

Abstract
Acetylation is a dynamic posttranslational modification that contributes to chromatin-regulated processes, including DNA replication, repair, recombination, and gene expression. Acetylation is controlled by complexes containing opposing lysine and histone acetyltransferase (KAT and HAT) and deacetylase (KDAC and HDAC) activities. The essential MYST family Esa1 KAT acetylates core histones and many nonhistone substrates. Phenotypes of esa1 mutants include transcriptional silencing and activation defects, impaired growth at high temperatures, and sensitivity to DNA damage. The KDAC Rpd3 was previously identified as an activity opposing Esa1, as its deletion suppresses growth and silencing defects of esa1 mutants. However, loss of Rpd3 does not suppress esa1 DNA damage sensitivity. In this work, we identified Hos2 as a KDAC counteracting ESA1 in the damage response. Deletion of HOS2 resulted in changes of esa1's transcriptional response upon damage. Further, loss of HOS2 or components of the Set3 complex (Set3C) in which it acts specifically suppressed damage sensitivity and restored esa1 histone H4 acetylation. This rescue was mediated via loss of either Set3C integrity or of its binding to dimethylated histone H3K4. Our results thus add new insight into the interactions of an essential MYST acetyltransferase with diverse deacetylases to respond specifically to environmental and physiological challenges.
INTRODUCTION
Chromatin regulates gene expression, recombination, and replication and DNA damage repair (1, 2). It is subject to multiple posttranslational modifications (3), including lysine acetylation, a dynamic modification that is established by lysine and histone acetyltransferases (KATs and HATs) and reversed by lysine and histone deacetylases (KDACs and HDACs). Acetylation partially neutralizes the basic charge of histone tails, relaxing nucleosome compactness. It also creates binding sites for proteins containing bromodomains and has been linked to an open chromatin conformation (4).
The Esa1 acetyltransferase acts in two different complexes, piccolo and NuA4 (5), in which it preferentially acetylates histones H4 and H2A and the histone variant Htz1 (6–11). Additionally, it acts on nonhistone substrates, such as the NuA4 subunits Epl1 and Yng2 (7, 12), the autophagy protein Atg3 (13), the RNA processing protein Nab3 (14, 15), and nearly 200 other proteins (12, 15, 16). Notably, the Tip60 human ortholog of Esa1 has been linked to multiple human diseases (17–19), thus increasing the relevance of gaining a deeper understanding of Esa1 functions.
ESA1 is an essential gene contributing to transcriptional regulation in response to growth stimuli that has been most extensively studied with conditional alleles (6–8, 20). Hypomorphic esa1 strains are temperature sensitive and have defects in progression through the G2/M phase of the cell cycle and transcriptional regulation, including failure to silence ribosomal DNA (rDNA) and telomere proximal genes (6, 21). ESA1 mutants are also defective in repairing DNA damage (22, 23).
DNA damage results from many environmental factors, such as UV and gamma irradiation or heavy metal toxins; it can also be introduced by intrinsic factors such as reactive oxygen species, DNA replication, and others (24–26). Among the many types of damage, DNA double-stranded breaks (DSBs) are deleterious lesions which if unrepaired can lead to mutation, cell death, and cancer in metazoans. Cells ordinarily respond to DNA damage by signal transduction cascades that lead to pauses in the cell cycle to allow repair, wide changes in gene expression, and direct action at the breaks, promoting rapid ligation of the broken DNA ends (27–30). Mutations can affect any and all stages of the repair processes.
Esa1 contributes to multiple aspects of the DNA damage response by regulating gene expression (31, 32) and by its direct recruitment to DSBs, where it promotes signaling to repair the breaks (22, 33). Multiple suppressors of esa1 phenotypes have been identified (14, 23, 34–36); however, suppression of DNA repair defects of esa1 has not yet been fully explored.
In a search to understand repair in esa1 mutants, we have now identified a role for an opposing deacetylase, Hos2. Hos2 is a class I KDAC necessary for induction of gene expression, perhaps by creating a permissive chromatin state for multiple rounds of transcription (37). More recently, however, it has been suggested that its activating role could be closely tied to repressing noncoding RNAs (ncRNAs) that overlap many Hos2-regulated genes (38). Hos2 is a component of the Set3 complex (Set3C), which includes the sirtuin deacetylase Hst1 (39). Set3C is important in regulating gene induction during the stress response, including changes in carbon sources (38), nitrogen starvation (39), and DNA damage (31). Set3C binds to the histone mark H3K4me2 (40), generally found in the 5′ region of the open reading frames (ORFs); however it can also be enriched in promoter regions of some genes, replacing H3K4me3 (38).
In this work, we report that Hos2 is the relevant activity opposing Esa1 in DNA damage repair. We found that esa1 had defects in transcriptional induction of DNA damage-regulated genes that were attenuated upon deletion of HOS2. Suppression by hos2Δ was in the context of Set3C, because deletion of other complex components also suppressed the DNA damage sensitivity of esa1 mutants. Loss of Set3C recruitment to the H3K4me2 mark rescued esa1's repair defects, supporting the concept that suppression was mediated through the DNA damage transcriptional response.
MATERIALS AND METHODS
Yeast strains and plasmids.
Strains, plasmids and oligonucleotides are listed in Tables S1 to S3 in the supplemental material. The esa1-414 and esa1-531 alleles have been previously characterized (6, 7). Both alleles are sensitive to DNA damage; however, esa1-531 is more defective than esa1-414 at 30°C, allowing isolation of damage effects from those introduced by temperature stress. The hst1Δ2::LEU2 (LPY18275) disruption was engineered into a wild-type BY strain (41). All other mutations were null alleles constructed using standard methods and backcrossed prior to use. Histone mutant strains had chromosomal deletions for both HHF-HHT loci and initially contained pJH33 (HTA HTB HHF2 HHT2 URA3 CEN) (42); these were transformed with TRP1 plasmids carrying relevant H4 (HHF2) mutations. The plasmid pJH33 was selected against by growth on 5-fluoroorotic acid (5-FOA). The catalytic mutant hos2-H195A,H196A was constructed with primers listed in Table S3. Strains were grown at 30°C in yeast extract-peptone-dextrose medium plus adenine (YPAD) or dropout medium for selection.
Growth dilution assays, silencing assays, and flow cytometry.
Unless otherwise noted, all dilution assays represent 5-fold serial dilutions, starting from an A600 of 0.5 after growth to saturation in YPAD. Growth and silencing assays were performed at 30°C as described previously (43, 44). For rDNA silencing, strains were grown in synthetic complete (SC) medium lacking adenine (Ade) and Arg (SC−Ade−Arg) to saturation, normalized as described above, and plated on SC−Ade−Arg and SC−Ade−Arg containing 32 μg/ml of canavanine. Telomeric silencing assays were conducted with plating on SC and SC with 0.1% 5-FOA. Camptothecin (CPT) sensitivity was assayed using CPT in dimethyl sulfoxide (DMSO) added to plates buffered with 100 mM potassium phosphate (pH 7.5) to maintain maximal drug activity (45). Growth control plates contained equal concentrations of DMSO and phosphate buffer. Images were captured after 2 to 6 days. Cells were processed for flow cytometry as described previously (14) and analyzed with Accuri (BD) after sonication.
Protein immunoblotting.
Whole-cell extracts were prepared from cells grown to an A600 of 0.8 to 1.0 at 30°C in YPAD. For DNA damage, cells were grown to an A600 of 0.5 and were exposed for 90 min to hydroxyurea at 0.2 M or to a carrier. Extracts were prepared as described previously (6) by vortexing cells with glass beads in phosphate-buffered saline (PBS) with protease inhibitors, denaturing in boiling sample loading buffer, and separating the insoluble pellet by centrifugation. Samples were separated on 18% SDS-polyacrylamide gels and transferred to 0.2-μm nitrocellulose. Primary antisera were anti-H4K5Ac (1:5,000 dilution; Serotec), anti-H4K8Ac (1:2,000; Serotec), anti-H4K12Ac (1:2,000; Active Motif), anti-H4K16Ac (1:2,000; Millipore), anti-H3K9 and -K14Ac (1:10,000; Upstate), anti-H3K14Ac (1:2,000; Upstate), anti-H4 (1:2,000; Active Motif 39269), and anti-H3ct (1:10,000; Millipore). The secondary reagent was horseradish peroxidase-conjugated goat anti-rabbit antibody (Promega, 1:10,000). Blots were quantified using the ImageQuant 5.2 program (Molecular Dynamics). The histogram peak function was applied to correct for background signal.
