

Abstract
V(D)J recombination is initiated by the binding of the RAG1 and RAG2 proteins to recombination signal sequences (RSSs) that consist of conserved heptamer and nonamer sequences separated by a spacer of either 12 or 23 bp. Here, we used RAG-inducible pro-B v-Abl cell lines in conjunction with chromatin immunoprecipitation to better understand the protein and RSS requirements for RAG recruitment to chromatin. Using a catalytic mutant form of RAG1 to prevent recombination, we did not observe cooperation between RAG1 and RAG2 in their recruitment to endogenous Jκ gene segments over a 48-h time course. Using retroviral recombination substrates, we found that RAG1 was recruited inefficiently to substrates lacking an RSS or containing a single RSS, better to substrates with two 12-bp RSSs (12RSSs) or two 23-bp RSSs (23RSSs), and more efficiently to a substrate with a 12/23RSS pair. RSS mutagenesis demonstrated a major role for the nonamer element in RAG1 binding, and correspondingly, a cryptic RSS consisting of a repeat of CA dinucleotides, which poorly re-creates the nonamer, was ineffective in recruiting RAG1. Our findings suggest that 12RSS-23RSS cooperation (the “12/23 rule”) is important not only for regulating RAG-mediated DNA cleavage but also for the efficiency of RAG recruitment to chromatin.
INTRODUCTION
The adaptive immune system relies on V(D)J recombination to assemble variable (V), diversity (D), and joining (J) antigen receptor gene segments in generating a diverse repertoire of immunoglobulins and T cell receptors. The process is initiated by the recombination-activating gene (RAG) recombinase, a protein complex consisting of the enzymes encoded by RAG1 and RAG2 (1, 2). RAG1 is 1,040 amino acids long, with a core region spanning amino acids 384 to 1008 that contains recombination signal sequence (RSS) and RAG2 binding activity as well as the catalytic residues responsible for DNA cleavage (3–7). RAG2 is 527 amino acids long, with a core region (amino acids 1 to 387) that interacts with and facilitates DNA binding and cleavage by RAG1 but which has no DNA binding activity on its own (3, 4, 8, 9). The RAG2 noncore region contains a plant homeodomain (PHD) finger that plays an important role in chromatin binding through interactions with the histone H3 N-terminal tail when lysine 4 is trimethylated (H3K4me3) (10–12).
The site of recombination is specified by the RSS that immediately flanks each V, D, and J gene segment (Fig. 1A). The RSS consists of a well-conserved 7-bp sequence called the heptamer (consensus sequence of 5′-CACAGTG-3′) and an AT-rich 9-bp sequence called the nonamer (consensus sequence of 5′-ACAAAAACC-3′) separated by a poorly conserved spacer whose length is either 12 or 23 bp (13–15) (Fig. 1B). An RSS with a spacer 12 bp long is defined as a 12RSS, while one with a spacer 23 bp long is defined as a 23RSS (13).
FIG 1.

RSSs and RAG proteins are essential components of V(D)J recombination. (A) An RSS (triangle) lies adjacent to the coding segment (rectangle) of an antigen receptor gene. (B) RSS showing consensus heptamer and nonamer sequences. (C) V(D)J recombination is initiated by binding of the RAG1-RAG2-HMGB1/2 complex (oval) to an RSS. The capture of a second RSS through synapsis leads to the formation of a paired complex, within which DNA cleavage is completed. Repair factors bring together the coding segments and RSSs to form coding and signal joints, respectively.
The consensus RSS is the most efficiently recombined sequence and therefore has been used for most in vitro experiments; however, endogenous RSSs often deviate from the consensus at multiple heptamer and nonamer positions (14). The first three nucleotides of the heptamer (CAC) exhibit almost perfect conservation and are required for cleavage (14, 16, 17); the cytosine at the second position and some nucleotides in the adenine tract of the nonamer are >85% conserved and are important for initial RAG binding (14, 16). Although the spacer is less conserved than the heptamer and nonamer, its sequence can influence recombination efficiency, and its length defines the functionality of the RSS, as efficient recombination occurs only between a 12/23RSS pair, a restriction known as the “12/23 rule” (18, 19).
RAG, along with the DNA binding and bending high-mobility-group protein B1 (HMGB1) or HMGB2, binds to one RSS to form a signal complex (20) (Fig. 1C). This complex then captures a second unbound RSS to form the synaptic (also known as paired) complex (21, 22). The RAG complex introduces a single-stranded nick between the RSS and the coding segment, leaving a hydroxyl group on the 3′ end of the coding flank that can then attack the opposite DNA strand, resulting in a hairpin coding end and a blunt-cut RSS signal end (23).
The “accessibility” of RSSs in chromatin contributes to the regulation of the initiation of V(D)J recombination. Accessibility correlates with features of open chromatin, such as germ line transcription, antisense transcription, activating histone modifications, nuclease accessibility, DNA hypomethylation, as well as the presence of transcription factors and transcriptional control elements (such as enhancers and promoters) (24). The presence of nucleosomes on RSSs has been shown to be inhibitory to RAG binding and cleavage but can be remedied by chromatin remodeling complexes that remove or reposition nucleosomes or lift a loop of chromatin off the nucleosome surface, thus making the RSSs accessible for RAG recruitment (24).
A recent study from our laboratory provided insights into the location of such accessible RSSs within the antigen receptor loci by revealing the in vivo pattern of RAG1 and RAG2 binding to antigen receptor loci in primary mouse lymphocytes (25). Chromatin immunoprecipitation (ChIP)-quantitative PCR (qPCR) was used to demonstrate that RAG binding was tightly regulated during lymphocyte development and associated with small, highly transcribed regions encompassing J, J-proximal D, and flanking RSSs in the Igh, Igκ, Tcrα, and Tcrβ loci. These regions, termed recombination centers, are rich in H3K4me3, acetylated histone H3 (H3Ac), and RNA polymerase II occupancy. In most loci, binding of RAG1 and RAG2 was roughly comparable in the presence and absence of the other RAG protein. At Igh, however, the level of binding of RAG1 was substantially lower in the absence of RAG2 than in its presence (25). This raised the question as to the extent to which and the circumstances under which RAG1 and RAG2 facilitate one another's recruitment to chromatin.
Our previous study also demonstrated RAG2 binding throughout the genome at promoter regions in a pattern very similar to that of H3K4me3, probably due to H3K4me3 recognition by the RAG2 PHD finger, while RAG1 binding was more restricted and not associated with all H3K4me3 sites. This finding, combined with data suggesting that RAG1 required its nonamer binding domain (NBD) and the presence of RSSs to bind to chromosomal recombination substrates, indicated RAG1's preference for direct interactions with RSSs. Intriguingly, RAG has the ability to bind and cleave certain cryptic RSSs (cRSSs) (26, 27), creating aberrant V(D)J recombination products and contributing to genome instability and tumorigenesis (28, 29). These results left unaddressed the issue of which components and properties of RSSs contribute to efficient RAG recruitment.
Here, we used RAG-inducible pro-B v-Abl cell lines in conjunction with ChIP-qPCR to better understand the protein and the RSS components for rapid and efficient RAG recruitment. Our results argue against strong synergy in recruitment between RAG1 and RAG2. We also report an enhanced efficiency of RAG1 recruitment to chromosomal substrates containing a complementary 12/23RSS pair compared to those of substrates containing a single RSS, a 12/12RSS or 23/23RSS pair, or a wild-type (WT) RSS paired with an RSS with a scrambled heptamer or nonamer, with the nonamer being particularly important. Our results suggest that the 12/23 rule applies not only to efficient V(D)J recombination but also to initial RAG binding.
(This paper represents work completed as part of K. Shetty's dissertation for a Ph.D. from Yale University, New Haven, CT, 2015.)
MATERIALS AND METHODS
Cell lines.
