

Abstract
Increased production of mitochondrion-derived reactive oxygen species (ROS) is characteristic of a metabolic shift observed during malignant transformation. While the exact sources and roles of ROS in tumorigenesis remain to be defined, it has become clear that maintaining redox balance is critical for cancer cell proliferation and survival and, as such, may represent a vulnerability that can be exploited therapeutically. STAT3, a latent cytosolic transcription factor activated by diverse cytokines and growth factors, has been shown to exhibit an additional, nontranscriptional function in mitochondria, including modulation of electron transport chain activity. In particular, malignant transformation by Ras oncogenes exploits mitochondrial STAT3 functions. We used mass spectrometry-based metabolomics profiling to explore the biochemical basis for the STAT3 dependence of Ras transformation. We identified the gamma-glutamyl cycle, the production of glutathione, and the regulation of ROS as a mitochondrion-STAT3-dependent pathway in Ras-transformed cells. Experimental inhibition of key enzymes in the glutathione cycle resulted in the depletion of glutathione, accumulation of ROS, oxidative DNA damage, and cell death in an oncogenic Ras- and mitochondrial STAT3-dependent manner. These data uncover a synthetic lethal interaction involving glutathione production and mitochondrial ROS regulation in Ras-transformed cells that is governed by mitochondrial STAT3 and might be exploited therapeutically.
INTRODUCTION
STAT3 is a latent cytosolic transcription factor activated by phosphorylation on tyrosine 705 in response to many growth factors and cytokines. In normal tissues, STAT3 target genes regulate proliferation, survival, angiogenesis, immune responses, inflammation, and self-renewal (1). STAT3 is also implicated in malignancy (2). Constitutively active STAT3 mutants facilitate experimental transformation (3), and STAT3 is aberrantly phosphorylated or overexpressed in many human tumors. Typically, enhanced STAT3 activation is due to the mutation of upstream tyrosine kinases or receptor tyrosine kinases (e.g., JAK2 in myeloproliferative disease, ALK in some lymphomas, or epidermal growth factor [EGFR] in head and neck cancer) or heightened secretion of cytokines (e.g., interleukin-6 [IL-6] in multiple myeloma or IL-11 in gastric cancer), and STAT3 often has been found to be essential for tumors driven by these stimuli (4). Intriguingly, STAT3 is also required for transformation driven by oncogenic mutations in the Ras-GTPase family in the absence of STAT3 Y705 phosphorylation (5–7). In the context of oncogenic Ras mutations, a mitochondrial pool of STAT3 regulates the activity of the electron transport chain, which is necessary for tumor formation (8). These varied involvements of STAT3 in human cancer have made it an attractive therapeutic candidate (9), although this promise has yet to be fulfilled (10).
Metabolic reprogramming is considered a hallmark of cancer. As cells adopt a transformed phenotype, they switch the major source of ATP synthesis from oxidative phosphorylation (OXPHOS) utilizing the electron transport chain (ETC) to aerobic glycolysis, a process named Warburg metabolism (11). Cancer cells rely heavily on aerobic glycolysis for ATP and biomolecule production, but they also maintain electron transport chain and tricarboxylic acid (TCA) cycle activity, which contribute to tumor cell anabolism (12). Indeed, Ras-driven tumors require complex I of the ETC to maintain aerobic glycolysis in support of tumor development (13). STAT3 has been found to be a key player in both aerobic glycolysis and ETC. Under conditions in which STAT3 is phosphorylated on Y705 to activate its transcriptional functions, it drives the expression of HIF1α and c-Myc, both of which can regulate the switch from OXPHOS to aerobic glycolysis (14). Additionally, mitochondrial STAT3 enhances ETC activity through a nontranscriptional mechanism, which is required for Ras transformation (5, 7). Therefore, there is a complex relationship between STAT3 and cancer metabolism that involves both nuclear transcriptional activities as well as mitochondrial nontranscriptional functions that may be distinct from actions in nontransformed cells.
Activating mutations in Ras GTPases occur in about 25% of human cancers (15), and the mitochondrial pool of STAT3 is critical for transformation by this family of oncogenes, due at least in part to regulation of metabolic processes (5–7). The genetic alterations that drive Warburg metabolism in cancer cells have provided new therapeutic avenues for cancer treatment (e.g., targeting IDH mutations in glioma and AML [16, 17]). The complex roles of STAT3 in the regulation of metabolism likely are dependent on the driving oncogene. However, the precise metabolic changes dependent on mitochondrial STAT3 in response to Ras oncogenes are largely unknown.
We used an unbiased mass spectrometry (MS) screen to explore mitochondrial STAT3-dependent metabolic processes. We analyzed metabolites in Ras-transformed mouse embryo fibroblasts (MEFs) and T24 human bladder carcinoma cells to identify substances whose abundance depended on the presence of mitochondrial STAT3. We found that metabolites generated by the γ-glutamyl cycle were altered in abundance depending on mitochondrial STAT3 status. The γ-glutamyl cycle is required for the synthesis of glutathione (GSH), which is the major cellular reactive oxygen species (ROS) scavenger. In the absence of STAT3, cellular glutathione concentrations were diminished, and blocking glutathione synthesis resulted in increased ROS and oxidative DNA damage, leading to the death of tumor cells but not of untransformed cells. Therefore, targeting the γ-glutamyl cycle or GSH accumulation kills cancer cells in a mitochondrial STAT3-dependent manner, which may prove to be an effective therapeutic approach.
MATERIALS AND METHODS
Antibodies and reagents.
The antibodies against the following proteins were obtained from commercial sources: phospho-S139-γH2A.X (Active Motif), Mc1-1 (Rockland), Bcl-X (Cell Signaling Technology), cleaved caspase 3 (Cell Signaling Technology), poly(ADP-ribose) polymerase (PARP) and tubulin (Santa Cruz Biotechnology), anti-rabbit Ig–IRDye800 and anti-mouse Ig–IRDye680 (LI-COR), and anti-rabbit Ig–Alexa Fluor 594 (Invitrogen). All chemicals were purchased from Sigma.
Mice.
All work with animals was approved by the Institutional Animal Ethics Committee. K-RasG12DLSL mice (18) were interbred STAT3-fl/fl mice (19). Bone marrow cells were harvested from femurs of adult mice.
Cell culture.
Spontaneously immortalized mouse embryo fibroblasts (MEFs) and HEK293T cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% bovine calf serum (BCS) at 37°C and 5% CO2. A549, T24, and SK147 cells were cultured in advanced RPMI medium supplemented with 1% FCS. Bone marrow was cultured at 2 × 106 cells/ml in Opti-MEM with 10% fetal calf serum (FCS), 100 μM β-mercaptoethanol, recombinant mouse TPO (20 ng/ml), and SCF (100 ng/ml). Recombinant retroviruses were generated in HEK293T cells (5).
Flow cytometry.
Bone marrow cells were treated with Fc block (BioLegend) and stained with Pacific Blue lineage cocktail or Ig control antibodies (133306; BioLegend) and sorted using an Influx cell sorter (BD Biosciences). Cells were incubated with tetramethylrhodamine, ethyl ester (TMRE), or monochlorobimane for 30 min, washed, and analyzed for mitochondrial membrane potential or GSH on a FACSCanto II (BD Biosciences). Fluorescence-activated cell sorter (FACS) data were analyzed using Cyflogic (CyFlo Ltd.) or FlowJo (FlowJo LLC).
Western blotting.
Cells were lysed in 150 mM NaCl, 50 mM Tris (pH 7.4), 1 mM EDTA, 0.5% Triton X-100, protease inhibitor cocktail (Sigma-Aldrich), 1 mM NaF, 1 mM β-glycerophosphate, 1 mM Na3VO4, and 1 mM dithiothreitol (DTT), resolved by SDS-PAGE, transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore), and probed with antibodies diluted in Tris-buffered saline (TBS)–0.1% Tween 20 overnight at 4°C. Blots were developed with a 1:15,000 dilution of fluor-conjugated secondary antibody (LI-COR).
GSH/GSSH measurement.
GSH and glutathione disulfide (GSSG) concentrations were measured using the GSH/GSSG-Glo assay (Promega) per the manufacturer's instructions.
ROS measurement.
ROS were measured by fluorescent flow cytometry or fluorescent spectroscopy following treatment of cells with 0.5 μM dihydroethidium (DHE; Sigma-Aldrich), 2.5 μM 2′,7′-dichlorofluorescein diacetate (DCFDA; Sigma-Aldrich), or 5 μM MitoSOX (Thermo-Fisher).
Immunofluorescence.
Cells were cultured in 24-well plates on sterile coverslips. Following appropriate treatment, cells were washed, fixed in 4% formaldehyde for 15 min, and permeabilized and blocked in Odyssey blocking buffer (LI-COR) with 0.1% Triton X-100. Primary antibody incubation was performed (1:100 phospho-S139-γH2A.X [Active Motif]) overnight at 4°C, and secondary antibody incubation (1:1,000; anti-rabbit antibody–Alexa 594 [Invitrogen]) was performed for 1 h at room temperature. Cells were mounted in VectaShield (Vector Laboratories) with 4′,6-diamidino-2-phenylindole (DAPI) and imaged using an API Deltavision deconvolution microscope (GE Healthcare), and images were analyzed using Image J software.
