

ABSTRACT
The c-Fos and c-Jun transcription factors, members of the activator protein 1 (AP-1) complex, form heterodimers and bind to DNA via a basic leucine zipper and regulate the cell cycle, apoptosis, differentiation, etc. Purified c-Jun leucine zipper fragments could also form stable homodimers, whereas c-Fos leucine zipper homodimers were found to be much less stable in earlier in vitro studies. The importance of c-Fos overexpression in tumors and the controversy in the literature concerning c-Fos homodimerization prompted us to investigate Fos homodimerization. Förster resonance energy transfer (FRET) and molecular brightness analysis of fluorescence correlation spectroscopy data from live HeLa cells transfected with fluorescent-protein-tagged c-Fos indicated that c-Fos formed homodimers. We developed a method to determine the absolute concentrations of transfected and endogenous c-Fos and c-Jun, which allowed us to determine dissociation constants of c-Fos homodimers (Kd = 6.7 ± 1.7 μM) and c-Fos–c-Jun heterodimers (on the order of 10 to 100 nM) from FRET titrations. Imaging fluorescence cross-correlation spectroscopy (SPIM-FCCS) and molecular dynamics modeling confirmed that c-Fos homodimers were stably associated and could bind to the chromatin. Our results establish c-Fos homodimers as a novel form of the AP-1 complex that may be an autonomous transcription factor in c-Fos-overexpressing tissues and could contribute to tumor development.
INTRODUCTION
Activator protein 1 (AP-1) is a transcriptional regulator composed of members of the Fos, Jun, and ATF families of DNA-binding proteins (1, 2). c-Fos and c-Jun regulate a variety of processes, including proliferation, differentiation, apoptosis, and oncogenesis (3). They function as dimers binding to the promoter/enhancer regions of numerous mammalian genes (4). Their DNA-binding domain is composed of a leucine zipper promoting dimerization and a basic region that binds with high affinity to a specific 8-bp-long DNA sequence (5, 6).
In addition to forming stable heterodimers with c-Fos (7–9), c-Jun can also homodimerize, as revealed in vitro by electrophoretic mobility shift assay (EMSA) (8), and bind to DNA as a homodimer, although with lower affinity than the heterodimer (8, 10). In contrast, the c-Fos homodimer was found to be unstable in vitro, and thus, c-Fos has been thought to interact with DNA only by forming heterodimers with c-Jun (9, 11, 12). The instability of the c-Fos dimer is thought to be due to repulsion between its negatively charged residues in the leucine zipper (6). The wild-type c-Fos zipper showed no homoassociation at a concentration of 0.1 μM according to EMSA (13). O'Shea and coworkers estimated the Kd of the c-Fos leucine zipper homodimer to be 3.2 and 5.6 μM at 0 and 25°C, implying that the failure of others to detect c-Fos dimerization was probably due to low protein concentrations (14). It was shown by EMSA that a single amino acid change in the leucine zipper is sufficient to allow a truncated c-Fos protein to homodimerize and bind to its DNA response element (15). Melting temperature analyses of different leucine zipper dimers revealed that thermal stability increases from c-Fos–c-Fos through c-Fos–c-Jun to c-Jun–c-Jun (16).
c-Fos expression and activation can be induced by growth factors, cytokines, or neurotransmitters via G-protein-coupled receptor-, mitogen-activated protein kinase-, cyclic AMP-, or Ca2+-dependent signaling pathways (17–19). c-Fos overexpression occurs in several pathological conditions, which can have both proliferative and antiproliferative effects. c-Fos was overexpressed in some tamoxifen-resistant human breast tumors (20) and highly overexpressed in malignant oral tissues (21). It could also contribute to hepatocarcinogenesis (22). In a murine skin carcinogenesis model, c-Fos was shown to be required for malignant tumor conversion (23). c-Fos can be upregulated via the thyroid hormone nuclear receptor α1, which is a tumor inducer in intestinal tumorigenesis (24). Conversely, c-Fos overexpression inhibited cell cycle progression and stimulated cell death in hepatocytes (25). It also activated apoptosis in colorectal carcinoma cells in a p53-dependent manner (26).
Because c-Fos, but not c-Jun, is overexpressed in many different types of tumors, we were interested whether c-Fos at higher concentrations could form stable homodimers and bind to DNA in live cells. Förster resonance energy transfer (FRET) can be used to assess distances between two fluorophores in the range of 2 to 10 nm (27, 28), whereas fluorescence cross-correlation spectroscopy (FCCS) can demonstrate the comobility of two molecules (29–31). Using these methods, we previously demonstrated heterodimerization and chromatin binding of c-Fos and c-Jun and described the conformation of their complex in live cells (7, 32). It was shown in our lab (German Cancer Research Center) by imaging FCCS that mobility and protein-protein interaction maps of c-Fos and c-Jun were correlated (33).
Here we performed FRET measurements of fluorescent protein-tagged c-Fos molecules by confocal microscopy and flow cytometry to examine whether c-Fos could form homodimers. We developed a method combining fluorescence correlation spectroscopy (FCS) and immunofluorescence to assess the concentrations of both fluorescently labeled and unlabeled endogenous c-Fos and c-Jun in cells. This allowed us to determine the Kd of c-Fos homodimers and c-Fos–c-Jun heterodimers in live HeLa cells by FRET titrations. We found that the Kd of the c-Fos homodimer is more than 1 order of magnitude higher than that of the heterodimer. To our knowledge, this is the first report on the determination of the Kd of transcription factors from FRET titrations in live cells. Imaging FCCS measurements revealed codiffusion of stable c-Fos homodimers and their binding to chromatin. Our molecular dynamics (MD) simulations support the notion that Fos homodimers can form, bind to DNA, and remain stable over the time span of the simulation (500 ns). This novel homodimeric form of c-Fos may act as an autonomous transcriptional regulator.
MATERIALS AND METHODS
Cell culture, plasmid construction, and transfection of HeLa cells.
Cell culture, plasmid construction, and transfection have been described elsewhere (34). For detailed information on these procedures and the plasmids and proteins used in this study, see the supplemental material and Fig. 1.
FIG 1.
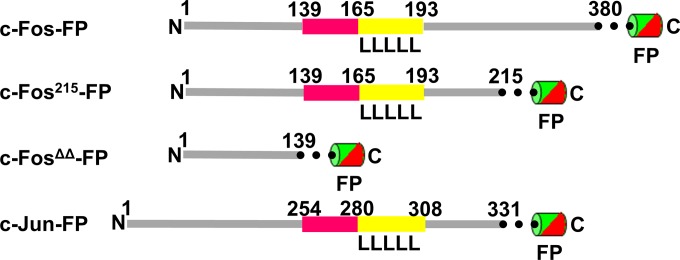
Schematic drawing of c-Fos, its mutant forms, and c-Jun. From the top: full-length Fos with fluorescent protein tag at the C terminus, C-terminally truncated Fos215 and FosΔΔ where the DNA-binding and dimerization domains were deleted, and Jun. Pink denotes the DNA-binding domain, yellow denotes the leucine zipper, and the dotted line denotes the linker between Fos/Jun and the fluorescent-protein tag (ECFP, EYFP, EGFP, or mRFP1).
Confocal microscopic and flow cytometric FRET analyses.
Confocal microscopic images were collected with a Zeiss LSM 510 microscope. Flow cytometric measurements were carried out with a Becton Dickinson FACSAria III instrument. For details of data acquisition and FRET analysis on a pixel-by-pixel or cell-by-cell basis, which have been described earlier (34), see the supplemental material.
FCS and calibration of fluorescence intensity as a measure of absolute concentration.
In FCS (35), molecules diffuse across a subfemtoliter (<1 μm3) detection volume defined by a focused laser beam. This causes fluorescence fluctuations, which are analyzed to derive dynamic parameters of the molecules studied. FCS measurements were performed with a modified Olympus FluoView 1000 confocal microscope mounted on an inverted IX-81 stand with an UPlanAPO 60× numerical aperture 1.2 water immersion objective. The FCS extension (Steinbeis Transfer Unit for Biophysical Analytics, Heidelberg, Germany) equipped with two avalanche photodiodes (Perkin-Elmer, Wellesley, MA) is attached to the confocal scanning unit. Fluorescence of enhanced green fluorescent protein (EGFP) was excited by the 488-nm line of an Ar ion laser and detected at 500 to 550 nm. To allow FCS measurements at high EGFP concentrations (up to 25 μM), laser illumination was dimmed by a neutral-density filter (optical density of 1) to yield P = 0.2 μW at the sample. FCS measurements of live HeLa cells were performed with eight-well chambered cover glass plates (Nunc). Points for FCS measurements were selected from confocal images. In each sample, ∼30 cells were measured at room temperature and six 8-s runs per cell were recorded. Fluorescence autocorrelation functions (ACFs) were calculated online by an ALV-5000E hardware correlator card (ALV Laser, Langen, Germany). Autocorrelation curves were fitted to a two-component three-dimensional (3D) diffusion model with triplet correction by the program QuickFit 3.0 (36) as follows:
| (1) |
where τ is the lag time, T denotes the triplet fraction, τtr is the triplet correlation time, τ1 and τ2 are the diffusion times of the fast and slow species (average dwell times of molecules in the detection volume), r1 and r2 = 1 − r1 are the fractional amplitudes of the two components, N is the average number of molecules in the detection volume, and S is the aspect ratio of the ellipsoidal detection volume. To assess the aggregation state of EGFP-labeled proteins, the molecular brightness or fluorescence per particle, F/N, was calculated and compared with that of monomeric EGFP.