RNA extraction and RT-PCR.
Strains were grown in 50 ml of 100 mM phosphate-buffered YPAD (pH 7.5) at 30°C. At an A600 of 0.4 to 0.5, cultures were split and treated with CPT (20 μg/ml) or a DMSO carrier. After 90 min at 30°C, RNA was extracted using the hot acid-phenol method (46), except that harvested cells were resuspended in sodium acetate buffer (50 mM sodium acetate [pH 5.3], 10 mM EDTA). After extraction, RNA was treated with the TURBO DNA-free kit (Ambion) and reverse transcribed using TaqMan reverse transcription reagents (Applied Biosystems) with random hexamer priming. The cDNA was then diluted 10-fold and analyzed by real-time PCR with a SYBR green PCR mix (Anaspec) on a DNA Engine Opticon2 (MJ Research). Oligonucleotides are listed in Table S3 in the supplemental material. Data shown in Fig. 4 are the averages of three separate RNA extractions analyzed in triplicate.
FIG 4.

Expression of DNA damage response genes was aberrant in esa1 cells. (A) Gene expression was analyzed in samples treated with DMSO or with 20 μg/ml of CPT for 90 min. The patterns of expression of the damage-activated genes HUG1, GRE2, and ERG5 and the control gene ACT1 are shown for wild-type (LPY6497), esa1-531 (LPY14757), esa1-531 hos2Δ (LPY14761), and hos2Δ (LPY14577) strains. The expression values were normalized to ACT1 expression. Three independent RNA samples were reverse transcribed and analyzed by qPCR with primers in Table S3 in the supplemental material. Student's t test was used to assess statistical significance, represented with asterisks as follows: *, P < 0.05, and **, for P < 0.01. (B) HUG1 and GRE2 expression levels upon damage were similar in esa1 and esa1 rpd3Δ strains but distinct from that in the esa1 hos2Δ strain. Gene expression was analyzed in samples treated with DMSO or with 20 μg/ml of CPT for 90 min. The expression values were normalized to ACT1 expression. Expression was analyzed in wild-type (LPY6496), esa1-531 (LPY14757), esa1-531 rpd3Δ (LPY21450), esa1-531 hos2Δ (LPY14761), and rpd3Δ (LPY13426) strains.
RESULTS
Genetic suppression of conditional alleles of ESA1 has provided insight into its regulation and roles in different cellular pathways. Deletion of RPD3, which encodes a global KDAC, suppresses the temperature and silencing defects of esa1 strains; however, esa1 rpd3Δ cells remain sensitive to DNA damage (23), suggesting the involvement of a different enzyme in opposing Esa1's function during response to DNA damage.
The DNA damage sensitivity of esa1 can be suppressed by deletion of HOS2.
As KDACs oppose acetylation established by KATs, we hypothesized that a KDAC other than Rpd3 could suppress the DNA damage sensitivity of esa1. Initial candidates tested included Hos1 and Hos2 (similar in sequence to Rpd3 and classified as type I KDACs), the type II KDAC Hda1 (47), and the sirtuin Sir2 (48). Growth of double esa1 mutants in combination with deletions of the candidate KDACs was tested by challenge with the DSB-inducing drug camptothecin (CPT) (Fig. 1). The RPD3 and HOS1 deletions increased sensitivity, whereas the HOS2, HDA1, and SIR2 deletions suppressed esa1 (Fig. 1A). As hos2Δ promoted the strongest growth, we focused on characterizing this suppression.
FIG 1.

Deletion of the histone deacetylase encoded by HOS2 suppressed the DNA damage sensitivity of esa1. (A) Deletion of HOS2 rescued esa1 DNA damage sensitivity, whereas deletion of HOS1 and RPD3 did not. Deletion of SIR2 and HDA1 partially rescued esa1. Shown are serial dilutions of wild-type (wt) (LPY5), esa1-414 (LPY4774), esa1-414 hos1Δ (LPY13712), esa1-414 hos2Δ (LPY13585), esa1-414 rpd3Δ (LPY12156), esa1-414 hda1Δ (LPY13478), esa1-414 sir2Δ (LPY11279) strains (top) and wild type (LPY5), esa1-414 (LPY4774), hos1Δ (LPY13706), hos2Δ (LPY13583), rpd3Δ (LPY12154), hda1Δ (LPY13472), and sir2Δ (LPY11) strains (bottom). Figure S1A in the supplemental material shows the phenotype of the same strains grown at 37°C. Note that some of these interactions overlap results from a genomewide study (7), yet others are distinct, an effect that we find is due to strain background differences. See Fig. S1B to D for more details. (B) Loss of the deacetylase activity of Hos2 was important for esa1 suppression. Strains in panel A were transformed with vector (pLP60), HOS2 (pLP2567), or hos2-H194A,H196A (hos2**; pLP2569) and tested for DNA damage sensitivity. CPT and DMSO plates were prepared without histidine to maintain the plasmid. (C) Suppression in esa1 hos2Δ strains transformed with a vector and with hos2** correlated with increased histone H4K8 acetylation. Quantification of H4K8Ac levels relative to histone H4 was performed with ImageQuant 5.2 (Molecular Dynamics). The histogram peak function was applied to correct for background. Representative immunoblots are shown in Fig. S1D.
To investigate whether suppression of esa1 by hos2Δ was dependent on loss of deacetylase activity, esa1 hos2Δ strains were transformed with HOS2, its catalytic mutant hos2-H195A,H196A (37), or a vector control. Transformants were tested for sensitivity to CPT. As shown in Fig. 1B, wild-type HOS2 expression in the esa1 hos2Δ strain mirrored the DNA damage sensitivity of the esa1 strain, whereas the esa1 hos2Δ strain transformed with hos2-H195A,H196A had decreased sensitivity, suggesting that loss of Hos2's catalytic activity was important for suppression. Both esa1 hos2Δ strains transformed with vector and with hos2-H195A,H196A also showed increased H4 acetylation levels relative to the esa1 strain transformed with vector (Fig. 1C). In contrast to hos2Δ, expression of catalytically dead Hos2 would not likely disrupt the integrity of the Set3 complex. This explains the partial suppression of esa1 when esa1 hos2Δ was transformed with hos2-H195A,H196A, as loss of other subunits of Set3C also have a role in suppressing esa1 (see below).
hos2Δ suppressed low histone H4 acetylation of esa1.
In addition to DNA damage sensitivity, ESA1 mutant strains are also characterized by defects in transcriptional silencing and progression through the cell cycle and by low levels of histone H4 acetylation (6, 21). Deletion of HOS2 was tested for suppression of these phenotypes.
Three transcriptionally silenced regions in Saccharomyces cerevisiae are the ribosomal DNA (rDNA) repeats, telomeres, and the silent mating-type loci (49). ESA1 mutants are defective in silencing the rDNA and telomeres (20) when assayed with reporter strains. The reporter for the rDNA has an ADE2-CAN1 cassette inserted in one of the rDNA repeats in chromosome XII (50). Defects in silencing lead to expression of the CAN1 gene, which encodes an arginine permease. Canavanine is a toxic arginine analog that is imported into cells only when CAN1 is expressed. When incorporated into proteins, it leads to reduced growth due to defects in protein folding. The telomeric reporter consists of a URA3 gene inserted on chromosome VR. Its expression inhibits growth on 5-FOA, which is toxic for cells expressing URA3 (50). The esa1 hos2Δ strain showed growth patterns similar to those of esa1 with both silencing reporters. Thus, HOS2 deletion could not suppress the silencing defects of esa1 (Fig. 2A and B).
FIG 2.

Deleting HOS2 improved histone H4 acetylation in esa1 but did not restore silencing and cell cycle regulation. (A) The esa1 hos2Δ strain is defective for rDNA silencing. The wild-type (LPY4908), esa1-414 (LPY4912), esa1-414 hos2Δ (LPY18074), hos2Δ (LPY18073), and sir2Δ (LPY5015) strains carry the rDNA::ADE2-CAN1 reporter. Defective rDNA silencing leads to expression of CAN1 and sensitivity to canavanine. (B) The esa1 hos2Δ strain had telomeric silencing defects. The wild-type (LPY4916), esa1-414 (LPY13520), esa1-414 hos2Δ (LPY18070), hos2Δ (LPY18071), and sir2Δ (LPY5034) strains carry the TELVR::URA3 reporter. Defective telomeric silencing results in 5-FOA sensitivity. (C) Cell cycle profiles showed a significant G2/M delay in cell cycle progression at 30°C in esa1-531 (LPY14757) cells that was modestly improved in the esa1-531 hos2Δ (LPY14761) strain. Control strains were the wild-type (LPY6497) and hos2Δ (LPY14577) strains. (D) Deletion of HOS2 increased acetylation of histone H4K5 in esa1 mutants. H3K9 and K14 acetylation was unaffected by mutation of ESA1 or hos2Δ. Whole-cell protein lysates from strains in panel C were immunoblotted as noted. The experiment was also performed with esa1-414 strains, with similar results. (E) Deletion of HOS2 improved acetylation of other H4 lysines in esa1 strains. Quantification of histone H4 acetylation at K5, K8, and K12 relative to histone levels was performed using two to four independent Western blots. ImageQuant 5.2 (Molecular Dynamics) and the histogram peak function were used as for Fig. 1. Representative immunoblots are shown in Fig. S2 in the supplemental material.
