

Abstract
Background and Purpose
Both pathogenic and regulatory immune processes are involved in the middle cerebral artery occlusion (MCAO) model of experimental stroke, including interactions involving the Programmed Death 1 (PD-1) receptor and its two ligands, PD-L1 and PD-L2. Although PD-1 reduced stroke severity, PD-L1 and PD-L2 appeared to play pathogenic roles, suggesting use of anti-PD-L monoclonal Ab (mAb) therapy for MCAO.
Methods
Male C57BL/6 mice were treated with a single dose of anti-PD-L1 mAb 4 h after MCAO and evaluated for clinical, histological and immunological changes after 96 h reperfusion.
Results
Blockade of the PD-L1 checkpoint using a single injection of 200μg anti-PD-L1 mAb given i.v. 4 h after occlusion significantly reduced MCAO infarct volumes and improved neurological outcomes after 96 h reperfusion. Treatment partially reversed splenic atrophy and decreased CNS infiltrating immune cells concomitant with enhanced appearance of CD8+ regulatory T cells in the lesioned CNS hemisphere.
Conclusions
This study demonstrates for the first time the beneficial therapeutic effects of PD-L1 checkpoint blockade on MCAO, thus validating proposed mechanisms obtained in our previous studies using PD-1 and PD-L deficient mice. These results provide strong support for use of available humanized anti-PD-L1 antibodies for treatment of human stroke subjects.
Keywords: MCAO, PD-L1, CD8+ Tregs, IL-10
Introduction
Ischemic stroke, characterized by the rapid development of an infarct upon disruption of cerebral blood flow 1, 2, is a leading cause of death and disability worldwide. Treatment options currently are limited and mainly involve restoration of blood flow (reperfusion) by intravenous administration of tissue plasminogen activator within 4.5 h after stroke onset. Although necessary, reperfusion can also enhance the inflammatory response and cause additional injury to adjacent brain tissue 3, in large part through the rapid transmigration of both innate (neutrophils and monocytes) and adaptive (T and B cells) immune cells from the periphery to the growing CNS infarct 4-9. Clearly, there is an urgent unmet need for new therapeutic approaches that can prevent or reverse this process.
Prior research from our and other laboratories has firmly established both pathogenic and regulatory immune processes involved in the middle cerebral artery occlusion (MCAO) model of experimental stroke in mice. Of particular interest are immune interactions involving the Programmed Death 1 (PD-1) receptor and its two ligands, PD-L1 and PD-L2 that regulate the function of inflammatory immune cells. Initially, we demonstrated that PD-1 played a protective role in stroke, since cortical, striatal, and hemispheric infarct volumes were significantly larger in PD-1-/- mice, with a marked recruitment of inflammatory cells from the periphery into the central nervous system (CNS) 10. Studies were then extended to investigate the role of PD-L1 and PD-L2 in modulating severity of ischemic brain injury and associated CNS inflammation. Contrary to our expectations, PD-L1-deficient (PD-L1-/-) and PD-L2-deficient (PD-L2-/-) mice that were similarly subjected to 60 min of MCAO followed by 96 h of reperfusion demonstrated smaller total infarct volumes compared to WT mice 11, suggesting a pathogenic rather than a regulatory role for both PD-ligands. The opposing roles of PD-1 and PD-L in stroke suggested alternative ligand/receptor binding partners for PD-L1 and PD-L2 besides PD-1. We thus assessed the contribution of several other co-stimulatory molecules that could promote or inhibit T-cell proliferation in combination with PD-L1 or PD-L2 as well as verifying the PD-L-expressing cell-types responsible for enhancing MCAO. We found that PD-L1 and PD-L2 have overlapping and/or distinct biological functions 12 that derive in part from the broad expression of PD-L1 in lymphoid and non-lymphoid organs versus the more restricted but overlapping expression of PD-L2 on DC and macrophages 13. Our studies demonstrated that PD-L1 played a dominant role in promoting CD8+ and CD4+ T-cell proliferation that contributed to increased infarct volumes in WT mice subjected to MCAO, whereas in the absence of PD-L1, inhibitory CTLA-4/CD80 interactions became prevalent and PD-1/PD-L2 interactions emerged to control CD4+ T-cell, APC and Breg cell responses.
These results indicated that PD-L1 and PD-L2 differentially control induction of T cell and Breg cell responses after MCAO and suggested that selective targeting of PD-L1 and/or PD-L2 might represent a valuable therapeutic strategy in stroke. Thus, in this study, we evaluated for the first time the effects of anti-PD-L1 mAb therapy on stroke severity in male WT mice treated 4 h after 60 min occlusion and 96 h reperfusion. Our results clearly demonstrate a significant reduction in cortical, striatal and hemispheric infarct volumes and neurological deficit scores as well as reduced infiltration of inflammatory cells but concurrent enhancement of CD8+CD122+ Treg cells but not Breg cells in the lesioned brain hemisphere. Of critical importance, the effects of anti-PD-L1 mAb antibody treatment mirrored results obtained in PD-L1 deficient mice 11, 12, thus indicating the potential for direct translation to the clinic using mAb blockade of the PD-L1 “checkpoint”. It is noteworthy that a human IgG1κ anti-PD-L1 mAb (MEDI4736) is currently being evaluated in 25 ongoing or planned clinical studies in multiple tumor types, with encouraging results in more than 800 treated patients 14. This antibody blocks PD-L1 binding to its receptors, has high affinity and selectivity for PD-L1 and sustained drug exposure for up to 1 year of dosing, is engineered to prevent collateral inflammatory damage and has no reported immunogenicity impacting its bioactivity, thus making it highly suitable for immediate testing as a novel treatment for stroke subjects.