To facilitate Kd determinations, we developed a method to assess absolute concentrations from fluorescence intensity similar to that described in reference 37. In the first step, we determined the detection volume, Veff, of the microscope by using a 130 nM Alexa Fluor 488 solution as a standard. From its ACF, τd and S were determined by fitting and the lateral radius, ωxy, and the axial radius, ωz, were calculated with the following equation:
| (2) |
where τd is the measured diffusion time of the dye, which measures mobility and is inversely proportional to the diffusion coefficient and D = 435 μm2/s is the diffusion coefficient of Alexa Fluor 488 at 22.5°C (38). The effective detection volume is calculated with the following equation:
| (3) |
From autocorrelation curves with EGFP, the particle numbers, N, were determined. From these, molar concentrations, c, were calculated as follows:
| (4) |
where NA is Avogadro's number. Before every FCS measurement, the fluorescence intensity, F, was measured with the imaging detector (photomultiplier tube) of the confocal microscope at the site of FCS measurement; thus, c-versus-F calibration lines were generated (see Fig. 6B).
FIG 6.

FCS-based concentration calibration and brightness analysis. (A) The EGFP concentration in HeLa cells was determined from the amplitude of the ACF. The curve was fitted to a two-component free-diffusion model with triplet correction. (B) Diffusing particle concentration (1/G0) as a function of the fluorescence intensity per pixel of EGFP, FosΔΔ-EGFP, Fos-EGFP, and Fos-EGFP coexpressed with Jun-mRFP1. Data were fitted with straight lines by Deming regression. (C) Fluorescence per particle or molecular brightness values characterizing the aggregation state plotted as a function of the concentration of the EGFP tag. Symbols are the same as in panel B. (D) Normalized ACFs fitted to a two-component free-diffusion model. (E) Diffusion constants and fractions of the second, slow component derived from the fits (n, number of cells).
To facilitate the comparison of measurements on different days, we used fluorescent beads. The fluorescence intensity of 6-μm green calibration beads (beads with 1% relative intensity from the InSpeck green microscope image intensity calibration kit [Molecular Probes, Life Technologies, Inc.]) was used to normalize EGFP fluorescence. Confocal sections in the equatorial plane of the beads were recorded on the same day as the FCS calibration with identical instrument settings. The average fluorescence intensity per pixel in the central area of the beads was obtained. With the calibration curve (see Fig. 6B, triangles) the local intensity of the bead at its center corresponded to an EGFP concentration of = ∼15.4 ± 0.7 μM (average ± standard error of the mean [SEM] of five experiments).
We could also transfer the concentration calibration to flow cytometric measurements. In the microscopic calibration described above, the local fluorescence intensities at a pixel of the sample and the bead are compared. In contrast, in flow cytometry, the total intensity of the whole cell and the bead is measured. Therefore, the ratio of the cellular and bead volumes had to be taken into account. The volume of HeLa nuclei (where Fos and Jun are localized) is ∼13.6 times larger than that of the beads, as determined by confocal microscopic 3D sectioning and the Imaris software (Bitplane AG, Zurich, Switzerland). In addition, the different detection efficiencies of the spectra of the bead and EGFP arising from different band-pass filters in the flow cytometer and the confocal microscope resulted in a factor of 13. Taking these factors into account, the total intensity of a bead corresponded to an EGFP concentration of = ∼1.2 μM (distributed in a HeLa cell nucleus) in flow cytometric experiments. If the localization of the protein is not perfectly nuclear, we can correct for this as well. From confocal microscopic sectioning, we determined the nuclear and cytoplasmic fractions of Fos215-EGFP (the protein we used for FRET titrations), which were 85% ± 5% and 15% ± 5%, independent of the expression level (see Fig. S3 in the supplemental material). With this correction, a bead unit of ∼1.0 ± 0.1 μM was used to calculate the nuclear concentration of Fos215-EGFP in flow cytometric experiments.
Determination of the absolute concentration of endogenously expressed Fos and Jun.
With regular immunofluorescence assays, only the relative amounts of endogenous and transfected proteins can be assessed. By knowing the absolute concentration of the transfected proteins, the endogenous concentration can also be determined. Therefore, we combined the immunofluorescence assay with the results of FCS-based EGFP concentration calibration to assess the endogenous concentrations of Fos and Jun in HeLa cells.
Immunofluorescence labeling was carried out as follows. After being washed three times with PBS, cells were fixed with 3.7% formaldehyde (4°C, 10 min), permeabilized with 0.25% Triton and 0.1% Tween–Tris-buffered saline (TBS) at room temperature for 30 min, and blocked with 2% bovine serum albumin with 0.1% Tween–TBS at room temperature for 30 min. Cells were incubated with a mouse anti-c-Fos (Merck, Whitehouse Station, NJ) or anti-c-Jun (Millipore, Billerica, MA) monoclonal antibody at 20 μg/ml for 1 h at room temperature and then incubated with the secondary polyclonal antibody NL-637-DAMIG (R&D Systems, Minneapolis, MN) at 50 μg/ml for 1 h at room temperature in the dark. Between consecutive steps, cells were washed three times with PBS. Titration of antibodies showed that these concentrations were close to saturating values (see Fig. S4A to C in the supplemental material).
Flow cytometric measurements were performed on a FACSAria III flow cytometer. The green EGFP signal (Igreen) was excited at 488 nm, and emission was detected at 515 to 545 nm, while the red NL637 signal (Ired) was exited at 633 nm and emission was detected through a 655-nm low-pass filter.
The amount of Fos-EGFP was determined by comparing its Igreen fluorescence signal to that of the green bead used for concentration calibration. The red signal, Ired, of the NL637-DAMIG antibody used for immunofluorescence labeling is proportional to the total amount of Fos, i.e., the endogenous Fos in the nontransfected sample and the endogenous Fos plus Fos-EGFP in the transfected one. The concentrations of endogenous Fos (cFos-endogenous) and Fos-EGFP (cFos-EGFP) were calculated from the green and red signals with transfected and nontransfected cells as follows:
| (5) |
| (6) |
| (7) |
with = ∼1.2 μM in our measurements. Ired signals were collected from fixed cells (required by the immunofluorescence labeling protocol), whereas Igreen signals were measured in nonfixed live cells (from the same transfected population) to avoid deterioration of EGFP fluorescence because of fixation. The concentrations of endogenous and EGFP-tagged Jun were determined by the same principle.
Calculation of dissociation equilibria from FRET data.
To assess the Kds of Fos homodimers and Fos-Jun heterodimers, we carried out FRET titration experiments. In the derivation of dissociation equilibria, we assumed that the heterodimer was more stable than the Fos homodimer. Therefore, in the case of Fos-Jun association, we neglected the presence of Fos homodimers (assuming that they were not present at lower concentrations). The law of mass action for heterodimer formation is calculated with the following equation:
| (8) |
where brackets denote concentrations of free monomers and heterodimers and is the dissociation constant of the heterodimeric complex. The total concentration of Fos or Jun can be written as the sum of the concentrations of free monomers F and J and heterodimers FJ as follows:
| (9) |
The concentration of the heterodimer is calculated with the following equation:
| (10) |
The measured FRET efficiency of EGFP- and mRFP1-labeled proteins is an average value stemming from FRET-producing and non-FRET-producing donor molecules. Free donors or donors associated with endogenous unlabeled protein give zero FRET. Only donors forming a complex with an acceptor make a positive contribution to FRET (Fig. 2). Thus, we need to calculate the concentration of Fos-Jun dimers labeled with both a donor and an acceptor. The total concentrations of Fos and Jun are calculated as follows:
| (11) |
where the indexes D, A, and e refer to donor-tagged, acceptor-tagged, and endogenous molecules. The fractions of donor-tagged Fos (pD) and acceptor-tagged Jun (pA) are calculated as follows:
| (12) |
The concentration of doubly labeled FosD-JunA dimers is calculated as follows:
| (13) |
We denote the FRET efficiency in the complex of a single donor-tagged Fos and an acceptor-tagged Jun by E0. The measured apparent FRET efficiency, Emeas, can be written as follows:
| (14) |
where [FDJA] is the concentration of complexes of donor-tagged Fos with acceptor-tagged Jun, and [FD]t is the total concentration of donor-tagged Fos (without respect to being monomeric or in a Fos-Jun complex). [FD]t − [FDJA] is the concentration of donor-tagged Fos not complexed with acceptor-tagged Jun (Fos-EGFP in monomeric form or complexed with endogenous Jun), contributing zero FRET efficiency. By introducing the acceptor-to-donor expression ratio NA/ND = [JA]/[FD] (see equation S8 in the supplemental material) and combining equations 10, 13, and 14, the measured FRET efficiency can be expressed as follows:
| (15) |
By substituting equation 11 and the expression for the NA/ND ratio into equation 15, we get the formula used in the nonlinear fit (see equation S12 in the supplemental material) with variables [FD]t and NA/ND.