Measuring apurinic/apyrimidinic sites.
Cells were cultured in complete media and treated with vehicle or with 1 μM azaserine or 1 μM azaserine in combination with 1 mM N-acetyl-l-cysteine or 10 μM MitoTempo. After 48 h of treatment, the number of apurinic/apyrimidinic sites per 100,000 bp was measured using the OxiSelect oxidative DNA damage kit per the manufacturer's instructions (Cell Biolabs).
Generating ρ0 cells.
Cells were cultured in complete media supplemented with 5 ng/ml ethidium bromide, 5 μg/ml uridine, and 10 mM sodium pyruvate for 2 weeks or until the loss of electron transport chain activity was confirmed by high-resolution respiritometry, as described below.
High-resolution respiritometry.
Oxygen consumption was measured using an Oxygraph-2k (Oroboros Instruments). Cells (2 × 106) were added to a closed chamber containing prewarmed DMEM supplemented with 4.4 mM glucose and 25 mM HEPES (pH 7.4) without added glutamine, pyruvate, or NaHCO3. Once O2 consumption stabilized (routine respiration), cells were treated with oligomycin (500 nM final concentration) to measure leak respiration, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP; 200 nM final concentration) to measure the maximal respiratory rate, and antimycin A (20 nM final concentration) to measure nonmitochondrial O2 consumption.
Metabolite identification.
Cells (4 × 106) in log-phase growth cultured in DMEM supplemented with 4.5 g/liter glucose and 5% BCS were harvested, and metabolite abundance was identified by Metabolon using their standard protocols. Briefly, metabolites were extracted from 6 independent replicates using organic and aqueous extractions to remove the protein fraction. Extracts were divided into two fractions for analysis by liquid and gas chromatography (LC and GC, respectively). Samples then were prepared for either LC-MS or GC-MS. LC-MS analysis was performed using a Waters Acquity ultraperformance liquid chromatograph (UPLC) and a Thermo-Finnigan LTQ mass spectrometer, which consisted of an electrospray ionization (ESI) source and linear ion trap (LIT) mass analyzer. The MS analysis alternated between MS and data-dependent MS2 scans using dynamic exclusion.
Samples for GC-MS analysis were redried under vacuum desiccation prior to being derivatized using bistrimethyl-silyl-trifluoroacetamide (BSTFA) under nitrogen. The GC column was 5% phenyl, and the temperature ramp was from 40° to 300°C in a 16-min period. Samples were analyzed on a Thermo-Finnigan Trace DSQ fast-scanning single-quadrupole mass spectrometer using electron impact ionization.
Statistical analysis.
For pairwise comparisons of metabolite data sets, statistical significance was tested using Welch's t tests and/or Wilcoxon's rank sum tests using the statistical analysis package within the R environment. Student's t tests were used for pairwise comparisons of smaller data sets.
RESULTS
STAT3 is required for glutathione synthesis.
To identify mitochondrial STAT3-dependent metabolites in Ras-transformed cells, we performed an unbiased mass spectrometry-based metabolomics screen using two separate panels of cells. The first set was an isogenic trio of H-RasV12-transformed mouse embryo fibroblasts (MEFs) expressing or lacking endogenous STAT3 following STAT3 gene ablation. These H-RasV12:STAT3−/− MEFs were engineered to express a mitochondrially restricted STAT3 mutant, such that all detectable STAT3 was localized to the mitochondria (5). The second set of cells was derived from the human T24 bladder carcinoma, which harbors a spontaneous H-RasV12 mutation. These cells were stably transduced with control or STAT3-targeting short hairpin RNA (shRNA) to ablate STAT3 expression. Biomolecules were extracted from cells cultured in identical media and conditions in log-phase growth and were identified using gas chromatography-mass spectrometry or liquid chromatography-tandem mass spectrometry. A total of 282 biomolecules were identified, of which 155 were significantly altered in the absence of STAT3 from MEFs and 90 were significantly altered in T24 cells depleted of STAT3. Of these biomolecules, 56 changed in abundance in both MEF and T24 cells, depending on STAT3 status. Importantly, 45 of these biomolecules were reverted toward wild-type abundance following the reexpression of mitochondrially restricted STAT3 in otherwise STAT3-null MEFs (Table 1).
TABLE 1.
Summary of metabolomics dataa
| Genotypes | No. of biomolecule changesb |
|
|---|---|---|
| Total | Up/down | |
| WT vs STAT3−/− | 155 | 77/78 |
| STAT3−/− vs MTS | 48 | 19/29 |
Summary of the number of biomolecules with significantly altered abundance between different H-RasG12V-transformed cells.
The numbers of biomolecules changing between STAT3 wild-type (WT) and STAT3−/− MEFs and between WT and mitochondrially restricted STAT3 (MTS) MEFs are shown, including a breakdown of those whose abundance increased (up) or decreased (down). Relative abundance was determined from 6 independent replicates, and statistical significance was calculated by Welch's t test (P < 0.05).
Pathway analysis of STAT3-dependent biomolecules, in particular those that were restored toward wild-type levels by mitochondrial STAT3 expression, identified the γ-glutamyl cycle as one of the highly affected biochemical pathways (Fig. 1A). The γ-glutamyl cycle contains 5 enzyme-catalyzed reactions required for the synthesis of the most abundant cellular reactive oxygen species scavenger, glutathione (20). In the absence of STAT3, the abundance of metabolites generated by 4 of the 5 γ-glutamyl cycle enzymes was altered. This included a significant elevation in the abundance of γ-glutamyl amino acids, 5-oxoproline, glutamine, and glycine in STAT3-null cells (Fig. 1B and data not shown). However, the abundance of the end product, glutathione, was reduced in STAT3-null cells, along with a significant decrease in the abundance of cysteine, a glutathione precursor. Under cysteine-limiting conditions, γ-glutamyl cysteine synthase substitutes 2-amino butyrate or alanine for cysteine, resulting in the production of ophthalmate and norophthalmate at the expense of glutathione (21). Consistent with this notion, we observed significantly elevated ophthalmate and norophthalmate in concert with reduced cysteine abundance in STAT3-null cells (Fig. 1B). Commensurate with the decrease in GSH in the absence of STAT3 was an increase in cellular ROS (Fig. 1B). Importantly, the expression of mitochondrially restricted STAT3 in otherwise STAT3-null cells restored the wild-type pattern of γ-glutamyl amino acids, 5-oxoproline, cysteine, glutamine, ophthalmate, norophthalmate, and glutathione abundance and restored normal ROS levels (Fig. 1B). Analysis of biomolecule abundance in T24 cells confirmed an increase in γ-glutamyl amino acids, ophthalmate, and norophthalmate, and a decrease in glutathione, when STAT3 expression was ablated by shRNA knockdown (Fig. 1C). Together, these data suggest that mitochondrial STAT3 impacts the γ-glutamyl cycle in H-Ras-transformed mouse and human cancer cells.
FIG 1.