Materials and Methods
Animals
8-12 week old male wild type C57BL/6J mice, weighing 20g to 25g, were obtained from The Jackson Laboratory (Sacramento, CA, USA). Naïve WT male mice were housed in the Animal Resource Facility at the Portland Veterans Affairs Medical Center in accordance with institutional guidelines. Animals were randomized to treatment groups and induction of transient focal cerebral ischemia. All WT male mice were housed in a climate-controlled room on a 12 h light/dark cycle. Food and water were provided ad libitum. All experiments were performed in accordance with National Institutes of Health guidelines for the use of experimental animals and the protocols were approved by the VA Portland Health Care System and Oregon Health & Science University Animal Care and Use Committees.
Middle Cerebral Artery Occlusion (MCAO) model
All surgeries were conducted under aseptic conditions by a surgeon. Transient focal cerebral ischemia was induced in male mice for 1 h by reversible MCAO in the right brain hemisphere under isoflurane anesthesia followed by 96 h of reperfusion as described previously15. Body temperature was controlled at 36.5 ± 1.0°C throughout MCAO surgery with warm water pads and a heating lamp. Occlusion and reperfusion were verified in each animal by laser Doppler flowmetry (LDF) (Model DRT4, Moor Instruments, Inc., Wilmington, DE, USA). The common carotid artery was exposed and the external carotid artery was ligated and cauterized. Unilateral MCAO was accomplished by inserting a 6-0 nylon monofilament surgical suture (ETHICON, Inc., Somerville, NJ, USA) with a heat-rounded and silicone-coated (Xantopren comfort light, Heraeus, Germany) tip into the internal carotid artery via the external carotid artery stump. Adequacy of MCAO was confirmed by monitoring cortical blood flow at the onset of the occlusion with a LDF probe affixed to the skull. Animals were excluded if mean intra-ischemic LDF was greater than 30% pre-ischemic baseline 15. At 1 h of occlusion, the occluding filament was withdrawn to allow for reperfusion and the incision was closed with 6–0 surgical sutures (ETHICON, Inc., Somerville, NJ, USA). One-half ml of prewarmed normal saline was given subcutaneously to each mouse after surgery. Mice were then allowed to recover from anesthesia and were survived for 96 h following initiation of reperfusion. The surgeon was blinded to treatment groups.
PD-L1 depletion
Mice were given 200μg of either monoclonal anti-PD-L1 antibody (clone 10F.9G2, Biolegend, San Diego, CA, USA) or an isotype matched control (anti-Keyhole Limpet Hemocyanin – KLH, clone LTF-2, BioXcell, West Lebanon, NH, USA) in 200μL PBS, 4 h following MCAO. Intravenous (i.v.) injection via the lateral tail vein and intraperitoneal (i.p.) injection were both utilized for administration of antibodies with similar results in PD-L1 depletion and reduced infarct volumes. Hence, data presented in this study may derive from mice treated by either route of injection. The researcher delivering treatment was not blinded to treatment groups. Depletion of PD-L1 on peripheral leukocytes was evaluated 96 h after reperfusion by flow cytometry.
Neurological deficit score
Neurological deficit scores were determined at 4, 24, 48, 72, and 96 h post-MCAO to confirm ischemia and the presence of ischemic injury using a 0 to 4 point scale as follows: 0, no neurological dysfunction; 1, failure to extend left forelimb fully when lifted by tail; 2, circling to the contralateral side; 3, falling to the left; and 4, no spontaneous movement or in a comatose state 16. Mice without a deficit after 1 h of reperfusion were excluded from the study.
Infarct volume analysis
The individual performing the infarct volume analysis was blinded to treatment group. Mice were euthanized and brains collected at 96 h of reperfusion for 2,3,5-triphenyltetrazolium chloride histology (TTC; Sigma, St. Louis, MO, USA) described previously 17. The 2-mm brain sections were incubated in 1.2% TTC for 15 min at 37°C, and then fixed in 10% formalin for 24 h. Infarction volume was measured using digital imaging and images were analyzed using Sigma Scan Pro 5.0 Software (Systat, Inc., Point Richmond, CA). To control for edema, infarct volume (cortex, striatum, hemisphere) was determined by subtraction of the ipsilateral non-infarcted regional volume from the contralateral regional volume. This value was then divided by the contralateral regional volume and multiplied by 100 to yield regional infarction volume as a percent of the contralateral region.
Isolation of leukocytes from spleen and brain
Spleens from individual control and B-cell recipient WT mice were removed and a single-cell suspension was prepared by passing the tissue through a 100μm nylon mesh (Fisher Scientific, Pittsburg, PA, USA). The cells were washed using RPMI 1640. Red cells were lysed using 1× red cell lysis buffer (eBioscience, Inc., San Diego, CA) and incubated for 3 min. Cells were then washed twice with RPMI 1640, counted and resuspended in stimulation medium (RPMI containing 2% FBS (GE Healthcare, Pittsburg, PA, USA), 1% sodium pyruvate (Life Technologies, Carlsbad, CA, USA), 1% L-glutamine (Life Technologies) and 0.4% β-Mercaptoethanol (Sigma-Aldrich, St. Louis, MO, USA)).
The brain was divided into the ischemic (right) and non-ischemic (left) hemispheres, dissociated enzymatically in RPMI supplemented with 3 U/mL recombinant DNase I (Roche, Indianapolis, IN, USA) and 1mg/mL collagenase from Clostridium histolyticum (Sigma-Aldrich), resuspended in 80% Percoll (GE Healthcare) overlaid with 40% Percoll and subjected to density gradient centrifugation for 30 min at 1600 rpm according to a described previously method 18. Inflammatory cells were removed from the interphase for further analysis. Cells were then washed twice with RPMI 1640, counted and resuspended in stimulation medium. Cells from individual brain hemispheres were evaluated by flow cytometry.