As esa1 mutants have defects in progression through G2/M (6), cell cycle profiles were evaluated by flow cytometry. The esa1 hos2Δ strain showed a delay in progression through G2/M similar to that of the esa1 strain; thus, hos2Δ cannot restore cell cycle regulation of esa1 (Fig. 2C).
Conditional esa1 mutants have low levels of histone H4 acetylation, especially for histone H4K5, a major target for Esa1. To test if deletion of HOS2 suppressed the global acetylation defect of esa1 strains, the status of Esa1 target lysines in histone H4 was assessed by immunoblotting. Deletion of HOS2 suppressed the low histone H4K5 acetylation levels of the esa1 strain (Fig. 2D), whereas H3 acetylation remained unchanged. Acetylation of H4K8 and H4K12 was also improved in the esa1 hos2Δ strain relative to that of the esa1 strain (Fig. 2E). To determine if DNA damage affected global histone acetylation levels, immunoblots were analyzed for samples after DNA damage was induced. The results were similar to those for cells without damage induction. The esa1 hos2Δ strain had improved acetylation of histone H4K5 compared to that of the esa1 strain (Fig. 2E; see also Fig. S2 in the supplemental material). Acetylation of H4K16 and H3K14 was unaffected by either hos2Δ or DNA damage in the esa1 background. The suppression of the esa1 global acetylation defect at H4K5 by hos2Δ thus appeared to be independent of DNA damage.
Loss of HOS2 suppressed esa1 through lysines 5, 8, and 12 of histone H4.
Because suppression of temperature sensitivity of esa1 by deletion of RPD3 is dependent on lysine 12 of histone H4 (23), we hypothesized that suppression of esa1 by hos2Δ could be mediated through one of the histone H4 lysines. To test this idea, we constructed esa1 and esa1 hos2Δ strains in combination with a series of histone mutants replacing lysines of histone H4 with alanines as proxies of nonmodifiable residues. As shown in Fig. 3A, the esa1 hos2Δ strains in combination with single H4K5A, H4K8A, or H4K12A mutants were sicker when grown on CPT than the strains expressing wild-type histones, although they still had improved growth relative to that of esa1 strains expressing the same histone mutants.
FIG 3.
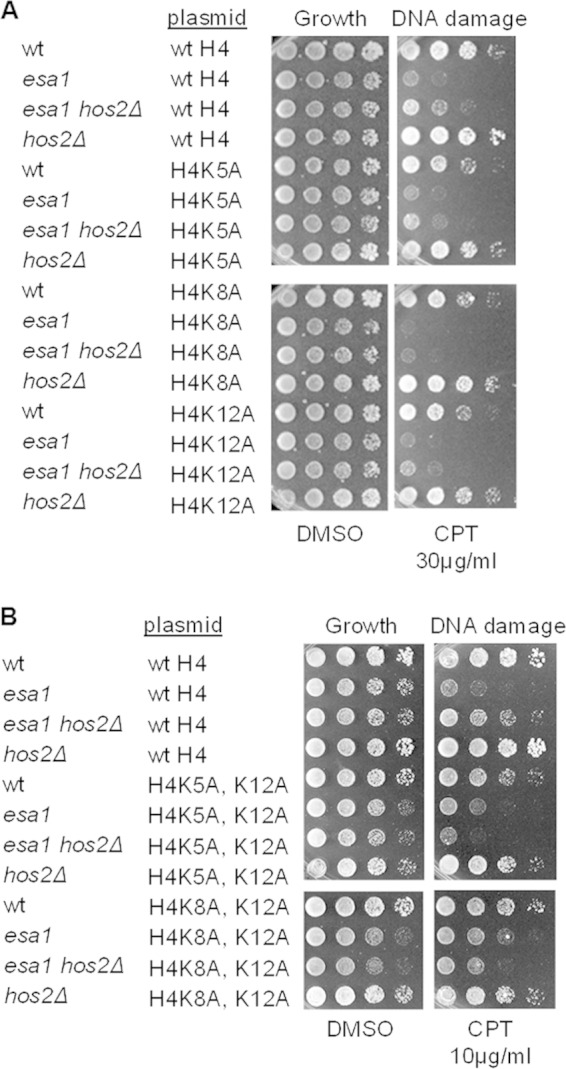
Suppression of esa1 by hos2Δ was dependent upon both individual and combined H4K5, K8, and K12 histone target residues. (A) Mutation of single H4K5, K8, or K12 residues to alanine did not fully disrupt suppression of esa1 DNA damage sensitivity by hos2Δ. Serial dilutions of strains with histone genes deleted and restored on plasmids included wild-type (LPY14161), esa1-414 (LPY14163), esa1-414 hos2Δ (LPY15906), and hos2Δ strains expressing wild-type histones from a plasmid; wild-type (LPY13656), esa1-414 (LPY13064), esa1-414 hos2Δ (LPY15911), and hos2Δ (LPY17911) strains expressing H4K5A from a plasmid; wild-type (LPY14162), esa1-414 (LPY14164), esa1-414 hos2Δ (LPY15912), and hos2Δ (LPY17912) strains expressing H4K8A from a plasmid; and wild-type (LPY13060), esa1-414 (LPY13063), esa1-414 hos2Δ (LPY15913), and hos2Δ (LPY17913) strains expressing H4K12A from a plasmid. Fivefold dilutions were plated as indicated. Note that suppression of esa1 DNA damage sensitivity was not as strong as in Fig. 1A, likely because the histone mutant background has altered histone dosage that can affect sensitivity (72). (B) The esa1 hos2Δ strains containing combined H4K5A and K12A and H4K8A and K12A mutations did not suppress esa1 DNA damage sensitivity and in some cases appeared more sensitive to damage. Wild-type (LPY19424), esa1-414 (LPY19406), esa1-414 hos2Δ (LPY19425) and hos2Δ (LPY19426) strains expressing H4K5A and K12A and wild-type (LPY19420), esa1-414 (LPY19421), esa1-414 hos2Δ (LPY19422), and hos2Δ (LPY19423) strains expressing H4K8A and K12A were compared to strains containing wild-type histones.
Lysines 5, 8, and 12 of H4 were previously reported to perform overlapping roles in vivo and to be modified by the same complexes (51). We hypothesized that the acetylation of multiple H4 lysines could contribute cooperatively to suppression of esa1 by hos2Δ. Strains containing combined H4 lysine mutants were tested. As shown in Fig. 3B, the esa1 hos2Δ H4K5A, K12A and esa1 hos2Δ H4K8A, K12A strains were more sensitive to CPT than the corresponding esa1 strains, suggesting that suppression of esa1 by HOS2 deletion required a combination of modifications of H4 lysines 5, 8, and 12.
Deletion of HOS2 modulated the transcriptional response of esa1 cells upon DNA damage.
Esa1 is important during DNA damage repair in at least two different pathways. The first is through modulation of gene expression (31, 32), and the second is through its direct recruitment to sites of damage to carry out specific modifications promoting repair signaling at the break sites (22, 52). Because Hos2 was reported to influence induction of gene expression under stress conditions (31, 38) but not to be recruited to sites occupied by Esa1 at the DNA double-strand breaks (7), we analyzed the transcriptional response to DNA damage in esa1 and esa1 hos2Δ strains. Multiple independent genomewide data sets were used to select candidates for analysis (53–56). As the Hos2-containing Set3C is recruited to H3K4me2-marked genomic areas and has a role in regulation of ncRNA expression, we identified genes with this histone mark and ncRNAs (54–58). Selected genes were reported to be induced upon methyl methanesulfonate (MMS)-induced DNA damage (59) and had global expression changes in hos2Δ or set3Δ mutants, even in the absence of damage (60). Table 1 summarizes published data for the genes tested. For example, the HUG1 locus has the H3K4me2 mark in its promoter region and is downregulated in hos2Δ and set3Δ strains.