The R1−/− (11-3-4), R2−/− (17-3-1), and D345 v-Abl pro-B mouse cell lines were previously prepared by infection of bone marrow from Bcl2 transgenic mice with the retroviral vector pMSCV-v-Abl (30). All cell lines were maintained at 37°C with 5% CO2 in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), penicillin-streptomycin, l-glutamine, and β-mercaptoethanol (50 μM).
Recombination substrates.
p12/23 was generated, as described previously (25), from pMX-RSS-GFP/IRES-hCD4 (31) by replacing the green fluorescent protein (GFP) cassette with a fragment containing the mouse CD90 (Thy1.1) gene. pNone was derived from p12/23 as described previously (25). p12 was generated by digestion of p12/23 with NcoI-SnaBI, blunting with Klenow DNA polymerase, and recircularization to remove the 23RSS. p23 was generated by digestion of p12/23 with NotI-EcoRI, blunting with Klenow DNA polymerase, and recircularization to remove the 12RSS. p12/12 was generated by digesting p12 with NcoI, amplifying the 12RSS with primers containing NcoI overhangs, and inserting the amplified fragment into the p12 vector via ligation. p23/23 was generated by digesting p12/23 with NotI and EcoRI, amplifying the 23RSS with primers containing NotI and EcoRI, and replacing the 12RSS with the amplified fragment in the p12/23 vector via ligation. pScrH/23, pScrN/23, and p(CA)38/23 were generated by digesting the p12/23 vector with NotI and EcoRI and inserting annealed oligonucleotides (Blue Heron Bio) with NotI and EcoRI overhangs into the digested vector via ligation. Nonamer, heptamer, spacer, and coding flank sequences of these mutant RSSs are listed in Table 1.
TABLE 1.
Synthesized oligonucleotides for mutant RSS substratesa
| Substrate | 12RSS region or 12RSS mutant oligonucleotide sequence |
|---|---|
| p12/23 | 5′-ATCTCGCGGCCGCACGCGTTCGAAGTCGAGGTTTTTGTTCCAGTCTGTAGCACTGTGCAGACGTTCCTATTATTTTTGACACCAGACAAGTTGGTAATGGTAGCGACCGGCGCTCAGTTGGAATTCTACG-3′ |
| pScrH/23 | 5′-ATCTCGCGGCCGCACGCGTTCGAAGTCGAGGTTTTTGTTCCAGTCTGTAGCGCTGCGCAGACGTTCCTATTATTTTTGACACCAGACAAGTTGGTAATGGTAGCGACCGGCGCTCAGTTGGAATTCTACG-3′ |
| pScrN/23 | 5′-ATCTCGCGGCCGCACGCGTTCGAAGTCGATCCGACTTATCCAGTCTGTAGCACTGTGCAGACGTTCCTATTATTTTTGACACCAGACAAGTTGGTAATGGTAGCGACCGGCGCTCAGTTGGAATTCTACG-3′ |
| p(CA)38/23 | 5′-ATCTCGCGGCCGCACACACACACACACACACACACACACACACACACACACACACACACACACACACACACACACACACACACACACACGTTGGTAATGGTAGCGACCGGCGCTCAGTTGGAATTCTACG-3′ |
The synthesized oligonucleotides (Blue Heron Bio) for the 12RSS sites of pScrH/23, pScrN/23, p(CA)38/23 are listed and compared to the sequence of the 12RSS region of p12/23. The nonamer and heptamer sequences are highlighted in boldface type, while the regions of mutagenesis are underlined.
Cell infections and RAG induction.
Retrovirus was prepared as described previously (32) and used to infect v-Abl cells. Infected cells were subjected to two rounds of sorting for hCD4+ cells. Cells were induced with 3 μM STI-571 (STI) for 0, 3, 6, 24, and 48 h, and RAG1 and RAG2 binding and H3K4me3 levels were assessed by ChIP.
ChIP.
The antibodies and procedures used for the ChIP assay were described in detail previously (25, 33). In brief, cells were harvested after 0, 3, 6, 24, and 48 h of STI-571 induction; cross-linked with 1% HCHO (formaldehyde) for 15 min at room temperature (RT); quenched with 0.125 M glycine; washed; and frozen as cell pellets. The pellets were then resuspended in radioimmunoprecipitation assay (RIPA) buffer containing 0.8 M NaCl and sonicated. The chromatin was incubated with anti-RAG1 or anti-RAG2 monoclonal antibody (33), anti-H3K4me3 antibody (Millipore), or rabbit IgG (Millipore), and immune complexes were isolated with protein A-agarose beads (Millipore). After reversal of cross-links and purification of the DNA (DNA Clean concentrator; Zymo), input and immunoprecipitated DNAs were quantitated by duplicate trials of TaqMan qPCR Qiagen HotStart Taq with a 3000 XP thermocycler (Stratagene). The immunoprecipitation (IP)/Inputcorr values were calculated as described previously (25). Briefly, IP/Inputcorr corresponds to [(IPsp − IPrIg)/Input] × 1,000, where IPsp and IPrIg are the amounts of DNA recovered in IPs with the specific antibody and background rabbit IgG, respectively. PCR primers (Sigma-Aldrich) and hydrolysis probes (Biosearch Technologies) for the gene segments of interest were designed as described previously (25). Table 2 provides primer and probe sequences for the qPCRs.
TABLE 2.
Oligonucleotide primers and probes used for detection of the RAG binding sequencea
| Region | Sequence |
||
|---|---|---|---|
| Forward primer | Hydrolysis probe | Reverse primer | |
| Jκ1 | 5′-TTGTACAGCCAGACAGTGGAG-3′ | 5′-TGGTGCCTCCACCGAACGTC-3′ | 5′-GCCACAGACATAGACAACGG-3′ |
| Jκ2 | 5′-CAGATTCTGGCACTCTCCAA-3′ | 5′-CAAAGAAGCAGGGTAGCCTGCCC-3′ | 5′-ACTGAGCATGGTCTGAGCAC-3′ |
| Jκ5 | 5′-CATAGTCCTCACTGTGGCTCA-3′ | 5′-CTTGGTCCCAGCACCGAACG-3′ | 5′-AGTGTACTTACGTTTCAGCTCCA-3′ |
| Pou2af1 | 5′-ATCTCCGATTCTGTGCATGA-3′ | 5′-AATCTGCCCTTCTGTCACCTCAGTGA-3′ | 5′-GTGAAGAACGGCTCCTAACC-3′ |
| γ-Actin | 5′-CCTCCTCCAATAAAGGGACA-3′ | 5′-AGAACTGGTGACCCTCTCCCACG-3′ | 5′-GCCATCACATCCCAGTCA-3′ |
| α-Actin | 5′-GGCACGTGACACTCTTGTCT-3′ | 5′-ATCGGTGGCTCCATCCTGGC-3′ | 5′-GTCGTACTCCTGCTTGGTGA-3′ |
| 12a | 5′-CCTCTAGACTGCCGGATCTC-3′ | 5′-TCCCTCCTCCAAGCCCTGGA-3′ | 5′-CATAAGCCTGCTGGTTCAGA-3′ |
| 23d | 5′-GCAAGACTGAGAGCAGGAGA-3′ | 5′-CCGTCCTCGAGGCACCATGA-3′ | 5′-ACACCGGCCTTATTCCAA-3′ |
Hydrolysis probes were labeled at the 5′ end with the reporter dye 6-carboxyfluorescein (FAM) and at the 3′ end with black hole quencher (BHQ) dye (Biosearch Technologies).
cRSS, heptamer, and nonamer density analysis.
The 1-kb regions flanking (on both sides) the Pou2af1 and γ-actin ChIP-qPCR amplicons were tested for cRSSs and RSS heptamer and nonamer motifs. The recombination information content (RIC) algorithm (34, 35) was used to identify 12- and 23-cRSSs (with cutoff values of −38 and −54, respectively). Scores for heptamers and nonamers were calculated by position weight matrices, which were generated by aligning the functional 12- and 23RSSs used to formulate the RIC algorithm. By using these matrices, the sequences were scanned for the corresponding motif using the FIMO tool (36). The threshold values for “high-scoring” heptamers and nonamers were selected to be 7.33 and 7.06, respectively.