Analysis of cell populations by flow cytometry
All antibodies were purchased from BD Biosciences (San Jose, CA) or eBioscience, Inc. (San Diego, CA) unless indicated otherwise. Four-color (FITC, PE, APC and 7AAD/PerCP/PECy7) fluorescence flow cytometry analyses were performed to determine the phenotype and cytokine production of splenocytes and brain leukocytes as previously published 19. Single-cell suspensions were washed with staining medium (PBS containing 0.1 % NaN3 and 0.5 % bovine serum albumin, Sigma, Illinois) and incubated with combinations of the following monoclonal antibodies for extracellular stains: CD4 (clone GK1.5), CD8a (clone 53–6.7), CD11b (clone M1/70), CD19 (clone 1D3), CD45 (clone Ly-5), CD122 (clone TM-β1 BD), PD-L1 (clone MIH5), CD80 (clone 16-10A1) and CD11c (clone HL3) for 20 min at 4°C prior to washing the cells. 7-Aminoactinomycin D (7AAD, BD Biosciences) was added to identify dead cells whenever only 3 channels on the flow cytometer were used for detection of fluorescent antibody staining. FACS data acquisition was performed using an Accuri C6 flow cytometer (BD Biosciences, San Jose, CA) and data were analyzed using FCS express software (De Novo Software, Los Angeles, CA).
Intracellular staining
Intracellular staining was visualized using a published immunofluorescence protocol 16. Briefly, isolated leukocytes were resuspended (2 × 106 cells/mL) in complete medium and cultured with LPS (10 μg/mL) in addition to Phorbol 12-myristate 13-acetate (PMA, 50 ng/mL), ionomycin (500 ng/mL) (all three from Sigma-Aldrich), and GolgiPlug (BD Biosciences) protein transport inhibitor for 4 h. Fc receptors were blocked with anti-FcR mAb (2.3G2, BD Biosciences) before cell surface staining and cells were fixed and permeabilized with Fixation/Permeabilization buffer (BD Biosciences) according to the manufacturer's instructions. Permeabilized cells were washed with 1× Permeabilization Buffer (BD Biosciences) and stained with antibodies specific for the following intracellular targets: TNF-α (clone MP6-XT22), IL-10 (clone JES5-16E3), PD-1 (clone J43) and FoxP3 (clone FJK-16s), then resuspended in staining buffer for acquisition. Isotype matched mAb served as negative controls.
RNA isolation and real-time PCR
Total RNA was isolated from the ischemic hemisphere from treated mice using the RNeasy mini kit protocol (Qiagen, Valencia, CA, USA) and converted into cDNA using oligo-dT primers and Superscript RT II (both Life Technologies). Quantitative real time PCR was performed on a StepOnePlus Real Time PCR System (Applied Biosystems, Foster City, CA, USA) using the following TaqMan Gene Expression Assays in Taqman Universal Master Mix (all Applied Biosystems): Mmp9, Il4 and Il10. ΔCt was calculated against the expression of the endogenous control GAPDH. Fold change in targeted transcript expression was determined using the formula 2-ΔΔCt.
Statistical Analysis
All values are reported as mean ± SEM. The infarct volume data were analyzed using the GraphPad Prism 6 software (version 6.00; GraphPad Software, San Diego, CA). Infarct volume data are presented as mean + SEM. Differences in regional infarct volumes were determined with Student's t-test. Functional outcomes for neurological deficit scores were analyzed by Mann Whitney Rank Sum test. For flow data analysis and representation of three and more groups, the one-way ANOVA followed by post hoc Tukey's test was applied. For RT-PCR, t tests with Welch's correction were used to compare anti-PD-L1 mAb conditions to isotype mAb treated controls. Statistical analyses were performed using GraphPad PRISM software version 5 (La Jolla, CA, USA). For all tests, p values ≤ 0.05 were considered statistically significant. Significant differences are denoted as *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
Results
Single dose of anti-PD-L1 mAb depletes PD-L1 expression without affecting cell composition in naive male WT mice
To test our central hypothesis that the use of anti-PD-L1 mAb in experimental stroke will ameliorate functional outcome and stroke-induced neuroinflammation, we first evaluated the effects of anti-PD-L1 mAb treatment on PD-L1 expression in naïve mice. Thus, naive WT male mice were injected i.p. with either 200μg anti-PD-L1 mAb or isotype control mAb to KLH dissolved in 200μL sterile phosphate buffered saline (PBS) and administered once (i.e. on D0) and evaluated 4 days later for PD-L1 expression. The results demonstrated that a single dose of anti-PD-L1 mAb was sufficient to deplete the expression of PD-L1 on different splenocyte subpopulations compared to the isotype control mAb (Figure 1A) without affecting their frequency (Figure 1B). Hence, for further stroke-related studies, a single dose of 200ug/200uL of mAb was used.
Figure 1. A single dose of anti-PD-L1 mAb depletes PD-L1 expression without affecting the cell composition in naive male WT mice.

A. Percentage of PD-L1 expression on CD11c+ dendritic cells, CD11b+ monocytes, CD19+ B-cells and CD4+ and CD8+ T-cells in spleens of anti-PD-L1 vs. isotype control mAb treated adult naïve C57BL/6J WT male mice as determined after 96 h by flow cytometry. B. Frequency of CD19+ B-cells, CD4+ and CD8+ T-cells, CD11b+ monocytes and CD11c+ dendritic cells in spleens of anti-PD-L1 vs. isotype control mAb treated naïve WT male mice as determined by flow cytometry. Values represent mean of 4 mice for the control group and 5 mice for the anti-PD-L1 mAb treated group. Statistical analysis was performed with Student's t test. Significant differences between control and anti-PD-L1 mAb-treated groups are indicated as *p≤0.05, **p≤ 0.01 and ***p≤ 0.001.
