn JM, Mackenzie GD, Oukrif D, et al. Image cytometry accurately detects DNA ploidy abnormalities and predicts late relapse to high-grade dysplasia and adenocarcinoma in Barrett’s oesophagus following photodynamic therapy. Br J Cancer. 2010;102:1608–17. doi: 10.1038/sj.bjc.6605688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fennerty MB, Sampliner RE, Way D, et al. Discordance between flow cytometric abnormalities and dysplasia in Barrett’s esophagus. Gastroenterology. 1989;97:815–20. doi: 10.1016/0016-5085(89)91483-2. [DOI] [PubMed] [Google Scholar]
- 74.Rygiel AM, Milano F, Ten Kate FJ, et al. Assessment of chromosomal gains as compared to DNA content changes is more useful to detect dysplasia in Barrett’s esophagus brush cytology specimens. Genes Chromosomes Cancer. 2008;47:396–404. doi: 10.1002/gcc.20543. [DOI] [PubMed] [Google Scholar]
- 75.Fritcher EG, Brankley SM, Kipp BR, et al. A comparison of conventional cytology, DNA ploidy analysis, and fluorescence in situ hybridization for the detection of dysplasia and adenocarcinoma in patients with Barrett’s esophagus. Hum Pathol. 2008;39:1128–35. doi: 10.1016/j.humpath.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Doak SH, Jenkins GJ, Parry EM, et al. Chromosome 4 hyperploidy represents an early genetic aberration in premalignant Barrett’s oesophagus. Gut. 2003;52:623–8. doi: 10.1136/gut.52.5.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Persons DL, Croughan WS, Borelli KA, et al. Interphase cytogenetics of esophageal adenocarcinoma and precursor lesions. Cancer Genet Cytogenet. 1998;106:11–7. doi: 10.1016/s0165-4608(98)00036-3. [DOI] [PubMed] [Google Scholar]
- 78.Beuzen F, Dubois S, Flejou JF. Chromosomal numerical aberrations are frequent in oesophageal and gastric adenocarcinomas: a study using in-situ hybridization. Histopathology. 2000;37:241–9. doi: 10.1046/j.1365-2559.2000.00887.x. [DOI] [PubMed] [Google Scholar]
- 79.Chaves P, Crespo M, Ribeiro C, et al. Chromosomal analysis of Barrett’s cells: demonstration of instability and detection of the metaplastic lineage involved. Mod Pathol. 2007;20:788–96. doi: 10.1038/modpathol.3800787. [DOI] [PubMed] [Google Scholar]
- 80.Rygiel AM, Milano F, Ten Kate FJ, et al. Gains and amplifications of c-myc, EGFR, and 20. q13 loci in the no dysplasia-dysplasia-adenocarcinoma sequence of Barrett’s esophagus. Cancer Epidemiol Biomarkers Prev. 2008;17:1380–5. doi: 10.1158/1055-9965.EPI-07-2734. [DOI] [PubMed] [Google Scholar]
- 81.El-Rifai W, Frierson HF, Jr, Moskaluk CA, et al. Genetic differences between adenocarcinomas arising in Barrett’s esophagus and gastric mucosa. Gastroenterology. 2001;121:592–8. doi: 10.1053/gast.2001.27215. [DOI] [PubMed] [Google Scholar]
- 82.Miller CT, Moy JR, Lin L, et al. Gene amplification in esophageal adenocarcinomas and Barrett’s with high-grade dysplasia. Clin Cancer Res. 2003;9:4819–25. [PubMed] [Google Scholar]
- 83.Frankel A, Armour N, Nancarrow D, et al. Genome-wide analysis of esophageal adenocarcinoma yields specific copy number aberrations that correlate with prognosis. Genes Chromosomes Cancer. 2014;53:324–38. doi: 10.1002/gcc.22143. [DOI] [PubMed] [Google Scholar]
- 84.Lai LA, Paulson TG, Li X, et al. Increasing genomic instability during premalignant neoplastic progression revealed through high resolution array-CGH. Genes Chromosomes Cancer. 2007;46:532–42. doi: 10.1002/gcc.20435. [DOI] [PubMed] [Google Scholar]
- 85.Riegman PH, Vissers KJ, Alers JC, et al. Genomic alterations in malignant transformation of Barrett’s esophagus. Cancer Res. 2001;61:3164–70. [PubMed] [Google Scholar]
