

Abstract
Reconstituting tissues from their cellular building blocks facilitates the modeling of morphogenesis, homeostasis, and disease in vitro. Here, we describe DNA Programmed Assembly of Cells (DPAC) to reconstitute the multicellular organization of tissues having programmed size, shape, composition, and spatial heterogeneity. DPAC uses dissociated cells that are chemically functionalized with degradable oligonucleotide “velcro,” allowing rapid, specific, and reversible cell adhesion to other surfaces coated with complementary DNA sequences. DNA-patterned substrates function as removable and adhesive templates, and layer-by-layer DNA-programmed assembly builds arrays of tissues into the third dimension above the template. DNase releases completed arrays of microtissues from the template concomitant with full embedding in a variety of extracellular matrix (ECM) gels. DPAC positions subpopulations of cells with single-cell spatial resolution and generates cultures several centimeters long. We used DPAC to explore the impact of ECM composition, heterotypic cell-cell interactions, and patterns of signaling heterogeneity on collective cell behaviors.
INTRODUCTION
The functional properties of tissues arise through interactions of numerous cell types1. In vivo, these interactions occur in a three-dimensional (3D) setting in the context of specific tissue structures. Tissue structure – defined here as tissue size, shape, composition, spatial heterogeneity (i.e. the relative location of multiple cell types), and the surrounding ECM – serves to organize the exchange of chemical, electrical, and mechanical information between neighboring and distant cells. An orderly exchange of signals allows cells to arrive at collective decisions and organize collective behaviors2. Defining the impact of a tissue’s structure on the behavior of its constituent cells remains a major goal of developmental biology and is a requirement for the successful application of tissue engineering to regenerative medicine3. However, directly connecting tissue structure to collective cell behaviors remains challenging – tissue structure is difficult to alter in vivo and the inherent structural complexity of tissues has so far precluded their de novo synthesis in vitro.
The challenges inherent to controlling tissue structure in vivo have motivated efforts to reconstitute, image, and perturb specific components of tissue structure in vitro to study collective cell behaviors. Common to all efforts is 3D cell culture, a requirement for proper tissue structure formation and cell behavior4. For example, 3D culture in mechanically and chemically defined ECM gels directs the morphogenesis of stem cells and cancer cells into organoids that model normal development and tumorigenesis, respectively5–7. However, rudimentary 3D culture methods lack key microenvironmental cues from surrounding tissue components that are necessary to specify tissue architecture over larger distances. Therefore, they provide limited control over ultimate tissue architecture. Dielectrophoretic patterning and micromolding have shown the effect of tissue size and shape on cell anabolic activity, differentiation, autocrine signaling, mechanics, and tissue outgrowth8,9. However, dielectrophoresis is limited to conditions with low ionic strength, and micromolding struggles when working with multiple cell types in precise arrangements or with ECM formulations having physiological stiffness such as Matrigel (<10 kPa). A variety of techniques have demonstrated that tissue composition, often referred to as cellular heterogeneity, contributes to a spectrum of collective cell behaviors absent from homogeneous tissues10–12.
While a number of methods have contributed to our understanding of tissue structure and its effect on collective cell behaviors, it remains challenging to control tissue size, shape, composition, and ECM systematically using a single experimental system. Moreover, spatial heterogeneity has proven especially difficult to reconstitute in vitro, particularly when positioning cells in cell-dense tissues. 3D printing techniques can reconstitute spatial heterogeneity for tissues with large features but suffer from low cell viability, can be limited in their ECM compatibility, and can not build cell-dense tissues with spatial features the size of single cells13. Advanced micromolding techniques have demonstrated patterning multiple cell types in 3D culture14 but typically use mechanically stiff hydrogels, have a maximum of two tissue compartments, and lack independent control over cellular composition and spatial heterogeneity within each tissue compartment.
To provide a rapid and modular means of reconstituting multiple aspects of tissue structure in 3D culture, we envisioned a bottom-up strategy that uses a DNA-patterned substrate as a template and temporary DNA-based cellular adhesions as synthetic linkages between cellular building blocks (Fig. 1a). Specific adhesive interactions between the template and building blocks are programmed by modifying different populations of cells with reactive or lipid-modified oligonucleotide sequences10,15–19 (Fig. 1b). Cells bearing complementary DNA sequences rapidly and specifically adhere according to the rules of Watson-Crick base pairing. Microtissue structure is thereby programmed through multiple synthetic steps above the DNA-patterned template prior to release of the microtissue from the template into a supporting ECM matrix for fully embedded 3D culture and imaging (Fig. 1c). Here, we describe DNA Programmed Assembly of Cells (DPAC) as a modular method for controlling 3D microtissue structure across multiple length scales, which can incorporate multiple cell types with high viability. We demonstrate the application of DPAC to study the impact of tissue size, shape, composition, spatial heterogeneity, and embedding ECM on individual and collective cell behaviors.
Figure 1. Programming the reconstitution of fully ECM-embedded 3D microtissues by DNA-programmed assembly (DPAC).

(a) Scheme showing the relationship between DNA spots (colored squares), DNA-programmed connectivity (colored lines), and multistep assembly. (b) Incubation of cells with lipid-modified oligonucleotides results in chemical remodeling of cell surfaces. Combining cells bearing complementary cell-surface oligonucleotides forms a temporary chemical adhesion. (c) 7 μm amino-modified DNA spots are patterned onto aldehyde-coated glass slides and covalently linked to the surface by reductive amination. Cells bearing complementary cell-surface oligonucleotides are introduced above the patterned substrate at high concentration and at controlled flow rate using a flow cell. Cells adhere to the appropriate DNA spot, and excess cells are removed by gentle washing. Iteration of this process assembles the microtissue into the third dimension. Addition of liquid ECM incorporating DNase releases the assembled microtissues from the template where they are trapped in the embedding ECM as it gels. The gel is peeled off the glass, releasing the tissues. Underlay of the gel with additional ECM results in a fully embedded 3D culture. Cells interact with each other and their microenvironment as they condense into 3D microtissues. (d) Implementation of the scheme described in Figure 1a–c using MCF10A mammary epithelial cells showing (i) DNA spots, (ii) cells in flow cell, and (iii) single cell array followed by additional rounds of programmed assembly. X,Z reconstructions show an unstained MCF10A cell aggregate embedded between Alexa Fluor-488 and Alexa Fluor 555-stained layers of Matrigel at (iv) 0 and (v) 24 hr. All scale bars are 100 μm.