Treatment with anti-PD-L1 mAb, 4 h after MCAO, reduces infarct volumes and improves neurological deficit scores in male WT mice
Based on our previous work demonstrating smaller infarct volumes in PD-L1-/- mice 11, we reasoned that treatment of MCAO with anti-PD-L1 mAb might improve stroke outcome. Indeed, WT male mice treated i.p. with anti-PD-L1 vs. isotype control mAb 4 h after 60 min MCAO followed by 96 h of reperfusion exhibited significantly reduced cortical (p=0.0013), striatal (p=0.0479) and total hemispheric (p=0.0013) infarct volumes (Figure 2A and 2B) and a significant improvement in median neurological deficit scores (p=0.0326, Table 1). These results demonstrate for the first time that post-MCAO treatment with anti-PD-L1 mAb can successfully ameliorate stroke-induced damage in the ischemic brain hemisphere.
Figure 2. Treatment with anti-PD-L1 mAb, 4 h after MCAO, reduces infarct volume in male WT mice.

Infarct volume (% corrected contralateral structure) in cortex, striatum and hemisphere were determined by 2,3,5-triphenyltetrazolium chloride staining in isotype control-treated vs. anti-PD-L1 mAb-treated adult male WT mice. All mice underwent 1 hour of middle cerebral artery occlusion (MCAO), with treatments at 4 h after MCAO followed by 96 hours of reperfusion. A. Infarct volumes in anti-PD-L1 Ab- treated (n=23) male WT mice were significantly reduced compared to the isotype control-treated (n=21) mice. Values represent mean ± SEM. *p<0.05, **p<0.01. B. Representative cerebral sections showing localization of the ischemic lesions in anti-PD-L1 vs. isotype control mAb treated mice. Note-Three mice from the anti-PD-L1 mAb treated group were excluded due to hemorrhagic transformation.
Table 1.
Distribution of neurological deficit scores at various reperfusion time points following 60 min of middle cerebral artery occlusion (MCAO) in C57BL/6J (wild-type, WT) treated intraperitoneally (i.p.) with PD-L1 Ab and vehicle at 4 h post-MCAO.
| 4 hours | 24 hours | 48 hours | 72 hours | 96 hours | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Experimental groups | 0 | 1 | 2 | 3 | 4 | 0 | 1 | 2 | 3 | 4 | 0 | 1 | 2 | 3 | 4 | 0 | 1 | 2 | 3 | 4 | 0 | 1 | 2 | 3 | 4 |
| Isotype control (n=21) | 0 | 1 | 18 | 2 | 0 | 0 | 0 | 9 | 12 | 0 | 0 | 4 | 9 | 7 | 1 | 0 | 4 | 10 | 4 | 3 | 5 | 10 | 3 | 3 | 0 |
| Anti-PD-L1 mAb (n=23) | 0 | 2 | 17 | 4 | 0 | 0 | 17 | 6 | 0 | 0 | 4 | 14 | 4 | 1 | 0 | 8 | 11 | 3 | 1 | 0 | 12 | 9 | 1 | 1 | 0 |
| p-value | 0.8617 | 0.0646 | 0.4864 | 0.1423 | 0.0328 a | ||||||||||||||||||||
Mann-Whitney test,
p <0.05 compared to Isotype control group
Mortality and exclusions
Overall mortality for all MCAO surgeries for infarct analysis and immunology studies was 11 mice out of a total of 133 mice, with mortality ranging from 4 to 7 mice within the experimental groups. Overall number of mice excluded due to intra ischemic LDF greater than 30% pre-ischemic baseline was 15 mice out of a total of 133 mice, with exclusions ranging from 7 to 8 mice within the experimental groups. Overall number of mice excluded due to failure in filament advancement was 2 mice out of a total of 133 mice, with exclusions ranging from 0 to 2 mice within the experimental groups. Overall, 5 mice were excluded due to severe hemorrhage after reperfusion in the anti-PD-L1 mAb-treated group of 73 mice. The five mice were excluded after 72 hours-96 hours reperfusion. One mouse died after 72 hours of reperfusion and 4 mice survived 96 hours of reperfusion but had high NDS (2-4). Severe hemorrhage of these 5 mice (6.8%) was detected when the brains were isolated after euthanization, suggesting that these 5 mice had delayed hemorrhagic transformation (HT) in the brain parenchyma.
Treatment with anti-PD-L1 mAb, 4 h after MCAO, reduces inflammatory responses but enhances accumulation of CD8+ Tregs in the ischemic brain hemisphere
Leukocytes are major effectors of inflammatory damage after MCAO. To determine if PD-L1 blockade with anti-PD-L1 mAb altered leukocyte composition in brain after MCAO, absolute numbers of total viable leukocytes were enumerated. As shown in Figure 3A, the ischemic (ipsilateral) hemisphere in mice treated with either anti-PD-L1 or isotype control mAb had a significant increase (p=0.017, p≤0.001, respectively) in the total number of viable leukocytes compared to the unaffected (contralateral) hemisphere, whereas no differences in total cell numbers were observed between treatment groups in either hemisphere (Figure 3A). However, treatment of MCAO mice with anti-PD-L1 vs. control mAb significantly reduced the percentage of activated TNF-α+ CD11b+ cells, likely microglia/monocytes (Figure 3B & 3C), but enhanced the percentages of total CD8+ and CD8+CD122+ Treg cells (Figure 3B & 3D) within the ischemic hemisphere, with a corresponding nominal increase in IL-10 production. No effects of anti-PD-L1 mAb treatment of MCAO mice were observed on CNS infiltrating CD4+ T cells, although a nominal reduction in CD5+CD1dhi CD19+ Breg cells was observed (not shown).
