

Abstract
We propose a method (GREML-LDMS) to estimate heritability for human complex traits in unrelated individuals using whole-genome sequencing (WGS) data. We demonstrate using simulations based on WGS data that ~97% and ~68% of variation at common and rare variants, respectively, can be captured by imputation. Using the GREML-LDMS method, we estimate from 44,126 unrelated individuals that all ~17M imputed variants explain 56% (s.e. = 2.3%) of variance for height and 27% (s.e. = 2.5%) for body mass index (BMI), and find evidence that height- and BMI-associated variants have been under natural selection. Considering imperfect tagging of imputation and potential overestimation of heritability from previous family-based studies, heritability is likely to be 60–70% for height and 30–40% for BMI. Therefore, missing heritability is small for both traits. For further gene discovery of complex traits, a design with SNP arrays followed by imputation is more cost-effective than WGS at current prices.
Introduction
Genome-wide association studies (GWAS) have identified thousands of genetic variants associated with hundreds of human complex traits and diseases1. However, genome-wide significant SNPs often explain only a small proportion of heritability estimated from family studies, the so-called missing heritability problem2. Recent studies show that the total variance explained by all common SNPs is a large proportion of the heritability for complex traits and diseases3,4. This implies that much of the missing heritability is due to variants whose effects are too small to reach genome-wide significance level. This conclusion is supported by recent findings that complex traits and diseases such as height, body mass index (BMI), age at menarche, inflammatory bowel diseases, and schizophrenia are influenced by hundreds or even thousands of genetic variants of small effects5–9. Nevertheless, the genetic variance accounted for by all common SNPs is still less than that expected from family studies and there has not been a consensus explanation to the ‘missing heritability’ problem2. There are three major hypotheses. The first hypothesis is that missing heritability is largely due to rare variants of large effect, which are neither on the current commercial SNP arrays nor well tagged by the SNPs on the arrays. Here we define rare variants as the variants with minor allele frequency (MAF) ≤ 0.01. To genotype rare variants with reasonably high accuracy, whole-genome sequencing (WGS) with sufficiently high coverage in a large sample is required. The second hypothesis is that the majority of heritability is attributable to common variants (MAF > 0.01) of small effect so that many variants are not detected at genome-wide significance level, and most of these common variants are either well tagged by the genotyped SNPs through linkage disequilibrium (LD) or can be imputed with reasonably high accuracy from WGS reference panels. If this is the case, increasing sample size is more important than extending variant coverage for continued progress in genetic association studies. The third hypothesis is that heritability estimates from family studies are biased upward, for instance, due to common environment effects. Therefore, quantifying the relative contributions of rare and common variants to trait variation is critical to inform the design of future experiments to disentangle the genetic architecture of complex traits and diseases. In this study, we seek to quantify the proportion of variation at common and rare sequence variants that can be captured by SNP array genotyping followed by imputation, and subsequently we estimate the proportion of phenotypic variance for the model complex traits height and BMI that can be explained by all imputed variants.
Results
Unbiased estimate of heritability using WGS data
Let denote the narrow-sense heritability (h2) for a complex trait captured by the sequence variants from WGS, and denote h2 captured by all variants from imputing the SNP array data to the 1000 Genome Project (KGP) reference panels10, where because of the loss of tagging due to imperfect imputation. We previously developed the single-component (based on a single genetic relationship matrix) GREML analysis (GREML-SC) as implemented in GCTA11, to estimate the proportion of variance explained by all common SNPs in a GWAS sample of unrelated individuals12. To quantify the amount of variation at sequence variants that can be captured by 1KGP imputation, we first needed to investigate whether this approach can provide an unbiased estimate of heritability using WGS data. We performed extensive simulations based on a WGS data set from the UK10K project13 (UK10K-WGS), which comprises 17.6M genetic variants (excluding singletons and doubletons) on 3,642 unrelated individuals after quality controls (QC) (Online Methods). The simulation results show that if causal variants are a random subset of all the sequence variants (52.7% rare), the GREML-SC estimate of using all variants (including the causal variants) is unbiased (Fig. 1), consistent with our theoretical derivation (Supplementary Note). By unbiased we mean that the mean estimate of from 200 simulation replicates is not significantly different from the h2 parameter used for simulation. We could also expect from the theoretical derivation that if causal variants have a different MAF spectrum from the variants used in the analysis, the GREML-SC estimate of will be biased. This is demonstrated using simulations (Online Methods): if we randomly sample disproportionally more rare (common) variants as causal variants, the estimate of is biased downwards (upwards) (Fig. 1). This problem has been discussed previously12 and can be solved by the MAF stratified GREML (GREML-MS) analysis14 (Online Methods). We show by simulations that the estimate of from GREML-MS is unbiased irrespective of the MAF spectrum of the causal variants (Fig. 1).
Figure 1.

Estimates of heritability using sequence variants under different simulation scenarios based on the UK10K-WGS data. Each column represents the mean estimate from 200 simulations. Each error bar is the s.e. of the mean. The true heritability parameter is 0.8 for the simulated trait (see Online Methods for the 4 simulation scenarios).