TABLE 1.
Characteristics of DNA damage-regulated genes testeda
| Gene | H3K4me2 | ncRNA | Expression in: |
Role | |
|---|---|---|---|---|---|
| hos2Δ | set3Δ | ||||
| HUG1 | Promoter | Meiotic ORF (AS) | — | —* | Involved in Mec1 checkpoint |
| ERG5 | Promoter and 5′ ORF | XUT (AS) promoter, CUT (S) promoter | —* | Oxidation reduction and lipid metabolism | |
| GRE2 | Promoter and ORF | ncRNA (AS) whole ORF | —* | —* | Methylbutanal and glyoxal reductase |
The H3K4me2 column indicates if dimethylation of H3K4 was identified in a previous global survey (56) and its localization within the gene body (Saccharomyces Genome Database). The ncRNA column specifies the type, localization, and orientation, sense (S) or antisense (AS), of ncRNAs overlapping the genes tested (54, 55, 57, 58). The hos2Δ and set3Δ columns show if expression was downregulated (—) in the corresponding null strains (60) and whether the change was statistically significant (*).
To test if transcripts of the selected genes are regulated by Esa1, cells from wild-type, esa1, esa1 hos2Δ, and hos2Δ strains were treated with CPT or with the vehicle control DMSO for 90 min. RNA was purified and cDNA for each sample was used to quantify the expression levels of candidate genes (Fig. 4).
Expression of HUG1 and GRE2 was increased in the wild-type strain treated with CPT, as previously reported for other damage-inducing drugs (Fig. 4A) (59). Although repressed by MMS-induced DNA damage (59), ERG5 expression was induced by CPT (Fig. 4A). This difference may reflect distinct effects on gene expression for MMS and CPT, for which mechanisms of damage response are known to be distinct (61, 62).
Mutant esa1 cells had lower levels of expression in DMSO than wild-type for HUG1 and ERG5. Both genes' expression upon damage was also lower than wild-type in esa1 cells. In contrast, GRE2 expression in esa1 cells had higher levels of expression than the wild type when treated with DMSO and CPT (Fig. 4A).
H3K4me2 marks are present at HUG1, ERG5, and GRE2 (Table 1). HUG1 and ERG5 are also marked by H3K4me3 at the promoter region, whereas GRE2 completely lacks this mark (56). We hypothesize that the variable pattern of expression of GRE2 in the esa1 strain compared to HUG1 and ERG5 reflects the possibility that GRE2 regulation is initially independent of the H3K4me3 mark present in HUG1 and ERG5.
The above-described results show that gene expression of DNA damage-responsive genes in esa1 cells is aberrant in DMSO- and CPT-treated samples. Expression for all three tested genes in the esa1 hos2Δ strain proved more uniform: lower than in wild-type and esa1 cells treated with either DMSO or CPT (Fig. 4A). When comparing DMSO- and CPT-treated samples for HUG1, GRE2, and ERG5, we found that the esa1 hos2Δ strain showed small changes in expression, whereas an increase in gene expression upon damage was clear in esa1 and wild-type cells.
As reported earlier, deletion of RPD3 can also suppress some esa1 mutant phenotypes through increased histone H4 acetylation, although not its DNA damage sensitivity (34). We hypothesized that the expression pattern in DNA damage-sensitive esa1 rpd3Δ cells would be different than the pattern found in the damage-resistant esa1 hos2Δ cells. As shown in Fig. 4B, when cells were treated with DMSO or CPT, expression of HUG1 and GRE2 in the esa1 rpd3Δ strain was similar to that of the esa1 strain, whereas in the esa1 hos2Δ strain, expression was low under all conditions relative to those in the wild-type, esa1, and esa1 rpd3Δ strains. This result shows that the esa1 hos2Δ expression pattern, although not fully equivalent to that of the wild type, is distinct from the pattern found in the damage-sensitive esa1 and esa1 rpd3Δ strains, consistent with the in vivo suppression we observed.
Loss of the Set3 complex suppressed esa1.
Hos2 is a centrally important component of Set3C, a complex with important roles in stress responses. The eponymous Set3 subunit has both PHD and SET domains (39). Other subunits of the complex include Snt1, Sif2, Cpr1, and Hos4, and catalytic subunits Hos2 and the class III KDAC sirtuin Hst1 (Fig. 5A). Hst1 is not a dedicated complex member, but it is also found in the Sum1 complex (Sum1C, containing Sum1, Rfm1, and Hst1), which functions mitotically to silence meiotic and sporulation genes (Fig. 5A) (63).
FIG 5.
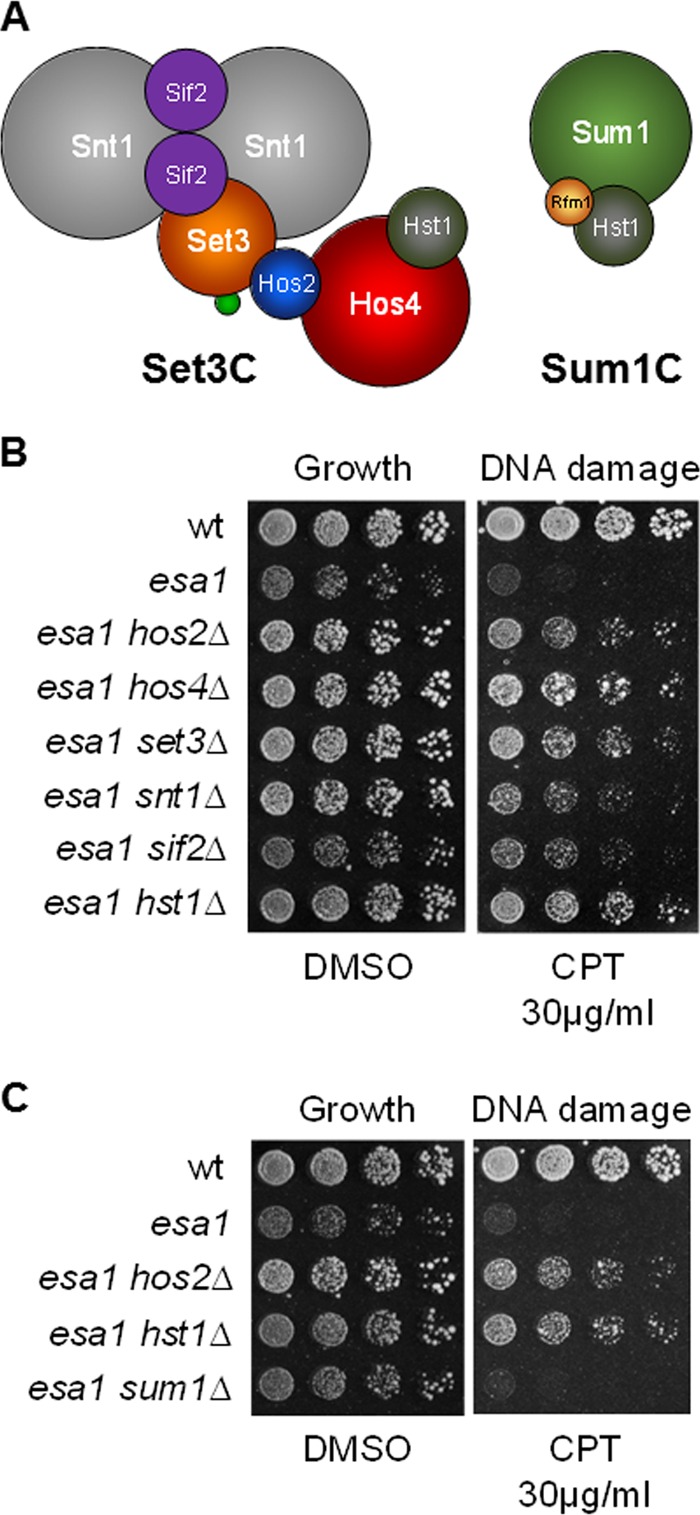
Deletion of Set3C subunits suppressed DNA damage sensitivity of esa1. (A) Subunit composition of Sum1C and Set3C (with data from reference 39). The relative size of the subunits is drawn to scale. The small green circle in Set3C represents the Cpr1 subunit. (B) Deletion of each Set3C subunit tested suppressed esa1 sensitivity to CPT. Wild-type (LPY6497), esa1-531 (LPY14757), esa1-531 hos2Δ (LPY14761), esa1-531 hos4Δ (LPY15865), esa1-531 set3Δ (LPY15869), esa1-531 snt1Δ (LPY15867), esa1-531 sif2Δ (LPY15863), and esa1-531 hst1Δ2 (LPY18266) strains were tested. (C) Suppression of esa1 DNA damage sensitivity was specific to Set3C. Deletion of SUM1 had no effect on esa1 sensitivity to CPT. Loss of HST1 was comparable to hos2Δ for suppression of esa1 DNA damage sensitivity. Wild-type (LPY6497), esa1-531 (LPY14757), esa1-531 hos2Δ (LPY14761), esa1-531 hst1Δ2 (LPY18266), and esa1-531 sum1Δ (LPY17505) strains were compared. Figure S3 in the supplemental material includes single mutant controls for Set3C subunits.