Figure 3. Treatment with anti-PD-L1 mAb, 4 h after MCAO, reduces inflammatory responses but leads to accumulation of CD8 Tregs in the ischemic brain.

Mononuclear cells were isolated, following 96 h reperfusion, from brains of WT male mice, that received either isotype control or anti-PD-L1 treatment, 4 h after MCAO and were analyzed for: A. Total cell count via hemocytometer. Values represent mean numbers (±SEM) of indicated cell subsets from 13-14 mice per group, from at least 4 separate experiments. B. CD11b+CD45high activated microglia (MG)/monocytes and CD8+ T-cells were obtained from the non-ischemic (left) and ischemic (right) hemispheres of the isotype or anti-PD-L1 mAb-treated mice. Values represent mean numbers (±SEM) of indicated cell subsets, from 12-13 mice per group, from at least 4 separate experiments. Determinations of: C. TNF-α production by CD11b+ cells, likely activated microglia/monocytes, and TNF-α+CD3+ T-cells in the non-ischemic (left) and ischemic (right) hemispheres of the isotype or anti-PD-L1 mAb-treated mice, 96 hours after MCAO. Values represent mean numbers (±SEM) of indicated cell subsets from 10-12 mice of each group, from at least 4 separate experiments. D. CD8+CD122+ T-cells and IL-10 production by gated CD8+CD122+ T-cells. Data are representative of 4 independent experiments with brains processed from 13-14 individual mice (mean ± SEM). Statistical analysis was performed with Student's t test with Welch's correction. Significant differences between sample means are indicated as #p≤0.05; ##p≤ 0.01; ###p≤ 0.001 as compared to their respective left hemisphere and *p≤0.05; **p≤ 0.01 compared to the ischemic right hemisphere of isotype-treated WT mice, post-MCAO.
To further assess the treatment effects of the anti-PD-L1 mAb on expression of pro- and anti-inflammatory cytokine/chemokine genes in the MCAO-affected hemispheres, Real time PCR was carried out. The expression level of the Mmp-9 gene implicated in cerebral ischemia 20, 21 was significantly decreased (p=0.016, Figure 4A) in the affected hemisphere of the PD-L1 vs. control mAb-treated MCAO mice. Moreover, the expression level of Il-10 in the ischemic hemispheres of the mAb-treated mice was significantly higher (p=0.038, Figure 4B), with an accompanying ∼3-fold, but not statistically significant, change in the expression level of Il-4 in the mAb-treated mice.
Figure 4. Treatment with anti-PD-L1 mAb, 4 h after MCAO, not only reduces the overall pro-inflammatory status but induces an anti-inflammatory milieu in the ischemic brain hemisphere.
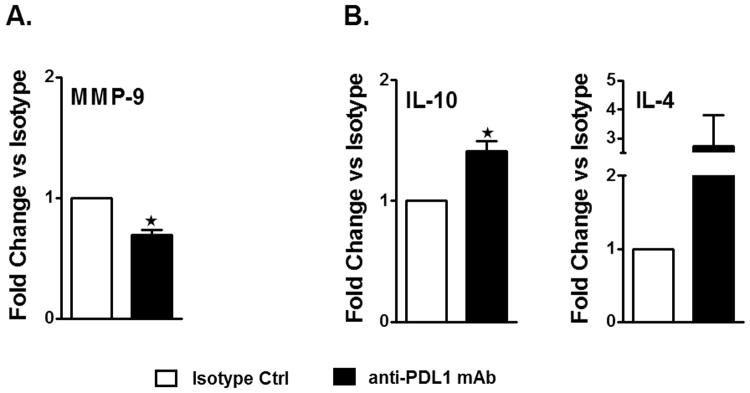
Brains were collected from isotype and anti-PD-L1 mAb-treated mice, 96 hours after occlusion, and mRNA prepared from ipsilateral (right) hemispheres of brain tissues for RT-PCR analysis. Fold change of mRNA levels is presented for A. inflammatory factor Mmp-9 and B. anti-inflammatory cytokines, Il-10 & Il-4. Values for A. and B. represent mean (±SEM) values for fold differences over the Ct values for isotype-treated ischemic brains. Significant differences between the right ischemic hemispheres of the isotype-treated and anti-PD-L1 mAb-treated groups were determined using Student's t-test. Significant differences between sample means are indicated *p≤0.05 compared to the ischemic right hemisphere of isotype-treated WT mice, post-MCAO.
Treatment with anti-PD-L1 mAb, 4 h after MCAO, rescues splenic atrophy, increases the expression of regulatory molecules on T-cells and decreases the expression of CD80 on APCs in spleens, post-MCAO
Effects of anti-PD-L1 mAb treatment were also evaluated on peripheral immunity (splenocytes) of MCAO induced mice. As anticipated from results in naïve WT mice, treatment with anti-PD-L1 vs. control mAb 4 hr after MCAO strongly inhibited expression of PD-L1 on CD19+ B cells, CD4+ and CD8+ T cells, CD11b+ monocytes/macrophages and CD11c+ DCs in the spleens of treated mice assessed after 96 h of reperfusion (Figure 5A). This modulation of PD-L1 was apparently not cytotoxic, since treatment with the anti-PD-L1 mAb significantly enhanced spleen cell numbers (Figure 5B), indicating partial rescue from MCAO-induced atrophy. We further evaluated possible regulatory cell types and found an increased frequency of CD8+CD122+ Tregs (p=0.016, Figure 5C) in anti-PD-L1 treated MCAO mice. However, no difference in the frequency of Foxp3+CD4+ Tregs was observed after treatment with anti-PD-L1 mAb (data not shown), suggesting the lack of involvement in our experiments of the PD-1/PD-L pathway in the generation of this Treg subpopulation 17, 22.