RESULTS
To assemble an array of epithelial microtissues embedded in ECM gels, we proceeded through a series of steps (Fig. 1c) that begin with patterning a series of ~7 μm amino-modified DNA spots on an aldehyde-coated glass slide using a Bioforce Nano eNabler20,21. Reductive amination results in a covalent linkage between the DNA and the slide. A 180 μm-tall PDMS flow cell was placed above the DNA pattern, allowing the addition of reagents and cell suspension in a minimized (30 μL) volume. The slide was passivated to background cell binding by treatment with hydrophobic silane and blocking with albumin. In parallel, two populations of human mammary epithelial cells (MCF10A) were labeled for five minutes with a 5 μM solution of either a lipid-modified oligonucleotide or its complement17,22. The cells labeled with DNA complementary to the template were introduced to the flow cell and incubated for five minutes. Single cells adhered to single DNA spots. After gentle washing, a pattern of cells matching the pattern of DNA spots on the template was revealed. Iterating with alternating populations of complementarily labeled cells assembled hemispherical microtissues, layer-by-layer, upward and outward from the single cells (Supplementary Fig. 1). Addition of Matrigel containing DNase cleaved the DNA, releasing the array of microtissues into the supporting ECM gel as it set at 37°C. Finally, the gel-encapsulated array was removed from the surface template, and an underlay of liquid ECM-gel resulted in a seamless and fully embedded 3D culture upon gelation.
To more clearly illustrate the 3D embedding process, we assembled microtissue arrays through two rounds of DPAC, but embedded the arrays in Matrigel containing covalently bound Alexa Fluor 555 and then underlaid the arrays with Matrigel containing Alexa Fluor 488. The unstained microtissues were observed at the interface of the two fluorescent gel layers. No voids were observed. The initially DNA-adherent cells were found to concentrate the gels at their surfaces over 24 hours in culture as they rearranged and condensed into microtissues (Fig. 1d). Cell viability exceeded 97% (n = 613) through assembly, transfer to ECM gels, and 24 hr culture. Microtissues polarized their cytoskeletal and adhesion machinery appropriately, consistent with previous reports10,23 (Supplementary Fig. 1).
Precise DNA surface patterning provides the opportunity for precise spatial arrangement of large numbers of single cells, and whole microtissues, for fullyembedded3Dculture. To quantify the capacity of DPAC to preserve spatial information when transferring patterns of cells from 2D to 3D, we first prepared arrays of DNA triangles with pitch of 18 or 38 μm. We used DPAC to render the DNA pattern as cells fully embedded in a Matrigel slab. Imaging of the embedded culture revealed a cell-to-cell spacing of 20 ± 2 and 40 ± 3 μm (mean ± s.d., n = 400; Fig. 2a–c). In another experiment, we varied cell spacing between two cell types in increments of several microns (Supplementary Fig. 3). To quantify the precision of cell positioning over larger distances and in less repetitive and biologically inspired arrangements, we generated a bitmap pattern from a whole mount image of a mouse mammary fat pad. We used DPAC to render the image as a 1.6 cm pattern of over 6000 single mammary epithelial cells fully embedded in Matrigel (Fig. 2d). The difference between cell positions on glass (2D) and embedded in Matrigel (3D) were visualized using a heat map (Fig. 2e–f). The majority of the differences occurred along the long, open axis of the flow cell (Supplementary Fig. 2). Expected cell-cell distances differed from actual cell-cell distances with a median of 22 μm across the whole pattern (n = 3.6 x 107 pairs) (Fig. 2g) and only 10 μm across cell pairs spaced less than 50 μm apart (n = 1.9 x 104 pairs) (Fig. 2h).
Figure 2. Cell position is preserved upon transfer of cell patterns from their template to ECM for fully embedded 3D culture.

(a) Scheme and (b) Matrigel-embedded cell triangles having a nominal cell-to-cell spacing of 18 and 38 microns, respectively. (c) Observed cell-to-cell spacing (mean ± s.d.) compared to the spacing of printed DNA spots (grey background) (n=200). (d) A whole mount image of a mouse mammary fat pad (reproduced with permission of Dr. William Muller) was digitized, used to print a pattern of DNA spots, and rendered as a 1.6 cm-long pattern of single cells fully embedded in Matrigel. (e) Globally aligned and superimposed images of the cell pattern while still attached to the glass template (green) and fully embedded in Matrigel (magenta). Global and relative differences in cell positioning were calculated using the indicated metrics. (f) Heat map illustrating differences in global cell position in 2D vs. 3D relative to the pattern center. (g) Graph generated from over 36 million cell pairs relating the difference from expected cell-to-cell distances for the pattern in (d). (h) Histogram showing deviations from expected cell-to-cell distances for all cell pairs patterned within 50 μm of one another. All scale bars are 100 μm.
We found that DPAC is compatible with varied cell types and extracellular matrices. Because cellular interactions are programmed with DNA, rather than genetically encoded adhesion molecules, the identity of the feedstock cells is arbitrary. For example, we successfully patterned primary or immortalized neuronal, epithelial, fibroblastic, endothelial, and lymphocytic cells with high resolution and yield (Supplementary Fig. 1). The choice of matrices is limited only by what can be added to the cellular pattern as a liquid and subsequently gel under biocompatible conditions. Thus, we transferred patterns of cells to Matrigel, collagen, fibrin, agarose, and their mixtures (Supplementary Fig. 1).
