

Abstract
Q fever is an uncommon but likely underreported zoonotic infection. Severe hyperferritinemia has been associated with hemophagocytic lymphohistiocytosis and other infectious diseases. In this study, we report a case of Coxiella burnetii infection in an asplenic patient complicated by severe hyperferritinemia and bone marrow infiltration. In this case, the marked ferritin elevation may have been an indicator of profound systemic macrophage activation due to preferential intracellular infection of this cell type by C burnetii, perhaps exacerbated by altered mononuclear phagocyte system function in the setting of asplenia.
Keywords: Coxiella burnetii, hemophagocytic lymphohistiocytosis, hyperferritinemia, macrophage activation, Q fever
CASE REPORT
A 53-year-old man was admitted with fever and chills for 10 days and new jaundice and encephalopathy. The patient had a remote history of splenectomy secondary to a motor vehicle accident but no prior liver disease and no history of infection with encapsulated organisms despite lapsed immunization status. Three weeks before symptom onset, he had cleaned out a barn that had previously been used for birthing sheep and other animals.
On admission, his temperature was 34.1°C. Other vital signs were normal. Physical examination was remarkable for scleral icterus, mild right upper quadrant tenderness, and an erythematous nonpetechial macular rash on the right forearm. Laboratory analysis revealed a white blood cell count of 8.7 k/µL, hemoglobin 15.9 g/dL, and platelets 98 k/µL. Peripheral smear showed reactive leukocytosis with left shift but no Howell-Jolly bodies. Aspartate aminotransferase and alanine aminotransferase levels were 336 µ/L and 246 U/L, respectively, and total bilirubin was 11.3 mg/dL (direct 9 mg/dL). Prothrombin time was normal. Serum ferritin was 29 000 ng/mL. Triglycerides and fibrinogen were both modestly elevated. Chest x-ray showed no infiltrates.
Initial work-up included negative blood cultures, negative serology for hepatitis A, B and C, prior immunity to Epstein-Barr and cytomegalovirus, and negative human immunodeficiency virus antibodies and p24 antigen. Serum ceruloplasmin, α-1-anti-trypsin, and markers for autoimmune hepatitis were all within normal range. The patient was briefly treated empirically with ciprofloxacin and metronidazole for ascending cholangitis until a right upper quadrant ultrasound showed no hepatobiliary abnormalities. Ultimately, a liver biopsy was performed and showed fibrin ring lipogranulomas (see Figure 1).
Figure 1.
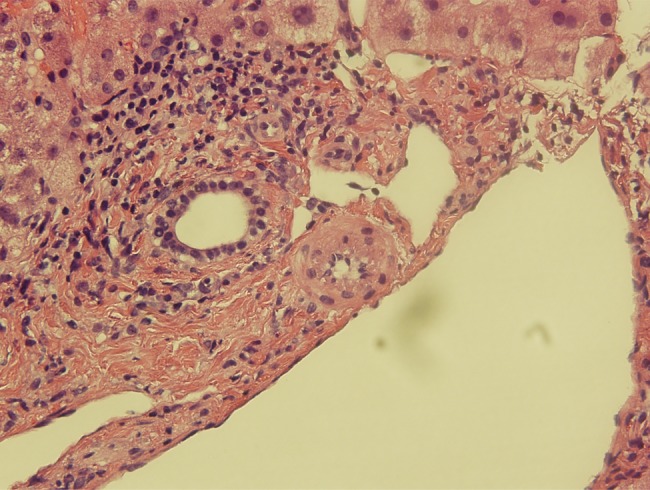
Liver biopsy (hematoxylin and eosin 400×) with “doughnut” lipogranuloma.