We know from the theoretical derivation (Supplementary Note) that GREML-SC is biased if causal variants have a different LD property from the variants used in the analysis. The difference in LD can be caused by the difference in MAF spectrum, which can be corrected for using the GREML-MS approach as shown above. However, GREML-MS is unable to correct for the region-specific LD heterogeneity across the genome as illustrated in Supplementary Fig. 1. That is, if causal variants tend to be enriched in genomic regions with higher or lower LD than average, the estimate of h2 from either GREML-SC or GREML-MS will be biased. This is confirmed using simulations where if all causal variants are sampled from the variants at the DNase I hypersensitive sites (DHSs) (Online Methods), which have systematically lower LD than average15, the GREML-MS estimate of is biased downwards (Fig. 1). There have been methods16,17 to adjust for the LD heterogeneity, e.g. the LDAK approach16 that gives each variant a weight according to the LD r2 between the variant and all the other variants in the region, and the LD residual (LDres) approach15,17 that uses residuals from a linear regression of each variant on a set of LD-pruned variants in the region. However, the LDAK adjustment resulted in a substantial overestimation of regardless of whether the variants were stratified by MAF or not (Fig. 1). This is because the LDAK adjustment created a strong negative correlation between the weights and MAF of the variants (Supplementary Fig. 2) such that rare variants, which tend to have lower LD with surrounding variants, receive too much weight. We also observed small biases using the LDres and the MAF stratified LDres (LDres-MS) approaches (Fig. 1). We propose a method, termed the LD and MAF stratified GREML (GREML-LDMS) approach, which corrects for differences in both MAF and LD between causal variants and the variants used in the analysis (Online Methods). We demonstrate by analysing simulated data, under four different scenarios, that the GREML-LDMS estimates of are unbiased regardless of the MAF and LD properties of causal variants (Fig. 1) and the number of LD and MAF groups (Supplementary Fig. 3). The heritability parameter used in all the simulations above was 0.8. We show in Supplementary Table 1 that all the conclusions hold irrespective of the size of heritability parameter used for simulation.
Variation at WGS variants captured by 1KGP imputation
We have quantified above the (un)biasedness of GREML methods in estimating under different simulation scenarios. In practice, however, there have not been WGS data available with sample size that is sufficiently large to estimate with useful precision. On the other hand, there are a large number of GWAS samples available that have been imputed to the 1KGP reference panels. We therefore addressed the question of how much variation at sequence variants can be captured by imputing GWAS genotype data to 1KGP. From UK10-WGS data we extracted the genotypes of variants represented on the Illumina CoreExome array, and then imputed the genotype data to the 1KGP reference panels (Online Methods). We used the GREML-MS approach (7 genetic components) to estimate the variance explained by the 1KGP-imputed variants ( ) for the simulated phenotype (1,000 causal variants randomly sampled from all sequence variants) (Online Methods). We know from the simulation results presented above that under this scenario (i.e. causal variants are completely randomly sampled), all the three GREML methods, GREML-SC, GREML-MS and GREML-LDMS, are unbiased. We chose GREML-MS because it is able to provide the estimates of variance explained for variants in different MAF groups with standard errors smaller than those from GREML-LDMS (Supplementary Table 1). The results show that the proportion of variation at WGS variants captured by 1KGP imputation decreases with more stringent imputation accuracy (the metric INFO from IMPUTE2) threshold used for variant filtering, and that all the 1KGP imputed variants (without filtering variants for IMPUTE-INFO) capture ~96% and ~73% of variation at common and rare WGS variants, respectively (Fig. 3a). We then repeated the imputation based on four other types of SNP arrays, namely Affymetrix 6.0, Affymetrix Axiom, Illumina OmniExpress and Illumina Omni2.5. The results are remarkably consistent irrespective of the types of SNP arrays used for baseline genotyping (Fig. 3b). On average across the five different SNP arrays used for baseline genotyping, ~97% and ~72% of variation at common and rare sequence variants can be captured by 1KGP-imputed variants, respectively. Surprisingly, despite the Illumina Omni2.5 array having ~933K and ~1.2M more variants than the OmniExpress and CoreExome arrays used for imputation (Supplementary Table 2), respectively, the proportions of variation at sequence variants captured by 1KGP-imputed variants based on these three types of SNP arrays were almost identical (Fig. 3b). We further performed simulations under 4 scenarios (Online Methods) and analysed the simulated data using the GREML-LDMS approach (28 genetic components). The result (Supplementary Fig. 4) shows that the proportion of variation at sequence variance captured by 1KGP imputation is almost independent of genetic architecture. On average across SNP arrays and simulation scenarios, ~97% of variation at common variants and ~68% at rare variants can be captured by 1KGP imputation.
Figure 3.

Proportion of variation at sequence variants captured by 1KGP imputation in the UK10K-WGS data. The results are the averages from 200 simulations (Online Methods). Panel (a): estimates of proportion of phenotypic variance explained by 1KGP-imputed variants in different MAF groups from GREML-MS. The 1KGP imputation was based on variants on Illumina CoreExome array extracted from the UK10K-WGS data. The column in purple represents the variance explained by the causal variants. The other four columns represent the estimates using 1KGP-imputed variants filtered at 3 levels of imputation accuracy (IMPUTE-INFO) threshold. The error bar is the s.e.m.. Without filtering variants for IMPUTE-INFO (columns in yellow), the sum of the estimate is 96.2% for common variants and 73.4% for rare variants. Panel (b): estimates of proportion of variation at sequence variants captured by 1KGP imputation (the estimate of phenotypic variance explained by the 1KGP-imputed variants summed over MAF groups divided by that explained by the causal variants) based on different types of SNP genotyping arrays. Common: MAF > 0.01; Rare: 0.01 ≥ MAF > 0.0003.
Quantifying the missing heritability for height and BMI
We next applied the GREML-LDMS approach to estimate the proportion of phenotypic variance explained by the 1KGP-imputed variants ( ) for height and BMI. We combined data from 7 GWAS cohorts (Online Methods), comprising ~17M 1KGP-imputed variants on 44,126 unrelated individuals after QC. Results from GREML-LDMS analyses (Online Methods) show that all 1KGP-imputed variants explain 55.5% (s.e. = 2.3%) of phenotypic variance for height and 27.4% (s.e. = 2.5%) for BMI, with common variants explaining 47.6% (s.e. = 1.2%) of variance for height and 25.0% (s.e. = 1.2%) for BMI (Supplementary Table 3 and Supplementary Fig. 5). The results also show that 1KGP-imputation captures a significant amount of genetic variation at rare variants for height, with (s.e. = 1.9%, P = 6.1×10−6) for variants with 0.001 < MAF ≤ 0.01, and for BMI, with (s.e. = 1.8%, P = 0.032) for variants with 2.5×10−5 < MAF ≤ 0.001 (Supplementary Table 3). The (co)variance matrix of the estimates of variance components from GREML-LDMS can be found in Supplementary Fig. 6. We also performed the analyses using GREML-MS. The results are similar, with (s.e. = 1.7%, P = 2.6×10−6) for variants with 0.001 < MAF ≤ 0.01 for height and (s.e. = 1.6%, P = 0.011) for variants with 2.5×10−5 < MAF ≤ 0.001 for BMI (Supplementary Fig. 7).