FIG 2.

Possible combinations of fluorescently tagged and endogenous Fos and Jun. (A) In the monomer-heterodimer equilibrium, fluorescently tagged and endogenous, unlabeled Fos and Jun molecules participate. The three species containing a donor tag contribute to the measured value of FRET efficiency, Emeas, i.e., the doubly labeled heterodimer having a FRET efficiency of E0 and donor-labeled Fos in complex with endogenous Jun or present as a monomer; the latter two species are characterized by zero FRET efficiency. The fractions of the different heterodimers follow a multinomial distribution. Emeas is a weighted average of the species-specific E values (see equation S12 in the supplemental material). (B) In the monomer-homodimer equilibrium, donor-tagged, acceptor-tagged, and endogenous Fos and endogenous Jun participate. Four heterodimeric species and the donor-tagged monomer contribute to Emeas (see equation S14 in the supplemental material).
To calculate Fos-Fos equilibria, we have to take Fos-Jun formation into account as well. Since the heterodimer is more stable, we make the simplifying assumption that all of the Jun molecules present are in complex with Fos at the high Fos concentrations where Fos homodimerization takes place, leaving no free Jun. We can write the following equilibrium equations:
| (16) |
where is the dissociation constant of the Fos homodimer and FF and FJ denote the homo- and heterodimer. The amount of Fos homodimer can be expressed as follows:
| (17) |
Fos molecules can be labeled with a donor or an acceptor or can be unlabeled, and only homodimers containing both a donor and an acceptor will produce FRET (Fig. 2). The fraction of such double-labeled pairs follows a multinomial distribution and is calculated as follows:
| (18) |
where pD = [FD]/([FD]t + [FA]t + [Fe]t) and pA = [FA]/([FD]t + [FA]t + [Fe]t) are the donor- and acceptor-tagged fractions of Fos. The measured FRET efficiency equals the following:
| (19) |
Combining equations 17 and 19 (see also equation S14 in the supplemental material) yields the following equation:
| (20) |
with
| (21) |
where [F]t is the total Fos concentration (including donor-tagged, acceptor-tagged, and endogenous Fos, without respect to a monomeric or dimeric state) and E0 is the FRET efficiency of a donor-acceptor pair (this may be different from the E0 of the heterodimer). These equations were used to determine the Kds of dimers from flow cytometric FRET experiments by nonlinear regression using Prism (GraphPad Software, Inc). In the analysis we also considered the presence of dark acceptor species due to imperfect maturation and a cytoplasmic fraction of Fos (see the supplemental material). These factors influence E0 but not the value of Kd.
SPIM-FCCS.
Single-plane illumination (light sheet) microscopy-based fluorescence cross-correlation spectroscopy (SPIM-FCCS) measurements were performed with an in-house-built SPIM setup based on the design described in references 39 and 40. Data were analyzed with the software QuickFit 3.0. For details of the experimental setup and analysis, see the supplemental material.
MD simulation of Fos-Jun and Fos-Fos complexes.
Two systems were submitted to MD simulation. The first one was constructed from the Fos (139-198)–Jun (257-313) protein fragments associated with the DNA fragment as deposited in the Protein Data Bank (41). It was completed by adding the missing hydrogen atoms and closing the N- and C-terminal residues by the acetyl and N-methyl groups, respectively. The second system, Fos (139-198)–Fos (139-198), was obtained from the first one by using the Jun fragment as a template in a proper position for homology modeling of the Fos protein. Each of these systems was put into a dodecahedral box, solvated by the TIP3P explicit water model, and neutralized by Na+ ions, and further, Na+ and Cl− ions were added to set the ionic strength to 0.15 M. They were then minimized, slowly heated to 310 K, and after an 80-ns equilibration period, submitted to 500-ns constant particle number (123,888 and 123,870 for the Fos-Jun and Fos-Fos systems, respectively), constant-pressure (105 Pa), constant-temperature (310 K) production dynamics. For the simulations, the AMBER99SB force field (42) and periodic boundary condition were used. Short-range electrostatic and van der Waals interactions were calculated explicitly within a 1-nm cutoff. For the long-range electrostatic interactions, the particle mesh Ewald method (43) was applied. A Berendsen barostat and thermostat (44) were used during this simulation. For the simulations, the GROMACS packages were used (45, 46).
As controls, MD simulations of the Leu zipper region only of the c-Fos–c-Jun and c-Fos–c-Fos dimeric structures were also carried out with the setup protocol detailed above. The Leu zipper region we considered consisted of amino acid residues 275 to 313 and 160 to 198 of the c-Jun and c-Fos fragments, respectively. Simulations were completed for both the wild-type Leu zipper regions and the corresponding structures by applying Leu280Asp and Leu294Asp virtual mutations in c-Jun protein fragments and Leu165Asp and Leu179Asp mutations in c-Fos protein fragments.
RESULTS
FRET microscopy implies Fos homodimerization.
FRET is the radiationless transfer of energy from a donor fluorophore to a nearby acceptor (27, 28), which is often used to assess molecular distances. To measure the association of c-Fos molecules (referred to as Fos in the following sections), we used Fos and the C-terminally truncated mutant form Fos215 tagged with enhanced cyan fluorescent protein (ECFP) (donor) or enhanced yellow fluorescent protein (EYFP) (acceptor) in confocal microscopic FRET experiments. Fos215 was prepared (32) to bring the fluorescent-protein-labeled C termini of Fos and Jun to similar distances from the leucine zipper to enhance FRET (Fig. 1). Images of donor, transfer, and acceptor signals were recorded, and FRET efficiencies (E values) of labeled proteins, as well as acceptor-to-donor molecular (NA/ND) ratios were calculated on a pixel-by-pixel or cell-by-cell basis.
Pixel-by-pixel FRET efficiency maps and histograms of representative cells are shown in Fig. 3. Cells cotransfected with full-length Fos-ECFP–Fos-EYFP (top row) yielded a mean FRET efficiency of E = 5.0% ± 0.5% (SEM; n ≈ 30 cells); for Fos215-ECFP–Fos215-EYFP (second row; n ≈ 30), it was 10.0% ± 0.5%. The higher E value of the truncated Fos215 molecules is probably due to the reduced distance between the shorter C-terminal regions (Fig. 1). We also measured the FRET efficiencies of the Fos-ECFP–Jun-EYFP and Fos215-ECFP–Jun-EYFP samples (third and fourth rows; n ≈ 30), which were 7.9% ± 0.4% and 15.0% ± 1.1%. For these pairs, we have shown heterodimer formation earlier by FCCS and FRET (7, 32, 34). Both Fos and Jun molecules showed strong nuclear enrichment. The negative control (ECFP and EYFP coexpressed as separate proteins) and the positive control (ECFP-EYFP fusion protein) had mean E values of 2.8% ± 0.4% and 48.6% ± 0.8% (fifth and sixth rows; n ≈ 20). These proteins had a diffuse distribution in the whole cell. The FRET efficiency of the Fos-Fos (or Fos215-Fos215) samples was lower than that of the Fos-Jun and Fos215-Jun heterodimers but significantly higher than that of the negative control, indicating that Fos formed homodimers in these cells.
FIG 3.