Because of concern for hemophagocytic lymphohistiocytosis (HLH), a bone marrow biopsy was performed. Histopathology also demonstrated fibrin ring “doughnut” lipogranulomas (see Figure 2). An extensive work-up was undertaken, including negative serum Histoplasma antibodies by complement fixation and immunodiffusion, Histoplasma urine antigen, serum Cryptococcus antigen, serum Bartonella polymerase chain reaction, serum Brucella antibodies by agglutination, and rapid plasma reagin. Serum Coccidioides immunoglobulin (Ig)G was weakly positive but negative by complement fixation; IgM was negative. However, Coxiella burnetii Phase II IgM was markedly positive at >1:4096, and Phase II IgG was positive at 1:256; Phase I IgM was negative and Phase I IgG was 1:16, consistent with acute Q fever. To assess for cardiac valvular abnormalities that might increase the patient's risk of chronic Q fever, transesophageal echocardiography was performed and was negative. The patient improved rapidly in the hospital. Because of concern about deep-seated marrow involvement, he was treated with a total of 8 weeks of oral doxycycline with no residual laboratory abnormalities or symptoms. Repeat C burnetii serology at 2 months showed Phase II IgM and IgG titers of 1:256 and a Phase I IgM of 1:256, Phase I IgG titer of 1:32. Repeat serology at 6 months to screen for evidence of chronic Q fever was planned [1, 2].
Figure 2.
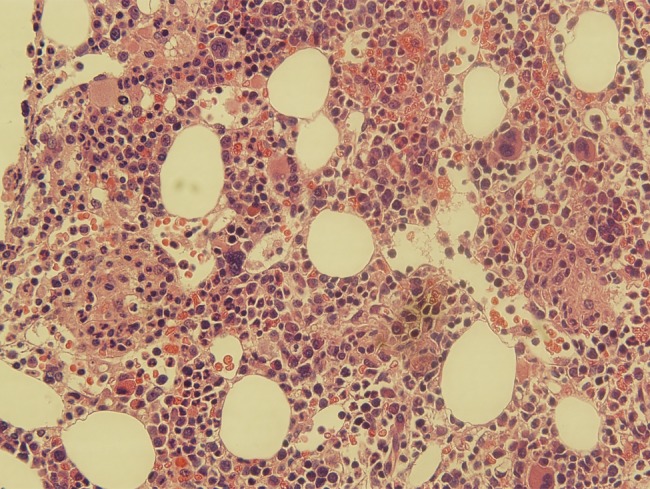
Bone marrow biopsy (hematoxylin and eosin 400×) “doughnut” lipogranuloma.
DISCUSSION
Q fever is the clinical manifestation of symptomatic C burnetii infection. Coxiella burnetii is an obligate intracellular, Gram-negative organism most frequently found in ruminants including sheep, cattle, and goats. The organism can survive for long periods outside of its host organism, and the infective dose is low [1]. Human infection most often occurs after inhalation of aerosolized animal waste or products of conception. Infection may be asymptomatic; acute Q fever is likely underrecognized and most commonly presents as a nonspecific febrile illness with pneumonia, mild hepatitis, or both [3]. Biopsy of solid organs may demonstrate “doughnut lipogranulomas”, a histologic pattern marked by granulomatous rings of dense fibrin surrounding a central lipid vacuole that is uncommon but considered pathognomonic for Q fever [4]. Diagnosis of acute Q fever is marked by elevated IgM and IgG against Phase II antigens. Chronic Q fever is rare (<2% of cases) and often manifests with rising Phase I antibody titers and endocarditis or vasculitis in patients with underlying valvular abnormalities [1].
In our patient, there was initial concern for HLH. Hemophagocytic lymphohistiocytosis is a rare inflammatory disorder characterized by hyperactivation of CD8+ lymphocytes and macrophages that migrate and proliferate systemically, causing the release of proinflammatory cytokines leading to multiorgan dysfunction and often death [5]. Hemophagocytic lymphohistiocytosis has 2 distinct clinical forms—inherited or primary HLH, which usually occurs in the pediatric population, and secondary HLH, which may be triggered by severe infection, malignancy, or rheumatologic disorders and is more common in adults. Although Epstein-Barr viral infection is the most common cause of infection-triggered HLH, viral, bacterial, fungal, protozoan, and helminthic infections have been reported to trigger HLH [5], including several reports associating C burnetii infection with HLH [6–9].