Under an evolutionary neutral model, most variants segregating in the population are rare whereas most genetic variation for a trait is due to common variants18. The neutral model predicts that the cumulative contribution of variants with MAF ≤ θ to the total genetic variance is linearly proportional to θ. However, our observed result for height strongly deviated from this model (Figure 4a), suggesting that height-associated variants have been under natural selection. Such deviation would be even stronger with WGS data because variation at rare sequence variants is less well captured by 1KGP imputation than that at common variants (Fig. 3 and Supplementary Fig. 4). The deviation was smaller for BMI and the s.e. of the estimate of cumulative contribution (see Supplementary Note for the method to calculate the s.e.) was much larger for BMI than that for height. Equivalently, the neutral model also predicts that variance explained is uniformly distributed as a function of MAF18, so that variance explained by variants with MAF ≤ 0.1 equals that of variants with MAF > 0.4. However, we observed that although variance explained per variant (defined as with m being the number of variants) for rare variants was much smaller than that for common variants for both height and BMI (Supplementary Fig. 8), all the variants with MAF ≤ 0.1 in total explained a significantly larger proportion of variance than those with MAF > 0.4 (21.0% vs. 8.8%, Pdifference = 9.2×10−7) for height (Fig. 4b and Supplementary Table 3), consistent with height-associated variants being under selection. The difference was much smaller (7.4% vs. 5.4%) and not significant (Pdifference = 0.45) for BMI. These results were not driven by population stratification (Supplementary Note and Supplementary Fig. 9).
Figure 4.

Evidence for height- and BMI-associated genetic variants being under natural selection. Results shown in panels (a) and (b) are from the GREML-LDMS analyses (Online Methods). Panel (a): the estimate of cumulative contribution of variants with MAF ≤ θ to the genetic variance, i.e. . The dash line represents that expected under a neutral model. Panel (b): the estimate of for variants in each MAF group. Error bar is s.e. of the estimate. Results shown on panel (c) are from genome-wide association analyses in the combined data set (Online Methods). bm is defined as the effect size of the minor allele of a variant. Variants are stratified into 100 MAF bins (100 quartiles of the MAF distribution). Plotted is the mean of b̂m against log10(mean MAF) in each bin. The correlation between mean b̂m and log10(mean MAF) is 0.77 (P < 1.0×10−6) for height and −0.39 (P = 8.0×10−6) for BMI. Shown on panel (d) are the results from the latest GIANT consortium meta-analyses for height5 and BMI22 (see Web Resources) for common SNPs (MAF > 0.01). There are ~2.5M SNPs stratified into 20 MAF bins. The correlation between mean b̂m and log10(mean MAF) is 0.89 (Ppermu < 1.0×10−6) for height and −0.87 (Ppermu < 1.0×10−6) for BMI. The mean b̂m seems smaller in panel (c) than that in panel (d) because of the smaller MAF range of each bin and larger number of variants in each bin in panel (c) than those in panel (d).
Theoretical studies on complex trait variation based on natural selection models suggest that rare variants only explain a substantial amount of variance under strong assumptions about the relationship between effect size and selection strength19–21. We performed genome-wide association analyses for height and BMI in the combined data set (Online Methods), and found that the minor alleles of variants with lower MAF tended to have stronger and negative effects on height and stronger but positive effects on BMI (Fig. 4c). The correlation between minor allele effect and MAF was highly significant for both height (P < 1.0×10−6) and BMI (P = 8.0×10−5), which was even stronger for both in the data from the latest GIANT consortium meta-analyses5,22 (Fig. 4d), and these correlations were not driven by population stratification (Supplementary Fig. 10). All these results suggest that height- and BMI-associated variants have been under selection. These results are consistent with the hypothesis that new mutations which decrease height or increase obesity tend to be deleterious to fitness and hence kept at low frequencies in the population by purifying selection.
Discussion
We have shown using simulations based on WGS data that the GREML-SC estimate of heritability is unbiased if causal variants are a random subset of the sequence variants used in the analysis, that GREML-MS is unbiased if there is no region-specific LD heterogeneity, and that GREML-LDMS is unbiased regardless of the MAF and LD properties of causal variants (Fig. 1). Although we described the methods for quantitative traits, the methods can also be applied to case-control studies by analysing the data on the observed disease status (0 or 1) scale and interpreting the estimates on an underlying liability scale by a linear transformation4,23. As suggested in previous studies4,24, more stringent quality controls than those used in GWAS are required to apply the GREML approaches in case-control data. Since GREML-LDMS fits a large number of components (e.g. we fitted 28 genetic components in the analyses of height and BMI data), the sampling variance of is much larger than that from GREML-SC. We calibrated empirically that the standard error (s.e.) of from GREML-LDMS was approximately 927 / n (Supplementary Fig. 11), i.e. inversely proportional to the sample size (n) and irrespective of the estimate of . For a GREML-LDMS analysis using either WGS or imputed-WGS data, a sample of at least 18,540 unrelated individuals is therefore required to get an s.e. < 0.05. We have further demonstrated that the GREML-LDMS approach is robust to the model assumption about the relationship between effect size and MAF (Supplementary Note, and Supplementary Figs. 12 and 13).