DPAC provides a flexible strategy for simultaneously controlling tissue size, shape, composition, spatial heterogeneity and ECM. We first demonstrated simultaneous control of tissue size and composition by showing that pairs of green and red fluorescent epithelial cells patterned closer than 18 μm apart condensed into single tissues upon transfer to Matrigel (Supplementary Fig. 3). Triangles comprising three uniquely stained epithelial cells behaved similarly (Fig. 3a). We prepared microtissues of equivalent size but different composition by performing multistep DPAC on cell triangles having two possible compositions (Fig. 3b–c). We prepared an array of over 700 microtissues containing a target of 8–13 total cells but containing either one or three fluorescent cells. For both compositions, 85% of microtissues contained the target number of total cells, and 79% of those microtissues also contained the target number of fluorescent cells. In comparision, the theoretical maximum yield for a Poisson-limited method, such as microwell molding, would be 26% or 16% for one or three fluorescent cells, respectively. We prepared larger microtissues by either increasing the area of the templating DNA pattern or further iterating layer-by-layer DPAC (Fig. 3d).
Figure 3. Reconstituting epithelial microtissues with programmed size, shape, composition, spatial heterogeneity, and embedding ECM.

(a) Scheme and images of magenta, green, and blue-stained MCF10A cells patterned with 18 and 38 μm spacing and fully embedded in Matrigel. (b) Scheme and images for Matrigel-embedded MCF10A microtissues programmed with two distinct compositions (one or three green cells) but similar average sizes. (c) Quantification of microtissue composition for data in (b). (d) Distribution of cross-sectional areas (mean ± s.d.) for microtissues assembled through each of five synthetic schemes (Supplementary Table) (for 3a, n=507. for 3b, n=640. for 4a, n=25. for S3f, n=40. for 3g, n=25.). Note that purple features (3a) come from single cell arrays, included to indicate the fundamental heterogeneity in the sizes of the cellular building blocks. (e) Scheme and average intensity projections for a multicellular assembly having three mutually perpendicular cell compartments. (f) Scheme and images of fully embedded aggregates of human luminal and myoepithelial cells. (g) Four-step synthetic scheme and images of MCF10A cells assembled into cylindrical microtissues and transferred to Matrigel/collagen mixtures. (h) Scheme, diagram, and images of cylindrical microtissues having defined patterns of spatial heterogeneity. Scale bars are 30 μm in (a), (b), and (f). Scale bars are 100 μm in (e), (g), and (h).
A unique capability of DPAC is the capacity to reconstitute cell-dense microtissues having tailored spatial heterogeneity. Unlike a printer, DPAC defines cell position by cell-cell connectivity rather than coordinates in a 3D Euclidean space. Therefore, the templating DNA pattern, and the order of addition of different DNA-functionalized populations of cells, determines the cell-to-cell connectivity of the assembled microtissue. To demonstrate this concept, we used DPAC to reconstitute microtissues consisting of three juxtaposed cellular compartments, one compartment boundary in the XY plane, the other in the XZ plane (Fig. 3e). This was accomplished using two orthogonal pairs of DNA sequences and a specific 8-step assembly scheme. We elaborated this strategy to form a microtissue having a core-shell topology similar to the human mammary gland. We assembled primary human luminal (LEP) and myoepithelial (MEP) cells using two orthogonal DNA sequences, a 6-step DPAC scheme, and a bull’s eye-shaped templating pattern. When released from the template and fully embedded in Matrigel, the microtissue retained the programmed topology, which was reinforced after 24 hr in culture (Fig. 3f). Some of these microtissues lumenized over 72 hr (Supplementary Fig. 3). A similar strategy was used to prepare arrays of either homogeneous or heterogeneous filled tubes of MCF10A cells having defined patterns of spatial heterogeneity (Fig. 3g–h).
DPAC provides a means to elucidate the effects of tissue structure on collective cell behavior by allowing simultaneous control of tissue size, shape, composition, spatial heterogeneity, and surrounding ECM. We explored this enabling capacity of DPAC in several model systems.
We first explored the impact of ECM composition on organoid tissue branching. We assembled microtissues from primary human luminal and myoepithelial cells, followed by embedding in either Matrigel or collagen I. Collagen I has previously been shown to influence the branching of mouse mammary organoids. Such organoids are prepared by mincing intact tissues, giving them a wide distribution of sizes and shapes24. To control for size and shape, we used DPAC to assemble similarly sized microtissues that were initially round upon transfer to Matrigel or collagen (Fig. 4a). After 24 hour culture, collagen-embedded microtissues had reduced circularity (mean ± s.d. of 0.36 ± 0.13, n = 25) compared to Matrigel-embedded microtissues (mean ± s.d. of 0.73 ± 0.11, n = 25, p = 2.8 x 10−14, two-tailed Welch’s t-test) (Fig. 4a–b). Qualitatively, the pattern of branching in collagen resembled previous reports for randomly minced mouse mammary organoids24.
Figure 4. Measuring the impact of microtissue size, shape, composition, spatial heterogeneity, and embedding ECM on collective cell behaviors.
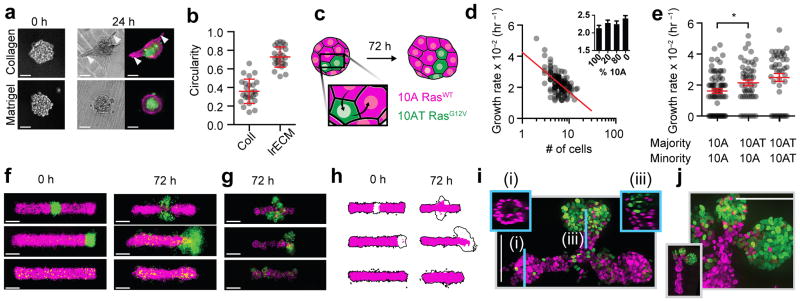
(a) Representative images of human mammary luminal and myoepithelial cells assembled through identical four-step synthetic schemes and then transferred to Matrigel or collagen-1. (b) Quantification (mean ± s.d.) of microtissue morphology for the experiment in (a) (n=25 for both conditions). (c) Scheme for assessing the impact of composition on the growth rate of 10A and H-RasG12V-expressing 10ATs. (d) The effect of initial microtissue size on cell growth rate for 10As (n=123). Inset shows growth rate (mean ± s.d.) for microtissues having different compositions. (e) Growth rates (mean ± s.d.) of single cells (minority) cultured in microtissues having the indicated majority cell-type (n=71, 49, 42). (f) Superimposed average intensity projections of 12–14 single confocal sections of 10As (magenta = H2B-mCherry) and 10ATs (green = H2B-eGFP) in Matrigel/collagen mixtures. (g) Representative epifluorescent microscopy images of microtissue after 72 hr culture. (h) 90% intensity contours of the collection of microtissues from (f). Black outline is the contour of the entire microtissue, and the magenta region is specifically the 10A component. (i) Maximum intensity projection of a center-patterned microtissue after processing using CLARITY. Insets are single confocal sections of the indicated region of the microtissue. (j) Maximum intensity projection showing detail from the branching region of an end-patterned tissue (inset) after processing using CLARITY. All scale bars are 100 μm.