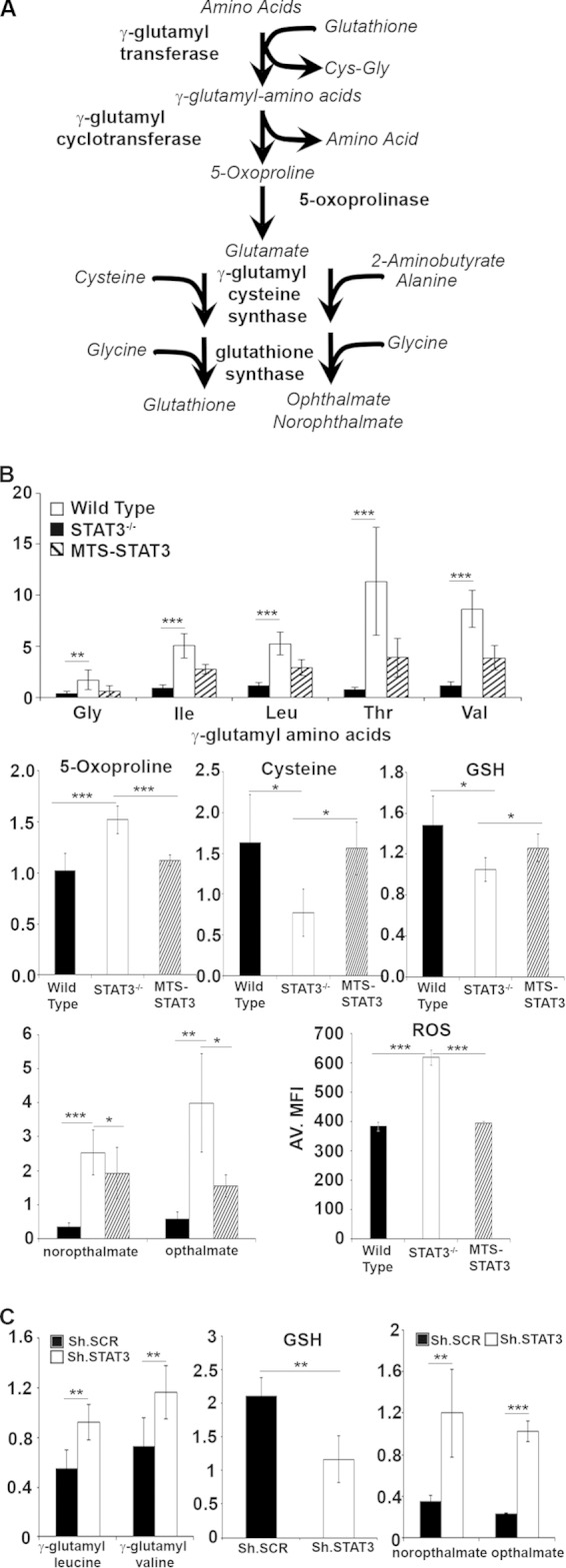
γ-Glutamyl cycle is dependent on STAT3 expression. (A) The γ-glutamyl cycle engages 5 enzymatically catalyzed reactions to generate glutathione (GSH). Metabolic intermediates in the γ-glutamyl cycle include the γ-glutamyl amino acids, 5-oxoproline, glutamine, cysteine, and glycine. (B) The abundance of various γ-glutamyl amino acids, 5-oxoproline, cysteine, GSH, norophthalmate, and ophthalmate were determined by mass spectrometry analysis of H-RasG12V-transformed STAT3 wild-type, STAT3−/−, and MTS-STAT3-reconstituted MEFs. ROS levels were measured by DCFDA fluorescence. (C) The relative abundance of γ-glutamyl cycle metabolites was deter mined in T24 cells stably expressing control shRNA (sh.SCR) or STAT3 targeting shRNA (sh.STAT3). Relative abundance was determined from 6 independent replicates (8 replicates for the determination of ROS levels), and statistical significance was calculated by Student's t test. *, P < 0.05; **, P < 0.01; ***, P < 0.0001.
Acute inhibition of γ-glutamyl transferase kills transformed cells in a STAT3-dependent manner.
To determine whether the mitochondrial STAT3 dependence of the γ-glutamyl cycle contributed to Ras transformation, we tested two independent and structurally distinct inhibitors (azaserine and acivicin) of γ-glutamyl transferase (GGT), the apical enzyme in this pathway. Treatment of H-RasV12-transformed MEFs with escalating doses of azaserine or acivicin resulted in significantly increased cell death, which was not observed in nontransformed MEFs treated with GGT inhibitors (Fig. 2A and B). Importantly, the azaserine- or acivicin-induced cell death observed in the H-RasV12-transformed MEFs was significantly reduced in the absence of STAT3 (Fig. 2A and B). Cell death was preceded by an accumulation of cleaved caspase 3 and of cleaved poly-ADP ribose polymerase (PARP) and by a loss of mitochondrial membrane potential, which are indicative of apoptotic cell death in response to GGT inhibition (Fig. 2C and D). Alteration of cellular ROS can affect the expression of antiapoptotic proteins, such as Mcl-1 and Bcl-X, leading to cell death (22), and expression of these proteins can be modulated by inhibition of STAT3 (23). However, treatment of K-Ras-transformed wild-type or STAT3-null MEFs with azaserine did not affect Mcl-1 or Bcl-X expression (Fig. 2E), suggesting that their expression was not sufficient to prevent azaserine-induced apoptosis. Nonetheless, azaserine-induced cell death was impaired by the pancaspase inhibitor QVD (Fig. 2F).
FIG 2.

Inhibition of γ-glutamyl transferase kills transformed cells in a STAT3-dependent manner. (A and B) Immortalized but not transformed STAT3 wild-type (WT) or STAT3−/− MEFs or H-RasG12V-transformed STAT3 wild-type or STAT3−/− MEFs were treated with the indicated doses of acivicin (A) or azaserine (B), and cell viability was measured after 72 h. (C and D) H-RasG12V-transformed STAT3 WT or STAT3−/− MEFs were treated with the indicated dose of azaserine for 6, 18, or 24 h, and the induction of apoptosis was measured by the increase of cleaved caspase 3 and PARP by Western blotting (C) or by the loss of mitochondrial membrane potential as measured by flow cytometry using tetramethylrhodamine ethyl ester (TMRE) (D). (E) H-RasG12V-transformed STAT3 WT or STAT3−/− MEFs were treated with 1 μM azaserine for 24 h, and the expression of Mcl-1 and Bcl-X was measured by Western blotting. (F) H-RasG12V- transformed STAT3 WT or STAT3−/− MEFs were treated with 1 μM azaserine in the presence or absence of 1 μM QVD, and cell death was quantified after 72 h. (G) Human cancer cell lines bearing mutant K-Ras (A549), H-Ras (T24), or N-Ras (SK147) were stably transduced with microRNA 30-based shRNA constructs targeting renilla luciferase (ShRen) or STAT3 (shSTAT3) and treated with 1 μM azaserine. Cell viability was determined after 72 h. (H) Bone marrow from LSLK-RasG12D mice was isolated and infected with adenoviral CreGFP to excise a lox-STOP-lox cassette, resulting in the expression of oncogenic K-RasG12D. Hematopoietic progenitor lineage-negative, GFP-positive cells were purified by fluorescence-activated cell sorting and treated with 1 μM azaserine. Cell viability was determined after 72 h. (I) H-RasG12V-transformed STAT3 wild-type or STAT3−/− MEFs were stably transduced with virus encoding an empty vector control (EV), WT STAT3, or a mitochondrially restricted STAT3 (MTS). SK147 cells stably expressing control (shRen) or STAT3-targeting shRNA (shSTAT3) were transduced with virus encoding WT STAT3 or mitochondrially restricted STAT3 (MTS STAT3). Cells were treated with 1 μM azaserine, and cell viability was determined after 72 h. In all experiments, cell death was plotted as the average ± 1 standard deviation of the percentage of death observed in three independent experiments. Statistical significance determined using the Student t test is indicated. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Enhanced differences in cell death between untreated and azaserine-treated cells depicted in panel I were statistically significant (P < 0.0001), with the exception of STAT3−/− plus EV (not significant [n.s.]).
To confirm that GGT inhibition-induced cell death was not restricted to experimental overexpression of oncogenic Ras, we tested the sensitivity of human cancer cell lines harboring spontaneous Ras mutations. When cells transformed by K-Ras (A549), H-Ras (T24), or N-Ras (SK147) were treated with azaserine, we observed significant cell death that was mitigated by stable knockdown of STAT3 by RNA interference (RNAi) (Fig. 2G). To extend this observation to primary transformed tissue, we tested GGT inhibition in malignant mouse hematopoietic cells derived from the activation of K-RasG12D from its endogenous promoter. Oncogenic Ras expression in the mouse hematopoietic compartment causes a disease similar to juvenile myelomonocytic leukemia (18, 24–27) that is in part dependent on STAT3 (7). We isolated the lineage-negative stem cell population of bone marrow leukocytes from K-RasG12DLSL mice (18) and infected them with adenoviral Cre-green fluorescent protein (GFP) to delete the Lox-STOP-Lox cassette and activate the expression of K-RasG12D from its endogenous promoter. As a control, cells were infected with adenoviral GFP control virus. Primary bone marrow expressing K-RasG12D displayed growth factor-independent growth, as expected (7). Treatment with azaserine resulted in significant cell death that was not observed in nontransformed leukocytes infected with control GFP adenovirus, expressing only wild-type K-Ras (Fig. 2H).
To determine whether the toxicity observed by GGT inhibition was due to the mitochondrial pool of STAT3, we reconstituted H-RasV12-transformed, STAT3-null MEFs with a mitochondrially targeted version of STAT3, so that STAT3 was restricted to the mitochondrial matrix (5). In a parallel panel of cells, we expressed mitochondrially restricted STAT3 in an N-Ras-transformed SK147 melanoma cell line in which endogenous STAT3 had been depleted by stable RNA interference. In both panels of cells, we observed that azaserine-induced toxicity was equivalent in cells expressing endogenous STAT3 or mitochondrially restricted STAT3 but was significantly reduced in cells lacking STAT3 (Fig. 2G). These results indicated that inhibition of γ-glutamyl aminotransferase was selectively toxic to Ras-transformed cells of human or mouse origin in a mitochondrial STAT3-dependent manner.
Inhibition of glutathione synthesis kills transformed cells in a STAT3-dependent manner.