Using the GREML-LDMS approach we estimated that all the 1KGP-imputed variants explained 56% (s.e. = 2.3%) and 27% (s.e. = 2.5%) of phenotypic variance for height and BMI, respectively. These estimates are still lower than the frequently quoted estimates of narrow-sense heritability (h2) for height (80%) and BMI (40%~60%) from family/twin studies. Therefore it seems that heritability is still “missing”. There are two possible explanations for the still missing heritability. The first is that there are a large number of extremely rare causal variants, not polymorphic in the 1KGP-imputed data, or removed by post-imputation QC. For example, there are > 40M and > 45M variants in 1KGP10 and UK10K13 data, respectively, whereas “only” 17M imputed variants were used in the GREML analyses for height and BMI. And also, complex DNA variations such as copy number variations are not well represented by current sequencing methods25,26. The second explanation is that heritability is overestimated in family studies due to effects such as common environment and assortative mating that are not properly modeled27. Result from a previous study shows that the phenotypic correlation for height between distant relatives (e.g. cousins) is larger than what we would expect given h2 = 0.8 under an additive model28, suggesting a substantial confounding in the family-based estimate of h2 but not supporting an important role for non-additive genetic variance. A recent study29 that used extended genealogy in a large sample (n = 38,167) provides very precise estimates of the heritability for height (ĥ2 = 0.69, s.e. = 0.016) and BMI (ĥ2 = 0.42, s.e. = 0.018). These estimates can be regarded as the upper limits of heritability for height and BMI because the common environmental effects were not explicitly fitted in the model and so that these estimates, to some extent, could still be inflated. Estimates from a within-family analysis that is free of confounding from common environmental effects are highly consistent with heritability being 0.69 (s.e. = 0.14) for height and 0.42 (s.e. = 0.17) for BMI but the standard errors are too large to draw strong inferences30. There has also been evidence suggesting that population-based heritability estimate is likely to be lower than that from pedigrees31. If we extrapolate from the GREML-LDMS estimates (Supplementary Table 3) by taking into account the imperfect tagging of 1KGP imputation (on average across five different types of SNP array and four simulation scenarios, ~97% and ~68% of variation at common and rare variants captured by 1KGP imputation), the adjusted estimate of heritability would be 0.61 (s.e. = 0.045) for height and 0.29 (s.e. = 0.47) for BMI (see Supplementary Note for the adjustment method). These estimates can be regarded as the lower limits of narrow-sense heritability for height and BMI. Our results suggest that heritability is likely between 0.6 and 0.7 for height and between 0.3 and 0.4 for BMI. Therefore, there is little missing heritability for these traits. These results also suggest there is little room for the other possible sources of missing heritability (Supplementary Note).
We know from the simulations (Fig. 1 and Supplementary Table 1) that GREML-SC and GREML-MS methods can be biased depending on MAF and LD properties of the causal variants. For completion of the analysis, we also performed the GREML-SC and GREML-MS analyses in the combined GWAS data set using 1KGP-imputed variants. The GREML-SC estimate of was 0.78 (s.e. = 0.017) for height and 0.40 (s.e. = 0.018) for BMI, larger than the estimate from GREML-LDMS for both traits. This is because the proportion of loss of tagging in 1KGP imputation for rare variants is larger than that for common variants (Fig. 3), analogues to the situation where common variants explain disproportionally more proportion of variance (simulation scenario II in Online Methods) resulting in an overestimation of (Fig. 1 and Supplementary Table 1). This is an important caveat. If we were to draw inference based on these results, we would conclude that all the heritability for height and BMI have been captured by the 1KGP imputation, which is obviously not true. The GREML-MS estimate of was 0.523 (s.e. = 0.021) for height and 0.261 (s.e. = 0.022) for BMI, which were not very dissimilar to those from GREML-LDMS especially for BMI. Therefore, it is more important to correct for the difference in MAF spectrum than LD heterogeneity (Supplementary Note and Supplementary Fig. 14). There are also other scenarios under which the GREML methods can be misused and thus may lead to wrong conclusions, e.g. the Morrison et al.32 WGS study (Supplementary Note).
More generally, we have shown by simulations that for variants with MAF > 0.0003 in the UK10K-WGS, 96% and 73% of variation at common and rare WGS variants, respectively, can be captured by 1KGP imputation using Illumina CoreExome arrays for baseline genotyping (Fig. 2). These percentages of variation need to be interpreted as multi-variant tagging, analogous to the multiple correlation squared ( ) between a sequence variant and all the 1KGP-imputed variants in the region, which are not comparable to the single-variant based imputation accuracy (variation at a sequence variant tagged by a single imputed variant) quantified in previous studies33,34. In GWAS, the analysis is usually performed based on a single-variant model. The statistical power of a single-variant analysis using imputed data depends on the squared correlation between a causal variant and its best-tagging imputed variant ( ). We show in Fig. 5 that, on average, 81% (s.e.m. = 0.4%) and 25% (s.e.m. = 0.4%) of variation at common and rare WGS variants, respectively, can be captured by their best-tagging variants from 1KGP imputation based on Illumina CoreExome arrays. These results are comparable with the single-variant imputation accuracy quantified previously. The single-variant tagging is slightly stronger for 1KGP imputation based on Illumina OmniExpress arrays, 88% (s.e.m. = 0.3%) for common and 29% (s.e.m. = 0.4%) for rare variants. These results suggest that for GWAS analyses using 1KGP imputed data, there is a great potential to gain power using a multi-variant or haplotype based association analysis approach, for rare variants in particular. Therefore, with the increasingly large amount of summary-level data from large-scale meta-analyses of 1KGP-imputed GWAS data that have been becoming available, there is a great demand to develop powerful and efficient methods for multi-variants or haplotype based association analysis using summary data, which take into account both common and rare variants.
Figure 2.