Figure 5. Treatment with anti-PD-L1 mAb, 4 h after MCAO, rescues splenic atrophy, increases the expression of regulatory molecules on T-cells and decreases the expression of CD80 on APCs in spleens, post-MCAO.

Ninety six hours after MCAO, mononuclear cells were isolated from spleens of isotype-treated or anti-PD-L1 mAb-treated mice and were analyzed for: A. Percentage of PD-L1 expression on CD11c+ dendritic cells, CD11b+ monocytes, CD19+ B-cells, CD4+ and CD8+ T-cells by flow cytometry. Values represent mean numbers (±SEM) of indicated cell subsets from 8-20 mice in each group, from at least 5 separate experiments B. Total cell count via hemocytometer. Values represent mean numbers (±SEM) of indicated cell subsets from 24-30 mice in each group, from at least 6 separate experiments; C. For CD8+CD122+ T-cells, data are representative of 4-5 independent experiments with spleens processed from 17-19 individual mice (mean ± SEM). For PD-1 expression on gated CD4+ T-cells and CD8+ T-cells, data are representative of 2 independent experiments with spleens processed from 4-5 individual mice (mean ± SEM). D. CD80 expression on gated CD11c+ and CD11b+ cells in the spleens. Values represent mean numbers (±SEM) of indicated cell subsets, from 8-12 individual mice from 4 separate experiments. Statistical analysis was performed with Student's t test with Welch's correction. Significant differences between isotype-treated and anti-PD-L1 mAb-treated groups, post-MCAO, are indicated as *p≤0.05, **p≤ 0.01 and ***p≤ 0.001.
Since our previous work 10, 11 implicated PD-1 signaling as a key component in limiting CNS inflammation in MCAO, we next sought to assess the expression of this co-inhibitory receptor on splenic T-cells. Although there were nominal increases in PD-1 expression in CD4+ and CD8+ T cells in anti-PD-L1 mAb-treated mice, the differences were not significant as compared to the isotype control mAb-treated mice post-MCAO (Figure 5C), indicating little effect of PD-L1 blockade on PD-1 expression and its possible regulatory effects after MCAO. Because PD-L1 can also bind to CD80 on APC to deliver inhibitory signals in T-cells 23, 24, it is possible that blockade of PD-L1 might obviate PD-L1/CD80 interactions and promote CNS inflammation through other co-stimulatory pathways (eg. PD-1/PD-L2). Consistent with this hypothesis, analysis of peripheral APCs demonstrated a significant reduction in CD80 expression on both CD11c+ dendritic cells (p=0.014) and CD11b+ monocytes (p=0.028) in PD-L1 mAb-treated mice as compared to the isotype control mice after MCAO (Figure 5D), suggesting reduced CD80 co-stimulatory potential after anti-PD-L1 mAb treatment.
Discussion
The results presented above demonstrate that blockade of the PD-L1 checkpoint using a single injection of anti-PD-L1 mAb given 4 h after occlusion can significantly reduce MCAO infarct volumes, partially reverse splenic atrophy, improve neurological outcome and enhance levels of CD8+CD122+ Tregs after 96 h reperfusion. The current results using mAb therapy to directly neutralize PD-L1 in vivo after MCAO validated results from our previous studies in genetically deficient PD-L1-/- mice 11,12 that also had reduced infarct volumes, a significant reduction in brain infiltrating pro-inflammatory cells, partial reversal of splenic atrophy and increased levels of IL-10-producing CD8+CD122+ T-suppressor cells in the ischemic brain hemisphere compared to WT MCAO mice. Our current study is the first to demonstrate the therapeutic potential of anti-PD-L1 therapy for treatment of MCAO and supports potential use of humanized anti-PD-L1 mAb for treatment of human stroke subjects.
Five mice were excluded due to severe hemorrhage after reperfusion in the anti-PD-L1 mAb-treated group of 73 mice. These hemorrhages corresponded were visually apparent intracerebral hemorrhages occurring in a small percentage of mice, but only in the anti-PD-L1 group. We also considered the possibility that this treatment could favor small hemorrhages. Micro-hemorrhages accompanying anti-PD-L1 mAb treatment would be difficult to rule out using the TTC stained slices that were analyzed. While there was no apparent RBC presence outside of noticeable hemorrhagic transformations, without having examined BBB permeability specifically, we can only say that using the current techniques, we could not detect any disruptions in BBB integrity large enough to allow substantial amounts of RBCs to enter the parenchyma. Studies examining the question of PD-L1 involvement in BBB permeability using endothelial cells derived from human brain tissue indicate that blockade of PD-L1 in tandem with PD-L2 results in increased susceptibility of endothelial cells to invasion by activated CD8+ T cells in vitro 25. In this model, blockade of PD-L1 alone did not produce statistically significant changes in the number of CD4+/8+ T cells or albumin able to translocate across an artificial BBB composed of human brain endothelial cells. Additional experiments would be necessary to conclusively determine tight junction integrity and permeability.