To determine whether toxicity due to GGT inhibition was specifically due to inhibition of γ-glutamyl aminotransferase or the resulting reduction in cellular glutathione abundance, we inhibited other enzymes in the GSH synthesis pathway or used domoic acid to efflux intracellular glutathione and reduce cellular glutathione levels (28). Treatment with escalating doses of l-butathione sulfoximine (BSO) to acutely inhibit γ-glutamyl cysteine synthase (29), a rate-limiting enzyme in glutathione synthesis, was significantly more toxic to H-RasV12-transformed cells that expressed STAT3 than corresponding STAT3-null cells (Fig. 3A). Cell death (30 to 50%) of STAT3 wild-type cells was observed with 0.25 to 1 mM BSO, while only 10 to 25% of STAT3-null MEFS were killed by BSO treatment. Similarly, H-RasV12-transformed STAT3 wild-type MEFs treated with domoic acid to efflux intracellular GSH displayed increased cell death relative to STAT3-null cells (Fig. 3B). Moreover, the simultaneous acute inhibition of new glutathione synthesis (azaserine treatment) with glutathione efflux (domoic acid) increased cell death in a STAT3-dependent manner (Fig. 3B). Together, these data confirmed that acute inhibition of glutathione synthesis or depletion of cellular glutathione following efflux preferentially kills Ras-transformed cells in a STAT3-dependent manner.
FIG 3.
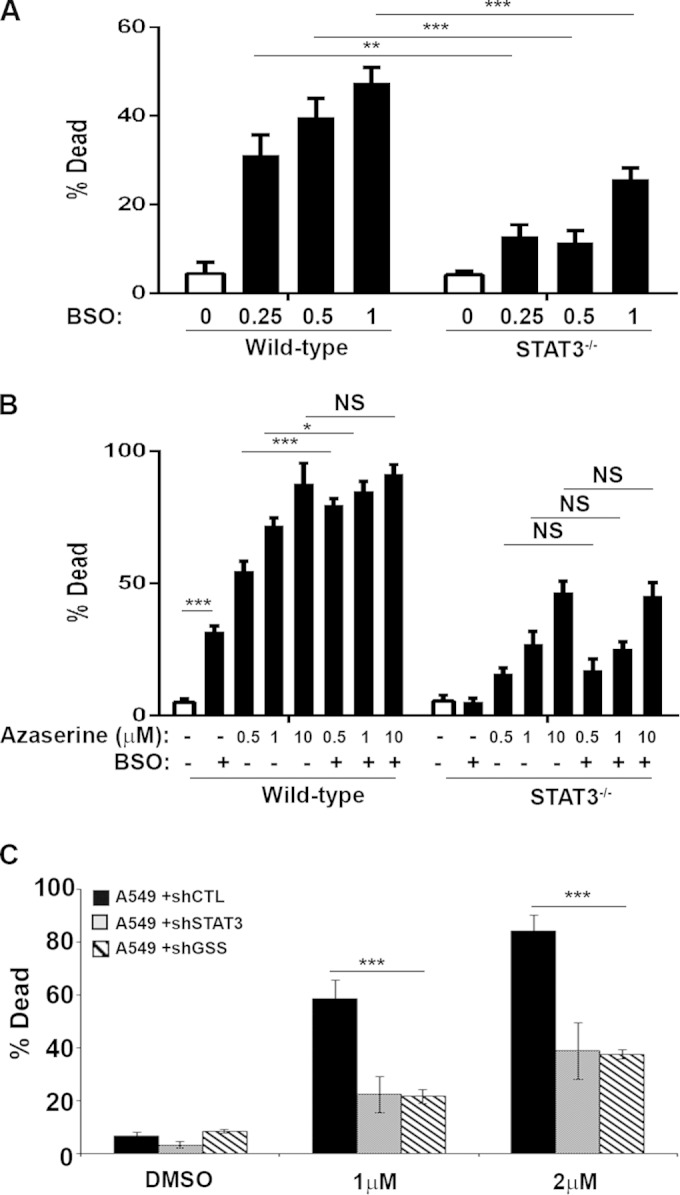
Depleting intracellular glutathione kills H-RasG12V-transformed cells in a STAT3-dependent manner. (A and B) H-RasG12V-transformed STAT3 wild-type or STAT3−/− MEFs were treated for 72 h with the indicated doses of l-butathione-sulfoximine (BSO) to block γ-glutamyl cysteine synthase (A) or 10 mM domoic acid to efflux intracellular GSH (B), in combination with the indicated doses of azaserine. (C) A549 cells transduced with control, GSS, or STAT3-targeting RNA interference hairpins were treated with azaserine for 72 h. In all experiments, cell death was determined by trypan blue exclusion and plotted as the average ± 1 standard deviation of the percentage of death observed in 3 or 4 independent experiments. Statistical significance was determined using the Student t test. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
We also tested stable loss of GSH synthesis by depleting cells for glutathione synthetase (GSS) by RNA interference. Depletion of GSS had no significant effect on K-Ras-transformed human A549 lung carcinoma cells (Fig. 3C). However, loss of GSS significantly protected cells from azaserine toxicity, similar to the degree of protection afforded by STAT3 depletion. Therefore, while acute loss of GSH induces cell death in K-Ras-transformed cells, stable depletion, either by removal of STAT3 or inhibition of GSH synthesis, removes the cellular dependence on reducing capacity, presumably through an adaptation process.
Acute inhibition of γ-glutamyl transferase decreases intracellular GSH concentration and increases reactive oxygen species.
Total GSH levels were reduced in STAT3-null cells, as measured by monochlorobimane fluorescent flow cytometry (Fig. 4A). Glutathione exists in two states, the abundant reduced form (GSH) and the less abundant oxidized form (GSSG). We measured the abundance of both forms using a luciferase-based assay (Fig. 4B to D) and high-performance liquid chromatography (data not shown). We observed a consistent decrease in both GSH and GSSG in STAT3-null cells. As expected, azaserine treatment further decreased the concentration of GSH independent of STAT3 (Fig. 4C) and led to a significant increase in the concentration of oxidized glutathione (GSSG) (Fig. 4D), consistent with a shift in cellular redox state.
FIG 4.
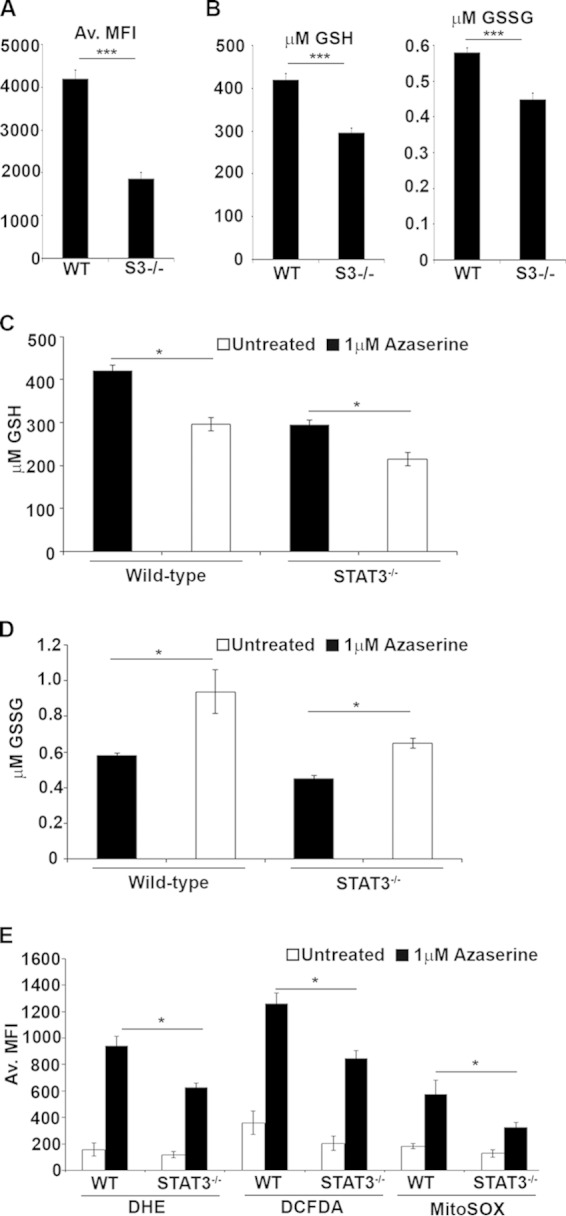
Azaserine treatment decreases reduced glutathione but increases superoxide and oxidized glutathione. (A and B) The concentration of total glutathione in untreated H-RasG12V-transformed STAT3 wild-type or STAT3−/− MEFs was measured by flow cytometry of monochlorobimane-stained cells (A), and reduced and oxidized glutathione abundance was measured by the GSH/GSSG glo luciferase assay (B). (C and D) Reduced (C) and oxidized (D) glutathione concentrations were determined after 24 h of treatment with 1 μM azaserine. (E) The production of reactive oxygen species was determined using specific probes for superoxide (DHE), mitochondrial superoxide (mitoSOX), or cytosolic hydroxyl peroxyl radicals (DCFDA) by flow cytometry. Data plotted are the means ± 1 standard deviation from three independent experiments. Statistical significance was determined using the Student t test. *, P < 0.05; **, P < 0.01.