- 86.Michael D, Beer DG, Wilke CW, et al. Frequent deletions of FHIT and FRA3B in Barrett’s metaplasia and esophageal adenocarcinomas. Oncogene. 1997;15:1653–9. doi: 10.1038/sj.onc.1201330. [DOI] [PubMed] [Google Scholar]
- 87.Meltzer SJ, Yin J, Huang Y, et al. Reduction to homozygostiy involving p53 in esophageal cancers demonstrated by the polymerase chain reaction. Biochemistry. 1991;88:4976–4980. doi: 10.1073/pnas.88.11.4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Prevo LJ, Sanchez CA, Galipeau PC, et al. p53-mutant clones and field effects in Barrett’s esophagus. Cancer Research. 1999;59:4784–7. [PubMed] [Google Scholar]
- 89.Reid BJ, Prevo LJ, Galipeau PC, et al. Predictors of progression in Barrett’s esophagus II: baseline 17p (p53) loss of heterozygosity identifies a patient subset at increased risk for neoplastic progression. Am J Gastroenterol. 2001;96:2839–48. doi: 10.1111/j.1572-0241.2001.04236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Blount PL, Galipeau PC, Sanchez CA, et al. 17p allelic losses in diploid cells of patients with Barrett’s esophagus who develop aneuploidy. Cancer Research. 1994;54:2292–5. [PubMed] [Google Scholar]
- 91.Dolan K, Garde J, Gosney J, et al. Allelotype analysis of oesophageal adenocarcinoma: loss of heterozygosity occurs at multiple sites. Br J Cancer. 1998;78:950–7. doi: 10.1038/bjc.1998.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gonzalez MV, Artimez ML, Rodrigo L, et al. Mutation analysis of the p53, APC, and p16 genes in the Barrett’s oesophagus, dysplasia, and adenocarcinoma. J Clin Pathol. 1997;50:212–7. doi: 10.1136/jcp.50.3.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Audrezet MP, Robaszkiewicz M, Mercier B, et al. Molecular analysis of the TP53 gene in Barrett’s adenocarcinoma. Hum Mutat. 1996;7:109–13. doi: 10.1002/(SICI)1098-1004(1996)7:2<109::AID-HUMU4>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 94.Dolan K, Walker SJ, Gosney J, et al. TP53 mutations in malignant and premalignant Barrett’s esophagus. Dis Esophagus. 2003;16:83–9. doi: 10.1046/j.1442-2050.2003.00302.x. [DOI] [PubMed] [Google Scholar]
- 95.Moore JH, Lesser EJ, Erdody DH, et al. Intestinal differentiation and p53 gene alterations in Barrett’s esophagus and esophageal adenocarcinoma. Int J Cancer. 1994;56:487–93. doi: 10.1002/ijc.2910560406. [DOI] [PubMed] [Google Scholar]
- 96.Schneider PM, Casson AG, Levin B, et al. Mutations of p53 in Barrett’s esophagus and Barrett’s cancer: a prospective study of ninety-eight cases. J Thorac Cardiovasc Surg. 1996;111:323–31. doi: 10.1016/s0022-5223(96)70441-5. discussion 331–3. [DOI] [PubMed] [Google Scholar]
- 97.Muzeau F, Flejou JF, Thomas G, et al. Loss of heterozygosity on chromosome 9 and p16 (MTS1, CDKN2) gene mutations in esophageal cancers. Int J Cancer. 1997;72:27–30. doi: 10.1002/(sici)1097-0215(19970703)72:1<27::aid-ijc3>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 98.Paulson TG, Galipeau PC, Xu L, et al. p16 mutation spectrum in the premalignant condition Barrett’s esophagus. PLoS One. 2008;3:e3809. doi: 10.1371/journal.pone.0003809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wong DJ, Paulson TG, Prevo LJ, et al. p16(INK4a) lesions are common, early abnormalities that undergo clonal expansion in Barrett’s metaplastic epithelium. Cancer Res. 2001;61:8284–9. [PubMed] [Google Scholar]