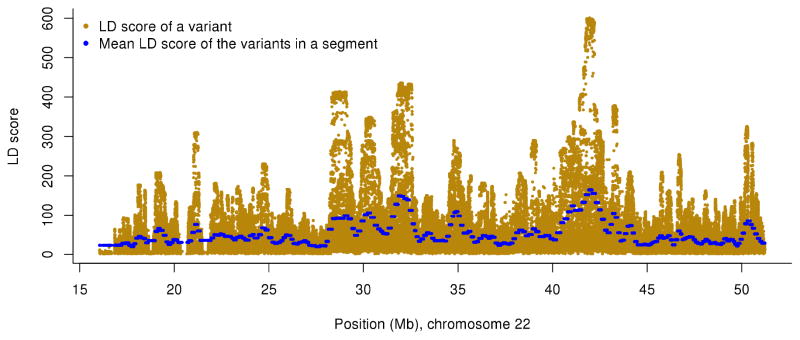
Fitting region-specific LD heterogeneity of the genome using a sliding-window approach. Shown are the results for chromosome 22 from the UK10K-WGS data as example. LD score of each variant is defined as the sum of LD r2 between the target variant and all variants (including the target variant) within ±10Mb distance. For the GREML-LDMS analysis, the region-specific LD heterogeneity is fitted by segments with average length of 100Kb (the dots in blue) using a sliding window approach as described in Online Methods.
Figure 5.
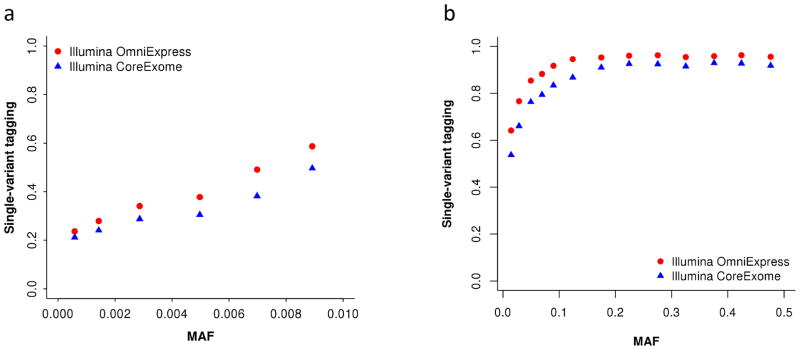
Single-variant tagging of sequence variants by 1KGP-imputed variants. Single-variant tagging is defined as the squared correlation (r2max) between a sequence variant and the best tagging variant from 1KGP imputation within ±1Mb distance. Shown are the average r2max of variants in MAF bins for 10,000 sequence variants randomly sampled from the UK10K-WGS data. The 1KGP imputation analyses are based on variants on Illumina OmniExpress (red) and Illumina CoreExome (blue) arrays extracted from the UK10K-WGS data (see Online Methods for details about the imputation analyses based on the UK10K-WGS data). Panel (a): rare variants. Panel (b): common variants.
With the advance of genome-sequencing technologies, we are now able to sequence a human genome at high depth for $1000, which, however, is still much more expensive than that using a SNP array (e.g. Illumina CoreExome). Given a fixed budget for genotyping and assuming genotyping cost using SNP arrays (e.g. $50 per sample) is 20 times cheaper than WGS (e.g. $1000 per sample), on average, 1KGP imputation is currently at least 13 times more powerful than WGS using a multi-variant association analysis approach (Supplementary Fig. 15). For a single-variant based association analysis, 1KGP imputation is still at least 13 and 4 times more powerful than WGS in detecting common and rare variant associations, respectively. These results suggest that SNP array based genotyping followed by imputation is now, and will continue to be in the near future, a more cost-effective strategy than WGS for GWAS of complex traits and diseases, even for rare variant associations. Nevertheless, the analyses above compared the average power for variants in a certain MAF range. As shown in Supplementary Fig. 16, there are a number of sequence variants (~10% rare and ~1% common) that are almost not tagged by any imputed variant individually (single-variant tagging ) from 1KGP imputation based on Illumina CoreExome array. For association analysis of such variants and those with extremely low frequency or unique to specific populations, high-coverage WGS or a haplotype based method will be a more efficient strategy. On the other hand, it has been suggested that extremely low-coverage WGS followed by imputation can be even more cost-effective than SNP-array based imputation for common variants35, an interesting strategy that is worth being followed by further investigation of its performance for rare variants.
With the latest imputation reference panel of large sample size36 (n = 31,000) and very large GWAS cohorts genotyped on the same type of SNP arrays (e.g. the UK Biobank has genotyped > 400,000 individuals using Affymetrix Axiom arrays) that are soon becoming available, we can expect a great improvement of imputation accuracy for rare variants. For complex traits and diseases that have a similar genetic architecture to that of height (enrichment of height associated variants with MAF < 0.1), we can expect to see a wave of discovery of trait- or disease-associated low-MAF variants in the near future, without the need for large-scale whole genome sequencing.
Online Methods
Simulating phenotypes based on whole genome sequencing data
We used WGS data from the UK10K project13 for simulations. The data set comprises 3,781 individuals from the TwinUK and ALSPAC cohorts and ~45.5M genetic variants called from whole genome sequencing after QC. Informed consent was obtained from all the subjects. Information on the cohorts, sequencing, variant calling, and QC have been detailed elsewhere13. We performed additional QCs of the data, excluding SNPs with missingness > 0.05, Hardy-Weinberg Equilibrium (HWE) test P < 1×10−6, or minor allele count (MAC) < 3 (equivalent to MAF < 0.0003) using PLINK37. We also excluded individuals with genotype missingness rate > 0.05, and one of each pair of individuals with estimated genetic relatedness > 0.05 using variants on HapMap phase 3 reference panels (HapMap3) after QC. We retained 3,642 unrelated individuals and 17.6M variants (see Supplementary Fig. 17 for the MAF distribution of the variants). We call this data set UK10K-WGS throughout the paper.