RESULTS
Experimental system for determining RAG binding dynamics in vivo.
We recently collected extensive data for RAG binding at antigen receptor loci in primary lymphocyte populations (25). However, this system was limited in that it did not capture the initial binding dynamics, providing only a snapshot of steady-state RAG-DNA interactions. To better explore the conditions needed for efficient and rapid RAG recruitment, we used an inducible expression system based on v-Abl-transformed pre-B cell lines, which, when treated with STI-571 (STI), induce RAG expression and are arrested in the G1 phase of the cell cycle (Fig. 2A) (37). In our experiments, we used v-Abl cell lines expressing inducible (i) RAG1 with a D-to-A mutation at position 708 (D708A RAG1) and RAG2 (cell line called D345), (ii) RAG1 alone (R2−/−), and (iii) RAG2 alone (R1−/−) (Fig. 2B). D708A RAG1 is a catalytically inactive mutant that retains normal DNA binding activity; its use prevents DNA cleavage and accumulation of recombination products that could interfere with the interpretation of the RAG binding data (25). Western blot analyses of the cell lines induced with STI over the course of 48 h showed similar levels of RAG1 expression in D345 and R2−/− cells and RAG2 expression in D345 and R1−/− cells (Fig. 2C).
FIG 2.
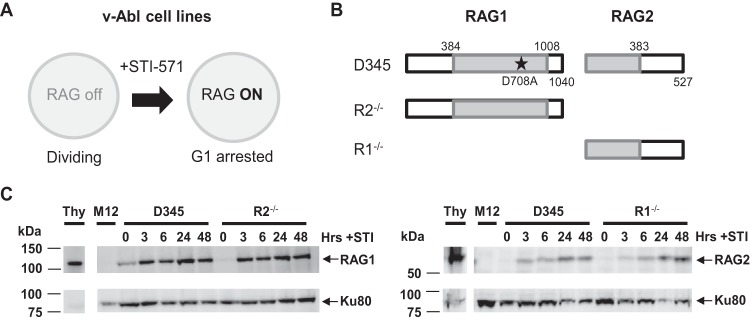
Experimental system for determining RAG binding dynamics in vivo. (A) Stimulation of v-Abl-transformed cell lines with STI-571 (STI) induces RAG expression and arrests cells in the G1 phase. (B) Diagram of RAG proteins expressed by the v-Abl cell lines used in this study. The D708A RAG1 active-site mutation is represented by a star. (C) Western blot analysis of D345, R1−/−, and R2−/− cell lines induced with STI for 0, 3, 6, 24, and 48 h. Whole-cell extracts from thymuses from 5-week-old mice (Thy) and RAG-deficient cells (M12) served as the positive and negative controls, respectively. Membranes were incubated with RAG1 monoclonal antibody 23 or RAG2 monoclonal antibody 39 (33) and then probed for Ku80 to assess protein loading.
Independent recruitment of RAG1 and RAG2 to the Jκ recombination center.
Our prior study suggested that although RAG1 and RAG2 interact with one another, they do not require the presence of the other for chromatin binding at steady state (25). However, it remained possible that the presence of one RAG protein increases the efficiency and rapidity of recruitment of the other protein to chromatin. To test this, D345, R2−/−, and R1−/− cell lines were induced with STI for 0, 3, 6, 24, and 48 h; harvested; and subjected to ChIP. STI treatment is known to stimulate rearrangement at the endogenous Igκ locus (37) and RAG binding to Jκ gene segments (25) in v-Abl-transformed cells, indicating that Jκ gene segments are accessible for RAG recruitment. The Igκ locus contains ∼140 Vκ gene segments and 5 Jκ genes, with Jκ1, Jκ2, Jκ4, and Jκ5 being functional (Fig. 3A). RAG binding was measured by qPCR using primer pairs that amplify a product spanning the RSSs of Jκ1, Jκ2, and Jκ5 (see Materials and Methods).
FIG 3.

Independent recruitment of RAG1 and RAG2 to the Jκ recombination center. (A) Binding of RAG proteins at the endogenous Jκ antigen receptor locus, which consists of 4 functional gene segments, Jκ1, Jκ2, Jκ4, and Jκ5, and one pseudogene, Jk3 (shaded), was assessed by ChIP-qPCR. The segments are separated from each other by 300 to 350 bp. The figure is not to scale. DNA recovery in immunoprecipitates was measured by qPCR using primers that span the RSSs of Jκ1, Jκ2, and Jκ5 (arrows). (B) Binding of RAG1 at Jκ1, Jκ2, and Jκ5 in the D345, R2−/−, and R1−/− v-Abl cell lines after 0, 3, 6, 24, and 48 h of STI-571 induction. (C) Binding of RAG2. Data are presented as described above for panel B. IP/Inputcorr values have been corrected for background signals and normalized to the input signal, as described in Materials and Methods. (D) H3K4me3 levels at the endogenous Jκ genes in the three cell lines. Bars and dot plots indicate the means of data from ≥3 independent experiments, and error bars represent the standard errors of the means.
RAG1 binding at Jκ1, Jκ2, and Jκ5 in the D345 cell line coexpressing RAG1 and RAG2 steadily increased over the 48-h time course, and notably, the kinetics and magnitude of RAG1 binding were quite similar in the R2−/− cell line (Fig. 3B). Binding was detected after 3 h of induction, which is when we detected protein expression (Fig. 2C). RAG2 binding also increased over the time course of the experiment, and ChIP signals were similar for D345 and R1−/− cells (Fig. 3C). RAG2 signals were higher than those for RAG1, consistent with data reported previously for primary pre-B cells (25). The signals for RAG1 binding in R1−/− cells and RAG2 binding in R2−/− cells were both very low, reflecting the low background signal for the RAG ChIP assays (Fig. 3B and C) (all ChIP data presented have had the control IgG background signal subtracted).
To interpret the RAG binding data, it was important to determine whether the Jκ region was equally accessible in the three cell lines. This was assessed by using H3K4me3 because of the strong links between this mark and transcription and RAG2 binding. H3K4me3 levels were similar in the D345 and R2−/− cell lines (Fig. 3D). Based on this finding and the similar RAG1 binding signals in the D345 and R2−/− cell lines (Fig. 3B), we conclude that RAG2 does not substantially alter the initial stages of RAG1 recruitment at Jκ gene segments. However, H3K4me3 levels were substantially higher in R1−/− than in D345 cells (Fig. 3D), perhaps due to clonal variation. This makes it difficult to rule out a possible contribution of RAG1 to initial RAG2 recruitment to the Jκ region.
RAG binding to non-antigen-receptor genes.
ChIP-sequencing (ChIP-seq) data from our laboratory revealed the ability of both RAG1 and RAG2 to bind at certain non-antigen-receptor genes, which do not harbor RSSs (25, 38). This result was surprising given that RAG1 has RSS binding domains and that the only known function of the RAG proteins is to assemble antigen receptor genes. To determine whether RAG binding to non-antigen-receptor genes followed the same rules and kinetics as those for RSS-containing antigen receptor genes, we measured RAG recruitment at the promoters of three genes: Pou2af1, which binds RAG1 and RAG2 (25, 38); γ-actin, which binds RAG2 but showed poor RAG1 binding previously (25), and α-actin, which is not expressed and not expected to bind either RAG protein. At Pou2af1, the kinetics and levels of RAG1 binding were similar in D345 and R2−/− cells (Fig. 4A), and the same was true for RAG2 binding in D345 and R1−/− cells (Fig. 4B). The time course of accumulation of RAG was similar to that seen at Jκ, but signals were lower at Pou2af1 than at Jκ1 or Jκ2 (Fig. 3). As expected, RAG2 binding was detectable at γ-actin, while RAG1 binding was weak, and no substantial RAG binding was seen at α-actin (Fig. 4A and B). These data suggest that the kinetics of RAG accumulation at non-antigen-receptor genes are similar to those seen in a bona fide recombination center. Furthermore, as for Jκ, we did not obtain evidence indicative of cooperation between the RAG proteins in their initial recruitment at non-antigen-receptor genes. We note that H3K4me3 levels were higher at Pou2af1 in D345 cells than in the R1−/− or R2−/− cell line (Fig. 4C), which, if anything, should have favored RAG recruitment in D345 cells compared to the single-RAG-expressing lines.