To examine if suppression of esa1 DNA damage sensitivity was mediated by the Set3C, we constructed double mutants combining esa1 with deletions of genes encoding Set3C components. Loss of any Set3C subunit tested suppressed esa1's DNA damage sensitivity (Fig. 5B). The suppression was not uniform: HOS4, SET3, HOS2, and HST1 deletions were more effective than deletions of SNT1 or SIF2.
Loss of Set3 and Hos2 leads to disassembly of the complex (39), whereas loss of Hos4 results in further loss of Hst1. Our results suggest that both Hos2 and Hst1 KDACs are important in opposing Esa1 during the DNA damage response. However, suppression of esa1 by hst1Δ was clearly mediated through Set3C and not through Sum1C, as loss of the Sum1 subunit did not suppress esa1 (Fig. 5C).
Impaired binding of Set3C to H3K4me2 suppressed esa1.
As Set3C binds H3K4me2 to influence induction of gene expression under stress conditions, we asked if suppression of DNA damage sensitivity was dependent on H3K4 methylation by deleting SET1, which encodes the H3K4 methyltransferase of the COMPASS complex (64). The esa1 set1Δ strain was extremely sick (Fig. 6A), with slow growth at 30°C and sensitivity to DMSO. We reasoned that reduced viability could be due to complete loss of H3K4 methylation. Consistent with this idea, the H3K4A mutant alone was very sensitive to damage in the esa1 background, and deletion of HOS2 could not suppress this phenotype (Fig. 6B).
FIG 6.

Loss of the methyl-binding domain of Set3 suppressed esa1 DNA damage sensitivity. (A) The esa1 set1Δ strain was synthetically sick. The esa1 set1Δ strain grew slowly at 30°C on rich medium and was inviable when grown on 0.2% DMSO or 10 μg/ml of CPT. (B) Mutation of H3K4 to alanine disrupted suppression of esa1 DNA damage sensitivity by hos2Δ, which was also sensitive to H3K4A. Shown are serial dilutions of wild-type (LPY14161), esa1-414 (LPY14163), esa1-414 hos2Δ (LPY15906), and hos2Δ strains expressing wild-type histones from a plasmid; wild type (LPY21480), esa1-414 (LPY21481), esa1-414 hos2Δ (LPY21482), and hos2Δ (LPY21483) strains expressing H3K4A from a plasmid. (C) esa1 strains are synthetically sick when combined with COMPASS complex deletions promoting simultaneous loss of H3K4 di- and trimethylation. The following strains were assayed: wild-type (LPY6497), esa1-531 (LPY14757), esa1-531 cps25Δ (LPY21498), esa1-531 cps40Δ (LPY21495), esa1-531 cps60Δ (LPY21503), esa1-531 hos2Δ (LPY14761), cps25Δ (LPY21499), cps40Δ (LPY21494), and cps60Δ (LPY21520) strains. (D) hos2Δ could not suppress esa1 when CPS25 or CPS40 were also deleted. Strains tested included wild-type (LPY6497), esa1-531 (LPY14757), esa1-531 hos2Δ cps25Δ (LPY21656), esa1-531 hos2Δ cps40Δ (LPY21661), and esa1-531 hos2Δ (LPY14761) strains. (E) Vector-transformed (pLP1358) wild-type, esa1-531, esa1-531 hos2Δ, esa1-531 set3Δ, set3Δ, and hos2Δ strains shown in Fig. 5B and Fig. S3 in the supplemental material were compared to set3-W140 (PHD domain mutant, pLP3020)-transformed esa1-531 set3Δ and set3Δ strains.
Loss of COMPASS subunits differentially affects di- or trimethylation of H3K4 (reviewed in reference 65). For example, deletion of CPS25 (SDC1) or CPS60 (BRE2) promotes loss of H3K4me3 and diminished levels of H3K4me2, whereas deletion of CPS40 (SPP1) is characterized by very low H3K4me3 levels (66–68). We considered the hypothesis that if H3K4me2 was reduced by deletion of CPS25 or CPS60, Set3C regulation would be impaired in esa1, perhaps promoting resistance to DNA damage. However, the double esa1 cps25Δ and esa1 cps60Δ mutants were very sick and extremely sensitive to DNA damage (Fig. 6C). In contrast, the esa1 cps40Δ strain, which should only affect H3K4me3, grew comparably to the esa1 single mutant (Fig. 6C), suggesting that H3K4me3 is not as critical as H3K4me2 in esa1 cells. This result supports a previous report that specific loss of H3K4me3 had no significant impact on gene expression in wild-type cells, whereas simultaneous loss of H3K4me2 and H3K4me3 led to greater changes in gene expression (68). We further tested if hos2Δ could suppress esa1 when H3K4me2 and H3K4me3 were impaired by deletion of CPS25. The triple esa1 hos2Δ cps40Δ mutant was also used as a control for specific loss of H3K4me3 that would have little effect on H3K4me2. Figure 6D shows that hos2Δ did not suppress esa1 when CPS25 or CPS40 was also deleted. Thus, deletion of COMPASS subunits proved insufficient to test the role of H3K4me2 in suppression of esa1 by hos2Δ for two reasons: because H3K4 methylation influences gene expression through independent, parallel pathways (69), and because COMPASS mutants cannot impair H3K4me2 without affecting H3K4me3, which was also shown to be important for suppression.
To evaluate the significance of binding of Set3C to the H3K4me2 mark in suppression, we used the set3-PHD domain mutant (set3-W140A) (40). This mutant retains Set3C integrity but loses the recognition of the H3K4me2 mark (40). We asked if a set3Δ strain transformed with a vector or with the set3-W140A allele could suppress CPT sensitivity in esa1. This set3-PHD mutant did suppress (Fig. 6E), validating a mechanism in which localized loss of Set3 and Hos2 recruitment suppressed the DNA damage sensitivity of esa1 strains.
DISCUSSION
The essential acetyltransferase Esa1 and its human ortholog Tip60 have key roles in responding to DNA damage. Esa1 participates in induction of gene expression and is recruited to sites of DNA damage, where it promotes ligation of broken DNA ends. Further, because Esa1 is involved in regulating cell cycle progression, it may also have a role in establishing cell cycle delays necessary for repair.
The powerful tool of genetic suppression has provided insight into the function of Esa1 through identification of specific conditions that relieve esa1 phenotypes. These include deletion of specific subunits of the Rpd3L and Rpd3S complexes (23, 34), as well as overexpression of the RNA binding protein Nab3 (14) and the amino acid biosynthetic protein Lys20 (33, 35). Each of these genetic manipulations suppresses a unique constellation of phenotypes of Esa1. For example, loss of Rpd3 rescues temperature sensitivity, silencing defects and diminished histone H4 acetylation (23), whereas NAB3 overexpression suppresses esa1's silencing defects and temperature sensitivity (14). In contrast to LYS20, neither rpd3Δ nor NAB3 overexpression improves the DNA damage response of esa1 mutants. In this work, we established that deletion of the deacetylase encoded by HOS2 suppressed DNA damage sensitivity of esa1, underscoring the diverse and intricate interactions of the chromatin-modifying activities.
Suppression of esa1 DNA damage sensitivity by hos2Δ correlated with enhanced acetylation of histone H4. A similar global increase in H4 acetylation correlates with suppression of a different set of phenotypes when the Rpd3L complex is removed. An important question, then, is this: why does enhanced global H4 acetylation in esa1 cells rescue independent phenotypes depending on the deacetylase removed?