We next explored the impact of tissue size on cell growth rate. We reconstituted over one thousand MCF10A (10A) microtissues ranging in size from 2–20 cells and tracked cell position over 72 hr. Analysis of the growth trajectories of individual microtissues revealed that growth rate was inversely proportional to initial microtissue size. This trend was also observed for microtissues assembled from more rapidly dividing cells expressing oncogenic H-RasV12 (10AT), as well as for microtissues bearing mixed populations of 10As and 10ATs (Fig. 4d and Supplementary Fig. 4). Proliferation rates fit a generalized logistic growth model25.
We next explored the impact of tissue composition on the growth of single cells, but in microtissues of fixed size. We synthesized an array of 5–8-cell microtissues from mixed populations of 10As and 10ATs (Fig. 4c). Expectedly, microtissues grew more rapidly as the proportion of 10ATs in the tissue increased (Fig. 4d). Unexpectedly, the rate of microtissue growth did not appear to be a linear combination of the growth rates of the two cell populations. Further investigation revealed that 10ATs triggered a statistically significant increase in the growth rate of neighboring 10As (mean 0.53 x 10−2 hr−1, p=0.04, one-way ANOVA with Holm-Sidak correction). Intriguingly, this effect appeared to require more than one 10AT cell in the tissue (p=0.03, one-way ANOVA with Holm-Sidak correction) (Supplementary Fig. 4), suggesting that even small compositional differences can alter the rate of tissue growth through cell-cell interactions.
We finally explored the impact of defined spatial heterogeneity on branching morphogenesis. During the branching morphogenesis of a variety of tissues, gradients of growth factors trigger the activation of their receptors and downstream pathways in distinct patterns of spatial heterogeneity26–29. Whether the heterogeneous patterns of pathway activation are sufficient to trigger branching tissue outgrowth, or additionally require guidance cues provided by external gradients, has not been explored. Therefore, we used DPAC to synthesize filled tubes of 10As incorporating 10% 10ATs. As 10ATs express the Ras oncogene at low levels, they simulate a population of cells with chronic stimulation of their growth factor receptors10. The 10ATs were patterned either randomly, in the middle, or at the end of the 10A filled tubes.
Cell dynamics and tissue morphology differed substantially between the three patterns over 72 hr. We visualized changes in microtissue morphology by capturing single confocal slices from at least 12 microtissues from each cell pattern, which were combined to generate average intensity maps of the fluorescent 10A and 10AT nuclei (Fi. 4f, left). 10ATs in randomly patterned tubes comingled with 10As but also extruded basally or capped local protrusions, consistent with previous reports (Fig. 4f–g and Supplementary Fig. 4)10,30. However, 10ATs patterned in the middle of tubes translated outward laterally and formed filled acini-like structures. Similar results were observed for tubes incorporating 10% 10ATs at their ends. Branching occurred along all three axes (Supplementary Fig. 4). End-patterned microtissues showed a statistically significant increase in length at 72 hours (mean ± s.d. 371 ± 38 μm, n=18, p = 7.6x10−6, two-tailed Welch’s t-test) compared to microtissues where the 10ATs were patterned in the center (mean ± s.d. 319 ± 28 μm, n=18). 90% intensity contours of 14 tissues indicated that the 10A component was also substantially longer in these tissues when compared to the microtissues having 10ATs patterned centrally or randomly (Fig. 4h). We examined the 3D structure of these microtissues qualitatively by CLARITY31(Fig. 4i and Supplementary Fig. 4). 10As formed necks connecting the filled 10AT-containing acini to the main 10A microtissue. Middle and end-patterned microtissues showed evidence of lumenization.
In vivo, epithelial tissues are supported by a variety of stromal cells, including fibroblasts, adipocytes, lymphocytes, and endothelial cells. Therefore, we explored the capacity of DPAC to reconstitute stromal cells into spatially organized 3D cultures. First, we prepared a branched pattern of endothelial cells (HUVECs) 5 mm long and fully embedded in a Matrigel/collagen mixture (Fig. 5a and Supplementary Fig. 5). After 24 hr, the pattern condensed into a continuous network of endothelial cells and formed side branches (Fig. 5a and Supplementary Fig. 5). Immunofluorescence of fixed 72 hr cultures showed evidence of phenotypic maturity including VE-cadherin localization to cell-cell junctions and exclusion from cell-ECM interfaces (Fig. 5b). To more closely mimic vasculature, we prepared microtissues of HUVECs with human brain vascular pericytes (HBVPs). At 72 hours, immunofluorescence staining revealed a subset of HBVPs stably associated with the HUVECs and extending cellular processes among endothelial cell-cell junctions (Fig. 5b)32.
Figure 5. DPAC control of stromal architecture.