FIG 4.

Kinetics of RAG accumulation at non-antigen-receptor genes are similar to those seen in a bona fide recombination center. (A) Binding of RAG1 at the non-antigen-receptor-endogenous genes Pou2af1, γ-actin, and α-actin was assessed by ChIP-qPCR. (B) Binding of RAG2. Data are presented as described above for panel A. (C) H3K4me3 levels at the non-antigen-receptor genes in the three cell lines. Bars and dot plots indicate the means of data from ≥3 independent experiments, and error bars represent the standard errors of the means.
Computational analysis (see Materials and Methods) showed the absence of any strong cRSSs in a ±1-kb region flanking Pou2af1 and γ-actin ChIP-qPCR amplicons. Four high-scoring heptamers and 11 high-scoring nonamers (see Materials and Methods) were detected in the 2-kb region surrounding the Pou2af1 amplicon, while 8 heptamers and 6 nonamers were found in the 2-kb region surrounding the γ-actin amplicon. The number of nonamers near Pou2af1 was higher than expected due to the presence of A-rich regions. The extent to which these isolated RSS components might contribute to RAG recruitment is not known (see Discussion).
A pair of RSSs improves RAG recruitment efficiency.
Seeing that regions lacking RSSs were able to recruit RAG with similar kinetics but at lower levels than those for regions containing RSSs, we decided to explore the RSS requirements for efficient RAG recruitment. With the 12/23 rule in mind, we used a retroviral recombination substrate containing a 12/23RSS pair as a benchmark (Fig. 5A; a simplified diagram with only RSSs and the location of PCR amplicons is depicted in Fig. 5B). Its variants included a substrate lacking RSSs (pNone), one containing a single 12RSS (p12), and one containing a single 23RSS (p23) (Fig. 5B). These substrates were introduced into the D345 cell line, and ChIP-qPCR was performed on polyclonal populations of infected cells after STI induction with primer pairs spanning the 12RSS site or the 23RSS site (Fig. 5B). Much stronger RAG1 binding was detected for p12/23 at both RSS sites at all postinduction time points than for pNone, p12, and p23 (Fig. 5C). This could not be attributed to differences in H3K4me3 levels, which showed a trend toward being somewhat lower at the 12RSS and higher at the 23RSS in p12/23-containing cells than in cells containing the other substrates (Fig. 5E). Notably, RAG1 binding signals were roughly 5-fold higher at the RSSs in p12/23-containing cells than at Jκ1 or Jκ2 (Fig. 3B [note the different y axis scales]), and the modest signals seen at the 12RSS of p12 and at the 23RSS of p23 were comparable to those seen at the Jκ segments (compare Fig. 3B and 5C, insets, which use the same y axis scales). RAG1 binding levels were somewhat higher at the 12RSS of p12 than at this same location when the 12RSS was absent in p23 (Fig. 5C, left), and a similar trend was observed for the 23RSS of p23 (Fig. 5C, right). The data also suggest that detectable RAG1 binding occurs at later time points at pNone. We conclude that initial RAG1 binding is strongly stimulated by a 12/23RSS pair and is sensitive to the presence of a single RSS.
FIG 5.

A pair of RSSs improves RAG recruitment efficiency. (A) The p12/23 retroviral recombination substrate. The 5′ and 3′ long terminal repeats (LTR), the mouse CD90 gene encoding Thy1.1 that lies in the opposite transcriptional orientation, the human CD4 (hCD4) gene, the internal ribosome entry site (IRES), and the 12RSS (12) and the 23RSS (23) are indicated (25). (B) Simplified schematic of p12/23 and its variants pNone (lacking the 12RSS and 23RSS), p12 (lacking the 23RSS), and p23 (lacking the 12RSS). Primer pairs 12a (gray underlining) and 23d (black underlining) span the locations of the 12RSS and 23RSS, respectively (25). (C and D) Binding of RAG1 (C) and RAG2 (D) in the D345 cell line over a 48-h time course of STI induction at the 12RSS and 23RSS sites was assessed by ChIP-qPCR. Insets show the RAG1 data for the 12RSS site of p12 and the 23RSS site of p23, presented with the same y axis scale as that for Jκ gene segments in Fig. 3B. (E) H3K4me3 levels at each RSS site for each substrate. Bars and dot plots indicate the means of data from ≥3 independent experiments, and error bars represent the standard errors of the means.
The level of RAG2 binding was also higher at p12/23 than at the other substrates (Fig. 5D) and 2- to 3-fold higher than that at Jκ segments (Fig. 3C [note the different y axis scales]). For the other three substrates, RAG2 binding exhibited a slight trend of being higher at RSSs, with signals generally being lowest at pNone, and higher in the presence of a single RSS than in its absence, although binding was readily detected at the 12RSS site of p23 and the 23RSS site of p12 (Fig. 5D). Preferential RAG2 binding to p12/23 is notable given the abundance of H3K4me3 at the amplified sites in all of the substrates (Fig. 5E) and comparable levels of expression, as assessed by levels of hCD4 surface expression, for the four substrates (data not shown). Overall, diminished RAG binding at non-p12/23 substrates indicates the requirement for an RSS pair for efficient RAG recruitment in this experimental system.
Preferential RAG1 recruitment to a 12/23RSS pair.
The data shown in Fig. 5 indicate that a pair of RSSs is more efficient in recruiting RAG than a single RSS. According to the 12/23 rule, a complementary 12/23RSS pair is required for efficient cleavage, but does this restriction also apply to RAG binding efficiency?
To address this question, we measured RAG binding in D345 cells using substrates containing either a 12/12RSS (p12/12) or a 23/23RSS (p23/23) pair (Fig. 6A) and compared the results with those obtained with p12/23. RAG1 binding levels were reduced at p12/12 and p23/23 for all postinduction time points at both the 12RSS and 23RSS sites compared to that at p12/23 (Fig. 6B). The differences did not appear to be due to differences in H3K4me3, as levels of this mark were similar for the three substrates (Fig. 6D). We also noted that at almost all time points, RAG2 showed little or no decrease in binding at p12/12 and p23/23 versus p12/23 (Fig. 6C). We conclude that the 12/23RSS pair is more efficient in recruiting RAG1 than are the symmetrical pairs.
FIG 6.

Preferential RAG1 recruitment to a 12/23RSS pair. (A) Simplified schematics of p12/23 and its variants p12/12 and p23/23. (B and C) Binding of RAG1 (B) and RAG2 (C) in the D345 cell line over a 24-h time course of STI induction at the 12RSS and 23RSS sites was assessed by ChIP-qPCR. Bars for p12/23 indicate the means of data from ≥3 experiments, while those for p12/12 and p23/23 indicate the means of data from 3 independent experiments, and error bars represent the standard errors of the means. (D) H3K4me3 levels at each RSS site for each substrate. Plots for p12/23 indicate the means of data from ≥3 experiments, while plots for p12/12 and p23/23 indicate the means of data from 2 independent experiments, and error bars represent the standard errors of the means. The RAG1, RAG2, and H3K4me3 data for p12/23 are shown for comparison and are the same as those shown in Fig. 5.
RAG1 recruitment is mediated primarily by the nonamer.