To begin to answer this question, it should be noted that the Rpd3L complex is targeted to promoter regions by the transcription factor subunits Ash1 and Ume6 (70). Through its PHD domain subunit, Pho23, Rpd3L is also capable of recognizing the H3K4me3 mark usually found at promoter regions. In contrast, Set3C binds genomic areas enriched with the H3K4me2 mark. Dimethylation of H3K4 is generally localized 5′ of ORFs (40); however, it can also replace H3K4me3 at specific promoters (38), including, for example, the promoter of GRE2 (56). We propose that RPD3 and HOS2 deletions suppress different phenotypes of esa1 because their loss promotes increased histone H4 acetylation at specific genomic areas or genes: loss of Rpd3L would promote increased acetylation at promoter regions, whereas loss of HOS2 would lead to enhanced acetylation at ORFs and select promoters regulated by H3K4me2. Indeed, dynamic relocalization of Hos2 has been reported upon MMS treatment to facilitate formation of noncanonical repair foci (71), hypothesized to be transcriptional factories.
Suppression of the DNA damage sensitivity of esa1 by hos2Δ could be mediated by the transcriptional response to damage or by direct recruitment of the complex to broken DNA ends. Because Hos2 was previously shown not to be recruited to areas enriched with Esa1 following induction of a single DSB by the HO endonuclease (7), it is likely that Hos2 opposes Esa1 through a different pathway.
We confirmed earlier results showing defective induction of HUG1 in hos2Δ and esa1 strains and tested other genes previously established as induced by DNA damage. The esa1 strain had an impaired transcriptional response to damage. The gene expression profile of hos2Δ cells was also abnormal compared to that of the wild type (Fig. 4A). This is in agreement with an impaired DNA damage response of hos2Δ and with its proposed role in acetylation dynamics involved in gene induction (31). The hos2Δ strain is not sensitive to DNA damage, suggesting that high induction of a specific set of genes during repair, such as HUG1, may function as a protective transcriptional response.
Upon damage induction, the gene expression profile in the esa1 hos2Δ strain was distinct from that of esa1. This response remained distinct from that of the wild type, suggesting that in a manner similar to that in the hos2Δ strain, the combined response in esa1 hos2Δ cells may be sufficient to promote resistance to DNA damage. Further supporting this idea, gene expression in the damage-sensitive esa1 rpd3Δ strain proved similar to that in the esa1 strain upon DNA damage. Additional insight into suppression of esa1's defective response to damage will ultimately be obtained with global analyses correlating gene expression and histone H4 acetylation with Rpd3L and Set3C occupancy at promoter and coding regions upon damage.
In defining suppression of esa1 by hos2Δ, we found that it could be mediated by removal of any Set3C subunit, with the strongest effects seen upon deletion of HOS2, HOS4, SET3, and HST1. Loss of Set3 and Hos2 disassemble Set3C, whereas deletion of HOS4 leads to loss of Hst1 association with the complex, suggesting that both KDACs, Hos2 and Hst1, have roles in opposing Esa1 during DNA damage repair. Since Hst1 and Hos2 have both shared and specific targets (38), future studies will define how deletion of HST1 affects esa1 upon DNA damage. In agreement with the expression data, genetic dissection revealed that suppression of esa1 by hos2Δ required chromosomal binding of Set3C to the H3K4me2 mark, a histone modification already implicated in cellular stress response.
Taking the results together, it appears that Esa1 and Hos2 oppose each other by promoting dynamic acetylation and deacetylation. In the absence of active Esa1 and Hos2, other HATs, such as Gcn5, and KDACs, such as Rpd3, can promote acetylation dynamics (Fig. 7A). In this scenario, the esa1 and hos2Δ single mutant strains have impaired acetylation dynamics and gene regulation in response to DNA damage (Fig. 7A). The dynamics would be reestablished in the esa1 hos2Δ strain by promoting an adjusted transcriptional response to allow growth following damage. In support of this idea, RPD3 and GCN5 proved necessary for suppression of esa1 by HOS2 deletion (Fig. 7B and C). Because of the conserved nature of these regulators of acetylation, continued dissection of their functional interactions will contribute to a deeper understanding of their fundamental roles. Understanding how Esa1 participates in DNA damage repair will ultimately point to mechanisms defining the role of human Tip60 in cancer progression associated with genomic instability and DNA damage.
FIG 7.

Potential impact of dynamic acetylation in regulation of gene expression upon DNA damage. (A) Wild-type and esa1 hos2Δ strains would maintain dynamic acetylation compared to esa1 and hos2Δ strains that have impaired dynamics. Acetylation is depicted by green circles on histone tails. (B) HOS2 deletion could not suppress esa1 in the absence of RPD3. Wild-type (LPY6497), esa1-531 (LPY14757), esa1-531 hos2Δ (LPY14761), esa1-531 hos2Δ rpd3Δ (LPY21428), esa1-531 rpd3Δ (LPY21450), and hos2Δ rpd3Δ (LPY21426) strains were plated on the indicated medium. (C) HOS2 deletion could not suppress esa1 in the absence of GCN5. Wild-type (LPY5), esa1-414 (LPY21400), esa1-414 hos2Δ (LPY21401), esa1-414 hos2Δ gcn5Δ (LPY21468), and gcn5Δ hos2Δ (LPY21399) strains were plated as indicated. The esa1-414 gcn5Δ strain was not included in the plating, as it was lethal when having esa1-414 expressed from a plasmid in the W303 background.
Supplementary Material
ACKNOWLEDGMENTS
We thank S. Buratowski, J. D. Boeke, N. Kurup, and S. Venkataramanan for providing strains. We also thank S. J. Jacobson, M. Melnik-Evpak, E. Petty, and B. X. Su for critical reading of the manuscript and members of the Pillus lab for helpful advice throughout the course of this study.
This work was initiated with prior support from the National Institutes of Health (GM5649). Additional funding has been obtained from the University of California Cancer Research Coordinating Committee and the UCSD Committee on Research. A.L.T-M. was supported by the University of California Institute for Mexico and the United States (UCMEXUS) and the National Council of Science and Technology of Mexico (CONACyT). C.S.C. was supported by T32-GM-007240 and GM033279 (to D. J. Forbes).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00298-15.
REFERENCES
- 1.Felsenfeld G, Groudine M. 2003. Controlling the double helix. Nature 421:448–453. doi: 10.1038/nature01411. [DOI] [PubMed] [Google Scholar]
- 2.Rando OJ, Winston F. 2012. Chromatin and transcription in yeast. Genetics 190:351–387. doi: 10.1534/genetics.111.132266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strahl BD, Allis CD. 2000. The language of covalent histone modifications. Nature 403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 4.Millar CB, Grunstein M. 2006. Genome-wide patterns of histone modifications in yeast. Nat Rev Mol Cell Biol 7:657–666. doi: 10.1038/nrm1986. [DOI] [PubMed] [Google Scholar]