Hemophagocytic lymphohistiocytosis is a clinical diagnosis confirmed by fulfilling 5 of 8 Henter criteria (2004): fever, splenomegaly, cytopenias affecting 2 or more lineages, hypertriglyceridemia and/or hyperfibrinogenemia, hemophagocytosis in bone marrow, spleen or lymph node, hyperferritinemia ≥500 ng/mL, impaired natural killer cell function, or elevated CD25 [10]. In our case, multiple supporting criteria for secondary HLH were present (fever, hyperferritinemia, mild cytopenia, hypertriglyceridemia, and hyperfibrinogenemia) and an additional feature, splenomegaly, was not evaluable. Although no hemophagocytosis was present on biopsy, this finding is rarely found on initial presentation of HLH [11]. Ultimately, the patient's rapid improvement was not consistent with HLH. However, the underlying cause for this presentation and extreme hyperferritinemia is intriguing.
Ferritin is a cytosolic protein found in many cell types but is primarily stored in macrophages, and it is a well known acute phase reactant [12]. In adults, modest elevations are nonspecific but most often associated with malignancy and iron-overload syndromes [13]. Severe hyperferritinemia (>10 000 µg/L) has been associated with adult-onset Still's disease and HLH, especially in the pediatric population. In adults, although severe hyperferritinemia has been associated with HLH as well as infectious conditions including disseminated histoplasmosis and tuberculosis, it is nonspecific [14, 15].
A common theme in the pathogenesis of these conditions is activation of macrophages. These cells play an important role in cellular homeostasis and innate and adaptive immunity. Depending on function, macrophages are polarized to 1 of 2 modes, M1 or M2. In the default M2 mode, macrophages continuously sample the local environment, ingesting senescent cells and debris and elaborating growth factors that maintain and heal tissue matrices. In the liver and spleen, M2 cells are the principal component of the monocyte phagocytic system (formerly called the reticuloendothelial system). Macrophages also recognize microbes via pathogen-associated molecular pattern receptors. This results in phagocytosis, lysosomal digestion, and conversion to the M1 mode. In this context, antigen is presented by M1 macrophages to T cells, stimulating potent cytokine production. These cytokines promote efficient intracellular killing, recruit circulating monocytes and neutrophils, and favor further M1 switching [16].
It is possible that this case represents an intermediate step along a spectrum of macrophage-driven inflammatory conditions, ranging from background M2 function to the overreactive M1 response characteristic of HLH or macrophage activation syndrome [17]. It is probably not coincidental that most bacteria associated with secondary HLH are intracellular organisms such as Mycobacterium tuberculosis and Salmonella spp that are able to evade lysosomal digestion, and whose clinical manifestations frequently include hepatosplenomegaly and leukopenia [18]. Coxiella burnetii preferentially infects monocytes and macrophages, where it too is able to evade immune destruction and persist (monocytes) or replicate (macrophage) in acid vacuoles [19]. In acute Q fever, the majority of macrophages are ultimately polarized toward a proinflammatory M1 response, stimulating release of interferon-γ and inflammatory cytokines [19]. In this setting, it is possible that the amount of ferritin released may be reflect the degree of macrophage activation. In contrast, chronic Q fever has been associated with a predominantly M2-type response in which macrophages are less able to clear the C burnetii bacteria [20].
CONCLUSION
In our case, we postulate that the severe hyperferritinemia was indeed a result of systemic overactivity of macrophages, perhaps exacerbated by impaired mononuclear phagocyte system function in the setting of asplenia. Although severe elevations in ferritin often trigger concern for secondary HLH, it is possible that a spectrum of macrophage activation exists and that ferritin may be useful to support the diagnosis of infection with Q fever and other intracellular pathogens.
Acknowledgments
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest.