(a) HUVEC cells assembled (scheme in Fig. 3h) into a 6.2 mm (corner-to-corner) network fully embedded in a Matrigel/collagen mixture. Detail shows the pattern immediately after transfer to gel and the same region after 24 hr culture. (b, top) Localization of VE-cadherin (green) at cell-cell interfaces and exclusion from cell-ECM interfaces (white arrowhead) in HUVEC networks, and (b, bottom) HUVEC networks incorporating peripheral pericytes (HBVP, magenta). (c) Morphology of HUVEC networks assembled with the indicated accessory cell type and cultured for 24 hr in a Matrigel/collagen mixture. (d) Quantification of branch length (mean ± s.d.) (n=7,9,9,5), and (e) branch density (mean ± s.d.) (n=36,59,36) in HUVEC networks incorporating the indicated accessory cell type. (f) Scheme for the assembly of a three-component microtissue incorporating epithelial and stromal cell types. (g) 3D tissue culture and detail of patterns containing perpendicularly oriented HUVEC networks and fibroblasts. (h) Analytical scheme and quantification (mean ± s.d.) of HUVEC extension in microtissues with HUVEC and fibroblast components (n=110). In (g) scale bars are 500 μm. All other scale bars are 100 μm.
Second, we explored the impact of mural cells on the frequency and length of HUVEC side branches (Fig. 5c–e). After 24 hr culture, HUVECs branched with a frequency of 1.1 mm−1 (s.d. 0.53, n=7) and an average branch length of 58 μm (s.d. 11 μm, n=36) (Fig. 5a, d-e). Smooth muscle cells (SMC) or HBVPs significantly increased the frequency (mean ± s.d. 2.7 ± 0.4 mm−1, n=9, p=0.0017, two-tailed Welch’s t-test, and mean ± s.d. 2.3 +/− 0.4 mm−1, n=5, p=0.0009, two-tailed Welch’s t-test for SMC and HBVP, respectively) (Fig. 5d) and the length (mean ± s.d. 89 ± 38 μm, n=59, p<0.0001, two-tailed Welch’s t-test and mean ± s.d. 94 ± 35 μm, n=36, p<0.0001, two-tailed Welch’s t-test) of HUVEC side branches (Fig. 5e) when assembled superficially to the HUVEC cords. Mesenchymal stem cells (MSCs) decreased the frequency of side branches (mean 0.041 mm−1, n=9, p<0.0001, two-tailed Welch’s t-test), yielding endothelial networks with remarkably smooth edges (Fig. 5c,e).
Finally, we generated a variety of microtissues having multiple and distinct epithelial and stromal compartments. These microtissues incorporated endothelial networks, fibroblasts, and epithelial cells using 6-step DPAC and three orthogonal pairs of DNA sequences (Fig. 5f–g and Supplementary Fig. 5). After 48 hr culture in collagen/Matrigel mixtures, some microtissues with perpendicularly oriented fibroblasts and HUVEC compartments distorted, with HUVECs forming extensions proximal to the patterned fibroblasts (Fig. 5g). We resynthesized microtissue arrays consisting only of correspondingly oriented fibroblasts and HUVECs, and measured increased extension of HUVEC into ECM near (mean ± s.d. 103 ± 47 μm, n=106) and far (mean ± s.d. 85 ± 38 μm, n = 106, p = 1.4x10−3, one-tailed Welch’s t-test) relative to the fibroblast compartment after 24 hr culture (Fig. 5h). These results demonstrate that the morphologies and behaviors of endothelial networks are altered by the proximity of networks of fibroblasts.
DISCUSSION
DPAC combines several unique features that provide unprecedented experimental control over microtissue structure, including size, shape, composition, spatial heterogeneity, and embedding ECM. DPAC functions as a rapid prototyping tool. A single cycle of pattern design, DNA printing, programmed assembly, and transfer into 3D ECM gels can be completed within eight hours. Moreover, hundreds of nearly identical microtissues can be assembled fully embedded within a slab of gel in a single optical plane, facilitating microscopy and statistical analysis. DPAC permits controlling the fine details of microtissue structure within multicomponent patterns spanning several centimeters. Unlike many cell-printing techniques, DPAC retains high cell viability because the rate-limiting step of the process (DNA printing) is performed in the absence of cells. Finally, DPAC can incorporate any combination of cell types because DNA-programmed adhesion is combinatorial and does not rely on the native adhesive or physical properties of cells. Together, these capabilities provide a means of exploring the aspects of tissue structure that are sufficient, and not merely necessary, for regulating specific cellular behaviors.
There remains numerous opportunities for improving DPAC. For example, delivery of structured chemical, physical, and hemodynamic signals to assembled microtissues, as well as the potential to perfuse embedded vasculature, could be achieved by merging DPAC with microfluidic technologies such as those used in organs-on-a-chip33. Merging DPAC with 3D printing could provide a means to control the spatial heterogeneity of ECM in addition to the spatial heterogeneity of cells. Combined with DPAC, stacking34 or rolling35 techniques could generate thicker microtissues. Finally, the incorporation of stem cells or organoids as building blocks could enable studying organoid development and disease processes in higher throughput and in a more reproducible 3D setting1. While DPAC provides substantial new capabilities for reconstituting 3D microtissues for culture and imaging, the method is fundamentally limited to cells that can survive dissociation and can be labeled by DNA.
As it stands, DPAC can deconvolute the consequences of tissue structure – including size, shape, composition, spatial heterogeneity, and embedding ECM – on collective cell behaviors. We found that, within single microtissues, the growth rate of human mammary epithelial cells increased with the fraction of Ras-expressing cells, indicating signal exchange between these neighboring populations. When patterned similarly to what is observed during branching morphogenesis, these cells develop into structures bearing a striking resemblance to the terminal ductal lobular units (TDLUs) of the mammary gland. In these structures, the Ras-expressing cells appeared to lead the surrounding cells as they grew into the surrounding ECM. We also demonstrated the quantitative and qualitative impact of ECM composition on the branching of reconstituted and bilayered human mammary epithelial organoids comprising both luminal and myoepithelial populations. Finally, we explored the effect of different mural cell types on the maturity and branching of patterned endothelial networks. Given the capacity of DPAC to directly link complex tissue structural features with specific single and collective cell behaviors, we anticipate that this method will find utility in a variety of contexts, both basic and applied.