Subcellular pixel-by-pixel analysis of dimerization by confocal microscopic FRET on HeLa cells. ECFP (donor channel) was excited at 458 nm and detected at 475 to 525 nm. In the transfer channel, excitation was at 458 nm and detection was at 530 to 600 nm. EYFP (acceptor channel) was excited at 514 nm and detected at 530 to 600 nm. Full-length Fos-ECFP–Fos-EYFP (top row), Fos215-ECFP–Fos215-EYFP (second row), Fos-ECFP–Jun-EYFP (third row), and Fos215-ECFP–Jun-EYFP (fourth row) showed nuclear localization. The negative control, ECFP and EYFP expressed independently, and the positive control, the ECFP-EYFP fusion protein (fifth and sixth rows), were evenly distributed in the whole cell. FRET efficiency (E) was calculated in each pixel. Histograms show the statistics of the subcellular distribution of E.
Dimer formation depends on the concentrations of the interacting partners; therefore, we analyzed FRET in cells expressing various amounts of the proteins. We calculated average intensities in the whole nucleus for Fos or Jun and in the whole cell for the positive and negative controls in single cells and determined FRET on a cell-by-cell basis. This method allowed a rapid analysis of several hundred cells. Figure 4A and B show the mean E value as a function of the acceptor-to-donor molecular NA/ND ratio for the Fos215-ECFP–Fos215-EYFP and Fos-ECFP–Fos-EYFP samples. Data were grouped into three subsets according to donor concentrations based on the fluorescence intensity of the donor (low, medium, high). For both protein pairs, E increases from low to high donor concentrations, in accordance with the higher probability of complex formation. E also increases with increasing NA/ND ratios because more acceptor-tagged Fos molecules are available for donor-tagged ones to form a complex. The plateau or, in its absence, the average of the highest E values is presented in Fig. 4C, which shows that at higher Fos concentrations, the extent of homodimerization increased as expected. Similar to the pixel-by-pixel analysis, the mean FRET efficiencies of the Fos-Fos and Fos215-Fos215 dimers are between those of the negative control and the respective Fos-Jun or Fos215-Jun dimers.
FIG 4.

Cell-by-cell analysis of dimerization by confocal microscopic FRET. (A, B) FRET efficiencies of donor (ECFP)- and acceptor (EYFP)-tagged Fos215 or full-length Fos molecules as a function of the acceptor-to-donor molecular ratio (NA/ND). Data from 300 cells were grouped into three subsets as a function of donor intensity (low, <800; medium, 800 to 1,200; high, >1,200 [arbitrary units]). Cellular data were binned in 0.25-wide intervals of the NA/ND values to reduce data scatter. FRET efficiencies increased with increasing NA/ND ratios. (C) Saturation values of FRET efficiencies at high NA/ND ratios (>0.95). ECFP-EYFP fusion protein served as a positive control, and independently expressed ECFP and EYFP served as a negative control. The FRET data of the Fos-Jun and Fos215-Jun pairs were previously published in reference 34.
The measured E values depend on the FRET efficiency E0 in a single donor-acceptor complex determined by the dye-to-dye distance and orientation and on the fraction of donors forming dimers with an acceptor. The length of the Fos215 molecule downstream of the dimerization domain is similar to that of Jun; thus, the dye-to-dye distances in the Fos215-Fos215 and Fos215-Jun complexes should be similar. However, in the case of Fos homodimers, only complexes of donor- and acceptor-tagged proteins yield FRET (Fig. 2). The measured mean E value is a weighted average of non-FRET-ting and FRET-ting dimers. Thus, the mean FRET efficiency of the homodimer is expected to be lower than that of the heterodimer. This was taken into account in our subsequent analyses.
Calibration of fluorescence intensity to measure absolute EGFP concentrations.
The above-described FRET titration curves demonstrated that FRET efficiency can be used to monitor the extent of homo- and heteroassociations quantitatively in our system. The stability of a complex is characterized by its dissociation constant, Kd. In Materials and Methods, we outlined a method to determine the Kd of interacting proteins in live cells from FRET titration curves. This requires knowledge of the absolute concentrations of all of the interacting molecules, i.e., the transfected fluorescent and endogenous nonfluorescent ones. In subsequent measurements, we used the EGFP-mRFP1 dye pair because of the higher photostability of EGFP than ECFP. First, we developed a calibration method to determine the concentration of fluorescent proteins. Confocal images of cells expressing free EGFP were taken, and ACF curves were recorded at selected points of the images (see Fig. 6A). From ACF curves, local dye concentrations were determined by nonlinear fitting, yielding a calibration curve of EGFP concentration versus fluorescence intensity per pixel, c(F) (see Fig. 6B) (37). To make the concentration calibration portable and facilitate the comparison of measurements on different days, we normalized EGFP fluorescence by using a fluorescent bead as an intensity standard. The EGFP concentration corresponding to one bead unit was ∼15.4 ± 0.7 μM for our confocal microscope and 1.0 ± 0.1 μM for the flow cytometer. For the latter calculation, we took the nucleus-to-bead volume ratio (13:1) and the nuclear fraction of Fos215-EGFP (85%) into account. In that way, the molar concentration of EGFP-tagged protein could be assessed by simply comparing its intensity to that of the bead measured on the same day in the microscopic or flow cytometric setup without having to repeat the FCS calibration.
Determination of the absolute concentrations of endogenous and transfected Fos and Jun.
To calculate the Kd of dimers, we also need to know the amount of endogenous Fos and Jun, since they can also form dimers with each other or with their fluorescent counterparts. First, we detected the green fluorescence signal of Fos-EGFP in transfected cells and compared it to that of the calibration bead to determine the absolute concentration of transfected protein (equation 5 in Materials and Methods; see Fig. S4D to G in the supplemental material). Then we used immunofluorescence labeling paired with far red channel flow cytometry to detect the total Fos pool in nontransfected and Fos-EGFP-transfected samples. The immunofluorescence signal of the nontransfected sample is proportional to the endogenous Fos concentration, while that of the transfected one corresponds to the sum of the endogenous and transfected amounts. Thus, using the known concentration of Fos-EGFP, we deduced the average concentrations of endogenous Fos (113 ± 11 nM) and Jun (94 ± 10 nM) in HeLa cells (equation 7 in Materials and Methods). Our procedure combining immunofluorescence and EGFP-tagged protein expression can be generally used to assess the absolute concentration of any endogenously expressed nonfluorescent protein.
Determination of the dissociation constant of Fos-Jun heterodimers and Fos homodimers in live cells with flow cytometric FRET data.
We wanted to determine the dissociation constants of homo- and heterodimers from FRET titrations. Therefore, we derived the expressions of FRET efficiency in terms of the concentrations of donor-tagged, acceptor-tagged, and unlabeled endogenous proteins of interest and the Kds (equations 15, 20, and 21; see equations S12 and S14 in the supplemental material). Flow cytometric FRET experiments were carried out with large cell populations expressing EGFP- and mRFP1-tagged proteins at various concentrations. Cell-by-cell FRET efficiency (E) values, donor concentrations [FD]t (derived from the FRET-corrected donor intensity [ID] by comparison to beads), and acceptor-to-donor molecular ratios (NA/ND) were determined. To characterize heterodimers, cells were cotransfected with Fos215-EGFP–Jun-mRFP1 or Jun-EGFP–Fos215-mRFP1 pairs. The extent of association depends on the concentrations of both molecules. To create functions with a single variable, cells were grouped into classes with approximately constant NA/ND ratios. E values of selected NA/ND ratio groups were plotted as a function of the donor concentration (Fig. 5A and C). The FRET efficiency increases with increasing NA/ND ratios, as expected. Figure 5B and D display the theoretical values of the maximal FRET efficiencies at various NA/ND ratios (see equation S13 in the supplemental material), which could be measured if all possible donor-acceptor complexes were formed (and association were complete). The function increases linearly up to NA/ND ratio of 1, where it reaches E0, the FRET efficiency of a single donor-acceptor pair, and remains constant at higher NA/ND ratios. E-versus-NA/ND data were fitted to the model function describing the heterodimerization process (see equation S12 in the supplemental material), taking the presence of fluorescent-protein-tagged and unlabeled endogenous Fos and Jun into account (Fig. 2A). The apparent Kd values derived from the fits varied with various NA/ND ratios for both pairs between 10 and 370 nM. We received lower apparent Kds where Fos was present in excess (larger Fos/Jun ratios) (Fig. 5E).
FIG 5.