References
- 1.Anderson A, Bijlmer H, Fournier PE et al. Diagnosis and management of Q fever--United States, 2013: recommendations from CDC and the Q Fever Working Group. MMWR Recomm Rep 2013; 62(RR-03):1–30. [PubMed] [Google Scholar]
- 2.Hartzell JD, Gleeson T, Scoville S et al. Practice guidelines for the diagnosis and management of patients with Q fever by the Armed Forces Infectious Diseases Society. Mil Med 2012; 177:484–94. [DOI] [PubMed] [Google Scholar]
- 3.Million M, Raoult D. Recent advances in the study of Q fever epidemiology, diagnosis and management. J Infect 2015; 71(suppl 1): S2–9. [DOI] [PubMed] [Google Scholar]
- 4.Fournier PE, Marrie TJ, Raoult D. Diagnosis of Q fever. J Clin Microbiol 1998; 36:1823–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weitzman S. Approach to hemophagocytic syndromes. Hematology Am Soc Hematol Educ Program 2011; 2011:178–83. [DOI] [PubMed] [Google Scholar]
- 6.Chen TC, Chang K, Lu PL et al. Acute Q fever with hemophagocytic syndrome: case report and literature review. Scand J Infect Dis 2006; 38:1119–22. [DOI] [PubMed] [Google Scholar]
- 7.Estrov Z, Bruck R, Shtalrid M et al. Histiocytic hemophagocytosis in Q fever. Arch Pathol Lab Med 1984; 108:7. [PubMed] [Google Scholar]
- 8.Harris P, Dixit R, Norton R. Coxiella burnetii causing haemophagocytic syndrome: a rare complication of an unusual pathogen. Infection 2011; 39:579–82. [DOI] [PubMed] [Google Scholar]
- 9.Hufnagel M, Niemeyer C, Zimmerhackl LB et al. Hemophagocytosis: a complication of acute Q fever in a child. Clin Infect Dis 1995; 21:1029–31. [DOI] [PubMed] [Google Scholar]
- 10.Henter JI, Horne A, Arico M et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007; 48:124–31. [DOI] [PubMed] [Google Scholar]
- 11.George MR. Hemophagocytic lymphohistiocytosis: review of etiologies and management. J Blood Med 2014; 5:69–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen LA, Gutierrez L, Weiss A et al. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood 2010; 116:1574–84. [DOI] [PubMed] [Google Scholar]
- 13.Moore C Jr, Ormseth M, Fuchs H. Causes and significance of markedly elevated serum ferritin levels in an academic medical center. J Clin Rheumatol 2013; 19:324–8. [DOI] [PubMed] [Google Scholar]
- 14.McKenzie SW, Means RT Jr. Extreme hyperferritinemia in patients infected with human immunodeficiency virus is not a highly specific marker for disseminated histoplasmosis. Clin Infect Dis 1997; 24:519–20. [DOI] [PubMed] [Google Scholar]
- 15.Schram AM, Campigotto F, Mullally A et al. Marked hyperferritinemia does not predict for HLH in the adult population. Blood 2015; 125:1548–52. [DOI] [PubMed] [Google Scholar]
- 16.Mills CD, Ley K. M1 and M2 macrophages: the chicken and the egg of immunity. J Innate Immun 2014; 6:716–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grom AA, Mellins ED. Macrophage activation syndrome: advances towards understanding pathogenesis. Curr Opin Rheumatol 2010; 22:561–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cascio A, Pernice LM, Barberi G et al. Secondary hemophagocytic lymphohistiocytosis in zoonoses. Eur Rev Med Pharmacol Sci 2012; 16:1324–37. [PubMed] [Google Scholar]
- 19.Amara AB, Bechah Y, Mege JL. Immune response and Coxiella burnetii invasion. Adv Exp Med Biol 2012; 984:287–98. [DOI] [PubMed] [Google Scholar]
- 20.Benoit M, Barbarat B, Bernard A et al. Coxiella burnetii, the agent of Q fever, stimulates an atypical M2 activation program in human macrophages. Eur J Immunol 2008; 38:1065–70. [DOI] [PubMed] [Google Scholar]