ONLINE METHODS
General Materials and Reagents
Aldehyde-silanized glass slides (Nexterion® Aldehyde AL, Schott), Sigmacote® (Sigma-Aldrich), Slygard® 184 (Fisher Scientific), sodium borohydride (NaBH4, ACROS, 98%), Pluronic® F108 NF (BASF), ethanol (Fisher Scientific), trypsin inhibitor from Glycine max (Sigma-Aldrich), Matrigel® (BD Biosciences), rat-tail collagen 1 (BD Biosciences), Turbo DNase (Life Technologies), amine-modified ssDNA (5′-amine-X20, Operon), PBS (UCSF Cell-Culture Facility), PBS-CMF (UCSF Cell-Culture Facility), trypsin (UCSF Cell-Culture Facility), 100x penicillin/streptomycin, heat-inactivated fetal bovine serum (UCSF Cell-Culture Facility), RPMI media (UCSF Cell-Culture Facility) were used as received without further purification. Lipid-modified ssDNA (5′-lipid-T80-X20) was synthesized as previously described17.
Cell Culture
MCF-10A and MCF-10AT cell lines were provided by J. Liu (University of California San Francisco). Finite lifespan HMECs and fibroblasts were provided by J. Garbe. HUVECs, MSCs, and SMCs were purchased from Lonza. HBVPs were purchased from Sciencell. CAD cells were provided by K. Monahan (University of California San Francisco). Bone marrow dendritic cells were provided by B. Boldajipour. Jurkats were provided by Z. Gartner.
MCF-10A and MCF-10AT cell lines were cultured as previously described23,36. Primary human mammary epithelial cells at passage 4 were established and maintained in M87A medium according as previously described37. CAD neuronal cells were cultured as previously described38. All other cells were cultured according to standard practices listed on American Type Culture Collection or Lonza.
No mycoplasma testing or cell authentication was performed for the experiments in this study.
Antibodies
For immunofluorescence, the following antibodies were used: anti-human keratin 19 (Sigma cat. #C6930) (clone A53-B/A2) (1:50 dilution), anti-human keratin 14 (Thermo cat. #RB-9020-P) (polyclonal) (1:50 dilution), and anti-human CD49f (Millipore cat. #MAB1378) (clone GoH3) (1:50 dilution).
Preparation of PDMS Flow Cells
Flow cells were cast with Sylgard 184 according to the specifications provided by Dow Corning. Briefly, the polymer and curing agent were mixed at a 10:1 ratio, degassed under vacuum, and cured over the flow cell master at 70 °C. The master was prepared with No. 1 thickness coverslips (Fisher Scientific) cut to the dimensions of 4.5mm x 18mm and attached to double-sided tape (3M, cat. 665) of .0762 μm thickness. The final dimensions of the flow cell master was 4.5 mm x 18 mm x 0.22 mm attached to a Nunclon® (Fisher Scientific) petri dish. Each PDMS flow cell was individually cut to have 1mm-thick side walls and to have a 4.5mm-wide inlet and outlet. Flow cells were treated with atmospheric plasma prior to use, as described below.
Preparation of DNA-patterned Surfaces
Cell and tissue patterns were designed as bitmap images in Microsoft Paint and translated into 6–8 μm-diameter droplets of 1.5 mM 5′-amine-modified ssDNA (5′-amine-X20, Operon) in a spotting solution of 225 mM NaCl, 22.5 mM sodium citrate, 5% w/v trehalose, 0.1 mg/mL N-octylglucoside, pH=9.5 onto aldehyde-silanized glass slides (Nexterion® Aldehyde AL, Schott) via the BioForce Nano eNabler. Upon completion of printing, ssDNA patterns were baked at 120 °C for 15 minutes and then stored in a vacuum desiccator until use.
Patterned slides were reduced in a solution of 0.25% NaBH4 in 25% ethanol, 75% PBS for 15 minutes. Slides were washed twice with 0.1% s.d.S, three times with dH2O, and then air-dried. For silanization, 150 μL of Sigmacote (Sigma-Aldrich) was pipetted onto the slide and a coverslip placed on top. After five seconds, the coverslip was removed and the slide submerged into a tube of 50 mL absolute ethanol. The slide was inverted ten times then transferred into a fresh tube of 50 mL absolute ethanol. The inverting was repeated, and the slide was transferred into a tube of ddH2O for a final set of inversions. The slide was removed from the tube and dried under a stream of air. A flow cell was cut for each pattern on the slide, cleaned of dust with tape, and subjected to atmospheric plasma in a Plasma Etch PE-50 for 35 seconds under 200 mTorr pressure with 15 cc/min gas flow and at intermediate power. Flow cells were immediately positioned over the patterned slide and secured with gentle finger pressure. The flow cells were primed with a solution of RPMI-1640, 10% FBS, 63.7 mg/L penicillin G, 100 mg/L streptomycin sulfate, and 1% Pluronic F108. The solution was left in the flow cell for 5 minutes at room temperature to block the surface, and then the flow cell was equilibrated with four flow-cell volumes of calcium/magnesium-free PBS (PBS CMF) and left undisturbed until ready for programmed assembly.
Preparation of DNA-labeled Cells
All cell-lines were labeled with lipid-ssDNA prepared according to a published procedure1. Briefly, cells were incubated for 5 min at room temperature with 5 _M of lipid–DNA. Sequences were chosen according to the requirements of each specific experiment. DNA-labeled cells were washed three times with PBS CMF and temporarily stored at 4 °C until required for programmed assembly.
Programmed Assembly and Tissue Embedding of Cell Patterns
DNA-labeled cells were resuspended to a concentration of 107 cells/mL, and 20 μL of these cells were introduced to one end of the flow cell. The cells were either allowed to settle to the surface by gravity for 5–10 minutes, or the slide was centrifuged for 3 min at 8 g in a Sorvall Legend RT+ centrifuge with acceleration and deceleration set to minimum. Ten flow cell volumes of PBS CMF were flowed into the flow cell to wash out unhybridized cells. The procedures in this paragraph were repeated for each assembly step desired, taking 5–15 minutes for each successive assembly step.