Determination of the dissociation coefficients of Fos-Jun heterodimers and Fos-Fos homodimers from flow cytometric FRET titrations. (A, C) FRET efficiency measured in cells cotransfected with Fos215-EGFP–Jun-mRFP1 and Jun-EGFP–Fos215-mRFP1 and plotted as a function of the donor-tagged Fos215 or Jun concentration. Data were grouped according to acceptor-to-donor molecular ratios (NA/ND) and fitted as described elsewhere (see equation S12 in the supplemental material) (solid lines), yielding the Kd value of the heterodimers and the FRET efficiency (E0) of individual donor-acceptor pairs. Endogenous Fos and Jun were also taken into account. (B, D) The solid lines represent the maximal theoretically attainable E values at different NA/ND ratios (assuming E0 values of 15 and 14.1% based on the fits) when all available Jun-mRFP1 molecules are engaged in heterodimers with Fos; the marked points correspond to the experimental NA/ND ratios (see equation S13 in the supplemental material). (E) Dependence of the Kd values from the fits on the Fos/Jun ratio. (F) FRET efficiency of Fos215-EGFP–Fos215-mRFP1 homodimers as a function of the donor-tagged Fos215 concentration with Kd and E0 yielded from a linked fit (see equation S14 in the supplemental material). (G) Maximal attainable FRET efficiencies at different NA/ND ratios (assuming an E0 value of 9.47% based on the fit) when all Fos molecules form homodimers (see equation S15 in the supplemental material).
Fos homodimerization was studied by measuring the FRET efficiency of Fos215-EGFP and Fos215-mRFP1 (Fig. 5F). Here, homodimers can contain donor-acceptor-, donor-donor-, and acceptor-acceptor-tagged protein pairs (plus dimers containing endogenous Fos) (Fig. 2B); thus, the dependence of E on the NA/ND ratio is different from that for heterodimerization (Fig. 5F). The theoretical Emax value approximates the value of E0 asymptotically as NA/ND tends to infinity (see equation S15 in the supplemental material). Curves were fitted by taking into account both the homoassociation of Fos (in all combinations of donor-tagged, acceptor-tagged, and endogenous molecules) and its heteroassociation with endogenous Jun (see equation S14 in the supplemental material). In the fits, the Kd and E0 values were linked for data sets with different NA/ND ratios, yielding a Kd of 6.7 ± 1.7 μM and an E0 of 9.5% ± 0.8% for the homodimerization process. When different NA/ND groups were fitted independently, Kd and E0 ranged from 5.4 to 9.7 μM and from 9.1 to 11.9%, respectively. As expected, the Kd of the Jun-Fos215 heterodimer is much lower than that of the Fos homodimer, which means that Fos homodimerization will be significant only in the case of its selective overexpression. At equal Fos and Jun concentrations, the formation of Fos-Jun heterodimers is more probable than the formation of Fos homodimers.
Fluorescence brightness and slow diffusion indicate Fos homoassociation and DNA binding.
FCS was used not only for concentration calibration but also as an additional tool to probe Fos homoassociation. Diffusing particle concentration-versus-fluorescence intensity curves were generated for EGFP, FosΔΔ-EGFP (a mutant form lacking the DNA-binding and dimerization domains, see Fig. 1), full-length Fos-EGFP, and Fos-EGFP–Jun-mRFP1 samples (Fig. 6B). The slopes of the fitted straight lines for EGFP and FosΔΔ-EGFP were 30.0 and 30.3 (nM/intensity unit). The similar slopes indicate that equal intensities of EGFP or FosΔΔ-EGFP correspond to equal particle numbers, suggesting that this mutant form contains one fluorophore per particle; i.e., it is monomeric. In contrast, full-length Fos-EGFP yielded a slope of 11.5, which is less than half of the previous values. Thus, an equal intensity of Fos-EGFP corresponds to a little less than half the particle concentration of the monomeric proteins, implying the formation of Fos homodimers. In fact, this curve is not expected to be linear in the low-concentration range because of the monomer-dimer transition. The slope of the Fos-EGFP–Jun-mRFP1 sample (with an average Jun/Fos ratio of 0.7) is between the monomeric and dimeric slopes. Therefore, Fos-EGFP is partially complexed with Jun-mRFP1, where the brightness of EGFP is similar to that of monomers, whereas the rest of Fos-EGFP may form homodimers. We also analyzed the specific particle brightness, F/N, defined as the ratio of the fluorescence intensity, F, to the number of particles, N. This parameter characterizes the association state of a labeled protein and is proportional to the number of fluorophores in a jointly diffusing complex. F/N-versus-EGFP concentration values are shown in Fig. 6C. Fos-EGFP is brighter than EGFP, FosΔΔ-EGFP, or Fos-EGFP–Jun-mRFP1, corroborating the conclusion that Fos-EGFP is homodimerized when there is not enough Jun present. The brightness of Fos-EGFP increases with its concentration, indicating that dimerization is enhanced at higher concentrations, whereas the brightness of EGFP or FosΔΔ-EGFP does not vary with its concentration, just as expected for monomers.
From the ACF curves, we also determined molecular diffusion properties (Fig. 6D). ACF curves from the EGFP, Fos-EGFP, FosΔΔ-EGFP, and Fos-EGFP–Jun-mRFP1 samples were fitted to a model assuming fast, freely diffusing and slowly moving components (7). Diffusion coefficients and the fractions of the species are shown in (Fig. 6E). The average fraction of the slow components was 0.35 ± 0.14 for Fos-EGFP expressed alone and 0.38 ± 0.10 for Fos-EGFP when coexpressed with Jun-mRFP1, whereas it was only 0.19 ± 0.12 for the nonbinding FosΔΔ-EGFP mutant form and 0.07 ± 0.05 for EGFP. The similarly increased slow fractions of Fos-EGFP expressed alone or together with Jun-mRFP1 suggest that Fos can bind to chromatin not only as a heterodimer but also as a homodimer.
SPIM-FCCS confirms stable homoassociation and chromatin binding of Fos proteins.
We used FCCS, the two-color version of FCS, to characterize the comobility of dimer-forming Fos molecules. In FCCS, the ACF and cross-correlation function (CCF) from two molecular species tagged with different colors are determined. A nonzero CCF amplitude indicates that the molecules are moving together. The ratio of the CCF and ACF amplitudes from a double-labeled sample is proportional to the fraction of molecules forming a complex. FCCS measurements were performed by SPIM with an electron-multiplying charge-coupled device camera as a sensor, which allows simultaneous measurements at many pixels in a cell. This improves the statistics and provides two-dimensional interaction and mobility maps (40).
Measurements were carried out with cells cotransfected with the following protein combinations: Fos215-EGFP–Fos215-mRFP1, Fos215-EGFP–Jun-mRFP1, FosΔΔ-EGFP–Fos215-mRFP1 (negative control), and EGFP-P30-mRFP1 (positive control, EGFP and mRFP1 connected by a 30-residue-long polyproline linker). Cells expressing about equal amounts of the green and red fluorophores were selected from the concentration range used in FRET experiments (0.3 to 10 μM). Figure 7A shows typical correlation curves obtained from the four samples. Amplitudes of the curves were low because of the high concentration required to see Fos homodimerization, but our data showed that quantitative measurements were possible even in this concentration range. At each pixel, we performed a global FCCS fit to the green and red ACF curves and the CCF (40) (see equations S16 to S18 in the supplemental material). The fit functions were parameterized by the concentrations of three diffusing species (green only, red only, green-red [GR] dimers), which were linked over all three curves. We assumed a two-component diffusion model for the ACF curves and a one-component model for the CCF. Diffusion coefficients were not linked. Figure 7B shows examples of the maps and histograms of the relative GR dimer concentration cGR/(cG-only + cR-only + cGR) obtained from these fits. Figure 7C shows the statistics from n >10 cells as the average ± the standard deviation (SD) of the medians extracted from the pixel distributions in each cell. As expected, the negative control had the smallest apparent dimer fraction and the positive control had the largest one, defining the dynamic range of the measurements (0.06 to 0.32). The upper limit is <1 because of the imperfect overlap of the green and red detection volumes, partial photobleaching of the dyes, and imperfect dye maturation/folding, producing green-only and red-only species besides doubly labeled molecules. The Fos-Jun heterodimer showed a large apparent dimer fraction (0.22 ± 0.07), whereas the value from the Fos215 homodimer was slightly smaller (0.16 ± 0.05) but still significantly larger than that of the negative control.
FIG 7.

SPIM-FCCS data analysis showing codiffusion and DNA binding of Fos homodimers. (A) ACFs and CCFs from SPIM-FCCS measurements. Green, EGFP ACF; red, mRFP1 ACF. Solid lines indicate the experimental data, whereas dashed lines are fits assuming two diffusing components (ACF curves) or one component (CCFs). The red horizontal line is the cross talk-corrected red ACF amplitude, and the blue horizontal line is the level of cross-correlation due to cross talk. Cross-correlation above this value is due to the codiffusion of green and red molecules. (B) The first two columns are fluorescence intensity maps of EGFP or mRFP1 from a selected cell. The third column is a map of the fraction of GR dimers among all of the molecules detected [cGR/(cG-only + cR-only + cGR)], determined from the fits, and the histograms show their distributions. (C and D) Average fractions of GR dimers (C) and diffusion coefficients, Dcross (D), from the cross-correlation fits (mean ± SD; n > 20 for each sample). Fits were carried out on a pixel-by-pixel basis, and the median of the respective parameter from each cell was then averaged. ***, P < 0.0001 (t test).