- 100.Galipeau PC, Li X, Blount PL, et al. NSAIDs modulate CDKN2A, TP53, and DNA content risk for future esophageal adenocarcinoma. PLoS Med. 2007;4:e67. doi: 10.1371/journal.pmed.0040067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Streppel MM, Lata S, DelaBastide M, et al. Next-generation sequencing of endoscopic biopsies identifies ARID1A as a tumor-suppressor gene in Barrett’s esophagus. Oncogene. 2014;33:347–57. doi: 10.1038/onc.2012.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang K, Yuen ST, Xu J, et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet. 2014;46:573–82. doi: 10.1038/ng.2983. [DOI] [PubMed] [Google Scholar]
- 104.The Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Weaver JM, Ross-Innes CS, Shannon N, et al. Ordering of mutations in preinvasive disease stages of esophageal carcinogenesis. Nat Genet. 2014;46:837–43. doi: 10.1038/ng.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Goh XY, Rees JR, Paterson AL, et al. Integrative analysis of array-comparative genomic hybridisation and matched gene expression profiling data reveals novel genes with prognostic significance in oesophageal adenocarcinoma. Gut. 2011;60:1317–26. doi: 10.1136/gut.2010.234179. [DOI] [PubMed] [Google Scholar]
- 108.Shackney SE, Shankey TV. Common patterns of genetic evolution in human solid tumors. Cytometry. 1997;29:1–27. [PubMed] [Google Scholar]
- 109.Finley JC, Reid BJ, Odze RD, et al. Chromosomal instability in Barrett’s esophagus is related to telomere shortening. Cancer Epidemiol Biomarkers Prev. 2006;15:1451–7. doi: 10.1158/1055-9965.EPI-05-0837. [DOI] [PubMed] [Google Scholar]
- 110.Risques RA, Vaughan TL, Li X, et al. Leukocyte telomere length predicts cancer risk in Barrett’s esophagus. Cancer Epidemiol Biomarkers Prev. 2007;16:2649–55. doi: 10.1158/1055-9965.EPI-07-0624. [DOI] [PubMed] [Google Scholar]
- 111.Xing J, Ajani JA, Chen M, et al. Constitutive short telomere length of chromosome 17p and 12q but not 11q and 2p is associated with an increased risk for esophageal cancer. Cancer Prev Res (Phila Pa) 2009;2:459–65. doi: 10.1158/1940-6207.CAPR-08-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lai LA, Kostadinov R, Barrett MT, et al. Deletion at fragile sites is a common and early event in Barrett’s esophagus. Mol Cancer Res. 2010;8:1084–94. doi: 10.1158/1541-7786.MCR-09-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li X, Galipeau PC, Sanchez CA, et al. Single nucleotide polymorphism-based genome-wide chromosome copy change, loss of heterozygosity, and aneuploidy in Barrett’s esophagus neoplastic progression. Cancer Prev Res (Phila Pa) 2008;1:413–23. doi: 10.1158/1940-6207.CAPR-08-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Greaves M. Does everyone develop covert cancer? Nat Rev Cancer. 2014;14:209–10. doi: 10.1038/nrc3703. [DOI] [PubMed] [Google Scholar]
- 115.Ciriello G, Miller ML, Aksoy BA, et al. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45:1127–1133. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Galipeau PC, Cowan DS, Sanchez CA, et al. 17p (p53) allelic losses, 4N (G2/tetraploid) populations, and progression to aneuploidy in Barrett’s esophagus. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:7081–4. doi: 10.1073/pnas.93.14.7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lee S, Han MJ, Lee KS, et al. Frequent occurrence of mitochondrial DNA mutations in Barrett’s metaplasia without the presence of dysplasia. PLoS ONE. 2012;7:e37571. doi: 10.1371/journal.pone.0037571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Miyazono F, Schneider PM, Metzger R, et al. Mutations in the mitochondrial DNA D-Loop region occur frequently in adenocarcinoma in Barrett’s esophagus. Oncogene. 2002;21:3780–3. doi: 10.1038/sj.onc.1205532. [DOI] [PubMed] [Google Scholar]