Once the desired cell populations were assembled in the flow cell, a mixture of liquid hydrogel (e.g. Matrigel) and DNase was flowed across the surface. One typical formulation was 6.1 mg/mL Matrigel, 2.1 mg/mL collagen I, 40 U/mL Turbo DNase, ice-cold. Another typical formulation was 9.0 mg/mL Matrigel, 40 U/mL Turbo DNase, ice-cold. The flow cell was put in an incubator at 37 °C for 30 minutes to allow for DNA cleavage and for the liquid gel to set as a solid hydrogel. Next, a border of 20 μL PBS CMF was applied all around the flow cell to reduce stiction, and then a sterile razor blade was used to slide the flow cell off the surface and onto a 20uL droplet of molten hydrogel waiting in a 3.5 cm culture dish. The dish was transferred to an incubator at 37 °C for 30 minutes to allow the underlying gel to set. 3 mL prewarmed culture media was added to the dish so as to completely submerge the flow cell. Sharp tweezers were used to carefully slide the flow cell off the set hydrogel. The released flow cell was then removed from the dish. The dish was returned to the incubator to begin tissue culture.
Immunofluorescence
All samples were fixed with 4% formaldehyde for 20 minutes and then incubated in blocking buffer (10% heat-inactivated goat serum in PBS+0.5% Triton X-100) at 4 °C for at least one day. Primary antibodies were then diluted in blocking buffer and added to the sample. After at least one day incubating at 4 °C with the primary antibodies, samples were washed several times with PBS+Triton X-100 for at least one day and incubated with fluorophore-conjugated secondary antibodies diluted at a concentration of 1:200 in blocking buffer for approximately one day. All sample were washed with PBS+1 μg/mL DAPI for at least one hour before imaging.
Image Acquisition
All confocal microscopy images were acquired using a temperature, atmosphere, and humidity controlled spinning disk confocal microscope (Zeiss Cell Observer Z1 equipped with a Yokagawa spinning disk and running Zeiss Zen Software). All other images were acquired using an inverted epifluoresence microscope (Zeiss Axiovert 200M running SlideBook software).
Cell Growth Measurements
Cell assemblies in 20x20 square arrays with pitch xy of 300 μm were imaged approximately every 24 hours by driving the Zeiss Cell Observer spinning disc confocal microscope to a pre-set list of nominal xy positions at 20x magnification with a z-slice spacing of 3 μm. Cell nuclei in red and green emission channels were counted manually from raw tiff z-stacks and maximum intensity projection images. Growth rates for each assembly were calculated as the slope of plots of log2 (N/No) vs. t where N is cell number at time t and No is initial cell number, assuming logarithmic growth of cells.
Supplementary Material
Acknowledgments
The authors thank K. Monahan (University of California San Francisco) for providing CAD cells, B. Boldajipour and the members of the Krummel lab (University of California San Francisco) for providing bone marrow dendritic cells, J. Liu (University of California San Francisco) for sharing MCF-10A and derivative cell lines expressing H2B-fluorescent proteins, C. Mosher for technical help with the Nano eNabler, and M. Riel-Mehan for help with illustration. This work was supported the Department of Defense Breast Cancer Research Program (W81XWH-10-1-1023 and W81XWH-13-1-0221 to ZJG); the National Institutes of Health common fund (DP2 HD080351-01 to ZJG); The Sidney Kimmel Foundation; The National Science Foundation (MCB-1330864 to ZJG) and the University of California San Francisco Program in Breakthrough Biomedical Research. ZJG is supported by the University of California San Francisco Center for Systems and Synthetic Biology (National Institute of General Medical Sciences Systems Biology Center grant P50 GM081879). AC was supported by the Department of Defense through the National Defense Science and Engineering program.
Footnotes
AUTHOR CONTRIBUTIONS
Z.J.G., N.Y.J., and M.E.T. conceived the study; Z.J.G., M.E.T., N.Y.J., M.C.C., and A.J.H designed experiments; N.Y.J., M.E.T., A.C., A.J.H., M.C.C., and J.C.G. performed experiments; M.E.T., N.Y.J., M.C.C., A.J.H., and J.F. analyzed and interpreted the data; and Z.J.G., M.E.T., N.Y.J, M.C.C., and A.J.H. wrote the manuscript. All authors discussed and commented on the manuscript.
STATEMENT OF COMPETING FINANCIAL INTERESTS
A provisional patent application has been filed on the basis of this work. Z.J.G. is a member of the scientific advisory board of Adheren, a company that is commercializing cell-tethering technology.
References
- 1.Sasai Y. Cytosystems dynamics in self-organization of tissue architecture. Nature. 2013;493:318–326. doi: 10.1038/nature11859. [DOI] [PubMed] [Google Scholar]
- 2.Nelson C, Bissell M. Of extracellular matrix, scaffolds, and signaling: tissue architecture regulates development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2006;22:287–309. doi: 10.1146/annurev.cellbio.22.010305.104315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bissell MJ, Rizki A, Mian IS. Tissue architecture: the ultimate regulator of breast epithelial function. Current opinion in cell biology. 2003;15:753–762. doi: 10.1016/j.ceb.2003.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmeichel KL, Bissell MJ. Modeling tissue-specific signaling and organ function in three dimensions. J Cell Sci. 2003;116:2377–2388. doi: 10.1242/jcs.00503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lancaster MA, Knoblich JA. Organogenesis in a dish: modeling development and disease using organoid technologies. Science. 2014;345:1247125–1247125. doi: 10.1126/science.1247125. [DOI] [PubMed] [Google Scholar]
- 6.van de Wetering M, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161:933–945. doi: 10.1016/j.cell.2015.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shamir ER, Ewald AJ. Three-dimensional organotypic culture: experimental models of mammalian biology and disease. Nature Reviews Molecular Cell Biology. 2014 doi: 10.1038/nrm3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Albrecht D, Underhill G, Wassermann T, Sah R, Bhatia S. Probing the role of multicellular organization in three-dimensional microenvironments. Nat Methods. 2006;3:369–375. doi: 10.1038/nmeth873. [DOI] [PubMed] [Google Scholar]