The crystal structure of the RAG1 nonamer binding domain (NBD) illuminated three residues (R391, R393, and R402) that make particularly critical contacts with the nonamer (39). Mutation of these residues in the context of D708A RAG1 dramatically reduced its recruitment to p12/23 (25), indicating the importance of nonamer contact for RAG1 binding. In addition, surface plasmon resonance (40) and in vivo one-hybrid (41) experiments showed that mutations in the nonamer reduced RAG1 binding, while mutations in the heptamer had little effect. The importance of the nonamer for RAG binding was also observed in subsequent biochemical studies (20). Here, we wanted to confirm that the RAG1 binding that we detected was strongly dependent on the nonamer and also test the contribution of the heptamer.
To investigate this, we designed substrates containing a consensus 23RSS paired with a 12RSS containing either a scrambled heptamer (p12ScrH/23) or a scrambled nonamer (p12ScrN/23) (Fig. 7A). RAG1 binding at both 12- and 23RSS sites of p12ScrH/23 was consistently reduced at the 6- and 24-h induction time points relative to that for p12/23 (Fig. 7B), suggesting a small but detectable role for the heptamer in RAG1 recruitment efficiency. In contrast, RAG1 binding was strongly reduced at both RSS sites of p12ScrN/23 (Fig. 7C), yielding results similar to those seen with substrates containing only a single RSS (Fig. 5C). This finding supports a critical role for the nonamer both in the direct recruitment of RAG1 and in facilitating RAG1 recruitment to a partner RSS and argues that an isolated heptamer makes a minor contribution. RAG2 binding was not as sensitive to heptamer or nonamer mutation as was RAG1 binding, although there was a clear trend toward weaker RAG2 binding at both RSS sites with the scrambled nonamer substrate (Fig. 7C).
FIG 7.

RAG1 recruitment is mediated primarily by the nonamer. (A) Simplified schematics of p12/23 and its variants pScrH/23, pScrN/23, and p(CA)38/23. (B) Alignment of the CA repeat with a consensus 12RSS. Residues in the CA repeat that match those in the consensus are underlined. (C and D) Binding of RAG1 (C) and RAG2 (D), presented as described in the legend to Fig. 6B and C. Bars for pScrH/23, pScrN/23, and p(CA)38/23 indicate the means of data from 3 independent experiments, and error bars represent the standard errors of the means. (E) H3K4me3 levels at each RSS site for each substrate. Plots for pScrH/23, pScrN/23, and p(CA)38/23 indicate the means of data from 2 independent experiments, and error bars represent the standard errors of the means. The RAG1, RAG2, and H3K4me3 data for p12/23 are shown for comparison and are the same as those shown in Fig. 5.
Finally, we designed an additional substrate with a consensus 23RSS paired with a cryptic RSS (cRSS) comprised of a CA dinucleotide repeated 38 times [p(CA)38/23] (Fig. 7A). Many thousands of CA repeats are found in the mouse and human genomes, and they can behave as cRSSs that produce aberrant recombination products at off-target sites (42, 43). Additionally, CA repeats recapitulate the invariant first 3 bp of the heptamer that are absolutely required for cleavage. The CACA sequence of the heptamer is conserved between the synthetic CA repeat and the consensus 12RSS, while the A tract of the nonamer, which is important for initial RAG binding, is interrupted in two positions (Fig. 7B). Little RAG1 binding was detected with p(CA)38/23 at either RSS site (Fig. 7C), consistent with the poor nonamer sequence of the CA repeat. Indeed, the data for p(CA)38/23 for both RAG1 and RAG2 binding were similar to those for p12ScrN/23 (Fig. 7C and D), suggesting that a CA repeat behaves much like a heptamer in regard to RAG recruitment. H3K4me3 levels were roughly similar for the four substrates and did not correlate in any obvious way with levels of RAG binding (Fig. 7E). Together, these data suggest that a nonamer has substantial potential to facilitate RAG recruitment and that the thousands of CA repeats in the vertebrate genome are unlikely to be strong sites of RAG recruitment.
DISCUSSION
Our previous study (25) showed that RAG binding is associated with recombination centers: regions enriched with germ line transcription, histone modifications, and highly accessible RSSs. However, that analysis was limited, as the primary lymphocyte populations could not capture the kinetics of the RAG-chromatin interactions. To address this, here we used an inducible RAG expression system combined with ChIP to measure the initial binding dynamics of the accumulated proteins. Our data indicate that (i) the presence of RAG2 does not increase the efficiency and rapidity of RAG1 binding during the 48 h of the assay, (ii) the most efficient RAG recruitment occurs at a substrate with more than one RSS, specifically a 12/23RSS pair, and (iii) efficient RAG1 recruitment at a 12/23RSS pair is strongly dependent on the nonamer and less so on the heptamer.
Kinetic analysis of RAG cross talk.
Our previous study (25) showed that each RAG protein can bind with roughly equal efficiency to recombination centers (with the exception of the Igh locus) in the presence or absence of the other protein. Here, we examined whether the RAG proteins could increase one another's accumulation at the Jκ recombination center (Fig. 3B and C) and at non-antigen-receptor genes (Fig. 4A and B), perhaps due to their ability to interact with each other. The data did not provide clear evidence of cooperation between the RAG proteins in their initial recruitment over a 48-h time period. RAG1 ChIP signals in the presence and in the absence of RAG2 were similar, as were H3K4me3 signals, except at the Pou2af1 and γ-actin promoters, where the higher H3K4me3 levels in D345 cells than in R2−/− cells might have been expected to favor RAG recruitment in the former. For RAG2, recruitment was similar in the presence and in the absence of RAG1, but the interpretation of this finding for the Jκ gene segments was obscured by the higher levels of H3K4me3 seen in R1−/− than in D345 cells (Fig. 3D).
Our failure to detect cooperation between the RAG proteins in initial binding at Jκ, while largely consistent with data from our previous ChIP-qPCR study (25), was notable in light of a recent in vitro kinetic analysis of RAG-mediated DNA cleavage, which predicted cooperation between RAG1 and RAG2 by virtue of their ability to recognize the RSS (RAG1) and H3K4me3 (RAG2) (44). This discrepancy might relate to differences in interactions with naked DNA in vitro and with chromatin in vivo as well as the need for RAG1 and RAG2 to associate with one another at their relatively low concentrations in vivo (45). This might be relevant for analyses of initial binding using an inducible expression system (as in this study), given that individually expressed RAG1 core and RAG2 core proteins interact with only a modest affinity, with a KD (equilibrium dissociation constant) value of ∼0.4 μM (45). We also note that our recent ChIP-seq analysis using mouse thymocytes revealed that RAG1 binding was reduced in intensity and less well focused at regions of high H3K4me3 levels in the absence of RAG2 than in its presence (38). These results (which represent a steady-state analysis) are more in keeping with the models derived from in vitro kinetic studies (44). This finding and our previous finding that RAG1 binding at the Igh recombination center was strongly dependent on RAG2 (25) suggest that there are circumstances under which RAG2 can enhance RAG1 binding.
Perhaps also relevant to the interpretation of RAG1 binding patterns is the finding that RAG1 can interact with histone 3 (46, 47). This emphasizes the point that our ChIP assay does not distinguish between interactions of the RAG proteins directly with DNA and interactions with the protein components of chromatin.
We note that an interpretation of the data reported here and in our previous study relies on the assumption, as yet unproven, that the RAG proteins do not influence one another's ability to be cross-linked to chromatin during the ChIP procedure. Also of note are the temporal limitations of our inducible system; we measured binding on a time scale of hours and hence did not address very early binding events, which would likely have been below the detection limits of the ChIP assay. Furthermore, our assay measures protein occupancy but not the underlying on and off rates. A technique that provides high temporal resolution, such as a modified ChIP assay that relies on short formaldehyde cross-linking incubation times (48) or single-molecule imaging (49), would be necessary to dissect such features of RAG binding.
RSS cooperation in RAG recruitment.