- 119.Suchorolski MT, Paulson TG, Sanchez CA, et al. Warburg and Crabtree effects in premalignant Barrett’s esophagus cell lines with active mitochondria. PLoS One. 2013;8:e56884. doi: 10.1371/journal.pone.0056884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tan BH, Skipworth RJ, Stephens NA, et al. Frequency of the mitochondrial DNA 4977bp deletion in oesophageal mucosa during the progression of Barrett’s oesophagus. Eur J Cancer. 2009;45:736–40. doi: 10.1016/j.ejca.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 121.Ek WE, Levine DM, D’Amato M, et al. Germline genetic contributions to risk for esophageal adenocarcinoma, Barrett’s esophagus, and gastroesophageal reflux. J Natl Cancer Inst. 2013;105:1711–8. doi: 10.1093/jnci/djt303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chak A, Faulx A, Kinnard M, et al. Identification of Barrett’s esophagus in relatives by endoscopic screening. Am J Gastroenterol. 2004;99:2107–14. doi: 10.1111/j.1572-0241.2004.40464.x. [DOI] [PubMed] [Google Scholar]
- 123.Cameron AJ, Lagergren J, Henriksson C, et al. Gastroesophageal reflux disease in monozygotic and dizygotic twins. Gastroenterology. 2002;122:55–9. doi: 10.1053/gast.2002.30301. [DOI] [PubMed] [Google Scholar]
- 124.Mohammed I, Cherkas LF, Riley SA, et al. Genetic influences in gastro-oesophageal reflux disease: a twin study. Gut. 2003;52:1085–9. doi: 10.1136/gut.52.8.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ochs-Balcom HM, Falk G, Grady WM, et al. Consortium approach to identifying genes for Barrett’s esophagus and esophageal adenocarcinoma. Transl Res. 2007;150:3–17. doi: 10.1016/j.trsl.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 126.Buas MF, Levine DM, Makar KW, et al. Integrative post-genome-wide association analysis of CDKN2A and TP53 SNPs and risk of esophageal adenocarcinoma. Carcinogenesis. 2014;35:2740–7. doi: 10.1093/carcin/bgu207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Berra Y. The Yogi Book. Workman Publishing Company; 1998. [Google Scholar]
- 128.Zeki SS, Haidry R, Graham TA, et al. Clonal selection and persistence in dysplastic Barrett’s esophagus and intramucosal cancers after failed radiofrequency ablation. Am J Gastroenterol. 2013;108:1584–92. doi: 10.1038/ajg.2013.238. [DOI] [PubMed] [Google Scholar]
- 129.Dua KS, Merrill J, Komorowski R. Neosquamous epithelium after ablation of Barrett’s epithelium: cause for concern? Gastrointest Endosc. 2012;76:1082–3. doi: 10.1016/j.gie.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 130.Allende D, Dumot J, Yerian L. Esophageal squamous cell carcinoma arising after endoscopic ablation therapy of Barrett’s esophagus with high-grade dysplasia. Report of a case Dis Esophagus. 2013;26:314–8. doi: 10.1111/j.1442-2050.2012.01411.x. [DOI] [PubMed] [Google Scholar]
- 131.Gorospe EC, Gupta M, Prasad GA, et al. Double trouble: two cases of squamous carcinoma arising from Barrett’s dysplasia after endoscopic mucosal resection. Am J Gastroenterol. 2012;107:1595–6. doi: 10.1038/ajg.2012.232. [DOI] [PubMed] [Google Scholar]
- 132.Orlando RC. How good is the neosquamous epithelium? Dig Dis. 2014;32:164–70. doi: 10.1159/000357185. [DOI] [PubMed] [Google Scholar]
- 133.Cairns J. Mutation Selection and the Natural History of Cancer. Nature. 1975;255:197–200. doi: 10.1038/255197a0. [DOI] [PubMed] [Google Scholar]