- 9.Nelson C, Vanduijn M, Inman J, Fletcher D, Bissell M. Tissue geometry determines sites of mammary branching morphogenesis in organotypic cultures. Science. 2006;314:298–300. doi: 10.1126/science.1131000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu JS, Farlow JT, Paulson AK, LaBarge MA, Gartner ZJ. Programmed cell-to-cell variability in Ras activity triggers emergent behaviors during mammary epithelial morphogenesis. Cell Reports. 2012;2:1461–1470. doi: 10.1016/j.celrep.2012.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leung CT, Brugge JS. Outgrowth of single oncogene-expressing cells from suppressive epithelial environments. Nature. 2012;482:410–413. doi: 10.1038/nature10826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boghaert E, et al. Host epithelial geometry regulates breast cancer cell invasiveness. Proc Natl Acad Sci USA. 2012;109:19632–19637. doi: 10.1073/pnas.1118872109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murphy SV, Atala A. 3D bioprinting of tissues and organs. Nat Biotechnol. 2014;32:773–785. doi: 10.1038/nbt.2958. [DOI] [PubMed] [Google Scholar]
- 14.Stevens KR, et al. InVERT molding for scalable control of tissue microarchitecture. Nat Commun. 2013;4:1847. doi: 10.1038/ncomms2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsiao S, et al. Direct cell surface modification with DNA for the capture of primary cells and the investigation of myotube formation on defined patterns. Langmuir. 2009;25:6985–6991. doi: 10.1021/la900150n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gartner ZJ, Bertozzi CR. Programmed assembly of 3-dimensional microtissues with defined cellular connectivity. Proc Natl Acad Sci USA. 2009;106:4606–4610. doi: 10.1073/pnas.0900717106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Selden NS, et al. Chemically programmed cell adhesion with membrane-anchored oligonucleotides. J Am Chem Soc. 2012;134:765–768. doi: 10.1021/ja2080949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bailey R, Kwong G, Radu C, Witte O, Heath J. DNA-encoded antibody libraries: a unified platform for multiplexed cell sorting and detection of genes and proteins. J Am Chem Soc. 2007;129:1959–1967. doi: 10.1021/ja065930i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teramura Y, Chen H, Kawamoto T. Control of cell attachment through polyDNA hybridization. Biomaterials. 2010;31:2229–2235. doi: 10.1016/j.biomaterials.2009.11.098. [DOI] [PubMed] [Google Scholar]
- 20.Birch HM, Clayton J. Cell biology: Close-up on cell biology. Nature. 2007;446:937–940. doi: 10.1038/446937a. [DOI] [PubMed] [Google Scholar]
- 21.Xu JT, et al. Microfabricated ‘Biomolecular Ink Cartridges’ - Surface patterning tools (SPTs) for the printing of multiplexed biomolecular arrays. Sensors and Actuators B-Chemical. 2006;113:1034–1041. [Google Scholar]
- 22.Weber RJ, Liang SI, Selden NS, Desai TA, Gartner ZJ. Efficient Targeting of Fatty-Acid Modified Oligonucleotides to Live Cell Membranes through Stepwise Assembly. Biomacromolecules. 2014;15:4621–4626. doi: 10.1021/bm501467h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 24.Nguyen-Ngoc KV, et al. ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc Natl Acad Sci USA. 2012;109:E2595–E2604. doi: 10.1073/pnas.1212834109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LAIRD AK. DYNAMICS OF TUMOR GROWTH. Brit J Cancer. 1964;13:490–502. doi: 10.1038/bjc.1964.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chi X, et al. Ret-Dependent Cell Rearrangements in the Wolffian Duct Epithelium Initiate Ureteric Bud Morphogenesis. Dev Cell. 2009;17:199–209. doi: 10.1016/j.devcel.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lecaudey V, Cakan-Akdogan G, Norton WHJ, Gilmour D. Dynamic Fgf signaling couples morphogenesis and migration in the zebrafish lateral line primordium. Development. 2008;135:2695–2705. doi: 10.1242/dev.025981. [DOI] [PubMed] [Google Scholar]
- 28.Ghabrial AS, Krasnow MA. Social interactions among epithelial cells during tracheal branching morphogenesis. Nature. 2006;441:746–749. doi: 10.1038/nature04829. [DOI] [PubMed] [Google Scholar]
- 29.Shaw AT, et al. Sprouty-2 regulates oncogenic K-ras in lung development and tumorigenesis. Genes & development. 2007;21:694–707. doi: 10.1101/gad.1526207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Slattum G, Gu Y, Sabbadini R, Rosenblatt J. Autophagy in oncogenic K-Ras promotes basal extrusion of epithelial cells by degrading S1P. Curr Biol. 2014;24:19–28. doi: 10.1016/j.cub.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chung K, et al. Structural and molecular interrogation of intact biological systems. Nature. 2013 doi: 10.1038/nature12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9:685–693. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 33.Bhatia SN, Ingber DE. Microfluidic organs-on-chips. Nat Biotechnol. 2014;32:760–772. doi: 10.1038/nbt.2989. [DOI] [PubMed] [Google Scholar]
- 34.Yang J, et al. Reconstruction of functional tissues with cell sheet engineering. Biomaterials. 2007;28:5033–5043. doi: 10.1016/j.biomaterials.2007.07.052. [DOI] [PubMed] [Google Scholar]
- 35.L’Heureux N, Pâquet S, Labbé R, Germain L, Auger FA. A completely biological tissue-engineered human blood vessel. FASEB J. 1998;12:47–56. doi: 10.1096/fasebj.12.1.47. [DOI] [PubMed] [Google Scholar]
- 36.Dawson PJ, Wolman SR, Tait L, Heppner GH, Miller FR. MCF10AT: a model for the evolution of cancer from proliferative breast disease. Am J Pathol. 1996;148:313–319. [PMC free article] [PubMed] [Google Scholar]
- 37.Stampfer MR, LaBarge MA, Garbe JC. An Integrated Human Mammary Epithelial Cell Culture System for Studying Carcinogenesis and Aging. Cell and Molecular Biology of Breast Cancer. 2013:323–361. [Google Scholar]
- 38.Qi YP, Wang J, McMillian M, Chikaraishi DM. Characterization of a CNS cell line, CAD, in which morphological differentiation is initiated by serum deprivation. J Neurosci. 1997;17:1217–1225. doi: 10.1523/JNEUROSCI.17-04-01217.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.