We also analyzed the protein mobility of the GR dimers that could be extracted from the fits of the CCFs. We used a single-component fit; a second component could not be fitted to the CCFs of the Fos-EGFP–Jun-mRFP1 and Fos215-EGFP–Fos215-mRFP1 samples, suggesting the presence of only a single slow species. The CCFs of the negative control could not be fitted reliably because of their very low amplitude. The diffusion coefficient (Dcross) of the EGFP-P30-mRFP1 fusion protein was ∼4.3 μm2/s (the mean value of two components present for this protein), whereas the average diffusion coefficients of the Fos215-Fos215 and Fos-Jun dimers were much lower, ∼0.3 and 0.4 μm2/s (Fig. 7D). The presence of a single slow component for Fos homodimers and Fos-Jun heterodimers indicated that these complexes could bind to slowly moving nuclear components, supposedly the chromatin.
Fos-Jun and Fos-Fos complexes form stable dimers, as revealed by MD modeling.
On the basis of the distance-related data from FRET measurements, we performed MD modeling to verify the stable formation of Fos homodimers. In Fig. 8, the first frames from the production dynamic trajectories are presented. During the 500-ns simulation interval (see Movies S5 and S6 in the supplemental material), not only the Fos-Jun heterodimer but also the Fos-Fos homodimer remained bound to DNA and the dimeric structures (coiled coil motifs) stayed strongly associated by their leucine zipper regions. It is noteworthy that the Fos-Fos homodimer exists despite the net negative charge of the leucine zipper regions. This indicates that in the dimeric form, hydrophobic interactions play a crucial role and electrostatic forces are largely shielded by counterions. While H bonds and even salt bridges can contribute to the stabilization of the dimeric structure as well, the H-bond networks between protein chains have variability in their connection patterns, which further supports the importance of hydrophobic interactions in the dimeric structures.
FIG 8.

Both Fos-Jun and Fos-Fos complexes form stable dimers and bind to DNA. (A and B) MD simulations were carried out on small Fos-Jun (A) and Fos-Fos (B) fragments bound to the DNA fragment. Ribbon representation (colored cyan) was applied for the helical secondary structure of the Fos protein fragment (A, B). The atomic details of constituent residues are shown by stick representation with the C, H, N, O, and S atoms in gray, white, blue, red, and yellow, respectively. For the Jun fragment (A) or the second Fos fragment (B), a solvent-excluded surface representation was applied by using the above-described color codes. (C to F) Visual representation of trajectories from MD simulations of the Leu zipper region of the Jun-Fos (C, E) and Fos-Fos (D, F) dimeric structures. Wild-type protein fragments (C, D) and virtually mutated (Leu280Asp and Leu294Asp in c-Jun and Leu165Asp and Leu179Asp in c-Fos) fragments (E, F) were considered. Mutant residues are shown by stick representation with the color scheme of the atoms as above. Jun is represented by the orange helix, and Fos is represented by the green and yellow helixes. From each 500-ns dynamic trajectory, 100 frames were saved equidistantly and superimposed (after removal of rotation and translation). Wild-type protein fragments (C, D) demonstrate stable coiled-coil motifs with relatively small fluctuations. The mutations in the Fos-Jun fragment (E) resulted in a somewhat distorted structure and larger fluctuations, indicating weakening of the interaction between the monomers. This is even more strongly expressed in the mutant dimeric Fos-Fos fragment (F), where the hydrophobic interaction between regions affected by the mutations is completely destroyed.
Simulations carried out solely for the leucine zipper region of c-Jun–c-Fos and c-Fos–c-Fos dimers indicated stable structures with coiled-coil motifs (Fig. 8C and D). These results are both in good accordance with former simulations carried out for the c-Jun–c-Fos leucine zipper region (47) and underline again the role of hydrophobic forces even in the stability of the c-Fos–c-Fos homodimer. However, when Leu-Asp virtual mutations were introduced into these dimeric structures (as a negative control), the contact between the corresponding regions of helices was either weakened (c-Jun–c-Fos) or even destroyed (c-Fos–c-Fos), as demonstrated in Fig. 8E and F.
DISCUSSION
Homodimer formation of short fragments, mainly the leucine zippers, of Fos proteins has been studied earlier. However, in vitro studies reported low stabilities of the homodimer, and it was assumed that it could not be present in live cells. By combining FRET, FCS, and imaging FCCS, we demonstrated that Fos proteins formed homodimers in live cells and presented a method of calculating their dissociation constant. The Kd of Fos homodimers in HeLa cells was 6.7 ± 1.7 μM, which is on the same order of magnitude as the value of 5.6 μM determined for its isolated leucine zippers in vitro by circular dichroism (14). Values reported for the heterodimers of the isolated leucine zippers (10, 48, 49) or longer polypeptides (50) in vitro varied between 1 and 140 nM. For the Fos-Jun heterodimer, we found a Kd range of 10 to 370 nM in live cells, which depended on the Fos/Jun ratio and on putting the donor and acceptor tags on one or the other protein. The variation of the Kd with different Fos/Jun ratios may be caused by the formation of Jun homodimers, which could interfere with the heterodimerization process. At lower Fos/Jun ratios, when there is excess Jun present, the relative amount of Jun homodimers is expected to be higher; thus, less free Jun is available and the heterodimerization process could shift toward higher concentrations (Fig. 5E), resulting in a higher apparent Kd. At higher Fos/Jun ratios, where the Jun homodimer is expected to be less abundant, we got a Kd of <100 nM for the heterodimer, in agreement with earlier in vitro results. The shift between the Fos-Jun and Jun-Fos curves in Fig. 5E might be due to dark states (51) and incomplete maturation of mRFP1, resulting in an error in the acceptor-to-donor ratios. Furthermore, the autofluorescence intensity of HeLa cells in the green channel corresponds to the specific intensity of ∼50 nM EGFP; therefore, the signal-to-noise ratio in the concentration range of the Kd is lower than in the case of the homodimer, making the Kd for the heterodimers less accurate.
Several groups have used FRET to determine the Kds of isolated proteins (50, 52). Other groups have used microscopic FRET to determine Kds in cells by utilizing prior estimates of protein copy numbers per cell (53) or applying in vitro concentration calibration with purified proteins (54). Here we present a method to calculate Kd values based on FRET titrations after concentration calibration by FCS, where the whole procedure was carried out with live cells. None of the earlier studies took into account the presence of endogenous, unlabeled proteins. With our method, the absolute concentrations of both overexpressed fluorescent and endogenous nonfluorescent proteins were determined and included in dissociation equilibria. Our concentration calibration method is transferable to measurements performed with different instruments or on different days with fluorescent beads as a standard. The procedure can be generally used to determine Kd values and absolute concentrations of proteins in live cells.
FRET reveals that a certain fraction of molecules is colocalized within Förster distance. We used FCS to assess the codiffusion of molecules, which is a direct indication of stable interaction. Our molecular brightness analysis of FCS data indicated that Fos-EGFP, when expressed alone, had a higher (≥2×) molecular brightness than its dimerization- and DNA-binding-deficient FosΔΔ mutant form or the free EGFP dye. This corroborated that at a concentration of a few micromolar, Fos was present mainly as a homodimer, which was stable at least for a few tens of milliseconds (the mean dwell time of particles in the focal volume setting the upper limit of the time scales observed in our FCS experiments). When fitted with slow and fast diffusion components, the slow fraction of Fos was about the same whether expressed alone or together with Jun; in contrast, the slow fraction of the FosΔΔ mutant form was significantly smaller, hinting at DNA binding of wild-type Fos either as a homodimer or as a heterodimer. The presence of a very small apparent slow fraction in the case of lone EGFP and FosΔΔ is probably due to molecular crowding in the nucleus leading to anomalous subdiffusion (37). This makes the autocorrelation curves less steep than for free diffusion, mimicking the presence of a second, slowly moving component with a longer diffusion time.
SPIM-FCCS allowed us to confirm the presence, visualize the distribution, and characterize the intranuclear mobility of Fos homodimers. These were stable for at least a few hundred milliseconds, the time window defined by the cross-correlation diffusion time. Their diffusion coefficient derived from the cross-correlation curve was ∼0.3 μm2/s, similar to that of Fos-Jun heterodimers (0.4 μm2/s). The measured diffusion coefficients are similar to those determined by confocal FCCS for the same proteins (7) and to values observed for other chromatin-binding proteins, e.g., nuclear receptors (55, 56) or HP1α (57).