We randomly sampled mq variants from UK10K-WGS as causal variants. We generated the phenotype using the model y = g + e, where with xi being the genotype variable of the i-th causal variant (coded as 0, 1 or 2) and pi being the frequency of the coded allele, and ui is the effect size per standardised genotype. This model assumes larger per-allele effect sizes for variants with lower MAF. We generated u from N(0, 1) and then generated the residual e from N[0, var(g)(1/ h2 − 1)]. We sampled the causal variants under four scenarios: I) Random: 1,000 causal variants randomly sampled from all the sequence variants (52.7% rare); II) More common: 1,000 random and additional 500 common (MAF > 0.01) causal variants; III) Rarer: 1,000 random and additional 500 rare (MAF < 0.01) causal variants; IV) Rarer & DHS: 1,000 random and additional 500 rare causal variants all sampled from the variants at the DNase I hypersensitive sites (DHSs) (Web Resources). In the UK10K-WGS data, mean LD score for variants at DHSs (59.3) is lower than that (80.3) for variants at non-DHSs, consistent with that observed from previous studies for common SNPs15, where LD score for a variant is defined as the sum of LD r2 between the target variant and all the variants (including the target variant itself) within ±10Mb distance (LD r2 threshold = 0.01). Given a simulated heritability of 0.8, the proportion of variance by the causal variants (stratified by MAF) in each of the four scenarios is presented in Supplementary Fig. 18. We repeated the simulations 200 times with the causal variants re-sampled in each replicate. We analyzed the simulated data using the GREML methods as described below. We also performed the analyses using LDAK16 v3.0 (options: minmaf = 1×10−6, minvar = 1×10−6, and maxiter = 4×105), and LDres as implemented in EIGENSTRAT38 v6.0.1 (options: ldposlimit = 5×105 and numoutlieriter = 0).
Quantifying the proportion of genetic variation captured by imputation
We extracted genotype data of the variants that are on the Illumina CoreExome array from the UK10K-WGS data, and imputed the genotype data to the 1KGP reference panels using IMPUTE2 (ref39). This was to mimic a GWAS where the subjects were genotyped using a SNP array followed by 1KGP imputation. We repeated the analysis for four other types of commonly used SNP arrays, i.e. Affymetrix 6.0, Affymetrix Axiom, Illumina OmniExpress and Illumina Omni2.5. The numbers of variants used in the 1KGP imputation analyses for these five types of arrays are listed in Supplementary Table 2. We converted the dosage scores (xdose) from imputation to hard genotype calls (x), i.e. x = 0 if xdose < 0.5, x = 2 if xdose > 1.5 and x = 1 otherwise), and removed those imputed variants with HWE p < 1×10−6 or MAC < 3 (equivalent to MAF < 2.5×10−5). We further removed variants with imputation accuracy (the metric INFO from IMPUTE2 output) < a threshold. We chose a range of IMPUTE-INFO threshold values (from 0 to 0.9 by 0.1) to investigate the loss of tagging by removing variants with lower imputation accuracy. We then used the MAF stratified GREML analysis (see below) to estimate the proportion of variance in the simulated phenotype (based on the UK10K-WGS data under scenario I as described above) that can be explained by the 1KGP-imputed variants for the 5 types of SNP arrays with the imputed variants filtered at 10 IMPUTE-INFO thresholds. This analysis is to quantify the proportion of genetic variance that can be captured by SNP array based genotyping followed by 1KGP imputation when causal variants are a random sample of the WGS variants.
GREML-MS and GREML-LDMS approaches
We have shown by theoretical derivation (Supplementary Note) that GREML-SC estimates of h2 are biased if causal variants have a different MAF spectrum from the variants used in analysis. This problem can be solved by the MAF stratified GREML (GREML-MS) analysis14, i.e. the variants are stratified into groups by MAF, and the genetic relationship matrices (GRMs) computed from the variants in each of these MAF groups are fitted jointly in a multi-component GREML analysis11. For a quantitative trait, the model of a multi-component GREML analysis can be written as
| [1] |
where y is a vector of phenotypes, c is a vector of the effects of the fixed covariates (e.g. the first 10 eigenvectors) with the corresponding coefficient matrix K, gt is a vector of genetic values of the individuals attributed to the variants in the t-th group with , and At = {Aij(t)} is the GRM between individuals at the variants in the t-th group, T is the number of groups (variance components), and e is a vector of residuals with . The variance-covariance matrix of the phenotypes is . For variants in the t-th group, the genetic relationship between individuals i and j is calculated as12
| [2] |
where mt is the number of variants, x is the genotype variable coded as 0, 1 or 2, and p is frequency of the coded allele. The variance components ( and ) can be estimated using the restricted maximum likelihood (REML) approach11,40. The variance explained by the variants in each group is defined as and variance explained by all the variants is defined as . Without loss of generality, we use the subscript “v” to represent a set of variants used in the analysis, for analysis using SNP array data, for analysis using WGS data, and for analysis using 1KGP imputed data. The standard errors (s.e.) of the estimates of and can be calculated by the delta method utilizing the sampling (co)variances of the estimates27. The multi-component GREML approach has been applied previously to partition genetic variance into the contributions from variants on 22 chromosomes, in 5 MAF groups24 or classified by functional annotation15 using common SNPs. For WGS or imputed data from WGS panels, there are a large number of rare variants, we therefore stratified the variants into 7 MAF groups, namely MAF ≤ 0.001, 0.001 < MAF ≤ 0.01, 0.01 < MAF ≤ 0.1, 0.1 < MAF ≤ 0.2, 0.2 < MAF ≤ 0.3, 0.3 < MAF ≤ 0.4 and 0.4 < MAF ≤ 0.5.