To determine whether treatment with γ-glutamyl aminotransferase inhibitors shift cellular redox potential due to an accumulation of reactive oxygen species (ROS), we measured ROS levels using three independent fluorescent ROS probes. We measured total cellular superoxide using dihydroethidium (DHE), cytosolic hydroxyl, or peroxyl radicals using 2′,7′-dichlorodihrdrofluorescein (DCFDA), and we measured mitochondrial superoxide using MitoSOX Red. Despite the higher basal ROS levels observed in STAT3-null relative to wild-type cells (Fig. 1B), azaserine treatment caused an acute increase in ROS accumulation in Ras-transformed wild-type cells relative to cells lacking STAT3 (Fig. 4E). These data document that γ-glutamyl cycle inhibition leading to the depletion of GSH caused a greater accumulation of ROS in Ras-transformed cells in the presence of STAT3, likely contributing to cell death.
Azaserine-induced mitochondrial ROS accumulation leads to cell death.
To determine whether the STAT3-dependent cell death induced by GGT inhibitors is due to the accumulation of ROS and, in particular, mitochondrial ROS, we cotreated cells with azaserine and the ROS scavengers N-acetylcysteine (NAC) and glutathione ethyl-ester (GSH-EE) or the mitochondrion-specific scavenger MitoTempo. The presence of ROS scavengers significantly rescued H-Ras-transformed STAT3-expressing MEFs from azaserine-induced cell death (Fig. 5A). Scavenging the mitochondrial pool of ROS was as effective at reducing azaserine-induced cell death as the general cellular scavenger NAC or GSH-EE, strongly suggesting that mitochondrial ROS production is responsible for the observed azaserine toxicity. Mitochondrial ROS are generated largely as a byproduct of the ETC, whose activity is augmented in a mitochondrial STAT3-dependent manner (5, 30). To definitively confirm that mitochondrial ROS production is required for azaserine-induced cell death, we generated cells lacking mitochondrial DNA (ρo); therefore, they lacked electron transport chain activity. ρo cells were derived from both H-Ras-transformed STAT3+/+ or STAT3−/− MEFs, and defective ETC function was confirmed for these ρo cells by high-resolution respiritometry (Fig. 5B). Loss of functional mitochondria led to a significant decrease in azaserine-induced toxicity in ρo cells relative to H-Ras-transformed STAT3+/+ parental cells (Fig. 5C). Significantly, loss of functional mitochondria reduced azaserine toxicity to levels similar to those observed in the absence of STAT3, suggesting that STAT3-dependent mitochondrial ROS production contributes to cell death following azaserine treatment.
FIG 5.
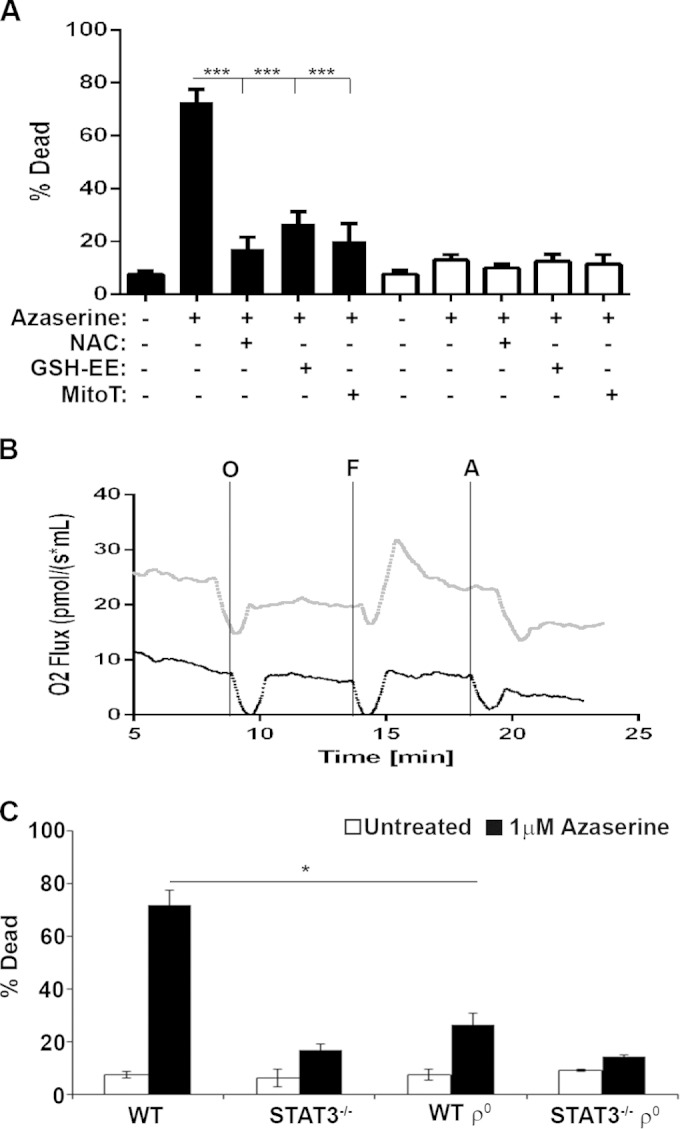
Mitochondrial reactive oxygen species are required for azaserine-induced cell death. (A) H-RasG12V-transformed STAT3 wild-type or STAT3−/− MEFs were treated with 1 μM azaserine for 72 h in the presence or absence of 1 mM N-acetylcysteine (NAC), 1 mM glutathione ethyl ester (GSH-EE), or 10 μM MitoTempo (MitoT), as indicated, and cell viability was determined. (B) Oxygen consumption was measured in H-RasG12V-transformed STAT3 wild-type (WT) or STAT3−/− ρ0 MEFs using the oroboros oxygraph 2K. O2 consumption was measured on intact cells in the absence of glucose, glutamate, and pyruvate (routine) or in the presence of 500 nM oligomycin (O), 200 nM carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (F), or 20 nM antimycin A (A). (C) ρ0 H-RasG12V-transformed STAT3 wild-type or STAT3−/− MEFs were treated with the indicated dose of azaserine, and cell death was determined after 72 h and plotted as the means ± 1 standard deviation of the percentage of death observed in three independent experiments. Statistical significance was determined using the Student t test. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Azaserine treatment leads to oxidative DNA damage and apoptosis.
Increased ROS results in oxidative stress and damage to protein, lipid, and DNA. To determine whether azaserine-induced increases in ROS lead to DNA damage, we assessed the accumulation of DNA double-strand breaks by phosphorylated γ-H2A.X (pγ-H2A.X) staining. We observed a rapid increase in the proportion of H-Ras-transformed MEF cells with greater than 20 pγ-H2A.X puncta per cell following azaserine treatment. This accumulation of pγ-H2A.X puncta was significantly reduced in H-Ras-transformed STAT3−/− MEFs (Fig. 6A and B). While DNA double-strand breaks are a consequence of oxidative stress, they are not a direct measure of DNA oxidation. The most common ROS-induced DNA lesion is the oxidation of guanine to form 8-oxo-2′-deoxyguanosine (8-oxo-dG). We measured oxidative DNA damage using an anti-8-OHdG monoclonal antibody-based enzyme-linked immunosorbent assay (ELISA). We found that the number of 8-oxo-dG residues per 100 kb of genomic DNA significantly increased following treatment of wild-type cells with azaserine (Fig. 6C). This increased oxidative DNA damage in response to azaserine was not observed in STAT3-null cells. Importantly, cotreatment of wild-type cells with azaserine and NAC or mitoTempo reduced the accumulation of 8-oxo-dG to that observed in untreated cells and in STAT3-null cells (Fig. 6C), confirming both the ROS and STAT3 dependence of the oxidative DNA damage. Taken together, these data support the hypothesis that inhibition of GGT activity in Ras-transformed cells results in enhanced toxicity due to accumulation of mitochondrial ROS derived from ETC activity that depends on the presence of STAT3 (Fig. 6D and E).
FIG 6.