Our MD modeling simulations also supported the possibility of homodimerization, showing that homodimers stayed together for the duration of the simulation.
The existence of stable Fos homodimers capable of chromatin binding brings up the possibility that they act as transcriptional regulators and may explain the importance of Fos overexpression in oncogenesis. Various complexes of different Fos and Jun variants occur in different cell types, contributing to cell proliferation or apoptosis (58–60). It is not clear yet whether the Fos homodimer could function as an autonomous transcription factor or, alternatively, occupy the binding sites of AP-1 heterodimers, preventing their normal function and interfering with their proliferative or antiproliferative effects.
ACKNOWLEDGMENTS
We thank Gabriele Müller for preparing the EGFP-P30-mRFP1 construct, László Nagy, Jörg Langowski, and Matt Peloquin for useful comments on the manuscript, and Edina Nagy for excellent technical assistance.
This work was supported by grants from the Hungarian Scientific Research Fund (OTKA K103965 to G.V.); the Magyar Ösztöndíj Bizottság and the Deutscher Akademischer Austauschdienst (MÖB/21-1/2013 to G.V. and K.T.); TÁMOP-4.2.2.A-11/1/KONV-2012-0023 VÉD-ELEM implemented through the New Hungary Development Plan cofinanced by the European Social Fund and the European Regional Development Fund (to G.V.); the European Union and the State of Hungary, cofinanced by the European Social Fund in the framework of the TÁMOP-4.2.4.A/2-11/1-2012-0001 National Excellence Program (to G.V.); and the Deutsche Forschungsgemeinschaft and the Heidelberg Graduate School for Mathematical and Computational Methods for the Sciences (HGS MathComp; GSC 220) in the framework of the German Excellency Initiative (to J.W.K.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00346-15.
[This article was published on 30 September 2015, but Fig. 1 and 2 were erroneously switched with each other and Fig. 5 and 6 were erroneously switched with each other in the full-text (HTML) version; the PDF version was correct. Figures 1 and 2 were properly placed in the full-text version on 20 June 2018; Fig. 5 and 6 were properly placed in the full-text version on 25 June 2018.]
REFERENCES
- 1.Angel P, Karin M. 1991. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta 1072:129–157. [DOI] [PubMed] [Google Scholar]
- 2.Zenz R, Eferl R, Scheinecker C, Redlich K, Smolen J, Schonthaler HB, Kenner L, Tschachler E, Wagner EF. 2008. Activator protein 1 (Fos/Jun) functions in inflammatory bone and skin disease. Arthritis Res Ther 10:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shaulian E, Karin M. 2001. AP-1 in cell proliferation and survival. Oncogene 20:2390–2400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- 4.Curran T, Franza BR Jr. 1988. Fos and jun: the AP-1 connection. Cell 55:395–397. doi: 10.1016/0092-8674(88)90024-4. [DOI] [PubMed] [Google Scholar]
- 5.Hurst HC. 1995. Transcription factors 1: bZIP proteins. Protein Profile 2:101–168. [PubMed] [Google Scholar]
- 6.Porte D, Oertel-Buchheit P, John M, Granger-Schnarr M, Schnarr M. 1997. DNA binding and transactivation properties of Fos variants with homodimerization capacity. Nucleic Acids Res 25:3026–3033. doi: 10.1093/nar/25.15.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baudendistel M, Müller G, Waldeck W, Angel P, Langowski J. 2005. Two-hybrid fluorescence cross-correlation spectroscopy detects protein-protein interactions in vivo. Chemphyschem 6:984–990. doi: 10.1002/cphc.200400639. [DOI] [PubMed] [Google Scholar]
- 8.Nakabeppu Y, Ryder K, Nathans D. 1988. DNA binding activities of three murine Jun proteins: stimulation by Fos. Cell 55:907–915. doi: 10.1016/0092-8674(88)90146-8. [DOI] [PubMed] [Google Scholar]
- 9.Smeal T, Angel P, Meek J, Karin M. 1989. Different requirements for formation of Jun:Jun and Jun:Fos complexes. Genes Dev 3:2091–2100. doi: 10.1101/gad.3.12b.2091. [DOI] [PubMed] [Google Scholar]
- 10.John M, Leppik R, Busch SJ, Granger-Schnarr M, Schnarr M. 1996. DNA binding of Jun and Fos bZip domains: homodimers and heterodimers induce a DNA conformational change in solution. Nucleic Acids Res 24:4487–4494. doi: 10.1093/nar/24.22.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chinenov Y, Kerppola TK. 2001. Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene 20:2438–2452. doi: 10.1038/sj.onc.1204385. [DOI] [PubMed] [Google Scholar]
- 12.Curran T. 1992. Fos and Jun: oncogenic transcription factors. Tohoku J Exp Med 168:169–174. doi: 10.1620/tjem.168.169. [DOI] [PubMed] [Google Scholar]
- 13.Porte D, Oertel-Buchheit P, Granger-Schnarr M, Schnarr M. 1995. Fos leucine zipper variants with increased association capacity. J Biol Chem 270:22721–22730. doi: 10.1074/jbc.270.39.22721. [DOI] [PubMed] [Google Scholar]
- 14.O'Shea EK, Rutkowski R, Stafford Iii WF, Kim PS. 1989. Preferential heterodimer formation by isolated leucine zippers from Fos and Jun. Science 245:646–648. doi: 10.1126/science.2503872. [DOI] [PubMed] [Google Scholar]
- 15.Nicklin MJH, Casari G. 1991. A single site mutation in a truncated Fos protein allows it to interact with the TRE in vitro. Oncogene 6:173–179. [PubMed] [Google Scholar]
- 16.Mason JM, Schmitz MA, Müller KM, Arndt KM. 2006. Semirational design of Jun-Fos coiled coils with increased affinity: universal implications for leucine zipper prediction and design. Proc Natl Acad Sci U S A 103:8989–8994. doi: 10.1073/pnas.0509880103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greenberg ME, Ziff EB. 1984. Stimulation of 3T3 cells induces transcription of the c-fos proto-oncogene. Nature 311:433–438. doi: 10.1038/311433a0. [DOI] [PubMed] [Google Scholar]
- 18.Piechaczyk M, Blanchard JM. 1994. c-fos proto-oncogene regulation and function. Crit Rev Oncol Hematol 17:93–131. doi: 10.1016/1040-8428(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 19.Sheng M, Thompson MA, Greenberg ME. 1991. CREB: A Ca2+-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science 252:1427–1430. doi: 10.1126/science.1646483. [DOI] [PubMed] [Google Scholar]
- 20.Johnston SRD, Lu B, Scott GK, Kushner PJ, Smith IE, Dowsett M, Benz CC. 1999. Increased activator protein-1 DNA binding and c-Jun NH2-terminal kinase activity in human breast tumors with acquired tamoxifen resistance. Clin Cancer Res 5:251–256. [PubMed] [Google Scholar]
- 21.Mishra A, Bharti AC, Saluja D, Das BC. 2010. Transactivation and expression patterns of Jun and Fos/AP-1 super-family proteins in human oral cancer. Int J Cancer 126:819–829. [DOI] [PubMed] [Google Scholar]
- 22.Güller M, Toualbi-Abed K, Legrand A, Michel L, Mauviel A, Bernuau D, Daniel F. 2008. c-Fos overexpression increases the proliferation of human hepatocytes by stabilizing nuclear cyclin D1. World J Gastroenterol 14:6339–6346. doi: 10.3748/wjg.14.6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saez E, Rutberg SE, Mueller E, Oppenheim H, Smoluk J, Yuspa SH, Spiegelman BM. 1995. c-fos is required for malignant progression of skin tumors. Cell 82:721–732. doi: 10.1016/0092-8674(95)90469-7. [DOI] [PubMed] [Google Scholar]
- 24.Kress E, Skah S, Sirakov M, Nadjar J, Gadot N, Scoazec J, Samarut J, Plateroti M. 2010. Cooperation between the thyroid hormone receptor TRα1 and the WNT pathway in the induction of intestinal tumorigenesis. Gastroenterology 138:1863–1874.e1861. doi: 10.1053/j.gastro.2010.01.041. [DOI] [PubMed] [Google Scholar]
- 25.Mikula M, Gotzmann J, Fischer AN, Wolschek MF, Thallinger C, Schulte-Hermann R, Beug H, Mikulits W. 2003. The proto-oncoprotein c-Fos negatively regulates hepatocellular tumorigenesis. Oncogene 22:6725–6738. doi: 10.1038/sj.onc.1206781. [DOI] [PubMed] [Google Scholar]
- 26.Preston GA, Lyon TT, Yin Y, Lang JE, Solomon G, Annab L, Srinivasan DG, Alcorta DA, Barrett JC. 1996. Induction of apoptosis by c-Fos protein. Mol Cell Biol 16:211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clegg RM. 1995. Fluorescence resonance energy transfer. Curr Opin Biotechnol 6:103–110. doi: 10.1016/0958-1669(95)80016-6. [DOI] [PubMed] [Google Scholar]