The GREML-MS approach corrects for the difference in MAF spectrum between causal variants and the variants used in the analysis. However, it does not take into account the region-specific LD heterogeneity as demonstrated in Supplementary Fig. 1. If causal variants tend to be enriched in genomic regions with lower or higher LD than average, the GREML-MS estimate will still be biased (Fig. 1 and Supplementary Table 1). We proposed a method called LD and MAF stratified multi-component GREML (GREML-LDMS) approach. We firstly calculated the LD score for each SNP in the data set used for analysis, where LD score of a variant is defined as the sum of LD r2 between the variant and all the variants (including the variant itself) within ±10Mb distance. We ignored LD r2 < 0.01 to avoid chance correlations between the SNPs that are not in LD. We used a sliding-window approach to fit the region-specific LD heterogeneity by a large number of segments (Fig. 2). There were 2ms variants in each segment and an overlap of ms variants between two adjacent segments, where ms is the average number of variants per 100Kb of a chromosome, i.e. ms = 100(m / L) with m being the total number of variants on a chromosome and L being the length chromosome in Kb. We calculated the mean LD score of the variants in each segment, and took the average of mean LD scores of two adjacent segments for the overlapped regions. Consequently, this process partitioned the genome into a large number of segments (ms variants in each segment with an average length of 100Kb) with different mean LD scores to fit the region-specific LD heterogeneity across the genome (Fig. 2). We then stratified the segments into 4 groups by their mean LD scores, i.e. the first, second, third and forth quartiles of the mean LD score distribution. We further stratified the variants in each of the 4 LD-stratified groups into 7 MAF-stratified groups as described in the GREML-MS analysis above, resulting in 28 groups in total. The number of variants in each group is illustrated in Supplementary Fig. 4c. All the methods described above have been implemented in GCTA11. For the analysis of real data, we performed the GREML-LDMS analysis with 28 variance components to investigate the variance explained as a function of MAF and LD. We have performed simulation to demonstrate the unbiasedness of the GREML-LDMS method to different genetic architectures (Fig. 1, Supplementary Table 1 and Supplementary Fig. 12) and to different number of MAF and LD groups (Supplementary Fig. 3).
Analysis of GWAS data for height and BMI
We accessed GWAS data from 7 cohorts, i.e. ARIC, NHS, HPFS, TwinGene, HRS, EGCUT and LifeLines. A summary description of the sample sizes, genotyping platforms and QC criteria of the genotype data is listed in Supplementary Table 4. Informed consent was obtained from all subjects. The genotype data of each cohort after QC were imputed to 1KGP using IMPUTE2, and the imputed dosage data were converted to hard genotype calls using the method as described above. We combined the imputed data of all the cohorts and excluded SNPs with HWE P < 1×10−6 or MAC < 3. We did not filter variants for IMPUTE-INFO because our simulation results showed that removing variants with an IMPUTE-INFO threshold reduced tagging (Fig. 3). To avoid including close relatives in the sample, we estimated the genetic relatedness between pairwise individuals in the combined data set using ~1.2M common variants on the HapMap3, and removed one of each pair of individuals with the estimated genetic relatedness > 0.05. We retained 44,126 unrelated individuals and 17,007,473 variants in the combined data set for further analysis. We performed principal component (PC) analyses38 in the combined data set i) using ~1.2M common variants on HapMap3 and ii) using all the ~17M variants. All the samples are of European descent, as demonstrated by the PC plot (Supplementary Fig. 19). In each sex group of each cohort, height phenotypes 5 standard deviations (SDs) and BMI phenotypes 7 SDs away from the mean were not included in the analyses. Height and BMI phenotypes were adjusted for age and standardized to z-scores in each sex group in each cohort, which removed the mean and variance difference between sexes and between cohorts. We then estimated the proportion of phenotypic variance that can be explained by all the imputed variants in the combined data set using the GREML-LDMS and GREML-MS approaches with GCTA11. We presented the main results from GREML analyses with the first 10 PCs computed from ~1.2M common variants on HapMap3 as fixed covariates, and also showed the results with PCs estimated from all the ~17M variants.
To investigate the relationship between minor allele effect (bm) and MAF, we performed genome-wide association analyses for height and BMI in the combined data set. The first 10 PCs computed from HapMap3 variants were fitted as covariates in the association analyses. Variants were stratified into 100 MAF bins (100 quartiles) with ~17,000 variants in each bin. We define bm as the effect size of the minor allele of a variant. We further define with p being the minor allele frequency, which is interpreted as the effect size per standardised genotype of a variant. We calculated the mean of b̂m, ûm and MAF in each bin, and then calculated the correlation between mean b̂m (or mean ûm) and log10(mean MAF) across bins. Since the MAF bins were not independent, we assessed the statistical significance of the correlation by comparing the observed value to the empirical distribution from 1 million resampling sets under the null hypothesis by randomly sampling the same number of variants for each MAF bin. In addition, we also performed the analyses above using ~2.5M common SNPs from the GIANT meta-analyses for height and BMI. For the GIANT data, we stratified the variants into 20 MAF bins because of the smaller MAF range and smaller number of variants.
Supplementary Material
Acknowledgments
This research was supported by the Australian National Health and Medical Research Council (grants 1052684, 1078037, 1050218), the Australian Research Council (grant 130102666), the National Institutes of Health (R01MH100141), the Sylvia & Charles Viertel Charitable Foundation, and the UQ Foundation. This study makes use of data from the database of Genotypes and Phenotypes (dbGaP) under accessions phs000090.v1.p1, phs000091.v2.p1 and phs000428.v1.p1, and the EGCUT, LifeLines, TwinGene and UK10K studies (see the Supplementary Note for the full set of acknowledgments for these data).
Footnotes
URLs
GCTA-GREML-LDMS: http://cnsgenomics.com/software/gcta/greml_ldms.html
DHS annotation: http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeRegDnaseClustered/
LD scores and GWAS summary data from the combined data set: http://cnsgenomics.com/software/data/yang_et_al_2015_ng.html
GIANT height and BMI summary data: http://www.broadinstitute.org/collaboration/giant/index.php/GIANT_consortium_data_files
Competing Financial Interests
The authors declare no competing financial interests.