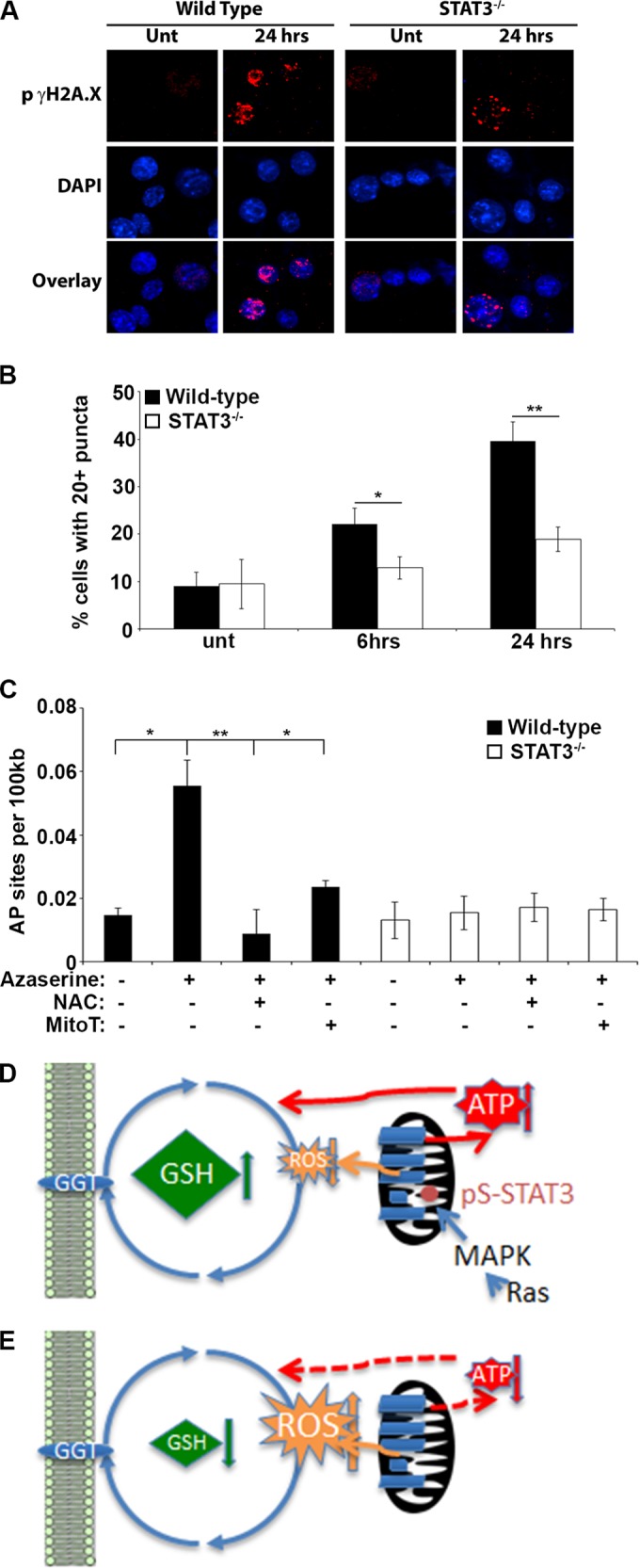
Azaserine treatment leads to oxidative DNA damage. (A) H-RasG12V-transformed STAT3 wild-type or STAT3−/− MEFs were treated with 1 μM azaserine for 24 h, and DNA double-strand breaks were identified by staining for phosphorylated γH2A.X. (B) The number of cells with greater than 20 phosphorylated γH2A.X puncta was counted from at least 4 microscopic fields containing a minimum of 50 cells/field after 24- and 48-h treatments with 1 μM azaserine. Average numbers from 3 independent experiments are shown. (C) The number of 8′-oxo-2′-deoxyguanosine DNA adducts (AP sites) per 100 kb of genomic DNA was determined following 24 h of incubation with 1 μM azaserine in the presence or absence of 1 mM N-acetylcysteine (NAC) or 10 μM MitoTempo (MitoT). Statistical significance was determined using the Student t test. *, P < 0.05; **, P < 0.01. (D and E) Model for synthetic lethality of GSH depletion and mitochondrial STAT3. Mitochondrial STAT3 phosphorylated on S727 in response to MAPK pathway activity in Ras-transformed cells enhances ETC activity and ATP production. (D) ROS production is balanced by GSH, which is enhanced in wild-type cells, possibly due to heightened ATP levels. (E) Acute loss of GSH would lead to imbalanced ROS production, oxidative stress, and cell death (not depicted). In the absence of STAT3, ETC activity and ATP production are impaired and GSH levels diminish, resulting in modestly increased ROS, possibly due to the increased proton motive force from reduced proton dissipation resulting from impaired complex V activity. Cell viability is maintained through adaptive responses.
DISCUSSION
A small pool of STAT3 accumulates in mitochondria and is critical for enhanced ETC activity, altered metabolism, increased ATP production, and oncogenic Ras-dependent transformation (5–7). The function of mitochondrial STAT3 is independent of its tyrosine phosphorylation and does not require intact DNA binding or SH2 domains. Nonetheless, its activity is dependent upon specific serine phosphorylation, a posttranslational modification that, at least in Ras-transformed cells, occurs in response to mitogen-activated protein kinase (MAPK) signaling. Importantly, the mitochondrial activity of STAT3 may be largely dispensable for normal biology, as mice containing a mutant form of the STAT3 gene lacking the ability to be serine phosphorylated are viable and outwardly normal (31), and the absence of STAT3 serine phosphorylation abrogates the ability of STAT3 to support Ras transformation (5). Notably, the activity of mitochondrial STAT3 is critical for transformed cell anchorage-independent growth in vitro and tumor growth in vivo, even though it is dispensable for anchorage-dependent cell growth. Therefore, the tumor-specific action of mitochondrial STAT3 could provide a transformation-selective vulnerability that could prove to be useful therapeutically.
The data from this study support a model in which the mitochondrial pool of STAT3 contributes to regulation of ROS and GSH that contributes to the redox balance of transformed cells (Fig. 6D). Mitochondrial STAT3 augments the activity of the electron transport chain in transformed cells, which could be expected to generate higher levels of cellular ROS. However, mitochondrial STAT3 also contributes, probably indirectly, to increased GSH levels, which likely buffers the total ROS. GSH synthesis is an ATP-consuming process (32), which could be impaired by the reduced ATP abundance observed in STAT3-null Ras-transformed cells (5). Elevated ROS is thought to be beneficial to, or even necessary for, cellular transformation (33). However, excessive ROS accumulation would result in oxidative stress and cell death. A common strategy to maintain acceptable cellular redox is to engage the γ-glutamyl cycle to generate GSH and scavenge ROS, thereby avoiding detrimental oxidative stress. We found that acute blockade of the GSH synthesis pathway, either by inhibition of GGT or GCS or by depletion of GSH, leads to excess accumulation of ROS, oxidative DNA damage, and cell death in Ras-transformed cells in a mitochondrial STAT3-dependent manner. In contrast, stable depletion of STAT3 allowed cells to adapt to decreased GSH levels, although this resulted in an increase in cellular ROS, albeit to lower levels than the toxic amounts observed following acute GSH ablation. The increased ROS production in STAT3-null Ras-transformed cells could be a direct consequence of an increased proton motive force resulting from decreased complex V activity (5, 34). These findings suggest a synthetic lethal interaction between mitochondrial STAT3 function and impaired GSH generation, since neither STAT3 ablation nor GSH inhibition alone is toxic, while GSH depletion becomes toxic in the presence of mitochondrial STAT3. Since this synthetic lethality was restricted to transformed cells, it could prove to be a therapeutically amenable tumor-specific vulnerability (Fig. 6D and E).
To define the metabolic consequences of mitochondrial STAT3 function, we undertook a global analysis of metabolites whose abundance depends on the presence of mitochondrial STAT3 in Ras-transformed cells. This analysis uncovered a dependence of the γ-glutamyl cycle and glutathione production on mitochondrial STAT3. A decreased trend and a significant decrease in the reduced form of the tripeptide glutathione were observed in STAT3-null and STAT3-depleted cells. This decreased trend in GSH was reversed when mitochondrial STAT3 was reexpressed in the absence of total cellular STAT3. Cysteine, which is utilized during glutathione synthesis, also decreased in STAT3-null cells. However, the small change in cysteine levels suggests that this is not the sole underlying cause for the observed decrease in GSH levels following STAT3 knockout and knockdown. Since glutathione functions as one of the major antioxidants in cells, one plausible explanation for the significant decrease in reduced glutathione is an increased demand for glutathione due to increased oxidative stress in these cells following loss of STAT3, which is consistent with the heightened ROS observed following STAT3 ablation. There were additional metabolite changes observed in the glutathione synthesis and utilization pathway that support the notion that increased glutathione utilization could account for the decreased GSH observed in the absence of STAT3. These findings included significantly increased levels of gamma-glutamyl dipeptides and significant increases in the tripeptides ophthalmate (gamma-glutamyl-alpha-aminobutyrylglycine) and norophthalmate (gamma-glutamyl-alanylglycine) in both mouse and human tumor cells lacking STAT3. Importantly, levels of ophthalmate and norophthalmate were returned to normal levels upon reintroduction of mitochondrial STAT3, commensurate with restored GSH levels and reduced ROS. Synthesis of ophthalmate involves both gamma-glutamylcysteine synthetase and glutathione synthetase, except cysteine is replaced with 2-aminobutyrate; thus, ophthalmate typically is synthesized when cysteine levels are depleted, as was observed in the absence of STAT3. Moreover, GSH also functions as an inhibitor of gamma-glutamylcysteine synthetase. Thus, reduced levels of GSH could drive increased gamma-glutamylcysteine synthetase activity, and the resulting increase in gamma-glutamylcysteine synthetase catalysis could increase its utilization of substrates such as 2-aminobutyrate and result in the observed increase in ophthalmate (21, 35). Increased gamma-glutamylcysteine synthetase activity acting on alanine as a substrate would result in the increased norophthalmate observed in STAT3-null and depleted cells. Although the underlying molecular mechanisms through which mitochondrial STAT3 regulates glutathione levels and cellular redox balance remain to be defined, these STAT3-dependent metabolic parameters encouraged investigation of the consequences of perturbing redox balance in Ras-transformed cells.