- 28.Jares-Erijman EA, Jovin TM. 2003. FRET imaging. Nat Biotechnol 21:1387–1395. doi: 10.1038/nbt896. [DOI] [PubMed] [Google Scholar]
- 29.Bacia K, Kim SA, Schwille P. 2006. Fluorescence cross-correlation spectroscopy in living cells. Nat Methods 3:83–89. doi: 10.1038/nmeth822. [DOI] [PubMed] [Google Scholar]
- 30.Liu P, Sudhaharan T, Koh RML, Hwang LC, Ahmed S, Maruyama IN, Wohland T. 2007. Investigation of the dimerization of proteins from the epidermal growth factor receptor family by single wavelength fluorescence cross-correlation spectroscopy. Biophys J 93:684–698. doi: 10.1529/biophysj.106.102087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tian Y, Martinez MM, Pappas D. 2011. Fluorescence correlation spectroscopy: a review of biochemical and microfluidic applications. Appl Spectrosc 65:115A–124A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vámosi G, Baudendistel N, Von Der Lieth CW, Szalóki N, Mocsár G, Müller G, Brázda P, Waldeck W, Damjanovich S, Langowski J, Tóth K. 2008. Conformation of the c-Fos/c-Jun complex in vivo: a combined FRET, FCCS, and MD-modeling study. Biophys J 94:2859–2868. doi: 10.1529/biophysj.107.120766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pernuš A, Langowski J. 2015. Imaging Fos-Jun transcription factor mobility and interaction in live cells by single plane illumination-fluorescence cross correlation spectroscopy. PLoS One 10:e0123070. doi: 10.1371/journal.pone.0123070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szalóki N, Doan-Xuan QM, Szöllosi J, Tóth K, Vámosi G, Bacsó Z. 2013. High throughput FRET analysis of protein-protein interactions by slide-based imaging laser scanning cytometry. Cytometry A 83:818–829. doi: 10.1002/cyto.a.22315. [DOI] [PubMed] [Google Scholar]
- 35.Haustein E, Schwille P. 2007. Fluorescence correlation spectroscopy: novel variations of an established technique. Annu Rev Biophys Biomol Struct 36:151–169. doi: 10.1146/annurev.biophys.36.040306.132612. [DOI] [PubMed] [Google Scholar]
- 36.Krieger JW, Langowski J. 2015. QuickFit 3.0 (compiled: 8 January 2015, SVN: 3697): a data evaluation application for biophysics. http://www.dkfz.de/Macromol/quickfit/. Accessed 8 January 2015. [Google Scholar]
- 37.Weidemann T, Wachsmuth M, Knoch TA, Müller G, Waldeck W, Langowski J. 2003. Counting nucleosomes in living cells with a combination of fluorescence correlation spectroscopy and confocal imaging. J Mol Biol 334:229–240. doi: 10.1016/j.jmb.2003.08.063. [DOI] [PubMed] [Google Scholar]
- 38.Petrásek Z, Schwille P. 2008. Precise measurement of diffusion coefficients using scanning fluorescence correlation spectroscopy. Biophys J 94:1437–1448. doi: 10.1529/biophysj.107.108811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Greger K, Swoger J, Stelzer EH. 2007. Basic building units and properties of a fluorescence single plane illumination microscope. Rev Sci Instrum 78:023705. doi: 10.1063/1.2428277. [DOI] [PubMed] [Google Scholar]
- 40.Krieger JW, Singh AP, Garbe CS, Wohland T, Langowski J. 2014. Dual-color fluorescence cross-correlation spectroscopy on a single plane illumination microscope (SPIM-FCCS). Opt Express 22:2358–2375. doi: 10.1364/OE.22.002358. [DOI] [PubMed] [Google Scholar]
- 41.Glover JNM, Harrison SC. 1995. Crystal structure of the heterodimeric bZIP transcription factor c-Fos–c-Jun bound to DNA. Nature 373:257–261. doi: 10.1038/373257a0. [DOI] [PubMed] [Google Scholar]
- 42.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. 2006. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins 65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Darden T, York D, Pedersen L. 1993. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 44.Berendsen HJC, Postma JPM, Van Gunsteren WF, Dinola A, Haak JR. 1984. Molecular dynamics with coupling to an external bath. J Chem Phys 81:3684–3690. doi: 10.1063/1.448118. [DOI] [Google Scholar]
- 45.Hess B, Kutzner C, Van Der Spoel D, Lindahl E. 2008. GRGMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput 4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 46.Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, Van Der Spoel D, Hess B, Lindahl E. 2013. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zuo ZL, Gandhi NS, Mancera RL. 2010. Calculations of the free energy of interaction of the c-Fos–c-Jun coiled coil: effects of the solvation model and the inclusion of polarization effects. J Chem Inf Model 50:2201–2212. doi: 10.1021/ci100321h. [DOI] [PubMed] [Google Scholar]
- 48.Heuer KH, Mackay JP, Podzebenko P, Bains NPS, Weiss AS, King GF, Easterbrook-Smith SB. 1996. Development of a sensitive peptide-based immunoassay: application to detection of the Jun and Fos oncoproteins. Biochemistry 35:9069–9075. doi: 10.1021/bi952817o. [DOI] [PubMed] [Google Scholar]
- 49.Pernelle C. 1993. An efficient screening assay for the rapid and precise determination of affinities between leucine zipper domains. Biochemistry 32:11682–11687. doi: 10.1021/bi00094a026. [DOI] [PubMed] [Google Scholar]
- 50.Patel LR, Curran T, Kerppola TK. 1994. Energy transfer analysis of fos-jun dimerization and DNA binding. Proc Natl Acad Sci U S A 91:7360–7364. doi: 10.1073/pnas.91.15.7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hendrix J, Flors C, Dedecker P, Hofkens J, Engelborghs Y. 2008. Dark states in monomeric red fluorescent proteins studied by fluorescence correlation and single molecule spectroscopy. Biophys J 94:4103–4113. doi: 10.1529/biophysj.107.123596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song Y, Madahar V, Liao J. 2011. Development of FRET assay into quantitative and high-throughput screening technology platforms for protein-protein interactions. Ann Biomed Eng 39:1224–1234. doi: 10.1007/s10439-010-0225-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mehta K, Hoppe AD, Kainkaryam R, Woolf PJ, Linderman JJ. 2009. A computational approach to inferring cellular protein-binding affinities from quantitative fluorescence resonance energy transfer imaging. Proteomics 9:5371–5383. doi: 10.1002/pmic.200800494. [DOI] [PubMed] [Google Scholar]
- 54.Erickson MG, Alseikhan BA, Peterson BZ, Yue DT. 2001. Preassociation of calmodulin with voltage-gated Ca(2+) channels revealed by FRET in single living cells. Neuron 31:973–985. doi: 10.1016/S0896-6273(01)00438-X. [DOI] [PubMed] [Google Scholar]
- 55.Brazda P, Krieger J, Daniel B, Jonas D, Szekeres T, Langowski J, Tóth K, Nagy L, Vámosi G. 2014. Ligand binding shifts highly mobile retinoid X receptor to the chromatin-bound state in a coactivator-dependent manner, as revealed by single-cell imaging. Mol Cell Biol 34:1234–1245. doi: 10.1128/MCB.01097-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brazda P, Szekeres T, Bravics B, Tóth K, Vámosi G, Nagy L. 2011. Live-cell fluorescence correlation spectroscopy dissects the role of coregulator exchange and chromatin binding in retinoic acid receptor mobility. J Cell Sci 124:3631–3642. doi: 10.1242/jcs.086082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Capoulade J, Wachsmuth M, Hufnagel L, Knop M. 2011. Quantitative fluorescence imaging of protein diffusion and interaction in living cells. Nat Biotechnol 29:835–839. doi: 10.1038/nbt.1928. [DOI] [PubMed] [Google Scholar]
- 58.Ameyar M, Wisniewska M, Weitzman JB. 2003. A role for AP-1 in apoptosis: the case for and against. Biochimie 85:747–752. doi: 10.1016/j.biochi.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 59.Eferl R, Wagner EF. 2003. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer 3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 60.Shaulian E, Karin M. 2002. AP-1 as a regulator of cell life and death. Nat Cell Biol 4:E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]