- 134.Thrift AP, Shaheen NJ, Gammon MD, et al. Obesity and risk of esophageal adenocarcinoma and Barrett’s esophagus: a Mendelian randomization study. J Natl Cancer Inst. 2014;106 doi: 10.1093/jnci/dju252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sager R, Gadi IK, Stephens L, et al. Gene amplification: an example of accelerated evolution in tumorigenic cells. Proc Natl Acad Sci U S A. 1985;82:7015–9. doi: 10.1073/pnas.82.20.7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Srivastava S, Gray JW, Reid BJ, et al. Translational Research Working Group developmental pathway for biospecimen-based assessment modalities. Clin Cancer Res. 2008;14:5672–7. doi: 10.1158/1078-0432.CCR-08-1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sullivan Pepe M, Etzioni R, Feng Z, et al. Phases of biomarker development for early detection of cancer. J Natl Cancer Inst. 2001;93:1054–61. doi: 10.1093/jnci/93.14.1054. [DOI] [PubMed] [Google Scholar]
- 138.Fleming TR, Prentice RL, Pepe MS, et al. Surrogate and auxiliary endpoints in clinical trials, with potential applications in cancer and AIDS research. Statistics in Medicine. 1994;13:955–68. doi: 10.1002/sim.4780130906. [DOI] [PubMed] [Google Scholar]
- 139.Prentice RL. Surrogate endpoints in clinical trials: definition and operational criteria. Stat Med. 1989;8:431–40. doi: 10.1002/sim.4780080407. [DOI] [PubMed] [Google Scholar]
- 140.Montgomery E, Bronner MP, Goldblum JR, et al. Reproducibility of the diagnosis of dysplasia in Barrett esophagus: a reaffirmation. Hum Pathol. 2001;32:368–78. doi: 10.1053/hupa.2001.23510. [DOI] [PubMed] [Google Scholar]
- 141.Reid BJ, Haggitt RC, Rubin CE, et al. Observer variation in the diagnosis of dysplasia in Barrett’s esophagus. Hum Pathol. 1988;19:166–78. doi: 10.1016/s0046-8177(88)80344-7. [DOI] [PubMed] [Google Scholar]
- 142.Odze RD. What the gastroenterologist needs to know about the histology of Barrett’s esophagus. Curr Opin Gastroenterol. 2011;27:389–96. doi: 10.1097/MOG.0b013e328346f551. [DOI] [PubMed] [Google Scholar]
- 143.Schnell TG, Sontag SJ, Chejfec G, et al. Long-term nonsurgical management of Barrett’s esophagus with high-grade dysplasia. Gastroenterology. 2001;120:1607–19. doi: 10.1053/gast.2001.25065. [DOI] [PubMed] [Google Scholar]
- 144.Weston AP, Sharma P, Topalovski M, et al. Long-term follow-up of Barrett’s high-grade dysplasia. Am J Gastroenterol. 2000;95:1888–93. doi: 10.1111/j.1572-0241.2000.02234.x. [DOI] [PubMed] [Google Scholar]
- 145.Rabinovitch PS, Reid BJ, Haggitt RC, et al. Progression to cancer in Barrett’s esophagus is associated with genomic instability. Laboratory Investigation. 1988;60:65–71. [PubMed] [Google Scholar]
- 146.Quante M, Bhagat G, Abrams JA, et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell. 2012;21:36–51. doi: 10.1016/j.ccr.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ordonez GR, Hillier LW, Warren WC, et al. Loss of genes implicated in gastric function during platypus evolution. Genome Biol. 2008;9:R81. doi: 10.1186/gb-2008-9-5-r81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Fleming TR, DeMets DL. Surrogate end points in clinical trials: are we being misled? Ann Intern Med. 1996;125:605–13. doi: 10.7326/0003-4819-125-7-199610010-00011. [DOI] [PubMed] [Google Scholar]
- 149.Fleming TR, Powers JH. Biomarkers and surrogate endpoints in clinical trials. Stat Med. 2012;31:2973–84. doi: 10.1002/sim.5403. [DOI] [PMC free article] [PubMed] [Google Scholar]