The dependence of GSH accumulation on mitochondrial STAT3 in Ras-transformed cells prompted us to investigate whether this represented a physiologic requirement. Acute inhibition of glutathione synthesis resulted in an accumulation of ROS, oxidative DNA damage, and death, but only in transformed cells that expressed mitochondrial STAT3. In contrast, stable depletion of GSH, either by inhibition of its synthesis through RNAi-mediated diminution of GSS or by removal of STAT3, protected cells from acute loss of GSS. These observations suggest that Ras-transformed cells become “addicted” to heightened GSH levels. Just as stable loss of STAT3 reverts many hallmark characteristics of transformed cells without altering viability under standard in vitro culture conditions (5), stable inhibition of GSH production allows cells to adapt by losing their acute dependence on glutathione. These observations support a model in which the mitochondrial pool of STAT3 increases ETC activity, resulting in increased mitochondrial ROS production that becomes balanced by cellular antioxidant buffering through an adaptive increase in GSH production. It is known that increased ROS production enhances tumorigenicity, but it must not be allowed to accumulate to levels that induce debilitating oxidative stress. The potentially toxic effects of ROS would be mitigated by the increased glutathione produced by wild-type cells. Thus, depleting glutathione through acute synthesis inhibition causes increased toxicity due to the unbalanced accumulated ROS in the absence of allowing adaptation under the selective pressure of stable depletion (Fig. 6D and E).
The role of ROS in tumorigenesis is complex. ROS is a driver of malignancy in the context of inflammation-induced tumors (e.g., gastric cancer following chronic Helicobacter pylori infection or colon cancer in animals repeatedly dosed with dextran sodium sulfate) (36). However, the importance of ROS to tumor development is not restricted to inflammation-induced cancer. Expression of oncogenic Ras increases ROS production, which is required for mitogenic signaling (37), and the incubation of Ras-transformed cells with ROS scavengers inhibits proliferation (38). Conversely, the ability to overwhelm endogenous ROS scavenging pathways leads to the death of Ras-transformed cells via apoptosis (39), and blocking of GSH synthesis can be toxic for Ras-transformed MEFs (40). These earlier observations are in close agreement with our data showing that pharmacological inhibition of the GSH synthesis pathway kills transformed cells in both mouse and human cell lines and primary mouse leukemia cells. Furthermore, this GSH depletion-induced cell death can be reversed by coincubation with ROS scavengers that specifically target mitochondrial ROS or total cellular ROS.
The data in this study suggest the use of compounds impeding GSH synthesis or increasing ROS (including azaserine, acivicin, or BSO) as anticancer agents, particularly in the presence of an active mitochondrial STAT3 pathway. Indeed, there have been a number of studies supporting the concept of targeting GSH production. Acivicin efficiently reduced metastatic disease in the B16F10 mouse melanoma model (41) and has been tested in a number of clinical trials for both solid and liquid tumors. The results from these studies show some patient responses in non-small-cell lung carcinoma (42, 43) and astrocytoma (44) but no response in relapsed or refractory acute leukemia (45), hepatocellular carcinoma (46), or ovarian carcinoma (47). Intermittent responses, severe central nervous system side effects, and myelosuppression and its reported ability to increase the formation of pancreatic cancer in rats (48) have limited the further development of this compound. Although azaserine and acivicin inhibit γ-glutamyl transferase and are effective at killing leukemic and melanoma cell lines in vitro (49), they have additional cellular targets. For instance, acivicin also inhibits other enzymes, including the aldehyde dehydrogenase ALDH4A1 (50). Our approach of using multiple pharmacologic and genetic approaches provides compelling evidence that the Ras- and mitochondrial STAT3-dependent cell death resulting from acute enzyme inactivation is due to GSH depletion rather than uncharacterized off-target effects. Nonetheless, the development of new and more specific compounds capable of impeding GSH synthesis is needed for an effective anticancer strategy. It also may be beneficial to stratify tumor cell lines and patient samples for GSH concentration and mitochondrial STAT3 activity in order to focus the use of redox modulators on appropriate cases. Targeting GSH synthesis pathways or the activity of mitochondrial STAT3 may prove to be an effective therapeutic strategy for a subset of human cancers.
ACKNOWLEDGMENTS
We thank members of the Levy and Gough laboratories for helpful discussions and comments on the manuscript. We also thank the Monash University FlowCore and the NYULMC Cytometry and Cell Sorting Core, supported in part by Cancer Center support grant NIH P30CA016087.
D. J. Gough is supported by a Career Development Fellowship (GNT1063914) and project grant from the National Health and Medical Research Council of Australia (GNT1024929) and grants from the Monash Comprehensive Cancer Centre and Australasian Sarcoma Study Group and the Victorian Government's Operational Infrastructure Support Program. M.A.-H. gratefully acknowledges support from the New York Community Trust and the Feinberg Lymphoma Foundation. D.E.L. acknowledges funding from the National Institutes of Health (R01AI28900 and U54AI057158).
T. J. Harris, C. L. White, M. Abdul-Hay, F. J. Rossello, D. J. Garama, and D. J. Gough performed experiments and analyzed and interpreted data; D. E. Levy and D. J. Gough designed experiments, analyzed and interpreted data, and wrote the manuscript.
We have no conflict of interest to disclose.
REFERENCES
- 1.Levy DE, Lee CK. 2002. What does Stat3 do? J Clin Investig 109:1143–1148. doi: 10.1172/JCI0215650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu H, Lee H, Herrmann A, Buettner R, Jove R. 2014. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer 14:736–746. doi: 10.1038/nrc3818. [DOI] [PubMed] [Google Scholar]
- 3.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE Jr. 1999. Stat3 as an oncogene. Cell 98:295–303. doi: 10.1016/S0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 4.Inghirami G, Chiarle R, Simmons WJ, Piva R, Schlessinger K, Levy DE. 2005. New and old functions of STAT3: a pivotal target for individualized treatment of cancer. Cell Cycle 4:1131–1133. doi: 10.4161/cc.4.9.1985. [DOI] [PubMed] [Google Scholar]
- 5.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. 2009. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 324:1713–1716. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Q, Raje V, Yakovlev VA, Yacoub A, Szczepanek K, Meier J, Derecka M, Chen Q, Hu Y, Sisler J, Hamed H, Lesnefsky EJ, Valerie K, Dent P, Larner AC. 2013. Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J Biol Chem 288:31280–31288. doi: 10.1074/jbc.M113.505057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gough DJ, Marie IJ, Lobry C, Aifantis I, Levy DE. 2014. STAT3 supports experimental K-RasG12D-induced murine myeloproliferative neoplasms dependent on serine phosphorylation. Blood 124:2252–2261. doi: 10.1182/blood-2013-02-484196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gough DJ, Sehgal PB, Levy DE. 2012. Nongenomic functions of STAT3, p 91–98. In Decker T, M̈uller M (ed), Jak-Stat signaling: from basics to disease. Springer-Verlag Gmbh, Vienna, Germany. [Google Scholar]
- 9.Yue P, Turkson J. 2009. Targeting STAT3 in cancer: how successful are we? Expert Opin Investig Drugs 18:45–56. doi: 10.1517/13543780802565791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wake MS, Watson CJ. 2015. STAT3 the oncogene–still eluding therapy? FEBS J 282:2600–2611. doi: 10.1111/febs.13285. [DOI] [PubMed] [Google Scholar]