Our previous study (25) defined recombination centers as 2- to 10-kb regions containing multiple J gene segments (and, in some cases, nearby D gene segments) and their respective RSSs but did not identify the minimum number of RSSs in these regions needed for RAG recruitment. Here, we demonstrate a clear hierarchy for RAG1 binding: it is weakest at sites lacking an RSS, increases at an isolated RSS, increases again if two similar RSSs (12/12- or 23/23RSS) are present, and is strongest with a 12/23RSS pair. We consider each of these situations in turn.
RAG1 binding was detected at the Pou2af1 promoter (Fig. 4A), which lacks strong RSS sequences, and appeared to be detected at later induction time points for substrate sites lacking RSSs (pNone, the 23RSS site of p12, and the 12RSS site of p23) (Fig. 5C). This finding is consistent with our recent RAG1 ChIP-seq data that revealed the binding of RAG1 at a wide range of intensities to thousands of active promoters in developing B and T cells (38). We conclude that RAG1 binding is not limited to regions containing RSSs, in contrast to the conclusions of our previous study (25), and that the association of RAG1 with chromatin might be mediated by various types of interactions, including sequence-specific and nonspecific DNA binding and interactions with nucleosomes. Isolated heptamer-like and nonamer-like sequences are abundant in the genome, and multiple such sequences are present in the vicinity of the Pou2af1 and γ-actin ChIP-qPCR amplicons. Our data reported here as well as our finding that high-scoring nonamer sequences are more prevalent than expected by chance at RAG1 binding sites genome wide (38) suggest that isolated nonamers might contribute to RAG1 binding at locations lacking discernible RSSs.
A single RSS provides a modest increase in binding over no RSS, but ChIP signals now resemble those seen at Jκ segments. V(D)J recombination is thought to be initiated when the RAG complex binds one RSS, so detection of binding to an isolated RSS would be expected, particularly in a region with high H3K4me3 levels (44). Interestingly, the addition of a second, similar RSS ∼660 bp away increased RAG1 ChIP signals both at the site of the added RSS and at the original RSS (compare data for p12/12 and p23/23 in Fig. 6B to those for p12 and p23 in Fig. 5C). Since DNA was sheared to an average size of 300 to 500 bp by the ChIP procedure, it seems unlikely that RAG1 bound at one RSS contributed substantially to the signal detected at the other RSS. Hence, we consider the possibility that having two binding sites in close proximity increases the local concentration of RAG, thereby increasing binding to both RSSs. Such a phenomenon could be important for RAG recruitment to recombination centers, particularly such as those at Tcrα and Igκ, which contain only 12RSSs and only 23RSSs, respectively.
Intriguingly, RAG1 binding was considerably stronger with a 12/23RSS pair than with a 12/12- or 23/23RSS pair (Fig. 6B). This finding cannot readily be explained by the 12- and 23RSSs acting completely independently of one another or by differences in levels of the substrate H3K4me3 (Fig. 6D). Instead, it argues for a form of cooperation in RAG1 recruitment that is specific for a 12/23RSS pair. An appealing mechanism for this is the formation of the synaptic, or paired, complex, which has been shown in biochemical experiments with truncated RAG proteins to be more efficient with an asymmetric 12/23RSS pair than with a symmetric 12/12- or 23/23RSS pair (21, 22, 50). While we do not have direct evidence for paired complex formation, the RSSs in our substrate support efficient recombination (30) and are separated by a distance similar to that found between certain D and J gene segments in the Igh and Tcrβ loci that recombine efficiently with one another. If 12/23RSS synapsis is the explanation for our findings, then it can be detected in vivo with full-length RAG proteins and in the absence of RSS nicking, since the D345 cell line expresses a catalytic mutant RAG1 protein. Such a mechanism could operate to enhance RAG recruitment to recombination centers that contain both types of RSS (e.g., Igh and Tcrβ) and conceivably also to those that contain only one type of RSS (e.g., Tcrα and Igκ), since the J RSSs in the recombination center might interact with V RSSs outside the recombination center. Superimposed on such RSS-driven mechanisms in some antigen receptor loci are transcription factor-mediated mechanisms for targeting RAG1 to specific gene segments: c-Fos at the Dβ1 23RSS (51) and RUNX1 at the human Dδ2 23RSS (52).
RAG2 binding to our recombination substrates paralleled that of RAG1 to some extent, with binding being strongest for substrates with 2 RSSs and weakest for pNone and with the single-RSS substrates exhibiting intermediate ChIP signals (Fig. 5D and 6C). However, a clear preference for p12/23 over p12/12 or p23/23 was not observed. The data are in agreement with those from our previous study that found poor RAG2 recruitment to pNone after 20 h of induction (25). These results are striking given that in primary thymocytes and pre-B cells, the pattern of RAG2 binding closely mirrors the pattern of H3K4me3 (25, 38), and H3K4me3 levels were high with all of the substrates. Our results are not compatible with a model in which initial RAG2 recruitment is driven solely by levels of H3K4me3. Instead, we consider the possibility that RAG2 recruitment during the first 24 to 48 h after induction, while dependent on H3K4me3, is enhanced by interactions with locally recruited RAG1. In contrast, in many primary lymphocytes, RAG2 has an opportunity to redistribute in a manner driven largely by H3K4me3.
Heptamer and nonamer contributions to RAG recruitment.
We showed previously that the nonamer binding domain of RAG1 was required for its binding to p12/23 (25), and this finding together with extensive biochemical data support the model that RAG1 recognition of the nonamer plays a critical role in stable RSS binding (53). Here, we showed that scrambling of the nonamer of a 12RSS paired with a 23RSS strongly reduced RAG1 binding, indicating that the nonamer is critical for the initial recruitment of RAG1 (Fig. 7C). RAG1 binding was depleted not only at the mutant 12RSS but also at the WT 23RSS, thus mimicking a single-RSS scenario. These results are consistent with the “nonamer first” model (24), in which RAG1 initially interacts with the nonamer and then, together with RAG2 and potentially other factors (such as RUNX1 at the human Dδ2 23RSS), binds to the coding/heptamer junction (51, 52). RAG2 stabilizes RAG1 binding by extending occupancy to the heptamer (54), so it was possible that scrambling of the heptamer would destabilize the complex. We observed that the loss of the heptamer had little effect on RAG2 binding and reduced RAG1 binding somewhat, yielding signals like those of p12/12 and p23/23 (Fig. 7C and D). This drop in RAG1 binding is plausibly due to a loss of the potential for 12/23RSS cooperation, which should be critically dependent on the heptamer (and RAG2) to read out the spacer length. This provides further (indirect) evidence for a role of 12/23RSS synapsis in mediating RAG recruitment.
RAG1 and RAG2 binding at p(CA)38/23 was detected (particularly after 24 h of induction) but was substantially reduced relative to that at p12/23. Binding at both the CA repeat and the accompanying intact 23RSS closely resembled that of pScrN/23 (Fig. 7C and D). This can be understood in light of the fact that CA repeats recapitulate the vital first 4 bp of the heptamer but interrupt the A5 portion of the nonamer in two locations, which should cripple nonamer function. An imperfect CA repeat in mouse Bcl11b functions as a cRSS and mediates an ∼65-kb internal deletion of the second and third exons of the gene. These events are readily detected in normal mouse thymocytes and contribute to the development of T cell lymphomas (55). Our data showing low-level RAG binding at p(CA)38/23 help explain the low level of activity detected for a CA repeat in a plasmid-based V(D)J recombination assay (42) and the ability of such repeats to contribute to RAG-mediated genome instability.
Our study characterizes basic parameters of the initial recruitment of RAG1 and RAG2 to chromatin. We identified features of the DNA substrate important for efficient RAG recruitment and obtained evidence consistent with known RSS communication and in accordance with the 12/23 rule. Most of the experiments were performed with consensus RSSs, which can behave differently from the endogenous ones; hence, there is likely more to be learned about the interactions that drive RAG binding in vivo. These findings have implications for the regulation of the targeting and mistargeting of RAG proteins to the genome.