- 5.Boudreault AA, Cronier D, Selleck W, Lacoste N, Utley RT, Allard S, Savard J, Lane WS, Tan S, Côté J. 2003. Yeast enhancer of polycomb defines global Esa1-dependent acetylation of chromatin. Genes Dev 17:1415–1428. doi: 10.1101/gad.1056603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clarke AS, Lowell JE, Jacobson SJ, Pillus L. 1999. Esa1p is an essential histone acetyltransferase required for cell cycle progression. Mol Cell Biol 19:2515–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin YY, Qi Y, Lu JY, Pan X, Yuan DS, Zhao Y, Bader JS, Boeke JD. 2008. A comprehensive synthetic genetic interaction network governing yeast histone acetylation and deacetylation. Genes Dev 22:2062–2074. doi: 10.1101/gad.1679508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith ER, Eisen A, Gu W, Sattah M, Pannuti A, Zhou J, Cook RG, Lucchesi JC, Allis CD. 1998. ESA1 is a histone acetyltransferase that is essential for growth in yeast. Proc Natl Acad Sci U S A 95:3561–3565. doi: 10.1073/pnas.95.7.3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keogh MC, Mennella TA, Sawa C, Berthelet S, Krogan NJ, Wolek A, Podolny V, Carpenter LR, Greenblatt JF, Baetz K, Buratowski S. 2006. The Saccharomyces cerevisiae histone H2A variant Htz1 is acetylated by NuA4. Genes Dev 20:660–665. doi: 10.1101/gad.1388106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Babiarz JE, Halley JE, Rine J. 2006. Telomeric heterochromatin boundaries require NuA4-dependent acetylation of histone variant H2A.Z in Saccharomyces cerevisiae. Genes Dev 20:700–710. doi: 10.1101/gad.1386306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Millar CB, Xu F, Zhang K, Grunstein M. 2006. Acetylation of H2AZ Lys 14 is associated with genome-wide gene activity in yeast. Genes Dev 20:711–722. doi: 10.1101/gad.1395506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitchell L, Huard S, Cotrut M, Pourhanifeh-Lemeri R, Steunou AL, Hamza A, Lambert JP, Zhou H, Ning Z, Basu A, Cote J, Figeys DA, Baetz K. 2013. mChIP-KAT-MS, a method to map protein interactions and acetylation sites for lysine acetyltransferases. Proc Natl Acad Sci U S A 110:E1641–E1650. doi: 10.1073/pnas.1218515110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yi C, Ma M, Ran L, Zheng J, Tong J, Zhu J, Ma C, Sun Y, Zhang S, Feng W, Zhu L, Le Y, Gong X, Yan X, Hong B, Jiang FJ, Xie Z, Miao D, Deng H, Yu L. 2012. Function and molecular mechanism of acetylation in autophagy regulation. Science 336:474–477. doi: 10.1126/science.1216990. [DOI] [PubMed] [Google Scholar]
- 14.Chang CS, Clarke A, Pillus L. 2012. Suppression analysis of esa1 mutants in Saccharomyces cerevisiae links NAB3 to transcriptional silencing and nucleolar functions. G3 2:1223–1232. doi: 10.1534/g3.112.003558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin YY, Lu JY, Zhang J, Walter W, Dang W, Wan J, Tao SC, Qian J, Zhao Y, Boeke JD, Berger SL, Zhu H. 2009. Protein acetylation microarray reveals that NuA4 controls key metabolic target regulating gluconeogenesis. Cell 136:1073–1084. doi: 10.1016/j.cell.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Downey M, Johnson JR, Davey NE, Newton BW, Johnson TL, Galaang S, Seller CA, Krogan N, Toczyski DP. 2015. Acetylome profiling reveals overlap in the regulation of diverse processes by sirtuins, Gcn5, and Esa1. Mol Cell Proteomics 14:162–176. doi: 10.1074/mcp.M114.043141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Squatrito M, Gorrini C, Amati B. 2006. Tip60 in DNA damage response and growth control: many tricks in one HAT. Trends Cell Biol 16:433–442. doi: 10.1016/j.tcb.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 18.Lafon A, Chang CS, Scott EM, Jacobson SJ, Pillus L. 2007. MYST opportunities for growth control: yeast genes illuminate human cancer gene functions. Oncogene 26:5373–5384. doi: 10.1038/sj.onc.1210606. [DOI] [PubMed] [Google Scholar]
- 19.Avvakumov N, Côté J. 2007. The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene 26:5395–5407. doi: 10.1038/sj.onc.1210608. [DOI] [PubMed] [Google Scholar]
- 20.Reid JL, Iyer VR, Brown PO, Struhl K. 2000. Coordinate regulation of yeast ribosomal protein genes is associated with targeted recruitment of Esa1 histone acetylase. Mol Cell 6:1297–1307. doi: 10.1016/S1097-2765(00)00128-3. [DOI] [PubMed] [Google Scholar]
- 21.Clarke AS, Samal E, Pillus L. 2006. Distinct roles for the essential MYST family HAT Esa1p in transcriptional silencing. Mol Biol Cell 17:1744–1757. doi: 10.1091/mbc.E05-07-0613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bird AW, Yu DY, Pray-Grant MG, Qiu Q, Harmon KE, Megee PC, Grant PA, Smith MM, Christman MF. 2002. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature 419:411–415. doi: 10.1038/nature01035. [DOI] [PubMed] [Google Scholar]
- 23.Chang CS, Pillus L. 2009. Collaboration between the essential Esa1 acetyltransferase and the Rpd3 deacetylase is mediated by H4K12 histone acetylation in Saccharomyces cerevisiae. Genetics 183:149–160. doi: 10.1534/genetics.109.103846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong LY, Recht J, Laurent BC. 2006. Chromatin remodeling and repair of DNA double-strand breaks. J Mol Histol 37:261–269. doi: 10.1007/s10735-006-9047-4. [DOI] [PubMed] [Google Scholar]
- 25.Zeman MK, Cimprich KA. 2014. Causes and consequences of replication stress. Nat Cell Biol 16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giglia-Mari G, Zotter A, Vermeulen W. 2011. DNA damage response. Cold Spring Harb Perspect Biol 3:a000745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Attikum H, Gasser SM. 2005. The histone code at DNA breaks: a guide to repair? Nat Rev Mol Cell Biol 6:757–765. doi: 10.1038/nrm1737. [DOI] [PubMed] [Google Scholar]
- 28.Fu Y, Pastushok L, Xiao W. 2008. DNA damage-induced gene expression in Saccharomyces cerevisiae. FEMS Microbiol Rev 32:908–926. doi: 10.1111/j.1574-6976.2008.00126.x. [DOI] [PubMed] [Google Scholar]
- 29.Branzei D, Foiani M. 2006. The Rad53 signal transduction pathway: Replication fork stabilization, DNA repair, and adaptation. Exp Cell Res 312:2654–2659. doi: 10.1016/j.yexcr.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 30.Sirbu BM, Cortez D. 2013. DNA damage response: three levels of DNA repair regulation. Cold Spring Harb Perspect Biol 5:a012724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharma VM, Tomar RS, Dempsey AE, Reese JC. 2007. Histone deacetylases RPD3 and HOS2 regulate the transcriptional activation of DNA damage-inducible genes. Mol Cell Biol 27:3199–3210. doi: 10.1128/MCB.02311-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robert F, Pokholok DK, Hannett NM, Rinaldi NJ, Chandy M, Rolfe A, Workman JL, Gifford DK, Young RA. 2004. Global position and recruitment of HATs and HDACs in the yeast genome. Mol Cell 16:199–209. doi: 10.1016/j.molcel.2004.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Torres-Machorro AL, Aris JP, Pillus L. 2015. A moonlighting metabolic protein influences repair at DNA double-stranded breaks. Nucleic Acids Res 43:1646–1658. doi: 10.1093/nar/gku1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Biswas D, Takahata S, Stillman DJ. 2008. Different genetic functions for the Rpd3(L) and Rpd3(S) complexes suggest competition between NuA4 and Rpd3(S). Mol Cell Biol 28:4445–4458. doi: 10.1128/MCB.00164-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scott EM, Pillus L. 2010. Homocitrate synthase connects amino acid metabolism to chromatin functions through Esa1 and DNA damage. Genes Dev 24:1903–1913. doi: 10.1101/gad.1935910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Torres-Machorro AL, Pillus L. 2014. Bypassing the requirement for an essential MYST acetyltransferase. Genetics 197:851–863. doi: 10.1534/genetics.114.165894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang A, Kurdistani SK, Grunstein M. 2002. Requirement of Hos2 histone deacetylase for gene activity in yeast. Science 298:1412–1414. doi: 10.1126/science.1077790. [DOI] [PubMed] [Google Scholar]
- 38.Kim T, Xu Z, Clauder-Munster S, Steinmetz LM, Buratowski S. 2012. Set3 HDAC mediates effects of overlapping noncoding transcription on gene induction kinetics. Cell 150:1158–1169. doi: 10.1016/j.cell.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pijnappel WW, Schaft D, Roguev A, Shevchenko A, Tekotte H, Wilm M, Rigaut G, Seraphin B, Aasland R, Stewart AF. 2001. The S. cerevisiae SET3 complex includes two histone deacetylases, Hos2 and Hst1, and is a meiotic-specific repressor of the sporulation gene program. Genes Dev 15:2991–3004. doi: 10.1101/gad.207401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim T, Buratowski S. 2009. Dimethylation of H3K4 by Set1 recruits the Set3 histone deacetylase complex to 5′ transcribed regions. Cell 137:259–272. doi: 10.1016/j.cell.2009.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith JS, Brachmann CB, Celic I, Kenna MA, Muhammad S, Starai VJ, Avalos JL, Escalante-Semerena JC, Grubmeyer C, Wolberger C, Boeke JD. 2000. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc Natl Acad Sci U S A 97:6658–6663. doi: 10.1073/pnas.97.12.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ahn SH, Cheung WL, Hsu JY, Diaz RL, Smith MM, Allis CD. 2005. Sterile 20 kinase phosphorylates histone H2B at serine 10 during hydrogen peroxide-induced apoptosis in S. cerevisiae. Cell 120:25–36. doi: 10.1016/j.cell.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 43.Renauld H, Aparicio OM, Zierath PD, Billington BL, Chhablani SK, Gottschling DE. 1993. Silent domains are assembled continuously from the telomere and are defined by promoter distance and strength, and by SIR3 dosage. Genes Dev 7:1133–1145. doi: 10.1101/gad.7.7a.1133. [DOI] [PubMed] [Google Scholar]