Author Contributions
J.Y. and P.M.V. conceived and designed the study. J.Y. performed statistical analyses and simulations. M.E.G., J.Y. and P.M.V. derived the theory. A.B., Z.H. and G.H. performed the imputation analysis. S.H.L, M.R.R., M.C.K. and N.R.W. provided statistical support. A.A.E.V., J.R.B.P., I.M.N, J.V.v.V.-O., H.S., T.L.C.S., T.E., L.M., R.M., A.M., A.H., P.K.E.M., N.L.P., E.I. and N.S. contributed to data collection. J.Y. wrote the manuscript with the participation of all authors.
References
- 1.Welter D, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–6. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manolio TA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang J, et al. Ubiquitous polygenicity of human complex traits: genome-wide analysis of 49 traits in Koreans. PLoS Genet. 2013;9:e1003355. doi: 10.1371/journal.pgen.1003355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee SH, Wray NR, Goddard ME, Visscher PM. Estimating missing heritability for disease from genome-wide association studies. Am J Hum Genet. 2011;88:294–305. doi: 10.1016/j.ajhg.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wood AR, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. 2014;46:1173–86. doi: 10.1038/ng.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Speliotes EK, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perry JR, et al. Parent-of-origin-specific allelic associations among 106 genomic loci for age at menarche. Nature. 2014;514:92–7. doi: 10.1038/nature13545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jostins L, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–24. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang J, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–9. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The UK10K Consortium. The UK10K project: rare variants in health and disease. Nature. 2015 doi: 10.1038/nature14962. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee SH, et al. Estimation of SNP heritability from dense genotype data. Am J Hum Genet. 2013;93:1151–5. doi: 10.1016/j.ajhg.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gusev A, et al. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am J Hum Genet. 2014;95:535–52. doi: 10.1016/j.ajhg.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Speed D, Hemani G, Johnson MR, Balding DJ. Improved heritability estimation from genome-wide SNPs. Am J Hum Genet. 2012;91:1011–21. doi: 10.1016/j.ajhg.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gusev A, et al. Quantifying missing heritability at known GWAS loci. PLoS Genet. 2013;9:e1003993. doi: 10.1371/journal.pgen.1003993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Visscher PM, Goddard ME, Derks EM, Wray NR. Evidence-based psychiatric genetics, AKA the false dichotomy between common and rare variant hypotheses. Mol Psychiatry. 2012;17:474–85. doi: 10.1038/mp.2011.65. [DOI] [PubMed] [Google Scholar]
- 19.Eyre-Walker A. Evolution in health and medicine Sackler colloquium: Genetic architecture of a complex trait and its implications for fitness and genome-wide association studies. Proc Natl Acad Sci USA. 2010;107:1752–6. doi: 10.1073/pnas.0906182107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simons YB, Turchin MC, Pritchard JK, Sella G. The deleterious mutation load is insensitive to recent population history. Nat Genet. 2014;46:220–4. doi: 10.1038/ng.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uricchio LH, Witte JS, Hernandez RD. Selection and explosive growth may hamper the performance of rare variant association tests. 2015 bioRxiv, 015917. [Google Scholar]
- 22.Locke AE, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee SH, Yang J, Goddard ME, Visscher PM, Wray NR. Estimation of pleiotropy between complex diseases using single-nucleotide polymorphism-derived genomic relationships and restricted maximum likelihood. Bioinformatics. 2012;28:2540–2. doi: 10.1093/bioinformatics/bts474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee SH, et al. Estimating the proportion of variation in susceptibility to schizophrenia captured by common SNPs. Nat Genet. 2012;44:247–250. doi: 10.1038/ng.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2012;13:36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet. 2014;15:121–32. doi: 10.1038/nrg3642. [DOI] [PubMed] [Google Scholar]
- 27.Lynch M, Walsh B. Genetics and analysis of quantitative traits. Sunderland, MA: Sinauer Associates; 1998. [Google Scholar]
- 28.Visscher PM, McEvoy B, Yang J. From Galton to GWAS: quantitative genetics of human height. Genet Res. 2010;92:371–9. doi: 10.1017/S0016672310000571. [DOI] [PubMed] [Google Scholar]
- 29.Zaitlen N, et al. Using extended genealogy to estimate components of heritability for 23 quantitative and dichotomous traits. PLoS Genet. 2013;9:e1003520. doi: 10.1371/journal.pgen.1003520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hemani G, et al. Inference of the genetic architecture underlying BMI and height with the use of 20,240 sibling pairs. Am J Hum Genet. 2013;93:865–75. doi: 10.1016/j.ajhg.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zaitlen N, et al. Leveraging population admixture to characterize the heritability of complex traits. Nat Genet. 2014;46:1356–62. doi: 10.1038/ng.3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morrison AC, et al. Whole-genome sequence-based analysis of high-density lipoprotein cholesterol. Nat Genet. 2013;45:899–901. doi: 10.1038/ng.2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang L, et al. Genotype-imputation accuracy across worldwide human populations. Am J Hum Genet. 2009;84:235–50. doi: 10.1016/j.ajhg.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44:955–9. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pasaniuc B, et al. Extremely low-coverage sequencing and imputation increases power for genome-wide association studies. Nat Genet. 2012;44:631–5. doi: 10.1038/ng.2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haplotype Reference Consortium. A Haplotype Reference Panel of over 31,000 individuals and Next Generation Imputation Methods; (Abstract/Program #176). the 64th Annual Meeting of The American Society of Human Genetics; San Diego, CA. October 20, 2014. [Google Scholar]
- 37.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 39.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patterson HD, Thompson R. Recovery of inter-block information when block sizes are unequal. Biometrika. 1971;58:545–554. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.