ACKNOWLEDGMENTS
We thank the members of the Schatz laboratory for their intellectual input. We also thank E. Corbett for her production of monoclonal antibodies and Y. Maman for computational analysis of cryptic RSSs and RSS components.
This work was supported by National Institutes of Health grant AI32524 (D.G.S.). K.S. was supported in part by NIH grant T32GM007223 and by the National Science Foundation Graduate Research Fellowship under grant DGE-1122492.
REFERENCES
- 1.Schatz DG, Oettinger MA, Baltimore D. 1989. The V(D)J recombination activating gene, RAG-1. Cell 59:1035–1048. [DOI] [PubMed] [Google Scholar]
- 2.Oettinger MA, Schatz DG, Gorka C, Baltimore D. 1990. RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science 248:1517–1523. doi: 10.1126/science.2360047. [DOI] [PubMed] [Google Scholar]
- 3.Kirch SA, Sudarsanam P, Oettinger MA. 1996. Regions of RAG1 protein critical for V(D)J recombination. Eur J Immunol 26:886–891. doi: 10.1002/eji.1830260425. [DOI] [PubMed] [Google Scholar]
- 4.Silver DP, Spanopoulou E, Mulligan RC, Baltimore D. 1993. Dispensable sequence motifs in the RAG-1 and RAG-2 genes for plasmid V(D)J recombination. Proc Natl Acad Sci U S A 90:6100–6104. doi: 10.1073/pnas.90.13.6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fugmann SD, Villey IJ, Ptaszek LM, Schatz DG. 2000. Identification of two catalytic residues in RAG1 that define a single active site within the RAG1/RAG2 protein complex. Mol Cell 5:97–107. doi: 10.1016/S1097-2765(00)80406-2. [DOI] [PubMed] [Google Scholar]
- 6.Kim DR, Dai Y, Mundy CL, Yang W, Oettinger MA. 1999. Mutations of acidic residues in RAG1 define the active site of the V(D)J recombinase. Genes Dev 13:3070–3080. doi: 10.1101/gad.13.23.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Landree MA, Wibbenmeyer JA, Roth DB. 1999. Mutational analysis of RAG1 and RAG2 identifies three catalytic amino acids in RAG1 critical for both cleavage steps of V(D)J recombination. Genes Dev 13:3059–3069. doi: 10.1101/gad.13.23.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cuomo CA, Oettinger MA. 1994. Analysis of regions of RAG-2 important for V(D)J recombination. Nucleic Acids Res 22:1810–1814. doi: 10.1093/nar/22.10.1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sadofsky MJ, Hesse JE, Gellert M. 1994. Definition of a core region of Rag-2 that is functional in V(D)J recombination. Nucleic Acids Res 22:1805–1809. doi: 10.1093/nar/22.10.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y, Subrahmanyam R, Chakraborty T, Sen R, Desiderio S. 2007. A plant homeodomain in Rag-2 that binds hypermethylated lysine 4 of histone H3 is necessary for efficient antigen-receptor-gene rearrangement. Immunity 27:561–571. doi: 10.1016/j.immuni.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matthews AGW, Kuo AJ, Ramon-Maiques S, Han SM, Champagne KS, Ivanov D, Gallardo M, Carney D, Cheung P, Ciccone DN, Walter KL, Utz PJ, Shi Y, Kutateladze TG, Yang W, Gozani O, Oettinger MA. 2007. RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature 450:1106–1118. doi: 10.1038/nature06431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramon-Maiques S, Kuo AJ, Carney D, Matthews AGW, Oettinger MA, Gozani O, Yang W. 2007. The plant homeodomain finger of RAG2 recognizes histone H3 methylated at both lysine-4 and arginine-2. Proc Natl Acad Sci U S A 104:18993–18998. doi: 10.1073/pnas.0709170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tonegawa S. 1983. Somatic generation of antibody diversity. Nature 302:575–581. doi: 10.1038/302575a0. [DOI] [PubMed] [Google Scholar]
- 14.Ramsden DA, Baetz K, Wu GE. 1994. Conservation of sequence in recombination signal sequence spacers. Nucleic Acids Res 22:1785–1796. doi: 10.1093/nar/22.10.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akira S, Okazaki K, Sakano H. 1987. Two pairs of recombination signals are sufficient to cause immunoglobulin V-(D)-J joining. Science 238:1134–1138. doi: 10.1126/science.3120312. [DOI] [PubMed] [Google Scholar]
- 16.Hesse JE, Lieber MR, Mizuuchi K, Gellert M. 1989. V(D)J recombination: a functional definition of the joining signals. Genes Dev 3:1053–1061. doi: 10.1101/gad.3.7.1053. [DOI] [PubMed] [Google Scholar]
- 17.Ramsden DA, McBlane JF, van Gent DC, Gellert M. 1996. Distinct DNA sequence and structure requirements for the two steps of V(D)J recombination signal cleavage. EMBO J 15:3197–3206. [PMC free article] [PubMed] [Google Scholar]
- 18.Feeney AJ, Goebel P, Espinoza CR. 2004. Many levels of control of V gene rearrangement frequency. Immunol Rev 200:44–56. doi: 10.1111/j.0105-2896.2004.00163.x. [DOI] [PubMed] [Google Scholar]
- 19.Lewis SM. 1994. The mechanism of V(D)J joining: lessons from molecular, immunological, and comparative analyses. Adv Immunol 56:27–150. doi: 10.1016/S0065-2776(08)60450-2. [DOI] [PubMed] [Google Scholar]
- 20.Swanson PC. 2004. The bounty of RAGs: recombination signal complexes and reaction outcomes. Immunol Rev 200:90–114. doi: 10.1111/j.0105-2896.2004.00159.x. [DOI] [PubMed] [Google Scholar]
- 21.Mundy CL, Patenge N, Matthews AG, Oettinger MA. 2002. Assembly of the RAG1/RAG2 synaptic complex. Mol Cell Biol 22:69–77. doi: 10.1128/MCB.22.1.69-77.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones JM, Gellert M. 2002. Ordered assembly of the V(D)J synaptic complex ensures accurate recombination. EMBO J 21:4162–4171. doi: 10.1093/emboj/cdf394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McBlane JF, Vangent DC, Ramsden DA, Romeo C, Cuomo CA, Gellert M, Oettinger MA. 1995. Cleavage at a V(D)J recombination signal requires only Rag1 and Rag2 proteins and occurs in 2 steps. Cell 83:387–395. doi: 10.1016/0092-8674(95)90116-7. [DOI] [PubMed] [Google Scholar]
- 24.Schatz DG, Ji Y. 2011. Recombination centres and the orchestration of V(D)J recombination. Nat Rev Immunol 11:251–263. doi: 10.1038/nri2941. [DOI] [PubMed] [Google Scholar]
- 25.Ji YH, Resch W, Corbett E, Yamane A, Casellas R, Schatz DG. 2010. The in vivo pattern of binding of RAG1 and RAG2 to antigen receptor loci. Cell 141:419–431. doi: 10.1016/j.cell.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang M, Swanson PC. 2008. V(D)J recombinase binding and cleavage of cryptic recombination signal sequences identified from lymphoid malignancies. J Biol Chem 283:6717–6727. doi: 10.1074/jbc.M710301200. [DOI] [PubMed] [Google Scholar]
- 27.Rahman NS, Godderz LJ, Stray SJ, Capra JD, Rodgers KK. 2006. DNA cleavage of a cryptic recombination signal sequence by RAG1 and RAG2—implications for partial V-H gene replacement. J Biol Chem 281:12370–12380. doi: 10.1074/jbc.M507906200. [DOI] [PubMed] [Google Scholar]
- 28.Lieber MR, Yu KF, Raghavan SC. 2006. Roles of nonhomologous DNA end joining, V(D)J recombination, and class switch recombination in chromosomal translocations. DNA Rep 5:1234–1245. doi: 10.1016/j.dnarep.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 29.Mills KD, Ferguson DO, Alt FW. 2003. The role of DNA breaks in genomic instability and tumorigenesis. Immunol Rev 194:77–95. doi: 10.1034/j.1600-065X.2003.00060.x. [DOI] [PubMed] [Google Scholar]