Results obtained by direct mAb blockade of PD-L1 function provide considerable support for proposed mechanisms of action in MCAO for PD-1, PD-L1, PD-L2 and various other co-stimulatory molecules belonging to the B7 family12. This study used clone 10F.9G2 anti-PD-L1 antibody to treat the mice. Literature suggests that this clone blocks both PD1:PD-L1 and B7-1:PD-L1 interactions 26. Although we cannot rule out cell surface down-regulation of PD-L1, the presence of a blocking Ab at PD-L1 on the cell could mean that the lack of binding of the detection Ab is due to occupation of PD-L1 binding site with the treatment Ab. Clinical studies have demonstrated high target occupancy for PD-L1 antibody on CD3+ T cells with a half-life of 15 days after injection 27. We, therefore, think that a 4 days effect of the PD-L1 Ab is very likely. As previously discussed 28, the affinity of the CD80 co-stimulatory molecule for CTLA-4, PD-L1 & CD28 are kd = 0.4 μM, 1.4 μM and 4.0 μM, respectively. Thus, at homeostatic levels in WT mice, PD-L1 competes with CTLA-4 for CD80 binding, thus providing in concert with MHC molecules a non-activating signal that promotes naïve T-cell survival, while simultaneously suppressing T-cell responses by outcompeting strong proinflammatory signaling through CD80/CD28. When stroke is induced in WT or PD-L2-/- mice that express PD-L1, the level of CD80 expression is significantly increased, thereby enhancing T-cell signaling through CD28 and further promoting T-cell activation that worsen stroke outcomes. However, after anti-PD-L1 blockade or in PD-L1 deficient mice with low CD80 expression and without PD-L1 to compete with CTLA-4, depressed T-cell signaling results through CD80/CTLA-4 interactions as well as inhibitory PD-1/PD-L2 interactions.
An important additional feature of PD-L1 blockade that likely contributed to better MCAO outcomes is the induction of IL-10-secreting CD8+CD122+ T-regulatory cells 29 that were significantly increased in both the spleen (Figure 5C) and the lesioned brain hemisphere (Figure 3D) after anti-PD-L1 therapy. The contribution of this CD8+ Treg population could account for the increased expression of Il-10 in the ischemic brain hemisphere (Figure 4B) and may be crucial for successful treatment of MCAO in the absence of increased levels of other protective anti-inflammatory cell populations, including CD4+FoxP3+ Tregs 17, 22 and IL-10+ Bregs 16, 30 that appear to be downregulated by PD-L2 in the absence of PD-L1 11.
Our understanding of the roles of PD-1, PD-L1 and PD-L2 in stroke has progressed concurrently with intense development of anti-PD-1 and anti-PD-L1 mAb for treatment of advanced solid tumors14. PD-L1 is expressed on many cancer cell types as well as on APCs and prevents tumor cell killing by blocking the function of tumor specific cytotoxic T cells. Treatment with anti-PD-L1 mAb releases this blocking effect and promotes T cell killing of tumor cells. Currently, a human IgG1κ anti-PD-L1 mAb (MEDI4736) is being evaluated in 25 ongoing or planned clinical studies in multiple tumor types, with encouraging results in more than 800 treated patients 14. This antibody blocks PD-L1 binding to its receptors, PD-1 and CD80, has high affinity and selectivity for PD-L1 but not PD-L2, produces sustained drug exposure for up to 1 year of dosing, is engineered to prevent antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity that cause off-target inflammatory side effects and has no reported immunogenicity impacting its pharmacokinetics/pharmacodynamics. This new and powerful clinical reagent is thus highly suitable for immediate testing as a novel treatment approach for stroke subjects.
Summary/Conclusions
This study demonstrates for the first time the beneficial therapeutic effects of PD-L1 checkpoint blockade on MCAO, thus validating proposed mechanisms obtained in our previous studies using PD-1 and PD-L deficient mice. Treatment of MCAO with anti-PD-L1 mAb 4 h after occlusion significantly reduced infarct volumes and improved neurological outcome after 96 h reperfusion, providing strong support for use of available humanized anti-PD-L1 antibodies for treatment of human stroke subjects.
Supplementary Material
Acknowledgments
SB designed and performed the immunology experiments, carried out statistical analyses, prepared graphics and wrote part of the manuscript; YC and JW performed the MCAO, carried out statistical analyses, prepared the graphics for the histology procedure; AL assisted in tissue preparations and acquisition of immunological data; AD assisted in performing the immunology experiments, carried out statistical analyses, prepared graphs and final Figure 1; AAV critiqued and edited the manuscript; JAS supervised the MCAO set-up and statistical analyses, processed the MCAO-related infarct volume, neurological deficit scores and mortality and morbidity data and edited the manuscript; HO directed the overall study and wrote the manuscript. All authors read and approved the final version of the manuscript. The authors wish to thank Gail Kent for assistance with manuscript submission.
Sources of Funding: This work was supported by NIH/NINDS 1RO1 NS075887 and 1RO1 NS047661. This material is based upon work supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development. The contents do not represent the views of the Department of Veterans Affairs or the United States Government.