- 11.Warburg O. 1956. On the origin of cancer cells. Science 123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 12.Lunt SY, Vander Heiden MG. 2011. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 13.Calabrese C, Iommarini L, Kurelac I, Calvaruso MA, Capristo M, Lollini PL, Nanni P, Bergamini C, Nicoletti G, Giovanni CD, Ghelli A, Giorgio V, Caratozzolo MF, Marzano F, Manzari C, Betts CM, Carelli V, Ceccarelli C, Attimonelli M, Romeo G, Fato R, Rugolo M, Tullo A, Gasparre G, Porcelli AM. 2013. Respiratory complex I is essential to induce a Warburg profile in mitochondria-defective tumor cells. Cancer Metab 1:11. doi: 10.1186/2049-3002-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demaria M, Giorgi C, Lebiedzinska M, Esposito G, D'Angeli L, Bartoli A, Gough DJ, Turkson J, Levy DE, Watson CJ, Wieckowski MR, Provero P, Pinton P, Poli V. 2010. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging 2:823–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Downward J. 2003. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 16.Schumacher T, Bunse L, Wick W, Platten M. 2014. Mutant IDH1: an immunotherapeutic target in tumors. Oncoimmunology 3:e974392. doi: 10.4161/2162402X.2014.974392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chaturvedi A, Araujo Cruz MM, Jyotsana N, Sharma A, Yun H, Gorlich K, Wichmann M, Schwarzer A, Preller M, Thol F, Meyer J, Haemmerle R, Struys EA, Jansen EE, Modlich U, Li Z, Sly LM, Geffers R, Lindner R, Manstein DJ, Lehmann U, Krauter J, Ganser A, Heuser M. 2013. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood 122:2877–2887. doi: 10.1182/blood-2013-03-491571. [DOI] [PubMed] [Google Scholar]
- 18.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. 2001. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raz R, Lee CK, Cannizzaro LA, d' Eustachio P, Levy DE. 1999. Essential role of STAT3 for embryonic stem cell pluripotency. Proc Natl Acad Sci U S A 96:2846–2851. doi: 10.1073/pnas.96.6.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meister A. 1974. The gamma-glutamyl cycle. Diseases associated with specific enzyme deficiencies. Ann Intern Med 81:247–253. [DOI] [PubMed] [Google Scholar]
- 21.Soga T, Baran R, Suematsu M, Ueno Y, Ikeda S, Sakurakawa T, Kakazu Y, Ishikawa T, Robert M, Nishioka T, Tomita M. 2006. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J Biol Chem 281:16768–16776. doi: 10.1074/jbc.M601876200. [DOI] [PubMed] [Google Scholar]
- 22.Glasauer A, Sena LA, Diebold LP, Mazar AP, Chandel NS. 2014. Targeting SOD1 reduces experimental non-small-cell lung cancer. J Clin Investig 124:117–128. doi: 10.1172/JCI71714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang X, Zhang J, Wei H, Tian Z. 2007. STAT3-decoy oligodeoxynucleotide inhibits the growth of human lung cancer via down-regulating its target genes. Oncol Rep 17:1377–1382. [PubMed] [Google Scholar]
- 24.Braun BS, Shannon K. 2008. Targeting Ras in myeloid leukemias. Clin Cancer Res 14:2249–2252. doi: 10.1158/1078-0432.CCR-07-1005. [DOI] [PubMed] [Google Scholar]
- 25.Chan IT, Kutok JL, Williams IR, Cohen S, Kelly L, Shigematsu H, Johnson L, Akashi K, Tuveson DA, Jacks T, Gilliland DG. 2004. Conditional expression of oncogenic K-ras from its endogenous promoter induces a myeloproliferative disease. J Clin Investig 113:528–538. doi: 10.1172/JCI20476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sabnis AJ, Cheung LS, Dail M, Kang HC, Santaguida M, Hermiston ML, Passegue E, Shannon K, Braun BS. 2009. Oncogenic Kras initiates leukemia in hematopoietic stem cells. PLoS Biol 7:e59. doi: 10.1371/journal.pbio.1000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J, Liu Y, Beard C, Tuveson DA, Jaenisch R, Jacks TE, Lodish HF. 2007. Expression of oncogenic K-ras from its endogenous promoter leads to a partial block of erythroid differentiation and hyperactivation of cytokine-dependent signaling pathways. Blood 109:5238–5241. doi: 10.1182/blood-2006-09-047050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giordano G, White CC, McConnachie LA, Fernandez C, Kavanagh TJ, Costa LG. 2006. Neurotoxicity of domoic acid in cerebellar granule neurons in a genetic model of glutathione deficiency. Mol Pharmacol 70:2116–2126. doi: 10.1124/mol.106.027748. [DOI] [PubMed] [Google Scholar]
- 29.Griffith OW. 1982. Mechanism of action, metabolism, and toxicity of buthionine sulfoximine and its higher homologs, potent inhibitors of glutathione synthesis. J Biol Chem 257:13704–13712. [PubMed] [Google Scholar]
- 30.Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, Moh A, Moghaddas S, Chen Q, Bobbili S, Cichy J, Dulak J, Baker DP, Wolfman A, Stuehr D, Hassan MO, Fu XY, Avadhani N, Drake JI, Fawcett P, Lesnefsky EJ, Larner AC. 2009. Function of mitochondrial Stat3 in cellular respiration. Science 323:793–797. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen Y, Schlessinger K, Zhu X, Meffre E, Quimby F, Levy DE, Darnell JE Jr. 2004. Essential role of STAT3 in postnatal survival and growth revealed by mice lacking STAT3 serine 727 phosphorylation. Mol Cell Biol 24:407–419. doi: 10.1128/MCB.24.1.407-419.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumar A, Bachhawat AK. 2010. A futile cycle, formed between two ATP-dependant gamma-glutamyl cycle enzymes, gamma-glutamyl cysteine synthetase and 5-oxoprolinase: the cause of cellular ATP depletion in nephrotic cystinosis? J Biosci 35:21–25. doi: 10.1007/s12038-010-0004-8. [DOI] [PubMed] [Google Scholar]
- 33.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. 2010. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A 107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murphy MP. 2009. How mitochondria produce reactive oxygen species. Biochem J 417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dello SA, Neis EP, de Jong MC, van Eijk HM, Kicken CH, Olde Damink SW, Dejong CH. 2013. Systematic review of ophthalmate as a novel biomarker of hepatic glutathione depletion. Clin Nutr 32:325–330. doi: 10.1016/j.clnu.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 36.Hussain SP, Hofseth LJ, Harris CC. 2003. Radical causes of cancer. Nat Rev Cancer 3:276–285. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- 37.Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, Sundaresan M, Finkel T, Goldschmidt-Clermont PJ. 1997. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 275:1649–1652. doi: 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- 38.Irani K, Goldschmidt-Clermont PJ. 1998. Ras, superoxide and signal transduction. Biochem Pharmacol 55:1339–1346. doi: 10.1016/S0006-2952(97)00616-3. [DOI] [PubMed] [Google Scholar]
- 39.Liou JS, Chen CY, Chen JS, Faller DV. 2000. Oncogenic ras mediates apoptosis in response to protein kinase C inhibition through the generation of reactive oxygen species. J Biol Chem 275:39001–39011. doi: 10.1074/jbc.M007154200. [DOI] [PubMed] [Google Scholar]
- 40.Chuang JI, Chang TY, Liu HS. 2003. Glutathione depletion-induced apoptosis of Ha-ras-transformed NIH3T3 cells can be prevented by melatonin. Oncogene 22:1349–1357. doi: 10.1038/sj.onc.1206289. [DOI] [PubMed] [Google Scholar]
- 41.Roy S, Maity P. 2007. Modulation of metastatic potential of B16F10 melanoma cells by acivicin: synergistic action of glutaminase and potentiation of cisplatin cytotoxicity. Asian Pac J Cancer Prev 8:301–306. [PubMed] [Google Scholar]
- 42.Maroun JA, Stewart DJ, Verma S, Evans WK, Eisenhauer E. 1990. Phase I study of acivicin and cisplatin in non-small-cell lung cancer. A National Cancer Institute of Canada study. Am J Clin Oncol 13:401–404. [DOI] [PubMed] [Google Scholar]
- 43.Bonomi P, Finkelstein D, Chang A. 1994. Phase II trial of acivicin versus etoposide-cisplatin in non-small cell lung cancer. An Eastern Cooperative Oncology Group study. Am J Clin Oncol 17:215–217. [DOI] [PubMed] [Google Scholar]
- 44.Taylor SA, Crowley J, Pollock TW, Eyre HJ, Jaeckle C, Hynes HE, Stephens RL. 1991. Objective antitumor activity of acivicin in patients with recurrent CNS malignancies: a Southwest Oncology Group trial. J Clin Oncol 9:1476–1479. [DOI] [PubMed] [Google Scholar]
- 45.Powell BL, Craig JB, Capizzi RL, Richards F Jr. 1988. Phase I-II trial of acivicin in adult acute leukemia. Investig New Drugs 6:41–44. [DOI] [PubMed] [Google Scholar]
- 46.Falkson G, Cnaan A, Simson IW, Dayal Y, Falkson H, Smith TJ, Haller DG. 1990. A randomized phase II study of acivicin and 4′deoxydoxorubicin in patients with hepatocellular carcinoma in an Eastern Cooperative Oncology Group study. Am J Clin Oncol 13:510–515. doi: 10.1097/00000421-199012000-00012. [DOI] [PubMed] [Google Scholar]
- 47.Earhart RH, Khandekar JD, Faraggi D, Schinella RA, Davis TE. 1989. Phase II trial of continuous drug infusions in advanced ovarian carcinoma: acivicin versus vinblastine. Investig New Drugs 7:255–260. [DOI] [PubMed] [Google Scholar]
- 48.Longnecker DS, Curphey TJ. 1975. Adenocarcinoma of the pancreas in azaserine-treated rats. Cancer Res 35:2249–2258. [PubMed] [Google Scholar]
- 49.Sava G, Giraldi T, Baldini L. 1982. Antitumor activity of N-diazoacetyl derivatives of glycine and phenylalanine against P388 leukemia and B16 melanoma in mice. Cancer Treat Rep 66:179–181. [PubMed] [Google Scholar]
- 50.Kreuzer J, Bach NC, Forler D, Sieber SA. 2014. Target discovery of acivicin in cancer cells elucidates its mechanism of growth inhibition. Chem Sci 6:237–245. doi: 10.1039/c4sc02339k. [DOI] [PMC free article] [PubMed] [Google Scholar]