- 30.Bredemeyer AL, Sharma GG, Huang CY, Helmink BA, Walker LM, Khor KC, Nuskey B, Sullivan KE, Pandita TK, Bassing CH, Sleckman BP. 2006. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature 442:466–470. doi: 10.1038/nature04866. [DOI] [PubMed] [Google Scholar]
- 31.Liang HE, Hsu LY, Cado D, Cowell LG, Kelsoe G, Schlissel MS. 2002. The “dispensable” portion of RAG2 is necessary for efficient V-to-DJ rearrangement during B and T cell development. Immunity 17:639–651. doi: 10.1016/S1074-7613(02)00448-X. [DOI] [PubMed] [Google Scholar]
- 32.Fugmann SD, Rush JS, Schatz DG. 2004. Non-redundancy of cytidine deaminases in class switch recombination. Eur J Immunol 34:844–849. doi: 10.1002/eji.200324418. [DOI] [PubMed] [Google Scholar]
- 33.Coster G, Gold A, Chen D, Schatz DG, Goldberg M. 2012. A dual interaction between the DNA damage response protein MDC1 and the RAG1 subunit of the V(D)J recombinase. J Biol Chem 287:36488–36498. doi: 10.1074/jbc.M112.402487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cowell LG, Davila M, Kepler TB, Kelsoe G. 2002. Identification and utilization of arbitrary correlations in models of recombination signal sequences. Genome Biol 3:RESEARCH0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davila M, Liu F, Cowell LG, Lieberman AE, Heikamp E, Patel A, Kelsoe G. 2007. Multiple, conserved cryptic recombination signals in VH gene segments: detection of cleavage products only in pro B cells. J Exp Med 204:3195–3208. doi: 10.1084/jem.20071224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grant CE, Bailey TL, Noble WS. 2011. FIMO: scanning for occurrences of a given motif. Bioinformatics 27:1017–1018. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muljo SA, Schlissel MS. 2003. A small molecule Abl kinase inhibitor induces differentiation of Abelson virus-transformed pre-B cell lines. Nat Immunol 4:31–37. doi: 10.1038/ni870. [DOI] [PubMed] [Google Scholar]
- 38.Teng G, Maman Y, Resch W, Kim M, Yamane A, Qian J, Kieffer-Kwon K-R, Mandal M, Ji Y, Meffre E, Clark MR, Cowell LG, Casellas R, Schatz DG. 2015. RAG represents a widespread threat to the lymphocyte genome. Cell 162:751–765. doi: 10.1016/j.cell.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin FF, Bailey S, Innis CA, Ciubotaru M, Kamtekar S, Steitz TA, Schatz DG. 2009. Structure of the RAG1 nonamer binding domain with DNA reveals a dimer that mediates DNA synapsis. Nat Struct Mol Biol 16:499–508. doi: 10.1038/nsmb.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spanopoulou E, Zaitseva F, Wang FH, Santagata S, Baltimore D, Panayotou G. 1996. The homeodomain region of Rag-1 reveals the parallel mechanisms of bacterial and V(D)J recombination. Cell 87:263–276. doi: 10.1016/S0092-8674(00)81344-6. [DOI] [PubMed] [Google Scholar]
- 41.Difilippantonio MJ, McMahan CJ, Eastman QM, Spanopoulou E, Schatz DG. 1996. RAG1 mediates signal sequence recognition and recruitment of RAG2 in V(D)J recombination. Cell 87:253–262. doi: 10.1016/S0092-8674(00)81343-4. [DOI] [PubMed] [Google Scholar]
- 42.Lewis SM, Agard E, Suh S, Czyzyk L. 1997. Cryptic signals and the fidelity of V(D)J joining. Mol Cell Biol 17:3125–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stallings RL, Ford AF, Nelson D, Torney DC, Hildebrand CE, Moyzis RK. 1991. Evolution and distribution of (GT)n repetitive sequences in mammalian genomes. Genomics 10:807–815. doi: 10.1016/0888-7543(91)90467-S. [DOI] [PubMed] [Google Scholar]
- 44.Askary A, Shimazaki N, Bayat N, Lieber MR. 2014. Modeling of the RAG reaction mechanism. Cell Rep 7:307–315. doi: 10.1016/j.celrep.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang YH, Shetty K, Surleac MD, Petrescu AJ, Schatz DG. 2015. Mapping and quantitation of the interaction between the recombination activating gene proteins RAG1 and RAG2. J Biol Chem 290:11802–11817. doi: 10.1074/jbc.M115.638627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grazini U, Zanardi F, Citterio E, Casola S, Goding CR, McBlane F. 2010. The RING domain of RAG1 ubiquitylates histone H3: a novel activity in chromatin-mediated regulation of V(D)J joining. Mol Cell 37:282–293. doi: 10.1016/j.molcel.2009.12.035. [DOI] [PubMed] [Google Scholar]
- 47.Deng Z, Liu H, Liu X. 2015. RAG1-mediated ubiquitylation of histone H3 is required for chromosomal V(D)J recombination. Cell Res 25:181–192. doi: 10.1038/cr.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Poorey K, Viswanathan R, Carver MN, Karpova TS, Cirimotich SM, McNally JG, Bekiranov S, Auble DT. 2013. Measuring chromatin interaction dynamics on the second time scale at single-copy genes. Science 342:369–372. doi: 10.1126/science.1242369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen J, Zhang Z, Li L, Chen BC, Revyakin A, Hajj B, Legant W, Dahan M, Lionnet T, Betzig E, Tjian R, Liu Z. 2014. Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell 156:1274–1285. doi: 10.1016/j.cell.2014.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Swanson PC. 2002. A RAG-1/RAG-2 tetramer supports 12/23-regulated synapsis, cleavage, and transposition of V(D)J recombination signals. Mol Cell Biol 22:7790–7801. doi: 10.1128/MCB.22.22.7790-7801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang X, Xiao G, Zhang Y, Wen X, Gao X, Okada S, Liu X. 2008. Regulation of Tcrb recombination ordering by c-Fos-dependent RAG deposition. Nat Immunol 9:794–801. doi: 10.1038/ni.1614. [DOI] [PubMed] [Google Scholar]
- 52.Cieslak A, Le Noir S, Trinquand A, Lhermitte L, Franchini DM, Villarese P, Gon S, Bond J, Simonin M, Vanhile L, Reimann C, Verhoeyen E, Larghero J, Six E, Spicuglia S, Andre-Schmutz I, Langerak A, Nadel B, Macintyre E, Payet-Bornet D, Asnafi V. 2014. RUNX1-dependent RAG1 deposition instigates human TCR-delta locus rearrangement. J Exp Med 211:1821–1832. doi: 10.1084/jem.20132585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schatz DG, Swanson PC. 2011. V(D)J recombination: mechanisms of initiation. Annu Rev Genet 45:167–202. doi: 10.1146/annurev-genet-110410-132552. [DOI] [PubMed] [Google Scholar]
- 54.Swanson PC, Desiderio S. 1998. V(D)J recombination signal recognition: distinct, overlapping DNA-protein contacts in complexes containing RAG1 with and without RAG2. Immunity 9:115–125. doi: 10.1016/S1074-7613(00)80593-2. [DOI] [PubMed] [Google Scholar]
- 55.Sakata J, Inoue J, Ohi H, Kosugi-Okano H, Mishima Y, Hatakeyama K, Niwa O, Kominami R. 2004. Involvement of V(D)J recombinase in the generation of intragenic deletions in the Rit1/Bcl11b tumor suppressor gene in gamma-ray-induced thymic lymphomas and in normal thymus of the mouse. Carcinogenesis 25:1069–1075. doi: 10.1093/carcin/bgh094. [DOI] [PubMed] [Google Scholar]