Footnotes
Disclosures: None
References
- 1.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 2.Lakhan S, Kirchgessner A, Hofer M. Inflammatory mechanisms in ischemic stroke: Therapeutic approaches. J Transl Med. 2009;7:97. doi: 10.1186/1479-5876-7-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaller B, Graf R. Cerebral ischemia and reperfusion: The pathophysiologic concept as a basis for clinical therapy. J Cereb Blood Flow Metab. 2004;24:351–371. doi: 10.1097/00004647-200404000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Iadecola C, Anrather J. The immunology of stroke: From mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Macrez R, Ali C, Toutirais O, LeMauff B, Defer G, Dirnagle U, et al. Stroke and the immune system: From pathophysiology to new therapeutic strategies. Lancet Neurol. 2011;10:471–480. doi: 10.1016/S1474-4422(11)70066-7. [DOI] [PubMed] [Google Scholar]
- 6.Hurn PD, Subramanian S, Parker SM, Afentoulis ME, Kaler LJ, Vandenbark AA, et al. T-and b-cell-deficient mice with experimental stroke have reduced lesion size and inflammation. J Cerebral Blood Flow & Metab. 2007;27:1798–1805. doi: 10.1038/sj.jcbfm.9600482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kleinschnitz C, Schwab N, Kraft P, Hagedorn I, Dreykluft A, Schwarz T, et al. Early detrimental t-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood. 2010;115:3835–3842. doi: 10.1182/blood-2009-10-249078. [DOI] [PubMed] [Google Scholar]
- 8.Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada L, et al. Pivotal role of cerebral interleukin-17-producing gammadelta t cells in the delayed phase of ischemic brain injury. Nat Med. 2009;15:946–950. doi: 10.1038/nm.1999. [DOI] [PubMed] [Google Scholar]
- 9.Yilmaz G, Arumugam T, Stokes K, Granger D. Role of t lymphocytes and interferon-gamma in ischemic stroke. Circulation. 2006;113:2105–2112. doi: 10.1161/CIRCULATIONAHA.105.593046. [DOI] [PubMed] [Google Scholar]
- 10.Ren X, Akiyoshi K, Vandenbark AA, Hurn PD, Offner H. Programmed death-1 pathway limits central nervous system inflammation and neurologic deficits in murine experimental stroke. Stroke. 2011;42:2578–2583. doi: 10.1161/STROKEAHA.111.613182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. Pd-l1 enhances cns inflammation and infarct volume following experimental stroke in mice in opposition to pd-1. J Neuroinflammation. 2013;10:111. doi: 10.1186/1742-2094-10-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bodhankar S, Chen Y, Lapato A, Vandenbark AA, Murphy SJ, Offner H. Targeting immune co-stimulatory effects of pd-l1 and pd-l2 might represent an effective therapeutic strategy in stroke. Front Cell Neurosci. 2014;8:228. doi: 10.3389/fncel.2014.00228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loke P, Allison J. Pd-l1 and pd-l2 are differentially regulated by th1 and th2 cells. Proc Natl Acad Sci USA. 2003;100:5336–5341. doi: 10.1073/pnas.0931259100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ibrahim R, SR, Shalabi A. Pd-l1 blockade for cancer treatment: Medi4736. Seminars in Oncology. 2015;42:474–483. doi: 10.1053/j.seminoncol.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 15.Chen Y, Bodhankar S, Murphy SJ, Vandenbark AA, Alkayed NJ, Offner H. Intrastriatal b-cell administration limits infarct size after stroke in b-cell deficient mice. Metab Brain Dis. 2012;27:487–493. doi: 10.1007/s11011-012-9317-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. Il-10-producing b-cells limit cns inflammation and infarct volume in experimental stroke. Metabolic brain disease. 2013;28:375–386. doi: 10.1007/s11011-013-9413-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab. 2006;26:654–665. doi: 10.1038/sj.jcbfm.9600217. [DOI] [PubMed] [Google Scholar]
- 18.Campanella M, Sciorati C, Tarozzo G, Beltramo M. Flow cytometric analysis of inflammatory cells in ischemic rat brain. Stroke. 2002;33:586–592. doi: 10.1161/hs0202.103399. [DOI] [PubMed] [Google Scholar]
- 19.Offner H, Subramanian S, Parker SM, Wang C, Afentoulis ME, Lewis A, et al. Splenic atrophy in experimental stroke is accompanied by increased regulatory t cells and circulating macrophages. J Immunol. 2006;176:6523–6531. doi: 10.4049/jimmunol.176.11.6523. [DOI] [PubMed] [Google Scholar]
- 20.Clark AW, Krekoski CA, Bou SS, Chapman KR, Edwards DR. Increased gelatinase a (mmp-2) and gelatinase b (mmp-9) activities in human brain after focal ischemia. Neurosci Lett. 1997;238:53–56. doi: 10.1016/s0304-3940(97)00859-8. [DOI] [PubMed] [Google Scholar]
- 21.Rosenberg GA, Navratil M, Barone F, Feuerstein G. Proteolytic cascade enzymes increase in focal cerebral ischemia in rat. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 1996;16:360–366. doi: 10.1097/00004647-199605000-00002. [DOI] [PubMed] [Google Scholar]
- 22.Schroeter M, Jander S, Witte OW, Stoll G. Local immune responses in the rat cerebral cortex after middle cerebral artery occlusion. J Neuroimmunol. 1994;55:195–203. doi: 10.1016/0165-5728(94)90010-8. [DOI] [PubMed] [Google Scholar]
- 23.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the b7-1 costimulatory molecule to inhibit t cell responses. Immunity. 2007;27:111–122. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park JJ, Omiya R, Matsumura Y, Sakoda Y, Kuramasu A, Augustine MM, et al. B7-h1/cd80 interaction is required for the induction and maintenance of peripheral t-cell tolerance. Blood. 2010;116:1291–1298. doi: 10.1182/blood-2010-01-265975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pittet CL, Newcombe J, Prat A, Arbour N. Human brain endothelial cells endeavor to immunoregulate cd8 t cells via pd-1 ligand expression in multiple sclerosis. J Neuroinflammation. 2011;8:155. doi: 10.1186/1742-2094-8-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paterson AM, Brown KE, Keir ME, Vanguri VK, Riella LV, Chandraker A, et al. The programmed death-1 ligand 1:B7-1 pathway restrains diabetogenic effector t cells in vivo. J Immunol. 2011;187:1097–1105. doi: 10.4049/jimmunol.1003496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-pd-l1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Francisco L, Sage P, Sharpe A. The pd-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–242. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bodhankar S, Chen Y, Lapato A, Vandenbark AA, Murphy SJ, Saugstad JA, et al. Regulatory cd8(+)cd122 (+) t-cells predominate in cns after treatment of experimental stroke in male mice with il-10-secreting b-cells. Metab Brain Dis. 2015;30:911–924. doi: 10.1007/s11011-014-9639-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. Treatment of experimental stroke with il-10-producing b-cells reduces infarct size and peripheral and cns inflammation in wild-type b-cell-sufficient mice. Metabolic brain disease. 2014;29:59–73. doi: 10.1007/s11011-013-9474-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.