- 44.van Leeuwen F, Gottschling DE. 2002. Assays for gene silencing in yeast. Methods Enzymol 350:165–186. doi: 10.1016/S0076-6879(02)50962-9. [DOI] [PubMed] [Google Scholar]
- 45.Nitiss J, Wang JC. 1988. DNA topoisomerase-targeting antitumor drugs can be studied in yeast. Proc Natl Acad Sci U S A 85:7501–7505. doi: 10.1073/pnas.85.20.7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Okomo-Adhiambo M, Nguyen HT, Abd EA, Sleeman K, Fry AM, Gubareva LV. 2014. Drug susceptibility surveillance of influenza viruses circulating in the United States in 2011-2012: application of the WHO antiviral working group criteria. Influenza Other Respir Viruses 8:258–265. doi: 10.1111/irv.12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang XJ, Seto E. 2008. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol 9:206–218. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brachmann CB, Sherman JM, Devine SE, Cameron EE, Pillus L, Boeke JD. 1995. The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev 9:2888–2902. doi: 10.1101/gad.9.23.2888. [DOI] [PubMed] [Google Scholar]
- 49.Rusche LN, Kirchmaier AL, Rine J. 2003. The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu Rev Biochem 72:481–516. doi: 10.1146/annurev.biochem.72.121801.161547. [DOI] [PubMed] [Google Scholar]
- 50.Roy N, Runge KW. 2000. Two paralogs involved in transcriptional silencing that antagonistically control yeast life span. Curr Biol 10:111–114. doi: 10.1016/S0960-9822(00)00298-0. [DOI] [PubMed] [Google Scholar]
- 51.Dion MF, Altschuler SJ, Wu LF, Rando OJ. 2005. Genomic characterization reveals a simple histone H4 acetylation code. Proc Natl Acad Sci U S A 102:5501–5506. doi: 10.1073/pnas.0500136102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tamburini BA, Tyler JK. 2005. Localized histone acetylation and deacetylation triggered by the homologous recombination pathway of double-strand DNA repair. Mol Cell Biol 25:4903–4913. doi: 10.1128/MCB.25.12.4903-4913.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gasch AP, Huang M, Metzner S, Botstein D, Elledge SJ, Brown PO. 2001. Genomic expression responses to DNA-damaging agents and the regulatory role of the yeast ATR homolog Mec1p. Mol Biol Cell 12:2987–3003. doi: 10.1091/mbc.12.10.2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yassour M, Pfiffner J, Levin JZ, Adiconis X, Gnirke A, Nusbaum C, Thompson DA, Friedman N, Regev A. 2010. Strand-specific RNA sequencing reveals extensive regulated long antisense transcripts that are conserved across yeast species. Genome Biol 11:R87. doi: 10.1186/gb-2010-11-8-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Neil H, Malabat C, d'Aubenton-Carafa Y, Xu Z, Steinmetz LM, Jacquier A. 2009. Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature 457:1038–1042. doi: 10.1038/nature07747. [DOI] [PubMed] [Google Scholar]
- 56.Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Lee TI, Bell GW, Walker K, Rolfe PA, Herbolsheimer E, Zeitlinger J, Lewitter F, Gifford DK, Young RA. 2005. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell 122:517–527. doi: 10.1016/j.cell.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 57.Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Munster S, Camblong J, Guffanti E, Stutz F, Huber W, Steinmetz LM. 2009. Bidirectional promoters generate pervasive transcription in yeast. Nature 457:1033–1037. doi: 10.1038/nature07728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Dijk EL, Chen CL, ubenton-Carafa Y, Gourvennec S, Kwapisz M, Roche V, Bertrand C, Silvain M, Legoix-Ne P, Loeillet S, Nicolas A, Thermes C, Morillon A. 2011. XUTs are a class of Xrn1-sensitive antisense regulatory non-coding RNA in yeast. Nature 475:114–117. doi: 10.1038/nature10118. [DOI] [PubMed] [Google Scholar]
- 59.Jaehnig EJ, Kuo D, Hombauer H, Ideker TG, Kolodner RD. 2013. Checkpoint kinases regulate a global network of transcription factors in response to DNA damage. Cell Rep 4:174–188. doi: 10.1016/j.celrep.2013.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lenstra TL, Benschop JJ, Kim T, Schulze JM, Brabers NA, Margaritis T, van de Pasch LA, van Heesch SA, Brok MO, Groot Koerkamp MJ, Ko CW, van LD, Sameith K, van H Sr, Lijnzaad P, Kemmeren P, Hentrich T, Kobor MS, Buratowski S, Holstege FC. 2011. The specificity and topology of chromatin interaction pathways in yeast. Mol Cell 42:536–549. doi: 10.1016/j.molcel.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pommier Y, Redon C, Rao VA, Seiler JA, Sordet O, Takemura H, Antony S, Meng L, Liao Z, Kohlhagen G, Zhang H, Kohn KW. 2003. Repair of and checkpoint response to topoisomerase I-mediated DNA damage. Mutat Res 532:173–203. doi: 10.1016/j.mrfmmm.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 62.Lundin C, North M, Erixon K, Walters K, Jenssen D, Goldman AS, Helleday T. 2005. Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Res 33:3799–3811. doi: 10.1093/nar/gki681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McCord R, Pierce M, Xie J, Wonkatal S, Mickel C, Vershon AK. 2003. Rfm1, a novel tethering factor required to recruit the Hst1 histone deacetylase for repression of middle sporulation genes. Mol Cell Biol 23:2009–2016. doi: 10.1128/MCB.23.6.2009-2016.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krogan NJ, Dover J, Khorrami S, Greenblatt JF, Schneider J, Johnston M, Shilatifard A. 2002. COMPASS, a histone H3 (lysine 4) methyltransferase required for telomeric silencing of gene expression. J Biol Chem 277:10753–10755. doi: 10.1074/jbc.C200023200. [DOI] [PubMed] [Google Scholar]
- 65.Shilatifard A. 2012. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem 81:65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schneider J, Wood A, Lee JS, Schuster R, Dueker J, Maguire C, Swanson SK, Florens L, Washburn MP, Shilatifard A. 2005. Molecular regulation of histone H3 trimethylation by COMPASS and the regulation of gene expression. Mol Cell 19:849–856. doi: 10.1016/j.molcel.2005.07.024. [DOI] [PubMed] [Google Scholar]
- 67.Dehé PM, Dichtl B, Schaft D, Roguev A, Pamblanco M, Lebrun R, Rodriguez-Gil A, Mkandawire M, Landsberg K, Shevchenko A, Shevchenko A, Rosaleny LE, Tordera V, Chavez S, Stewart AF, Geli V. 2006. Protein interactions within the Set1 complex and their roles in the regulation of histone 3 lysine 4 methylation. J Biol Chem 281:35404–35412. doi: 10.1074/jbc.M603099200. [DOI] [PubMed] [Google Scholar]
- 68.Margaritis T, Oreal V, Brabers N, Maestroni L, Vitaliano-Prunier A, Benschop JJ, van HS, van LD, Dargemont C, Geli V, Holstege FC. 2012. Two distinct repressive mechanisms for histone 3 lysine 4 methylation through promoting 3′-end antisense transcription. PLoS Genet 8:e1002952. doi: 10.1371/journal.pgen.1002952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shilatifard A. 2008. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr Opin Cell Biol 20:341–348. doi: 10.1016/j.ceb.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carrozza MJ, Florens L, Swanson SK, Shia WJ, Anderson S, Yates J, Washburn MP, Workman JL. 2005. Stable incorporation of sequence specific repressors Ash1 and Ume6 into the Rpd3L complex. Biochim Biophys Acta 1731:77–87. doi: 10.1016/j.bbaexp.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 71.Tkach JM, Yimit A, Lee AY, Riffle M, Costanzo M, Jaschob D, Hendry JA, Ou J, Moffat J, Boone C, Davis TN, Nislow C, Brown GW. 2012. Dissecting DNA damage response pathways by analysing protein localization and abundance changes during DNA replication stress. Nat Cell Biol 14:966–976. doi: 10.1038/ncb2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Libuda DE, Winston F. 2010. Alterations in DNA replication and histone levels promote histone gene amplification in Saccharomyces cerevisiae. Genetics 184:985–997. doi: 10.1534/genetics.109.113662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.